
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA thioether-decorated {Mn11Tb4} coordination cluster with slow magnetic relaxation†
Sebastian
Schmitz
a,
Jan
van Leusen
a,
Arkady
Ellern
b,
Paul
Kögerler
 *ac and
Kirill Yu.
Monakhov
*a
*ac and
Kirill Yu.
Monakhov
*a
aInstitut für Anorganische Chemie, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de; kirill.monakhov@ac.rwth-aachen.de
bChemistry Department, Iowa State University, 1711 Gilman Hall, Ames, IA 50011, USA
cJülich-Aachen Research Alliance (JARA-FIT) and Peter Grünberg Institute (PGI-6), Forschungszentrum Jülich, 52425 Jülich, Germany
First published on 13th October 2015
Abstract
The oxidation reaction of manganese(II) acetate tetrahydrate with terbium(III) nitrate hexahydrate in acetonitrile in the presence of 4-(methylthio)benzoic acid (Hbza·SMe) afforded a polynuclear [MnIII11TbIII4O9(OH)5(bza·SMe)18(NO3)2(H2O)6(OMe)2] complex (1) with a {Mn5Tb2O6}-bridged double-{TbMn3O4}-cubane structure. This antiferromagnetically coupled, neutral molecule is decorated in the periphery with structurally exposed thioether groups which can be used as anchors to metallic surfaces. Compound 1 is potentially a single-molecule magnet (SMM), exhibiting slow relaxation of the magnetisation up to 3 K. While 1 is not thermally stable, the coordination cluster remains stable in various Lewis base-type organic solvents like THF and acetone.
Introduction
Single-molecule magnets (SMMs) based on the 3d transition metals and/or 4f lanthanides have attracted much attention1 due to the variety of their physical phenomena, including quantum tunneling of magnetisation, quantum coherence, magnetic relaxation, etc.2 To use these metalorganic spin structures as functional units in the molecular electronic and spintronic devices for digital storage and quantum computing,3 it is crucial to gain the understanding of the physisorption and chemisorption on conducting surfaces4,5 of individual molecules or thin films, to control the molecule–electrode interfaces, and to explore the feasibility of creating charge and spin transport measurements or devices. From this perspective, tailoring the structural and electronic properties of the ligand environments in these SMMs, in particular their peripheral functionalisation6 with specific N, P, O or S-grafting groups, is appealing and worthwhile. From the chemical point of view, this can be regarded as a tool to attach individual molecules intact to the solid support and to prevent the damage of molecular redox and spin states.For deposition from the solid phase on a metallic substrate the SMM must possess high thermal stability as well as high vapor pressure under vacuum, whereas deposition from the liquid phase requires preferably a low or no molecular charge and a moderate solubility (low concentration) of the molecular material in anhydrous solvents. For instance, Powell and colleagues reported on adsorption of high-spin (S = 83/2), ferromagnetic, mixed-valence {MnIII12MnII7} coordination complexes onto highly oriented pyrolytic graphite (HOPG) surfaces using deposition from MeOH solutions.7 Interestingly, the solution-driven deposition of the methylthio-functionalised Mn19-nuclearity analogues on the Au(111) surface has revealed reduction of MnIII to MnII ions and a total ground-state spin much lower than the parent S = 83/2.8 Remarkably, a central MnII cation in these complexes can be virtually replaced with DyIII, thus resulting in a quasi-isotopological {MnIII12MnII6DyIII} SMM.9
In 2004, Christou and coworkers described mixed manganese–lanthanide SMMs10 containing the [Mn11Ln4]45+ core (Ln = Nd, Gd, Dy, Ho, and Eu). We now provide a facile synthesis of the as-of-yet elusive Tb member of this family, however furnished with structurally exposed, peripheral thioether groups to facilitate the eventual attachment of this magnetic coordination complex to substrate surfaces. Herein, we present the synthesis of the charge–neutral complex [Mn11Tb4O9(OH)5(bza·SMe)18(NO3)2(H2O)6(OMe)2] (1) that is of potential interest for liquid phase deposition and epitaxial growth of thin films on single-crystal substrates. Compound 1 was characterised by sulfur elemental and metal analyses, infrared (IR) spectroscopy, and thermogravimetric analysis (TGA). The solid-state molecular structure of the compound was determined via single-crystal X-ray diffraction. It is noteworthy that the Cambridge Crystallographic Data Center (CCDC) contains 38 entries for the mixed manganese–terbium complexes, although the composition {Mn11Tb4} with eleven MnIII and four magnetically anisotropic11 TbIII ions has been observed for the first time. Notably, manganese–terbium complexes usually offer very high magnetic axial anisotropy.12 A study of the magnetism of compound 1 by the dc and ac magnetic susceptibility is discussed.
Results and discussion
The one-pot reaction of Mn(OOCMe)2·4H2O with 4-(methylthio)benzoic acid and Tb(NO3)3·6H2O in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.5 ratio in MeCN under reflux resulted in the formation of the dark-brown [Mn11Tb4O9(OH)5(bza·SMe)18(NO3)2(H2O)6(OMe)2] complex (1) with the well-known [Mn11Ln4]45+ mixed-metal core in ca. 30% yield (Scheme 1). We want to stress that in contrast to the synthetic procedure described in ref. 10, using a [Mn3O(O2CPh)6(py)2(H2O)] complex and the different Ln(NO3)3 salts (Ln = Nd, Gd, Dy, Ho, and Eu) as starting materials, we utilised the simple salts manganese(II) acetate tetrahydrate and terbium(III) nitrate hexahydrate.
:1:0.5 ratio in MeCN under reflux resulted in the formation of the dark-brown [Mn11Tb4O9(OH)5(bza·SMe)18(NO3)2(H2O)6(OMe)2] complex (1) with the well-known [Mn11Ln4]45+ mixed-metal core in ca. 30% yield (Scheme 1). We want to stress that in contrast to the synthetic procedure described in ref. 10, using a [Mn3O(O2CPh)6(py)2(H2O)] complex and the different Ln(NO3)3 salts (Ln = Nd, Gd, Dy, Ho, and Eu) as starting materials, we utilised the simple salts manganese(II) acetate tetrahydrate and terbium(III) nitrate hexahydrate.
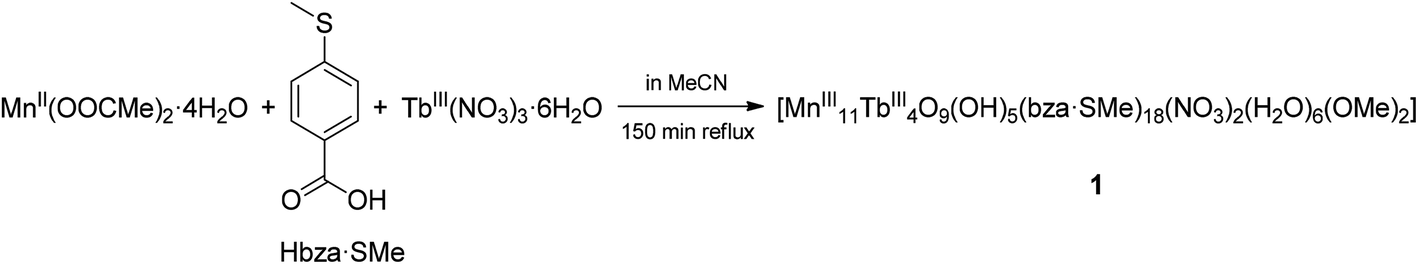 | ||
| Scheme 1 Synthesis of the polynuclear mixed-metal complex 1. For its IR spectrum, see Fig. S1 in the ESI.† Note that FTIR spectroscopic measurements of the complex in THF or acetone solution revealed the presence of the main bands observed in the solid-state (KBr pellet) IR spectrum of the polycrystalline sample. Compound 1 retains structural integrity in the solid state after evaporation of the specified solutions under an ambient atmosphere. | ||
Although oxidation by air cannot be excluded in such reaction procedures, in the present case we anticipated that nitrates NO3− could act as oxidising agents13 (Mn2+→Mn3+) during the synthesis of compound 1. As an indication for this, the reaction mixture does not display its characteristic colour change from light rose to dark brown when replacing Tb(NO3)3·6H2O with TbCl3·6H2O. Apart from charge neutrality arguments, the formal oxidation states of the metal atoms in compound 1 were confirmed by bond valence sum calculations 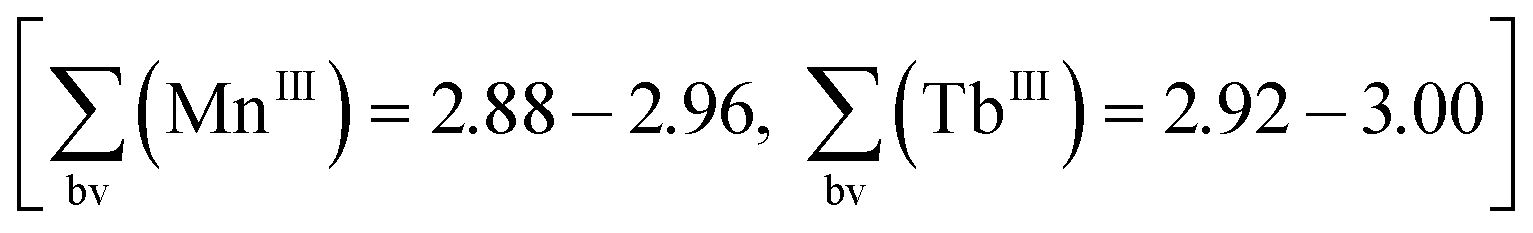 (see discussion of the X-ray crystal structure below).
(see discussion of the X-ray crystal structure below).
Although methanol was not used in the course of the reaction, we found the presence of two coordinated methoxide anions (MeO−) in the structure of compound 1. This in situ formation of deprotonated methanol molecules can be the result of oxidative decarboxylation of acetic acids formed upon exchange of acetate ligands in Mn(OOCMe)2·4H2O for bulkier 4-(methylthio)benzoate ligands. A tentative scheme for proposed formation of MeO− in compound 1 is given in the ESI (see Scheme S1†). The carboxylate substitution is very well known in the chemistry of homoleptic manganese14 or palladium15 carboxylates. Oxidative decarboxylation of carboxylic acids catalysed, e.g. by manganese(III) porphyrin16 or manganese(III) Schiff-base complexes17 was described in the literature. In this regard, it will be interesting to assess compound 1 as a precatalyst for such type of catalytic reactions. It is noteworthy that the production of the [Mn11Ln4]45+ core for Ln = Tb (1) in the mixture of MeOH/MeCN (1:20) has failed, in contrast to the synthetic protocol given in ref. 10 for Ln = Nd, Gd, Dy, Ho, and Eu.
Complex 1 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Tables S1–S4 in the ESI†). Its molecular structure (Fig. 1a) displays eleven octahedrally coordinated MnIII ions, two eight-coordinated TbIII ions, two TbIII ions in nine-coordination environments including two chelating nitrate (NO3−) ligands, four tridentate bridging thioether-functionalised benzoate μ3-bza·SMe− groups, twelve bidentate bridging μ2-bza·SMe− groups, two monodentate coordinated μ1-bza·SMe− groups, two μ2-bridging methoxido (MeO−) ligands, six terminal water ligands, nine O2− ligands, and five hydroxyl (OH−) groups. Interestingly, the latter fourteen oxygen atoms (9O2− + 5OH−) constitute the {Mn11Tb4O14} skeleton of compound 1 shown in Fig. 1b. Five protons are disordered over six μ3-oxygen atoms [Mn–μ3-O 1.945(7) Å–2.300(8) Å; OMn2Tb type] due to the very typical positional disorder of H in high symmetrical, polynuclear metal complexes (Fig. 1b). The two remaining μ3-oxygen atoms (OMn3 type) are assumed to be not protonated because of the shorter Mn–μ3-O bond distances ranging from 1.870(7) Å to 1.901(6) Å. No protonation is indeed expected for the six four-fold coordinated O atoms with the Mn–μ4-O bond lengths of 1.860(7)–2.440(7) Å. Complex 1 can be viewed as being composed of two strongly distorted {TbMn3O4}4+ cubane units bridged by a {Mn5Tb2O6}9+ fragment (Fig. 1b). It is noteworthy that the previously described octanuclear chloro-bridged nickel(II) double cubane, which is a SMM with the formula [({NiII4(μ3-OH)Cl3(HL)3}μ2-Cl)2], was deposited onto the HOPG surface from CH2Cl2 solution and investigated by scanning tunneling microscopy.18
(Tables S1–S4 in the ESI†). Its molecular structure (Fig. 1a) displays eleven octahedrally coordinated MnIII ions, two eight-coordinated TbIII ions, two TbIII ions in nine-coordination environments including two chelating nitrate (NO3−) ligands, four tridentate bridging thioether-functionalised benzoate μ3-bza·SMe− groups, twelve bidentate bridging μ2-bza·SMe− groups, two monodentate coordinated μ1-bza·SMe− groups, two μ2-bridging methoxido (MeO−) ligands, six terminal water ligands, nine O2− ligands, and five hydroxyl (OH−) groups. Interestingly, the latter fourteen oxygen atoms (9O2− + 5OH−) constitute the {Mn11Tb4O14} skeleton of compound 1 shown in Fig. 1b. Five protons are disordered over six μ3-oxygen atoms [Mn–μ3-O 1.945(7) Å–2.300(8) Å; OMn2Tb type] due to the very typical positional disorder of H in high symmetrical, polynuclear metal complexes (Fig. 1b). The two remaining μ3-oxygen atoms (OMn3 type) are assumed to be not protonated because of the shorter Mn–μ3-O bond distances ranging from 1.870(7) Å to 1.901(6) Å. No protonation is indeed expected for the six four-fold coordinated O atoms with the Mn–μ4-O bond lengths of 1.860(7)–2.440(7) Å. Complex 1 can be viewed as being composed of two strongly distorted {TbMn3O4}4+ cubane units bridged by a {Mn5Tb2O6}9+ fragment (Fig. 1b). It is noteworthy that the previously described octanuclear chloro-bridged nickel(II) double cubane, which is a SMM with the formula [({NiII4(μ3-OH)Cl3(HL)3}μ2-Cl)2], was deposited onto the HOPG surface from CH2Cl2 solution and investigated by scanning tunneling microscopy.18
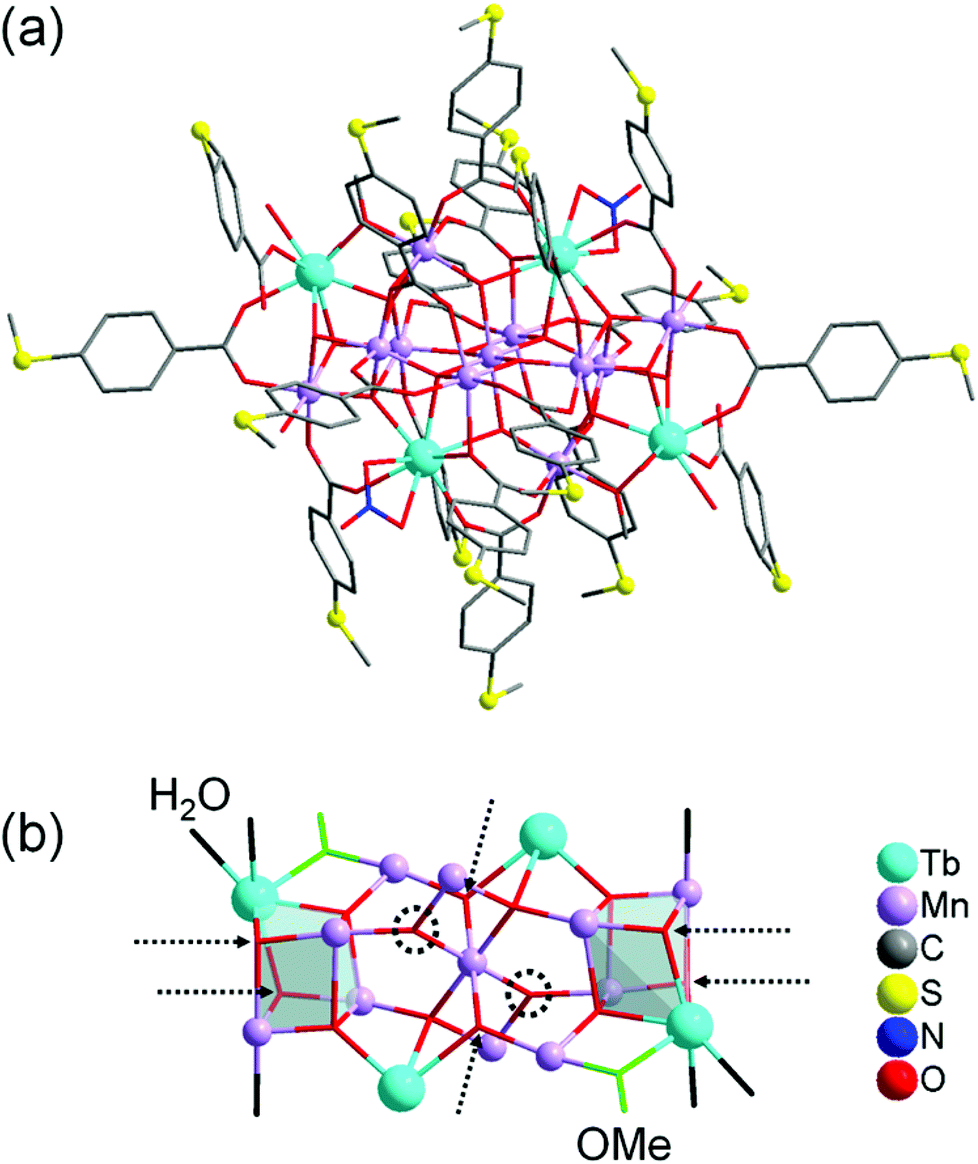 | ||
| Fig. 1 (a) Molecular structure of compound 1. (b) Metal–oxide skeleton {Mn11Tb4O14} of compound 1 incorporating two methoxide anions and six water molecules (eighteen bza·SMe− ligands and two NO3− ligands are not shown). Six possible μ3-O protonation sites are indicated by dashed arrows and the two remaining, nonprotonated μ3-oxygen atoms by dashed circles. Hydrogen atoms are omitted for clarity. The metal and sulfur atoms are represented as ball-and-stick models. Selected bond lengths [Å]: Tb–O (of H2O) 2.371(10) and 2.427(9), Tb–O (of MeO−) 2.282(8), Tb–μ3-O 2.366(8)–2.492(8), Tb–μ4-O 2.388(7)–2.565(7), Mn–O (of H2O) 2.212(9), Mn–O (of MeO−) 1.891(8), Mn–μ3-O 1.870(7)–2.300(8), Mn–μ4-O 1.860(7)–2.440(7), nearest-neighbor Mn–Tb 3.4045(18), nearest-neighbor Mn–Mn 2.855(2). All bond lengths and angles are reported in the ESI.† | ||
Although the structure of complex 1 resembles that of the reported {Mn11Dy4} complex,10 there are key differences in the coordination environment of LnIII ions (eight- and nine-coordinated Tb atoms in compound 1vs. only nine-coordinated Dy atoms in ref. 10), metal oxide skeleton ({Mn11Tb4O14} in compound 1vs. {Mn11Dy4O18} in ref. 10), coordination modes of benzoate ligands (μ1,μ2,μ3-bza·SMe− in compound 1vs. μ2,μ3-bza− in ref. 10), and the type of these ligands (methylthio-functionalised bza− in compound 1vs. non-functionalised bza− in ref. 10). Note that the peripheral SMe groups of the bza·SMe ligands are not involved in any intermolecular coordination bond. According to preliminary spectroscopy data (Fig. S6†), the replacement of terbium with dysprosium in the synthesis of compound 1 results in an isostructural {Mn11Dy4}-type coordination cluster (see the ESI†).
The TGA curve reveals an extremely low thermal stability of compound 1 up to only 50 °C against degradation under air. The gradual destruction of the complex occurs in one big undefined step (see Fig. S2 in the ESI†), with a total weight loss (Δmfound) of 55.3% in the range 25–570 °C. This indicates that compound 1 releases eighteen bza·SMe ligands without their oxygen atoms (Δmcalc = 51.0%), six water molecules (Δmcalc = 2.3%), two methyl groups of the methoxide anions (Δmcalc = 0.6%), two NO of the nitrates (Δmcalc = 1.3%), and five hydrogen atoms of the OH-groups (Δmcalc = 0.1%); calculated total weight loss: Δmcalc = 55.3%. TGA measurements performed under a N2 atmosphere did not result in a considerable change of the overall thermal stability of compound 1. The thermal stability of sulfur-free {Mn11Ln4}-type complexes (Ln = Nd, Gd, Dy, Ho, and Eu)10 was not reported.
The static magnetic behaviour of compound 1 is illustrated by the temperature-dependence of χmT at B = 0.1 T and the field-dependence of the molar magnetisation at T = 2 K in Fig. 2. χmT reaches 65.2 cm3 K mol−1 at 290 K, which is less than the expected 80.1–82.4 cm3 K mol−1 (ref. 19) for eleven MnIII and four TbIII non-interacting ions. χmT continuously decreases with decreasing temperature down to 6.5 K, exhibiting a local minimum of 51.1 cm3 K mol−1. Further cooling beyond 6.5 K reveals an increase of χmT up to 54.2 cm3 K mol−1 at 2 K. The low χmT value at 290 K and the shape of χmT vs. T between 6.5 K and 290 K are consistent with predominant antiferromagnetic exchange interactions within compound 1 (see Fig. S3 in the ESI†). This observation is in line with the magnetic behaviour10 of the 3d–4f coordination complex exhibiting the [Mn11Dy4]45+ core.
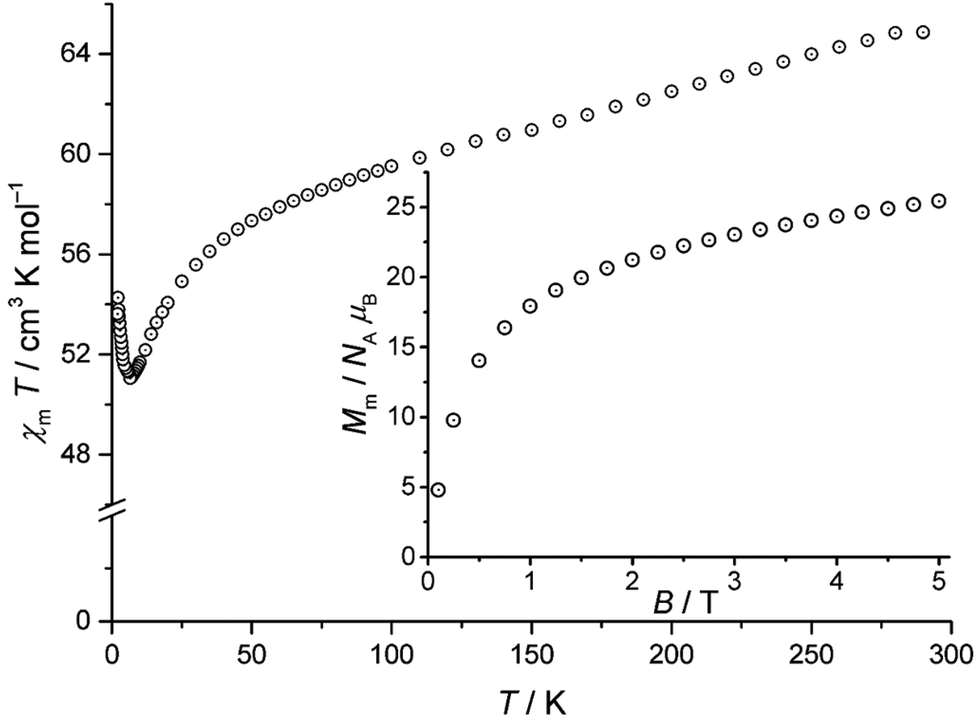 | ||
| Fig. 2 Temperature dependence of χmT at 0.1 Tesla for compound 1; inset: associated molar magnetisation Mmvs. B at 2 K. | ||
Since both χmT vs. T curves differ by a constant amount that can be directly linked to the substitution of TbIII by DyIII, it is safe to assume that the observation of strong antiferromagnetic exchange interactions results from the interactions within the MnIII centers. For some unknown reasons, χmT is presented in ref. 10 only for T ≥ 5 K. A potential increase below this temperature, as reported in the present work, was not taken into consideration. The observed behaviour of compound 1 at T ≤ 6.5 K indicates additional minor contributions of ferromagnetic exchange interactions within the compound. Because both types of metal centers in compound 1 are characterised by different quantum numbers (MnIII: S = 2 (5D0); TbIII: J = 6 (7F6)), only a qualitative estimation of the ground state can be made from the field-dependence of the molar magnetisation (Fig. 2, inset). Because MnIII and TbIII are characterised by even quantum numbers, the lowest possible value of Mm is 0 NAμB, whilst the highest one is (11gSS + 4gJJ) NAμB = (11 × 2 × 2 + 4 × 3/2 × 6) NAμB = 80NAμB. Although 2 K may be sufficient thermal energy to significantly populate higher energy levels besides the total ground state and the corresponding steps in Mmvs. B could thus be smeared out, it is highly improbable that the ground state is characterised by a singlet due to Mm > 20 NAμB for B > 2 T. Note that the shape of the Mm curve for B ≤ 5 T hints at a saturation value in the interval 30–50 NAμB, therefore suggesting a medium ground-state multiplet value.
The dynamic magnetic susceptibility at the zero field as in-phase χ′m and out-of-phase χ′′m component vs. T is shown in Fig. 3. In analogy with the work of Mishra et al.10 only “tails” of peaks are observed in the χ′′mvs. T plane, therefore revealing slow relaxation for compound 1 at temperatures (T < 3 K) close to the operation limit of the used SQUID magnetometer. In lieu of more sophisticated experimental instrumentation such as a micro-SQUID20 to directly measure the dc hysteresis of the magnetisation, a static external field was applied to analyse the slow relaxation in more detail. Unfortunately, the out-of-phase signals never reveal maxima at various applied static bias fields. The in-phase and out-of-phase ac susceptibility components display a slightly more pronounced divergence at a static bias field of 0.015 T, though insufficient for a reliable Cole–Cole analysis (within the frequency limits of our experimental setup). At higher bias fields, the in-phase/out-of-phase divergence gradually vanishes with no out-of-phase signal remaining above 0.3 T at the discussed frequencies (see Fig. S4 and S5†). For this reason, a more specific analysis of the dynamic behaviour of compound 1e.g. in terms of a relaxation constant τ0 and an effective energy barrier Ueff could not be accomplished with the available experimental method. Nevertheless, if a coordination compound containing DyIII is identified to be an SMM, the TbIII analogue typically also represents an SMM, provided that slow relaxation is detected for the Tb analogue.21 Since the [Mn11Dy4]45+ core of Mishra et al. is a single-molecule magnet (with hysteretic behavior evident at mK temperatures),10 compound 1 possibly might display SMM characteristics at sub-Kelvin temperatures as well.
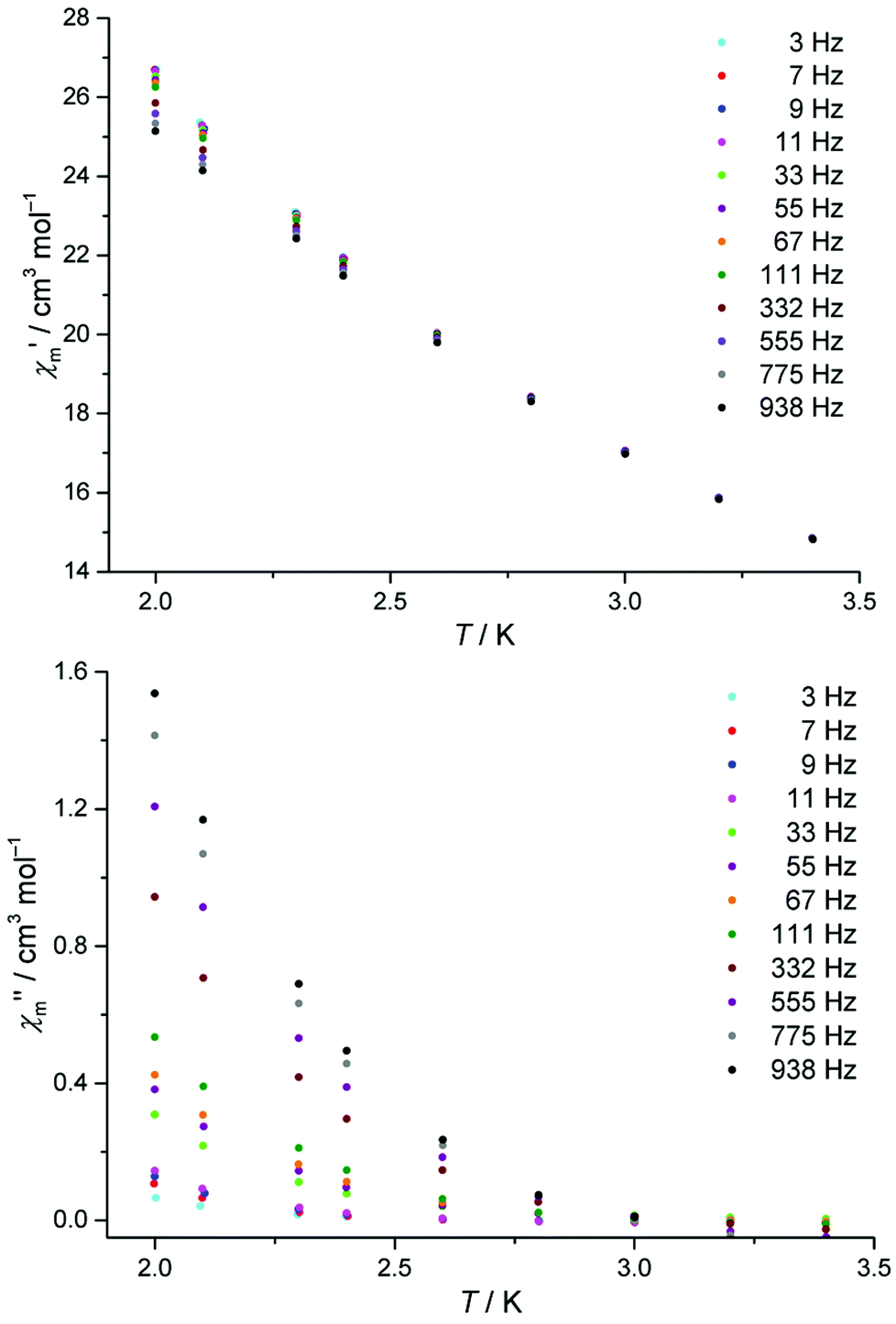 | ||
Fig. 3 Temperature-dependence of the in-phase 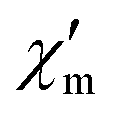 and out-of-phase ac susceptibilities χ′′m of compound 1 at zero static bias field. and out-of-phase ac susceptibilities χ′′m of compound 1 at zero static bias field. | ||
Conclusion
We synthesised the thioether-functionalised MnIIITbIII coordination complex (1) with a medium total ground-state spin. 1 represents the missing member of the family of {Mn11Ln4}-type coordination clusters. Despite the fact that this charge–neutral, antiferromagnetically coupled heterometallic 3d–4f complex is not thermally stable and thus cannot be deposited intact on the metallic surface from the solid phase via sublimation, its solubility in THF and acetone opens possibilities to study surface deposition processes from solution using, e.g. drop-casting approaches. The latter may grant the possibility of the formation of molecular self-assembled monolayers. The structurally exposed methylthioether functionalities in compound 1 may provide effective anchoring to Au, Ag or HOPG surfaces. Finally, we note that 1 might be a suitable precursor for the (pyrolysis) production of nanosized manganese–terbium oxide materials with interesting physical characteristics.22Experimental section
Materials and methods
The synthesis of compound 1 was carried out under aerobic conditions. All starting materials were from commercial sources and used as received. Solvents were used without further purification. Elemental analysis was performed to determine the sulfur composition (deviation ± 0.05) in the polycrystalline sample of compound 1, by using a Vario EL elemental analyser. Inductively coupled plasma optical emission spectrometry (ICP-OES) analysis of compound 1 was used to determine the manganese and terbium composition (deviation ± 0.7%). The IR spectrum of compound 1 was recorded on a Nicolet Avatar 360 FTIR spectrometer by using KBr pellets (mKBr ≈ 250 mg) in the range v = 4000–400 cm−1. The FTIR spectra of compound 1 in the solid state and in solution were also recorded on a Bruker Vertex 70 FTIR spectrometer equipped with a Platinum ATR unit A225 (on diamond) in the range v = 4000–400 cm−1. TG/DTA analysis of compound 1 was performed under air and under a nitrogen atmosphere with a heating rate of 5 K min−1 in the temperature range 25–800 °C by using a Mettler Toledo TGA/SDTA 851e instrument.Synthesis
Mn(OOCMe)2·4H2O (0.245 g, 1.0 mmol), 4-(methylthio)benzoic acid (0.168 g, 1.0 mmol) and Tb(NO3)3·6H2O (0.227 g, 0.5 mmol) were dissolved in 7 mL of MeCN and stirred for 2.5 h under reflux. The colour change from light rose to dark brown was observed. The MeCN solution was filtered off and the filtrate was stored in a capped vial at ambient temperature. Dark-brown single crystals of compound 1 were obtained after ca. two weeks. Yield: 0.076 g (29% based on bza·SMe). Elemental analysis, calcd for Mn11Tb4C146H149N2O64S18 (4773.4 g mol−1; disregarding solvent): Mn, 12.67; Tb, 13.32; S, 12.09%. Found: Mn, 13; Tb, 13; S, 11.47%. IR, νmax(KBr pellet)/cm−1: 3423 (br, m), 2982 (vw), 2919 (w), 1593 (s), 1543 (m), 1491 (sh, vw), 1407 (s), 1385 (sh, w), 1299 (vw), 1186 (m), 1149 (w), 1116 (vw), 1091 (m), 1014 (w), 967 (w), 845 (w), 815 (vw), 768 (m), 740 (w), 724 (w), 707 (w), 691 (w), 631(vw), 606 (m), 536 (m), 478 (w).X-ray crystallography
Single-crystal X-ray diffraction data for compound 1 were collected on a SuperNova (Agilent Technologies) diffractometer with Mo Kα radiation (λ = 0.71073 Å) at 153 K. A crystal was mounted on a Hampton cryoloop with Paratone-N oil. Absorption corrections were applied numerically based on the multifaceted crystal model using ChrysAlis software.23 A summary of the crystal data and structure refinements for compound 1 is given in Tables S1–S4 in the ESI.† Details are described in the CIF file. CCDC 1062401 contains the supplementary crystallographic data for compound 1.Acknowledgements
K. Y. M. thanks the Excellence Initiative of the German federal and state governments for an RWTH Start-Up grant.References
- See special issues of J. Mater. Chem., 2006, 16, 2501; Chem. Soc. Rev., 2011, 40, 3053; Dalton Trans., 2012, 44, 13555 and the ACS virtual issue on “Quantum Molecular Magnets”, ed. K. R. Dunbar, Inorg. Chem., 2012, 51, 12055.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS PubMed.
- (a) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed; (b) W. Wernsdorfer, N. Aliaga-Acalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406–409 CrossRef PubMed; (c) J. Tejada, E. M. Chudnovsky, E. del Barco, J. M. Hernandez and T. P. Spiller, Nanotechnology, 2001, 12, 181–186 CrossRef CAS; (d) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed; (e) M. Mannini, E. Tancini, L. Sorace, P. Sainctavit, M.-A. Arrio, Y. Qian, E. Otero, D. Chiappe, L. Margheriti, J. C. Cezar, R. Sessoli and A. Cornia, Inorg. Chem., 2011, 50, 2911–2917 CrossRef CAS PubMed.
- (a) S. Voss, M. Burgert, M. Fonin, U. Groth and U. Rüdiger, Dalton Trans., 2008, 499–505 RSC; (b) D. Gatteschi, A. Cornia, M. Mannini and R. Sessoli, Inorg. Chem., 2009, 48, 3408–3419 CrossRef CAS PubMed; (c) A. Saywell, G. Magnano, C. J. Satterley, L. M. A. Perdigão, A. J. Britton, N. Taleb, M. del Carmen Giménez-López, N. R. Champness, J. N. O'Shea and P. H. Beton, Nat. Commun., 2010, 1, 75 Search PubMed.
- (a) M. Mannini, P. Sainctavit, R. Sessoli, C. Cartier dit Moulin, F. Pineider, M.-A. Arrio, A. Cornia and D. Gatteschi, Chem. – Eur. J., 2008, 14, 7530–7535 CrossRef CAS PubMed; (b) M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia and D. Gatteschi, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed; (c) M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M.-A. Arrio, E. Otero, L. Joly and J. C. Cezar, Nature, 2010, 468, 417–421 CrossRef CAS PubMed; (d) A. Scheurer, A. M. Ako, R. W. Saalfrank, F. W. Heinemann, F. Hampel, K. Petukhov, K. Gieb, M. Stocker and P. Müller, Chem. – Eur. J., 2010, 16, 4784–4792 CrossRef CAS PubMed; (e) M. J. Rodriguez-Douton, M. Mannini, L. Armelao, A.-L. Barra, E. Tancini, R. Sessoli and A. Cornia, Chem. Commun., 2011, 47, 1467–1469 RSC; (f) A. Cornia, M. Mannini, P. Sainctavit and R. Sessoli, Chem. Soc. Rev., 2011, 40, 3076–3091 RSC; (g) V. Heß, F. Matthes, D. E. Bürgler, K. Yu. Monakhov, C. Besson, P. Kögerler, A. Ghisolfi, P. Braunstein and C. M. Schneider, Surf. Sci., 2015, 641, 210–215 CrossRef PubMed.
- See, e.g.: A. Ghisolfi, K. Yu. Monakhov, R. Pattacini, P. Braunstein, X. López, C. de Graaf, M. Speldrich, J. van Leusen, H. Schilder and P. Kögerler, Dalton Trans., 2014, 43, 7847–7859 RSC.
- (a) A. M. Ako, M. S. Alam, S. Mameri, Y. Lan, M. Hibert, M. Stocker, P. Müller, C. E. Anson and A. K. Powell, Eur. J. Inorg. Chem., 2012, 4131–4140 CrossRef CAS PubMed; (b) S. Mameri, A. M. Ako, F. Yesil, M. Hibert, Y. Lan, C. E. Anson and A. K. Powell, Eur. J. Inorg. Chem., 2014, 4326–4334 CrossRef CAS PubMed.
- J. Dreiser, A. M. Ako, C. Wäckerlin, J. Heidler, C. E. Anson, A. K. Powell, C. Piamonteze, F. Nolting, S. Rusponi and H. Brune, J. Phys. Chem. C, 2015, 119, 3550–3555 Search PubMed.
- A. M. Ako, V. Mereacre, R. Clérac, W. Wernsdorfer, I. J. Hewitt, C. E. Anson and A. K. Powell, Chem. Commun., 2009, 544–546 RSC.
- A. Mishra, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2004, 126, 15648–15649 CrossRef CAS PubMed.
- (a) V. Mereacre, Angew. Chem., Int. Ed., 2012, 51, 9922–9925 CrossRef CAS PubMed; (b) Lanthanides and Actinides in Molecular Magnetism, ed. R. Layfield and M. Murugesu, Wiley-VCH, 2015 Search PubMed.
- L. R. Piquer and E. C. Sañudo, Dalton Trans., 2015, 44, 8771–8780 RSC and references cited therein.
- L. Zhang and W. Schmitt, J. Am. Chem. Soc., 2011, 133, 11240–11248 CrossRef CAS PubMed.
- J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 2450–2460 CrossRef CAS.
- See, e.g.: T. A. Stromnova, K. Yu. Monakhov, J. Cámpora, P. Palma, E. Carmona and E. Alvarez, Inorg. Chim. Acta, 2007, 360, 4111–4116 CrossRef CAS PubMed and references cited therein.
- V. Mirkhani, S. Tangestaninejad, M. Moghadam and Z. Karimian, Bioorg. Med. Chem. Lett., 2003, 13, 3433–3435 CrossRef CAS.
- V. Mirkhani, S. Tangestaninejad, M. Moghadam and M. Moghbel, Bioorg. Med. Chem., 2004, 12, 903–906 CrossRef CAS PubMed.
- A. Scheurer, K. Gieb, M. S. Alam, F. W. Heinemann, R. W. Saalfrank, W. Kroener, K. Petukhov, M. Stocker and P. Müller, Dalton Trans., 2012, 41, 3553–3561 RSC.
- H. Lueken, Magnetochemie, Teubner, Stuttgart, 1999 Search PubMed.
- W. Wernsdorfer, Adv. Chem. Phys., 2001, 118, 99–190 CrossRef CAS PubMed.
- (a) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (b) L. Zhao, J. Wu, H. Ke and J. Tang, Inorg. Chem., 2014, 53, 3519–3525 CrossRef CAS PubMed.
- I. I. Steblevskaya, M. A. Medkov and M. V. Belobeletskaya, Theor. Found. Chem. Eng., 2010, 44, 517–520 CrossRef CAS.
- CrysAlisPro, Agilent Technologies, Version 1.171.36.21 (release 14-08-2012 CrysAlis171.NET).
Footnote |
| † Electronic supplementary information (ESI) available: IR spectrum, crystal data and structure refinement, TGA thermogram, and additional SQUID data of compound 1 as well as synthesis of the dysprosium analogue, including a comparison of their IR spectra. CCDC 1062401. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00172b |
| This journal is © the Partner Organisations 2015 |