
Cation reduction and comproportionation as novel strategies to produce high voltage, halide free, carborane based electrolytes for rechargeable Mg batteries†
Scott. G.
McArthur‡
a,
Linxiao
Geng‡
b,
Juchen
Guo
*bc and
Vincent
Lavallo
*a
aDepartment of Chemistry, University of California Riverside, Riverside, CA 92521, USA. E-mail: vincent.lavallo@ucr.edu
bDepartment of Chemical and Environmental Engineering, University of California Riverside, Riverside, CA 92521, USA. E-mail: jguo@engr.ucr.edu
cMaterials Science and Engineering Program, University of California Riverside, Riverside, CA 92521, USA
First published on 15th October 2015
Abstract
Here we describe the cation reduction and comproportionation as novel routes to synthesize electrolytes for rechargeable Mg-ion batteries. Reduction of the ammonium cation in [HNMe31+][HCB11H111−] with metallic Mg affords the halide free carborane salt [Mg2+][HCB11H111−]2. Comproportionation of [Mg2+][HCB11H111−]2 with MgPh2 affords the novel monocationic electrolyte [MgPh1+][HCB11H111−], which reversibly deposits/strips Mg with a remarkable oxidative stability of 4.6 V vs. Mg0/+2.
Over the last decade there has been an explosion of technological advances in rechargeable portable devices and electric vehicles. However, innovations that reduce the cost, improve the sustainability, and increase the storage capacity offered by the state-of-the-art Li-ion technology have not kept pace with this revolution.1 Li-ion batteries are also disadvantageous, since lithium is expensive, not earth abundant (Li+, 0.0017% of the earth's crust), very pyrophoric, and prone to hazardous dendrite formation.2 To avoid dendrite formation, intercalation type anodes (e.g. graphite) are used in current Li-ion batteries, significantly reducing the possible energy capacity both gravimetrically and volumetrically. Mg-based batteries3 are an attractive alternative to Li-ion systems because Mg is less expensive, much more abundant (4% of the earth's crust), more tolerant of air, and does not form dendrites. The absence of dendrite formation during Mg deposition allows the utilization of pure Mg anodes, which drastically increases the energy storage capacity of the battery. In addition, since Mg is a small divalent atom it can store twice the amount of electrons compared to Li.
In contrast to Li, Mg has a unique chemistry that limits suitable solvents to aprotic and relatively non-polar ethers (e.g. dimethoxyethane (DME), and higher glymes).3b,c Mg salts that are analogous to Li salts, such as magnesium hexafluorophosphate ([Mg][PF6]2), are not suitable solutes for Mg-ion electrolytes because the anions of these salts degrade during the required electrochemical processes and form a solid organic/inorganic film covering the Mg anode. Due to its divalency, Mg ions cannot penetrate this solid layer, hence reversible Mg deposition and stripping cannot be achieved utilizing these common salts.
While Grignard reagents (RMgX) were observed to reversibly deposit/strip Mg in as early as the 1920s,4 they are not suitable electrolytes due to their reducing power and poor electrochemical stability. The first breakthrough in electrolyte development was made by Gregory5 in 1990 who reported that non-reducing Mg organo-borates/aluminates, such as [Mg][BBu2Ph2]2, would reversibly deposit/strip Mg; however, these systems suffer from low oxidative stability (<2 V vs. Mg0/+2). Based on this observation Aurbach and coworkers3a,c introduced a transmetalation strategy between Lewis basic diorganomagnesium reagents (MgR2) and Lewis acidic aluminum species (AlCl2R), to produce Mg organo-haloaluminate electrolytes (Fig. 1, top). The inorganic chloride was found to be the only suitable anionic supporting ligand for Mg ions in these systems. The solutions of these electrolytes are complex equilibrium mixtures of multiple neutral and ionic compounds. The optimal mixtures have an enhanced electrochemical stability (up to 3.3 V vs. Mg0/+2 on a Pt current collector)3c and lead to the first example of a working rechargeable Mg battery.3a One drawback of this transmetalation strategy is that it prohibits the exploration of organic anionic supporting ligands, which might produce distinct or perhaps superior monocationic Mg fragments. In addition, the inherent requirement for the introduction of other metals can result in unwanted side reactions, such as the electrochemical deposition of aluminium.3c Moreover, the presence of halide ions is undesirable in practical Mg batteries because they lead to corrosion of the non-noble metal battery components.6 Therefore, over the last several years there has been a significant effort to discover suitable electrolyte systems that are both halide free and have feature enhanced electrochemical performance.7
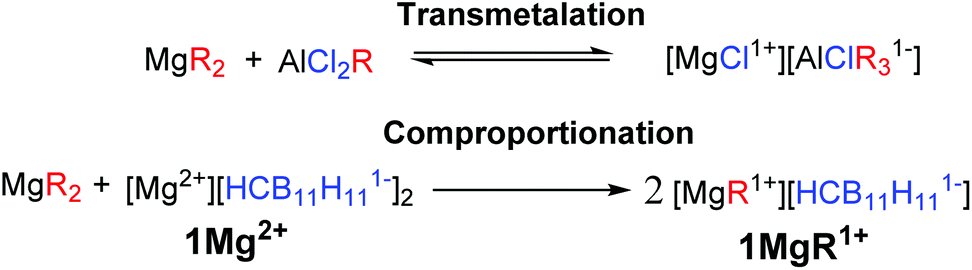 | ||
| Fig. 1 A simplified chemical equation representing a transmetalation reaction to form a generic organo-haloaluminate electrolyte (top). A general equation for a comproportionation reaction between a diorganomagnesium reagent and a simple magnesium salt of a weakly coordinating anion (bottom). R = alkyl or aryl. | ||
Here we describe a unique and economical approach to produce halide free electrolytes for Mg batteries, namely the chemical reduction of reactive cations with the Mg metal. In addition, we introduce a novel comproportionation strategy between a diorganomagnesium reagent and a simple halide free Mg salt of a weakly coordinating carborane anion 1[Mg2+] to produce a highly electrochemically stable monocationic electrolyte 1[MgR1+], featuring an organic ligand. This strategy is adventitious to transmetalation, since only Mg reagents are used.
One of the most inert and weakly coordinating polyatomic anions is the icosahedral carborane anion HCB11H111− (1) (Fig. 2).8 Classically, derivatives of this cluster have been utilized as spectator anions for reactive molecular species9 and more recently as ligand substituents.10 Elegant work by Boeré and Knapp11 has shown that the measured oxidation potential of 1 is +2.35 V versus Fc0/+ (approx. +5.36 V vs. Mg0/+2) in liquid SO2. The reduction potential of 1 is estimated to be well below −4 V versus Fc0/+ (below −1 V vs. Mg0/+2), but has not been measured, since no suitable solvent has been found. Such carborane anions should be ideal components for Mg battery electrolytes. Very recently Mohtadi and coworkers7a at the Toyota Research Center reported the preparation of halide free 1[Mg2+]via salt metathesis of MgBr2 with non-commercially available 1[Ag1+] in THF (Fig. 2, left). From an economic perspective this strategy is not practical since two equivalents of a precious metal salt by-product (AgBr) are produced per molar equivalent of Mg2+. Moreover, the crude 1[Mg2+] product is not sufficiently pure for electrochemistry without an unusual purification procedure. Sometime ago, we independently prepared 1[Mg2+] by a superior and much simpler method namely the direct chemical reduction of 1[HNMe31+] with the Mg metal (Fig. 2, right). Here the metallic Mg transfers two electrons to two HNMe31+ molecules with subsequent evolution of volatile H2 and NMe3 as the only by-products. This method results in quantitative conversion of commercially available 1[HNMe31+] to 1[Mg2+] in a single simple step and avoids the formation of precious metal by-products. Moreover, the material is sufficiently pure for electrochemical studies, vide infra. As observed by Mohtadi,7a when produced in THF, 1[Mg2+] precipitates as a completely insoluble white powder, which is only appreciably soluble in triglyme and tetraglyme. In these solvents this electrolyte shows electrochemical stability up to the limit of the solvent (3.8 V vs. Mg0/+2) but only moderate conductivity. The modest conductivity of 1[Mg2+], is likely due to the strong coulombic attraction between the dipositively charged Mg2+ ion and the carborane anions.
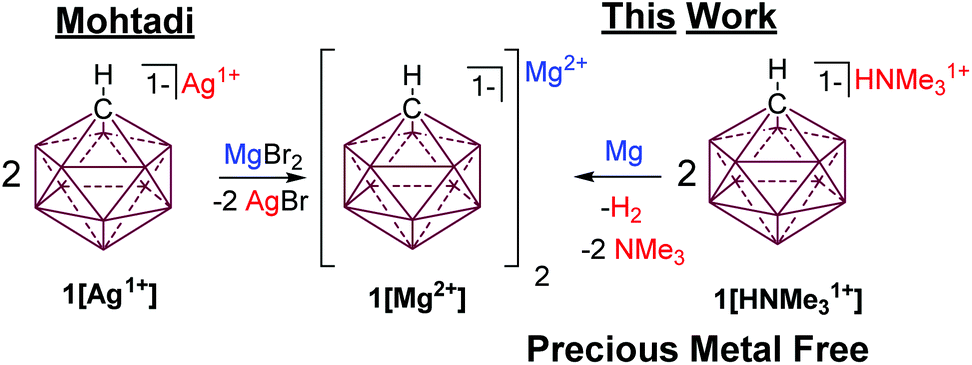 | ||
| Fig. 2 Mohtadi's route to 1[Mg2+] requires the use of a precious metal (left). Halide and Ag free synthesis of 1[Mg2+] by cation reduction with Mg. | ||
Seeking to improve the solubility and conductivity of this electrolyte we envisioned designing a system with reduced coulombic interactions. Furthermore, enhanced solubility might offer the possibility of utilizing more oxidatively stable solvents, which could lead to higher voltage electrolyte systems. We hypothesized that attaching a suitable organic ligand (R−) to the Mg2+ ion in 1[Mg2+] would produce unique carborane electrolytes 1[MgR1+]. We also desired a method that could potentially be used to rapidly create a library of electrochemically distinct Mg electrolytes without the introduction of potential contaminants, as in transmetalation reactions. It was reasoned that treatment of Lewis acidic 1[Mg2+] with an equal molar amount of a halide free Lewis basic diorganomagnesium reagent MgR2 would result in comproportionation to afford 1[MgR1+] species. Inspired by Aurbach's all phenyl complex electrolyte (APC),3c which features an oxidation resistant phenyl substituent, we predicted that readily available MgPh2 would be an ideal reactant. Indeed, treatment of a suspension of 1[Mg2+] in DME with a DME solution of MgPh2 instantly solubilizes the mixture suggesting that a reaction occurred (Fig. 3). The analysis of the solution by 1H NMR spectroscopy (ESI†) shows the disappearance of resonances associated with MgPh2 and the formation of a single new product with aromatic resonances, consistent with the formation of 1[MgPh1+].
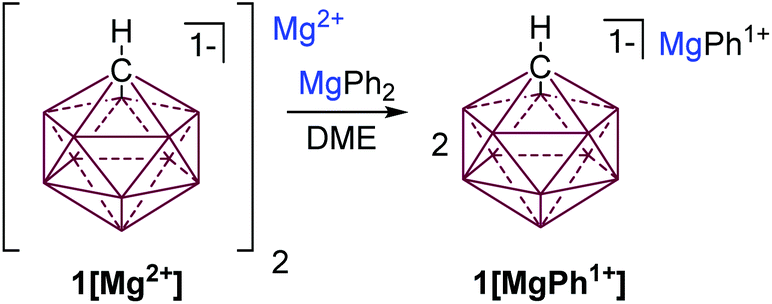 | ||
| Fig. 3 Comproportionation of 1[Mg2+] and MgPh2 to produce 1[MgPh+1] (top). | ||
In addition, the 13C NMR spectrum (ESI†) shows a distinct quaternary aryl resonance at 169.8 ppm, which is 10 ppm upfield with respect to MgPh2 and suggests that the aryl substituent is bound to a more electrophilic Mg center.12 Variable temperature NMR experiments to a temperature as low as −50 °C, show the presence of only single molecular species, which demonstrates the absence of an observable Schlenk-like equilibrium on the NMR time scale. The apparent absence of an observable Schlenk equilibrium highlights the poor nucleophilicity of the carborane anion compared to the halide ions present in Grignard reagents and organo-haloaluminates. The 11B NMR of 1[MgPh1+] (ESI†) is identical to 1[Mg2+], which suggests that the carborane anion 1 has no interaction with the [MgPh1+] cation. Although the data is not of sufficient quality to accurately discuss bond lengths and angles, a single crystal X-ray diffraction study confirms the structure of 1[MgPh1+] (Fig. 4). In the solid-state, the Mg center is octahedrally coordinated to the Ph group, two molecules of chelating DME, and a single molecule of THF. The closest approach between the carborane anion 1 and the Mg center is approximately 6.7 angstroms, which is out of the range for both covalent and van der Waals interactions.
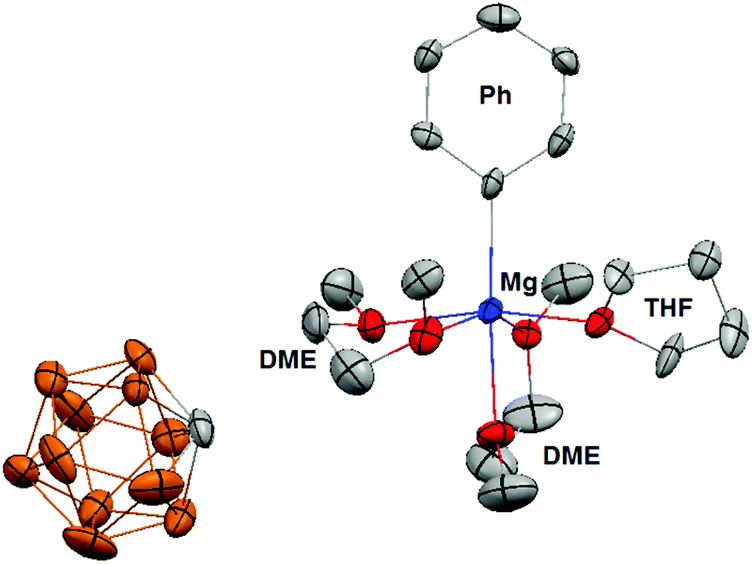 | ||
| Fig. 4 Solid state-structure of 1[MgPh1+] with a THF and two coordinated DME molecules. Hydrogen atoms and two molecules of cocrystallized toluene are omitted for clarity and thermal ellipsoids are drawn at the 50% probability level (color code: blue = Mg; grey = carbon; brown = boron; red = oxygen). | ||
The 1[MgPh1+] electrolyte in DME demonstrates an excellent room temperature conductivity. As shown in the ESI,† the conductivity of the 1[MgPh1+] electrolyte in DME increases as a function of salt concentration. At 0.4 M, the conductivity is 1.24 × 10−2 S cm−1, which is more than 4 times higher than the maximum conductivity of 1[Mg2+] reported by Mohtadi (0.75 M in triglyme, 2.9 × 10−3 S cm−1).7a As demonstrated by cyclic voltammetry (CV), 1[MgPh1+] also shows reversible Mg deposition/stripping and excellent anodic stability on various metal surfaces. As shown in Fig. 5a, CV scans show facile Mg deposition and stripping on all working electrodes (WE) including platinum (Pt), glassy carbon (GC), titanium (Ti), nickel (Ni), 316 stainless steel (SS) with low overpotentials (250 to 300 mV depending on the surface). Metallic Mg deposition was confirmed by scanning electron microscopy and X-ray diffraction studies (ESI†). Impressively, anodic scans of the electrolyte (inset in Fig. 5a) demonstrate the unprecedented oxidative stability of 1[MgPh1+] with various WEs using a scan rate of 5 mV s−1. Enlargement of the 3.5 to 5.0 V region of the anodic scan shows the oxidative onset potentials (inset). This electrolyte is stable up to 4.6 V vs. Mg0/+2 on both Pt and GC, rendering it by far the most oxidatively stable Mg-ion electrolyte reported to date. The anodic stability is 4.2 V on Ti and 3.5 V on both SS and Ni. While the oxidative stability of the carborane anion 1 is not surprising given the data reported by Boeré and Knapp, the electrochemical stability of the novel cation [MgPh1+] containing an organic ligand is truly remarkable. The Mg deposition/stripping CV cycling curves and cycling coulombic efficiency are shown in Fig. 5b. The efficiency of the first cycle on Pt WE is 93.0%, which improves to 95.0% in the following 50 cycles. The average coulombic efficiency of Mg deposition/stripping on Ni, Ti, and SS is also measured with a galvanostatic cycling method (ESI†) as 95.3%, 93.7%, and 91.7%, respectively. The feasibility of 1[MgPh1+] in DME as an electrolyte in rechargeable Mg-ion batteries was demonstrated using coin cells with a Mg metal anode and the standard Mo6S8 cathode (ESI†). The Mg-Mo6S8 batteries demonstrate performance consistent with other reported studies.13
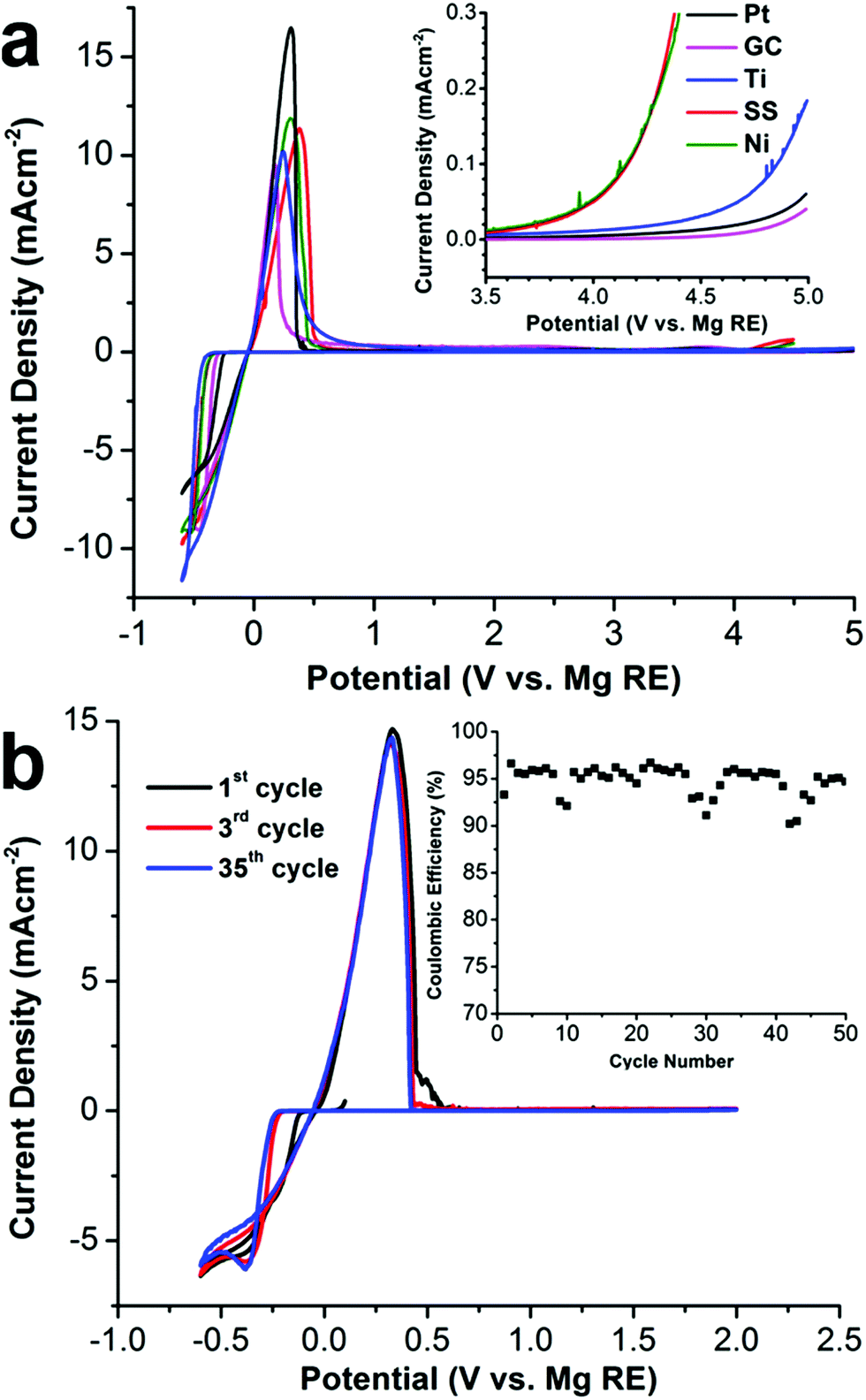 | ||
| Fig. 5 (a) First CV scan of 0.4 M 1[MgPh1+] in DME on various WEs using scan rate of 5 mV s−1. Enlargement of 3.5 to 5.0 V region of the anodic scan shows the oxidative onset potentials (inset). (b) Selected CV cycling curves of Mg deposition/stripping on a Pt working electrode with a 5 mV s−1 scan rate. The inset shows the coulombic efficiency as a function of the CV cycle number. | ||
Conclusions
The manuscript above introduces several key advances that pave the way for the development of practical high capacity Mg batteries. The reduction of reactive cations to form halide free electrolytes should be a broadly applicable method for the preparation of any electrolytes that will be suitable for Mg batteries. Since the anionic component of competent electrolytes must be chemically inert towards Mg, this method also serves as a chemical test to ensure electrolyte compatibility with the anode. The comproportionation strategy serves as an entry way into sophisticated highly oxidatively stable monocationic carborane salts 1[MgR1+] containing organic ligands, as exemplified by the preparation of 1[MgPh1+]. Lastly, access to electrolytes that have oxidative stability beyond 4.5 V creates a novel paradigm for discovering entirely new high voltage cathode materials for the development of practical high capacity Mg-ion batteries.Acknowledgements
This material is based on work supported in part by the National Science Foundation (DMR-1508537).Notes and references
- R. Van Noorden, Nature, 2014, 507, 26 CrossRef CAS PubMed.
- H. Kim, G. Jeong, Y.-U. Kim, J.-H. Kim, C.-M. Park and H.-J. Sohn, Chem. Soc. Rev., 2013, 42, 9011 RSC.
- (a) D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724 CrossRef CAS PubMed; (b) R. Mohtadi and F. Mizuno, Beilstein J. Nanotechnol., 2014, 5, 1291 CrossRef PubMed; (c) H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265 RSC; (d) J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941 RSC; (e) D. Aurbach, G. S. Suresh, E. Levi, A. Mitelman, O. Mizrahi, O. Chusid and M. Brunelli, Adv. Mater., 2007, 19, 4260 CrossRef CAS PubMed.
- (a) L. W. Gaddum and H. E. French, J. Am. Chem. Soc., 1927, 49, 1295 CrossRef CAS; (b) W. V. Evans and F. H. Lee, J. Am. Chem. Soc., 1934, 56, 654 CrossRef CAS.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur, A. G. Oliver, J. Zajicek, G. D. Allred and W. C. Boggess, Energy Environ. Sci., 2013, 6, 482 CAS.
- (a) O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900 CrossRef CAS PubMed; (b) S. Su, Z. Huang, Y. NuLi, F. Tuerxun, J. Yang and J. Wang, Chem. Commun., 2015, 51, 2641 RSC; (c) J. Zhu, Y. Guo, J. Yang, Y. Nuli, F. Zhang, J. Wang and S.-i. Hirano, J. Power Sources, 2014, 248, 690 CrossRef CAS PubMed; (d) T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173 CrossRef CAS PubMed; (e) S.-Y. Ha, Y.-W. Lee, S. W. Woo, B. Koo, J.-S. Kim, J. Cho, K. T. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 4063 CrossRef CAS PubMed; (f) R. Mohtadi, M. Matsui, T. S. Arthur and S.-J. Hwang, Angew. Chem., Int. Ed., 2012, 51, 9780 CrossRef CAS PubMed.
- (a) C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179 CrossRef PubMed; (b) D. Olid, R. Nunez, C. Vinas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318 RSC; (c) P. Farras, E. J. Juarez-Perez, M. Lepsik, R. Luque, R. Nunez and F. Teixidor, Chem. Soc. Rev., 2012, 41, 3445 RSC; (d) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035 CrossRef CAS PubMed; (e) Y. Li, P. J. Carroll and L. G. Sneddon, Inorg. Chem., 2008, 47, 9193 CrossRef CAS PubMed.
- (a) C. A. Reed, Acc. Chem. Res., 2009, 43, 121 CrossRef PubMed; (b) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188 CrossRef CAS PubMed.
- (a) A. R. Popescu, F. Teixidor and C. Viñas, Coord. Chem. Rev., 2014, 269, 54 CrossRef CAS PubMed; (b) A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903 CrossRef CAS PubMed; (c) J. Estrada, D. H. Woen, F. S. Tham, G. M. Miyake and V. Lavallo, Inorg. Chem., 2015, 54, 5142 CrossRef CAS PubMed; (d) M. J. Asay, S. P. Fisher, S. E. Lee, F. S. Tham, D. Borchardt and V. Lavallo, Chem. Commun., 2015, 51, 5359 RSC; (e) J. Estrada, S. E. Lee, S. G. McArthur, A. El-Hellani, F. S. Tham and V. Lavallo, J. Organomet. Chem., 2015 DOI:10.1016/j.jorganchem.2015.05.008; (f) A. El-Hellani, C. E. Kefalidis, F. S. Tham, L. Maron and V. Lavallo, Organometallics, 2013, 32, 6887 CrossRef CAS; (g) A. El-Hellani and V. Lavallo, Angew. Chem., Int. Ed., 2014, 53, 4489 CrossRef CAS PubMed; (h) V. Lavallo, J. H. Wright, F. S. Tham and S. Quinlivan, Angew. Chem., Int. Ed., 2013, 52, 3172 CrossRef CAS PubMed; (i) M. Asay, C. E. Kefalidis, J. Estrada, D. S. Weinberger, J. Wright, C. E. Moore, A. L. Rheingold, L. Maron and V. Lavallo, Angew. Chem., Int. Ed., 2013, 52, 11560 CrossRef CAS PubMed.
- R. T. Boeré, C. Bolli, M. Finze, A. Himmelspach, C. Knapp and T. L. Roemmele, Chem. – Eur. J., 2013, 19, 1784 CrossRef PubMed.
- Similar to aryl Li reagents, aryl Mg reagents display very downfield shifted resonances for the carbon bound to the metal. This can be explained by a reduction in π-delocalization as a result of electrostatic repulsion between the carbanion “like” center and the π-electrons. A cationic metal center reduces this effect. For a discussion, see: J. A. Ladd, Spectrochim. Acta, 1966, 22, 1157 CrossRef CAS.
- (a) Y. Cheng, L. R. Parent, Y. Shao, C. Wang, V. L. Sprenkle, G. Li and J. Liu, Chem. Mater., 2014, 26, 4904 CrossRef CAS; (b) T. Liu, Y. Shao, G. Li, M. Gu, J. Hu, S. Xu, Z. Nie, X. Chen, C. Wang and J. Liu, J. Mater. Chem. A, 2014, 2, 3430 RSC; (c) P. Saha, P. H. Jampani, M. K. Datta, C. U. Okoli, A. Manivannan and P. N. Kumta, J. Electrochem. Soc., 2014, 161, A593 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, NMR spectra of the synthesized organo-Mg compounds, and electrochemical characterization. CCDC 1403836 for 1[MgPh1+]. See DOI: 10.1039/c5qi00171d |
| ‡ Designates S.G.M. and L.G. have equal author contributions. |
| This journal is © the Partner Organisations 2015 |