
Dual nature of the excited state in organic–inorganic lead halide perovskites†
Kevin G.
Stamplecoskie
 a,
Joseph S.
Manser
ab and
Prashant V.
Kamat
*abc
a,
Joseph S.
Manser
ab and
Prashant V.
Kamat
*abc
aRadiation Laboratory, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: pkamat@nd.edu
bDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana 46556, USA
cDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA
First published on 3rd November 2014
Abstract
The rapid increase in efficiency of methylammonium lead halide perovskite solar cells necessitates further investigation into the nature of perovskite absorption features and optical properties. Films obtained from the deposition of solutions containing lead halides and the CH3NH3+ organic cation is known to yield the CH3NH3PbI3 perovskite structure upon annealing. In examining the precursor solution used in the processing of CH3NH3PbI3 solar cells, we find that Pb2+ readily forms plumbate complexes in the presence of excess iodide ions and exhibits characteristic absorption bands at 370 (PbI3−) and 425 nm (PbI42−). Through comparative spectral analysis of the absorption features of charge transfer complexes in the solution phase and the final solid-state perovskite films, we are able to fully classify the absorption features in the excited state of CH3NH3PbI3 across the transient absorption spectrum recorded following laser pulse excitation. In particular, we attribute the broad photoinduced absorption to a charge-transfer excited state, and show correlation between the photoinduced absorption and 480 nm bleach signals. These observations lead us to propose a band structure composed of two distinct transitions that is consistent with the various spectral features and kinetic behavior of the CH3NH3PbI3 excited state. Characterization of this unique dual excited state nature provides further insight into the optoelectronic behavior of hybrid lead halide perovskite films and thus aids in elucidating their exceptional photovoltaic properties.
Broader contextThe development of thin film solar cells with efficiencies comparable to commercial PV technology is attractive due to simplicity of fabrication and lower carbon footprint. The recent breakthroughs in developing organic–inorganic metal halide perovskite-based solar cells have delivered efficiencies in the range of 17–19%. Whereas the majority of recent efforts have focused towards fabrication of high-efficiency perovskite solar cells, less is known about the nature of the excited state that leads to charge separation and ultimately charge extraction. Using femtosecond transient absorption spectroscopy, we have now identified the dual nature of the CH3NH3PbI3 excited state. We find that both a charge separated state and charge transfer state explain the overall behavior of the excited state. The spectral characterization and kinetic profiles presented here provide insight into the excited state behavior of CH3NH3PbI3 films. These results have important implications in understanding the photoinduced processes in perovskite solar cells. |
The unusually high photovoltaic performance of methylammonium lead halide perovskites has drawn considerable attention in understanding the primary photoinduced events that lead to charge separation.1–3 The excited state properties of these hybrid materials are of particular interest considering their efficacy when coupled with both electron accepting and insulating metal oxides and in planar thin film photovoltaic architectures.4–7 Additionally, ambipolar carrier diffusion within lead halide perovskites further highlights the unique excited state character of this peculiar class of semiconductors.8–10 The quest to design high efficiency (>20%) perovskite solar cells demands better understanding of the nature of interactions in the ground state as well as the excited state behavior leading to efficient charge separation and extraction.11,12
One of the most promising attributes of hybrid perovskites for device applications is their ability to be processed from solution and at low temperatures (∼100 °C). Despite these low-energy requirements, organic–inorganic perovskites display surprisingly excellent semiconductor properties, as evidenced by the limited contribution from trap states to the overall excited state dynamics and the significant deviation from simple Langevin recombination kinetics.13–16 A wide variety of methods for depositing high-quality perovskite thin films have been developed, including solution-based sequential deposition and co-evaporation of the solid precursor components.2,6,7,17,18 However, the most facile technique involves a single-step deposition of the perovskite precursors from solution. This method entails dissolving both the PbX2 and RNH3X compounds in a common solvent such as dimethylformamide, dimethyl sulfoxide, and/or γ-butyrolactone. The solution is then cast onto a planar or mesostructured film by spin coating followed by drying/annealing, during which time the precursor components self-assemble into the final perovskite phase with the general AMX3 stoichiometry.1,19–22 For the 3-dimensional hybrid perovskites discussed here, A is a small organic cation (e.g., CH3NH3+), M is a divalent group 14 metal (Sn2+, Pb2+), and X is a halide ion (I−, Br−, Cl−).
The solution-based processing of hybrid perovskites necessarily leads to an intriguing question: what is the nature and identity of the species that comprise the precursor solution, and can characterizing their properties help us better understand the perovskite excited state? It is well established that transition metals with an s2 electron configuration (i.e. Ti+, Sn2+, Pb2+, Sb3+, Bi3+, Te4+) form labile complexes with halogens in a range of solvents.23 For Pb2+, these complexes are referred to as ‘plumbates’ (e.g., triiodoplumbate (PbI3−) and tetraiodoplumbate (PbI42−)) and are investigated herein.23 It is difficult to isolate complexes with an exact number of halogens for these metals due to equilibrium between multiple halogenated complexes. Though these complexes are kinetically labile, varying the concentration of I− allows one to spectroscopically characterize different forms of the iodoplumbate complex.23
Initial investigations into the methylammonium lead halide excited state focused primarily on charge mobility and transfer rates to selective electron- and hole-accepting materials.24–26 Since then, additional pump-probe spectroscopic studies in both the visible and terahertz spectral regions have revealed the intrinsic nature of photoinduced charge carriers and their recombination mechanisms within these materials.14,15,25,27–30 These studies have provided insight into the optoelectronic properties of hybrid perovskites, including the large fraction of free carriers generated under non-resonant (Eexcitation > Egap) room temperature photoexcitation. This observation, of course, is important for photovoltaic applications. Yet given this growing body of spectroscopic information, full characterization of the broad and complex spectral features across the visible region of the lead halide perovskite excited state remains elusive. In particular, questions remain regarding the transient photoinduced bleaching at 480 nm and induced absorption extending from approximately 550 to 680 nm.
Initially, Xing, et al. attributed the two photoinduced bleaches that arise following laser pulse excitation of CH3NH3PbI3 to a dual valence band structure,25 although they recognized that this type of configuration could not fully account for the broad photoinduced absorption. Recently it has been suggested that the bleaching at 480 nm stems from a PbI2 passivation layer at the interface between oxide support and lead halide perovskite.31 In both the methylammonium lead iodide structure and the PbI2 rhombus structure (i.e. plumbous iodide), Pb2+ is coordinated to six I− ligands. The obvious question is how the excited state chemistry of the lead iodide influences the overall excited state behavior of the organic–inorganic metal halide framework under visible excitation. Using both steady-state and time-resolved absorption spectroscopy, we have now probed the formation of iodoplumbate complexes in the CH3NH3PbI3 perovskite precursor solution and established the role of charge transfer states in dictating the net photoinduced charge separation in CH3NH3PbI3.
Results and discussion
Complexation between PbI2 and iodide ions
PbI2 is a well-known semiconductor with limited solubility in water,32–34 and colloidal particles of PbI2 have been shown to exhibit semiconducting properties. Interestingly, in the presence of excess iodide ions, PbI2 undergoes complexation to produce soluble PbI3−, PbI42−, and higher coordinated plumbate species. In order to probe the complexation between PbI2 and CH3NH3I, we monitored the changes in the absorption spectra of a PbI2 solution at different concentrations of CH3NH3I (Fig. 1A). With increasing concentration of CH3NH3I, two new absorption bands appear with maxima at 370 and 425 nm, respectively. The growth of absorbance monitored at these two maxima (Fig. 1B) show different dependence on the methylammonium iodide concentration. The growth of absorption at 425 nm saturates at lower concentration than at 370 nm. The appearance of concentration dependent absorption bands suggests the presence of two separate complexation equilibria. These observations are consistent with the reported complexation between PbI2 and I− and calculated absorption spectra for each of the tri- and tetra-iodoplumbate complexes (equilibria (1) and (2)).23| PbI2 + I− ⇐ K1 ⇒ PbI3− | (1) |
| PbI3− + I− ⇐ K2 ⇒ PbI42− | (2) |
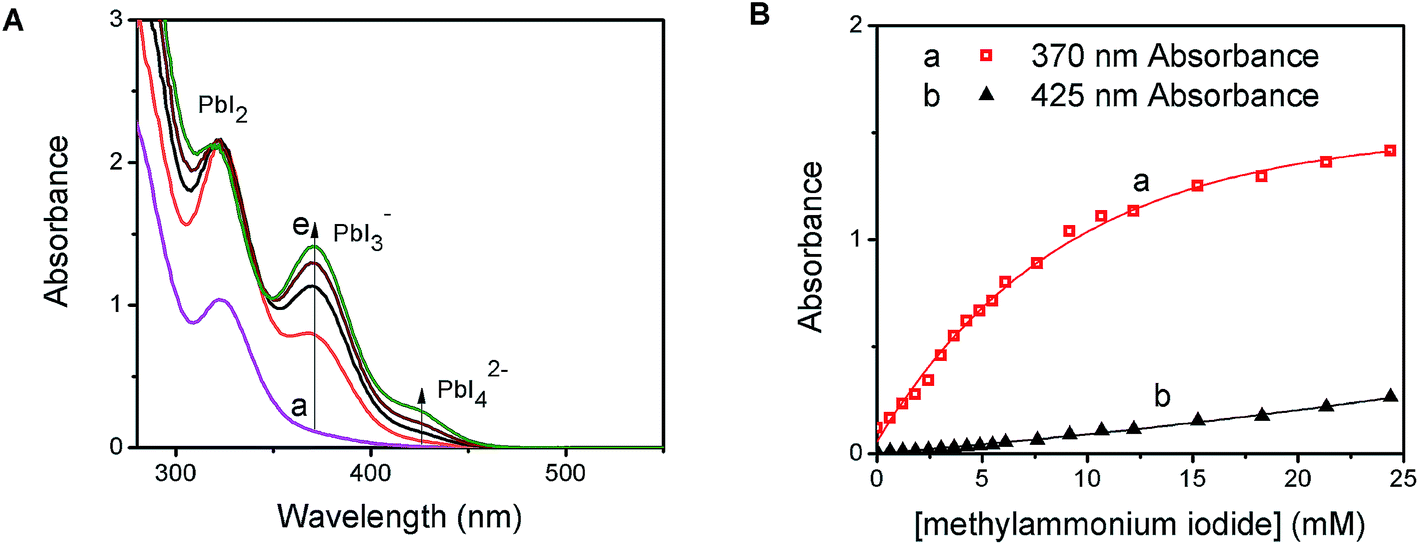 | ||
| Fig. 1 (A) Absorption spectra of 250 μM PbI2 solution in dimethylformamide (a) with increasing concentration of MAI from 6 mM (b) to 24 mM (e), and (B) the change in absorbance monitored at the 370 nm peak for PbI3− and 425 nm for PbI42− as a function of the MAI/Pb2+ ratio. | ||
We further evaluated the complexation process by analyzing the absorption change using Benesi–Hildebrand plots.35–37 The linear relationship of the double reciprocal plot (1/ΔA vs. 1/[I−]) (Fig. S1 in the ESI†) for two separate concentration ranges of iodide confirms the presence of the two equilibria illustrated in (1) and (2). From the slope and intercept of the linear plot we obtain equilibrium constants of 54 M−1 for K1 and 6 M−1 for K2. The average association constant for the tri- and tetra-iodoplumbate in acetonitrile observed in the present experiments agrees well with the qualitative estimate of 27 M−1 obtained as a combined value for the two equilibria.20 The present study enables determination of two separate equilibrium constants.
The presence of the organic cation (viz., CH3NH3+) is integral in determining the final hybrid perovskite structure upon drying and annealing of the iodoplumbate film.27 Geometries ranging from 1-D polymeric chains to 3-D isotropic crystal structures can form depending on the size and identity of the organic component.38 The obvious question is whether the organic cation plays a role in the initial complexation process. We probed the cation effect by repeating the experiment in Fig. 1, replacing CH3NH3I with KI. The absorption changes seen with increasing KI concentration are shown in Fig. S2 (ESI†). The appearance of absorption bands at 370 nm and 425 nm, similar to Fig. 1A and B, confirms that PbI2 undergoes complexation with iodide ions irrespective of the nature of the cation. Thus, the yellow-colored precursor solution employed in the preparation of organometal halide perovskite films for solar cell devices is essentially a lead halide complex (PbIxn−). The ratio of PbI2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :I− determines the type of initial complex formed in this system and hence careful control of this complex is important to obtain perovskite structure in the annealed films. Given its ability to coordinate with six ligands, we can expect up to six I− ions to associate with a single Pb2+ ion in the film. Because of the solubility limitation of iodide, it is not possible to probe higher order complexes in solution.
:I− determines the type of initial complex formed in this system and hence careful control of this complex is important to obtain perovskite structure in the annealed films. Given its ability to coordinate with six ligands, we can expect up to six I− ions to associate with a single Pb2+ ion in the film. Because of the solubility limitation of iodide, it is not possible to probe higher order complexes in solution.
Excited state behavior of PbIxn− complex in solution
The photochemical activity of PbI2 semiconductor colloids as well as lead halide complexes is well documented in the literature.34 The absorption properties of plumbate complexes are determined by the octahedrally coordinated Pb and have been associated with the halide-to-metal charge transfer absorptions.23,39 Despite the intense efforts to fabricate high efficiency solar cells with varying composition of lead and halide counterparts, relatively few studies have focused on elucidation of the fundamental properties of these perovskite films in the excited state.14,24–26,39The transient absorption spectra recorded following 387 nm laser pulse excitation of the PbIxn− precursor solution are shown in Fig. 2A. The transient bleaching below 410 nm is accompanied by an induced absorption with a maximum around 460 nm. The absorption-time profile recorded at 500 nm shows a quick decay of the transient with a lifetime of 3.6 ± 0.2 ps, followed by a longer lived component with a lifetime of 698 ± 92 ps (Fig. 2B). In order to establish the identity of the transient absorption peak at 460 nm, we compared the induced absorption spectrum with various iodide species (I−, I3−, I2etc.). It is interesting to note that the induced absorption has close spectral match with the spectrum of I2 in dimethylformamide solution (see Fig. S3 in ESI†). Given this spectral similarity, we assign this transient formed following the excitation of the PbIxn− complex to a species similar to I2.
| PbIxn− + hν → (PbIx−2n−⋯I2) | (3) |
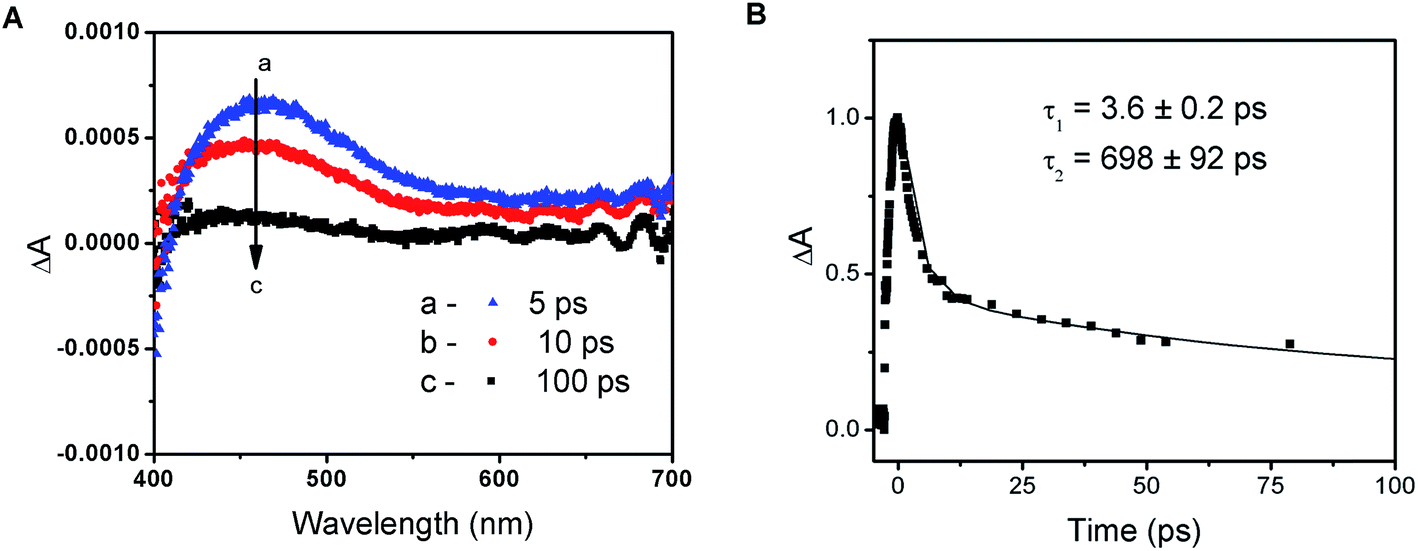 | ||
| Fig. 2 (A) Time-resolved transient absorption spectra recorded following 387 nm laser pulse excitation of methylammonium lead iodide precursor solution with 250 μM PbI2 and 24 mM MAI in dimethylformamide and (B) absorption-time profile of the induced absorption for the same precursor solution monitored at 500 nm (the difference absorption maximum is normalized to value 1). | ||
As shown previously,23,39 ligand-to-metal charge transfer in the excited state leads to transfer of electrons from I3− to Pb2+. The formation of I2 species formed within the complex is short-lived, as it decays to regenerate the complex. The appearance of this transient following laser pulse excitation indicates that the excited state is photoactive and is capable of undergoing charge transfer within the complex. The charge transfer characteristics of PbIxn− and its ability to undergo photochemical charge separation remains an important feature of the unannealed perovskite films.
Excited state behavior of perovskite films
In designing CH3NH3PbI3 thin films, one usually casts a layer of the yellow-colored precursor solution prepared with PbI2 and CH3NH3I on a conducting electrode surface or mesoscopic metal oxide film. Fig. 3A shows the absorption spectra recorded following the deposition of the precursor solution onto a mesoporous alumina scaffold, along with the spectrum of the precursor solution. The wide band gap alumina support prevents unwanted charge transfer events that could convolute the intrinsic excited state properties of CH3NH3PbI3 (see ESI† for experimental details on perovskite film preparation).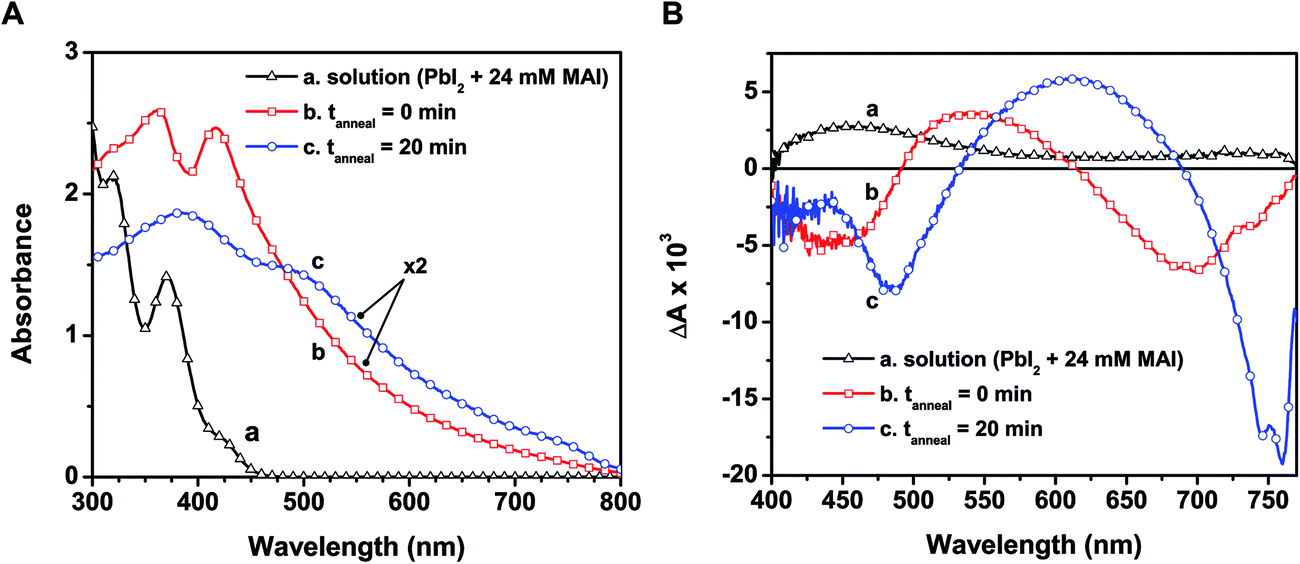 | ||
| Fig. 3 (A) Ground state absorption spectra of (a) the perovskite precursor solution in DMF, (b) as-deposited film from a 1:1 CH3NH3I:PbI2 solution on alumina, and (c) after annealing the deposited film at 100 °C for 20 min. (B) Excited state spectra of the same samples recorded 5 ps after 387 nm femtosecond laser pulse excitation in an evacuated cell. | ||
The absorption spectrum of the as-deposited film before annealing (spectrum b in Fig. 3A) shows absorption maxima at 365 and 420 nm that closely resemble the plumbate complexes seen in solution (spectrum a). Given the anionic nature of the complex, the cation (CH3NH3+) is expected to be associated with the complex in the film as the solvent molecules are removed during the drying process. The absorption of the unannealed film extends into the visible as a result of partial conversion of the plumbate complex to the perovskite structure upon drying. The X-ray diffraction (XRD) pattern of the solid-state film before annealing indeed reveals reflections in good agreement with the tetragonal phase of CH3NH3PbI3 (see Fig. S4 in ESI†), indicating some degree of perovskite crystallite formation within the mesoporous support.2,40 This initial perovskite formation prior to annealing is expected given the stoichiometric ratio of the precursor components (CH3NH3I:PbI2) and the tendency of the precursors to rapidly crystallize when deposited from a relatively weak coordinating solvent such as DMF (compared to the stronger coordination between PbI2 and DMSO).7 It has been reported previously that increasing the CH3NH3I:PbI2 molar ratio requires longer annealing times and/or higher temperatures to drive mass loss and initiate perovskite crystal formation.15,41
Though we observe partial perovskite formation upon thin film deposition, the similarity of spectral features at wavelengths below 500 nm in spectra a and b (Fig. 3A) and the lack of features near the band edge transition (760 nm, spectrum b) indicate that the electronic transitions in both cases are dictated primarily by the PbIxn− complex. Thus the organic cation acts largely as a spectator (viz., charge neutralizer) at this early stage, with minimal influence on the ground state absorption characteristics. Earlier reports have shown other plumbate complexes possess optical properties that are dominated by the hexagonally coordinated lead, and that the various cations balance charges but have little effect on the light absorption properties.38,42
When annealed at 100 °C for 20 min, the color of the film changes from light yellow-brown to dark brown, indicating structural transformations to produce the perovskite morphology. The absorption peaks seen at 365 and 420 nm disappear and broadened peaks now appear at 480 and 760 nm (spectrum c in Fig. 3A). The changes seen in the absorption features following the annealing process reflect full transformation of the [PbIxn− (CH3NH3)+] complex into the CH3NH3PbI3 perovskite. Additionally, the characteristic CH3NH3PbI3 X-ray reflections become more intense with annealing (Fig. S4, ESI†). Although we cannot discern various degrees of order in the XRD analysis reported here, previous X-ray scattering measurements have revealed an interplay between crystalline and amorphous hybrid perovskite phases within mesoporous supports.43 While annealing of perovskite films increases the fraction of crystalline domains,27 further studies are required to pinpoint the specific absorption characteristics of both disordered and ordered CH3NH3PbI3 regions. We can however rule out a transition from amorphous to crystalline perovskite as the sole cause for the change upon annealing, as the unannealed sample exhibited no characteristic band edge peak and closely resembled the absorbance of the precursor solution.
Several studies employing XRD, grazing incidence wide-angle X-ray scattering, and scanning electron microscopy have probed the structural transformation from a lead halide complex to the final perovskite phase.1,44,45 In particular, Tan, et al. identified a crystalline precursor phase following deposition of a mixed halide precursor solution containing CH3NH3I and PbCl2 that appears prior to perovskite crystal formation.44 Manifestation of this unknown crystalline precursor was shown to be a function of processing conditions such as annealing atmosphere and time/temperature. It was posited that this precursor compound influences the final perovskite thin film morphology and ultimately the device performance. These recent results suggest that characterizing the individual components and precursors that precede hybrid lead halide perovskite formation can enable improved control over material and optoelectronic properties.
To investigate the excited state properties of the iodoplumbate complex in the solid state, the as-deposited and annealed films were transferred into a spectroscopic cell, evacuated, and subjected to laser pulse excitation. The transient spectra recorded following 387 nm laser pulse excitation are shown in Fig. 3B. The excited state spectrum of the as-deposited film displays bleaching at 440 and 680 nm, whereas the annealed film exhibits bleaching at 480 and 760 nm. The bleaching centered at 680 nm of the as-deposited film arises most likely as a result of formation of a small amount of CH3NH3PbI3 upon drying (vide supra). Both samples exhibit a broad photoinduced absorption. Shown also in Fig. 3B is the excited state spectrum of the precursor solution, which has a similar photoinduced absorption in the visible and a probable bleaching below 400 nm. The red-shifted spectral features in the as-deposited film compared to the precursor solution are expected considering higher order coordination between Pb2+ and I− dominates in the solid-state films. Qualitative comparison of these spectral signals suggests that a charge transfer complex nature persists in the excited state of the as-deposited and annealed films.
Time-resolved transient absorption spectra recorded following 387 nm laser pulse excitation of the annealed film are shown in Fig. 4A. Two bleaching bands prominently seen at 480 and 760 nm were attributed to the transitions seen in the ground state absorption spectrum. A previous study attributed the origin of these two bands to transitions between two valence bands (VB1 & VB2) and a common conduction band minimum (CB1).25 As discussed in our earlier study, the lower energy transition (760 nm bleach) arises from intrinsic charge separation corresponding to the band edge transition (reaction (4)).14
| CH3NH3PbI3 + hν → CH3NH3PbI3 [h + e] → CH3NH3PbI3 | (4) |
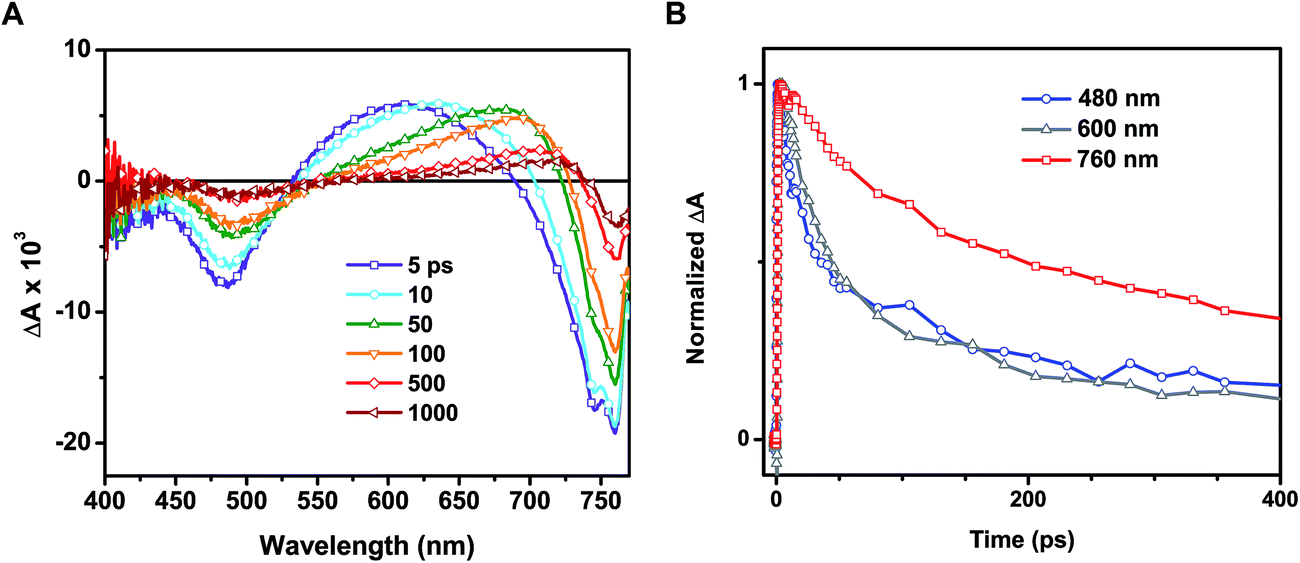 | ||
| Fig. 4 (A) Transient absorption spectra recorded following 387 nm excitation of annealed CH3NH3PbI3 on a mesoporous alumina support at various pump-probe delay times. (B) Difference absorption-time profiles recorded at 480 (blue trace), 600 (grey trace), and 760 nm (red trace). | ||
Based on the observed second order recovery of the 760 nm bleach, we suggested that charge recombination proceeds principally through nongeminate bimolecular recombination across a broad range of excitation intensities.14 The band filling model established in this study further corroborated this conclusion and revealed the limited contribution from excitonic states in CH3NH3PbI3 following room temperature laser pulse excitation. The relatively low exciton binding energy (30–50 meV) estimated for CH3NH3PbI3 facilitates the direct formation of a large fraction of photogenerated free charge carriers in CH3NH3PbI3.12,46 Furthermore, theoretical investigations into possible dielectric screening by the organic cation suggest the binding energy may be well below the 26 meV of thermal energy at room temperature.47
One of the distinguishable features of the two bleaching bands seen in the transient spectra (Fig. 4A) is the narrowing of the 760 nm bleaching band with time. The spectral narrowing, which is also reflected as a red-shift in the zero absorbance cross over point with time, can be well described by the dynamic Burstein–Moss or band filling model.14 As a result of this shift, we fail to observe a clean isosbestic point between the bleaching band in the 680–740 nm region and induced absorption. This spectral behavior in the 680–800 nm region fails to correlate with the other parts of the transient absorption spectrum, indicating the presence of additional transient species.
In addition to examination of the excited state spectral features, we also observed significant discrepancies in the excited state recovery kinetics at 480, 600, and 760 nm. Interestingly, the induced absorption seen in the 550–680 nm region and the bleaching at shorter wavelengths (<500 nm) in the annealed films are closely associated. We base this argument on two facts: (1) the presence of a 530 nm isosbestic point between the two bands and (2) similar decay lifetimes at 480 and 600 nm. A set of transient decay profiles are compared in Fig. 4B. Fitting the kinetics to a biexponential decay reveals that the recovery at 480 and induced absorption at 600 nm match well with an average lifetime of 97 ± 7 and 105 ± 6 ps, respectively (see ESI† for details on data fitting). These average lifetimes deviate markedly from that seen at 760 nm (313 ± 37 ps). It should be noted that these lifetimes are shorter than those determined in other studies utilizing time-resolved spectroscopic techniques due to the relatively high excitation fluence employed here (∼19 μJ cm−2).24,25 As we have demonstrated previously, the intrinsic bimolecular recombination kinetics are a strong function of excitation intensity in CH3NH3PbI3.14
Based on the complete analysis of the spectral data for the precursor solution and solid-state samples, we propose the existence of a dual excited state composed of a charge transfer band (480 nm) and charge separated band gap state (760 nm) in CH3NH3PbI3 (Fig. 5). We assign the spectral features below 680 nm (induced absorption and bleach) in the fully formed CH3NH3PbI3 perovskite to those of the excited complex that undergoes charge transfer to produce an I2-like species. Furthermore, the charge transfer excited state can be generated with lower energy (590 nm) excitation as well (Fig. S5†), indicating that the absorption is broad. However, the relative intensities of the 760 and 480 nm bleaches differ when using 590 nm laser excitation from that of 387 nm excitation, which excites with energy above the absorption maximum for both transitions. Following photoexcitation, this species recombines to regenerate the ground state according to reactions (4) and (5).
| CH3NH3PbI3 + hν → [CH3NH3PbI⋯I2] | (5) |
| [CH3NH3PbI⋯I2] → CH3NH3PbI3 | (6) |
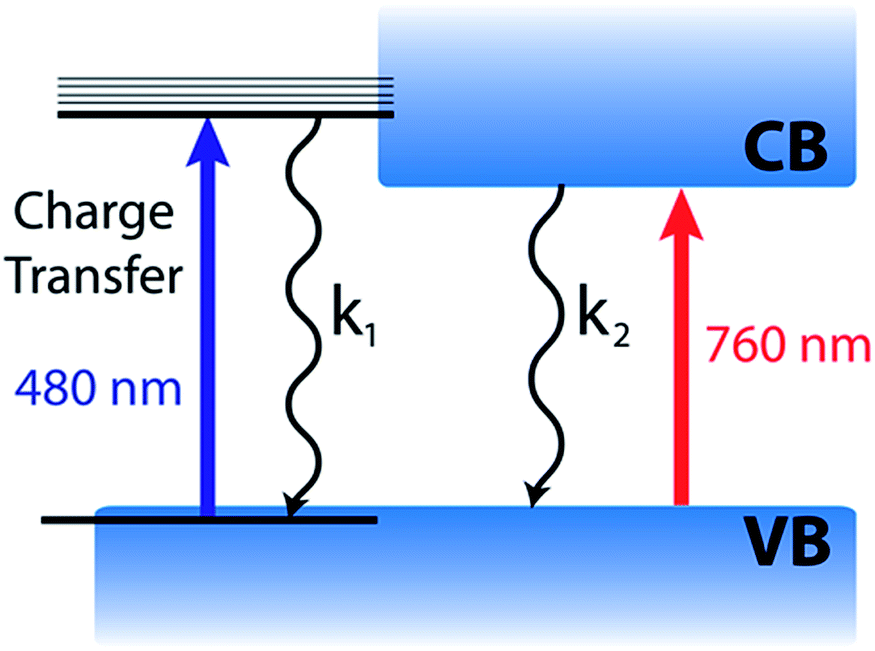 | ||
| Fig. 5 Scheme illustrating the dual nature of the organic–inorganic perovskite excited state. | ||
The similarity of the kinetic traces recorded at the 480 nm bleach and 600 nm photoinduced absorption (Fig. 4B) represent the regeneration of the charge transfer ground state via reaction (6). The generation of an I2-like species in the excited state may have interesting effects on the long term stability and photovoltaic performance of CH3NH3PbI3-based devices. In the present study, we observe full recovery of this excited state and no indication of photodegradation of the films with repeated experiments.
The dual valence band model considered previously was derived to account for behavior of the two photoinduced bleaches at 480 and 760 nm in CH3NH3PbI3.25 However, this model cannot account for the discrepancy in recovery kinetics at the two spectral positions. Given a common conduction band minimum, one would expect the transient kinetics to be homogeneous across the visible spectrum, assuming the difference absorbance signal stems principally from state filling. The kinetics of the bleaching recovery profiles below 500 nm and identification of the transient species responsible for the induced absorption need to be reconciled in order to obtain a complete understanding of the excited state.
It is evident from the present investigation that the annealed CH3NH3PbI3 perovskite film retains the charge transfer complex character that is inherited from the parent lead halide complex. Despite the larger extinction coefficient of the transition at 480 nm (as compared to the 760 nm transition), the magnitude of the observed bleaching is relatively small. This suggests that the contribution of the charge transfer complex to the overall excited state is relatively small. The model depicted in Fig. 5 also helps in understanding why the photoinduced bleaches at 480 and 760 nm have different recovery dynamics, since the charge transfer (480 nm) and charge separated (760 nm) excited states can decay at different rates.
Such a dual excited state with diverse characteristics could serve as a model for explaining the unusual semiconductor properties of organic–inorganic perovskites. The 760 nm charge separated state is well understood based on the semiconductor band gap model.14 The 480 nm charge transfer excited state, however, does not display the same simple free carrier kinetics previously reported for the 760 nm bleach. Although kinetically we observe no evidence for carrier cooling from the 480 nm charge transfer state to the band edge, these two states remain in equilibrium. Furthermore, these two states are active in solar cell operation as evidenced by the photocurrent action spectrum (quantum efficiency at various excitation wavelengths), with external quantum efficiency of approximately 80% across the visible spectrum.48
Gottesman et al. have provided insight into the charge transfer nature (I → Pb) of the interband transitions in CH3NH3PbI3 and the important role it plays in the low binding energy for electrons.39 Both of the lowest lying unoccupied bands in CH3NH3PbI3 are composed of Pb (6p) orbitals as the accepting states in what are referred to as the 480 nm charge transfer and 760 nm charge separated transitions here.39,46 The low binding energy is an important factor in realizing highly efficient perovskite photovoltaics.11,12
The charge transfer nature of the interband transitions makes it possible for methylammonium ions to align in the electric field in working photovoltaic devices and contribute to the high open circuit voltages and photocurrents achieved.39 Moreover, weak localization of charges within the hybrid perovskite lattice has been proposed to explain the surprisingly low recombination rates in perovskite thin films.15 The charge transfer nature described here corroborates these postulations and spatial separation of charges within organic–inorganic perovskites could help explain the significant deviation from Langevin recombination kinetics observed previously.
To summarize, the spectroscopic investigation of methylammonium lead halide perovskite films reveals two different excited state behaviors, viz., charge separated state (reaction (4)) and an excited charge transfer state (reaction (5)). These two states remain coupled to form a dual excited state. Both these states undergo charge recombination to regenerate the ground state. The unusual dual nature of the excited state presented in this work provides further insight into the initial photoinduced processes leading to charge separation in organic–inorganic metal halide perovskite films.
Acknowledgements
We thank the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy for their support through award DE-FC02-04ER15533. We also thank the Center for Sustainable Energy at Notre Dame (cSEND) for partial funding of the project. This work was completed with the use of the cSEND Material Characterization Facility's Bruker D8 Advance Davinci powder X-ray diffractometer. We thank Barry Reid for his assistance in recording XRD spectra. This is contribution number NDRL no. 5036 from the Notre Dame Radiation Laboratory.References
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- J.-W. Lee, D.-J. Seol, A.-N. Cho and N.-G. Park, Adv. Mater., 2014, 26, 4991–4998 CrossRef CAS PubMed.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H.-S. Duan, H.-H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2013, 136, 622–625 CrossRef PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- Y. Wu, A. Islam, X. Yang, C. Qin, J. Liu, K. Zhang, W. Peng and L. Han, Energy Environ. Sci., 2014, 7, 2934–2938 CAS.
- L. Etgar, P. Gao, Z. Xue, Q. Peng, A. K. Chandiran, B. Liu, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2012, 134, 17396–17399 CrossRef CAS PubMed.
- G. Giorgi, J.-I. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. Lett., 2013, 4, 4213–4216 CrossRef CAS.
- W. A. Laban and L. Etgar, Energy Environ. Sci., 2013, 6, 3249–3253 CAS.
- N.-G. Park, J. Phys. Chem. Lett., 2013, 4, 2423–2429 CrossRef CAS.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- F. Deschler, M. Price, S. Pathak, L. E. Klintberg, D.-D. Jarausch, R. Higler, S. Hüttner, T. Leijtens, S. D. Stranks, H. J. Snaith, M. Atatüre, R. T. Phillips and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 1421–1426 CrossRef CAS.
- J. S. Manser and P. V. Kamat, Nat. Photonics, 2014, 8, 737–743 CrossRef CAS.
- C. Wehrenfennig, G. E. Eperon, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Mater., 2014, 26, 1584–1589 CrossRef CAS.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- K. Liang, D. Mitzi and M. T. Prikas, Chem. Mater., 1998, 10, 403–411 CrossRef CAS.
- P. Pistor, J. Borchert, W. Fränzel, R. Csuk and R. Scheer, J. Phys. Chem. Lett., 2014, 5, 3308–3312 CrossRef CAS.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739–1743 CAS.
- V. M. Burlakov, G. E. Eperon, H. J. Snaith, S. J. Chapman and A. Goriely, Appl. Phys. Lett., 2014, 104, 091602 CrossRef PubMed.
- J. A. Christians, R. C. M. Fung and P. V. Kamat, J. Am. Chem. Soc., 2013, 136, 758–764 CrossRef PubMed.
- G. E. Eperon, V. M. Burlakov, P. Docampo, A. Goriely and H. J. Snaith, Adv. Funct. Mater., 2014, 24, 151–157 CrossRef CAS.
- O. Horváth and I. Mikó, J. Photochem. Photobiol., A, 1998, 114, 95–101 CrossRef.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. Lim, Y. Lam, M. Gratzel, S. Mhaisalkar and T. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- H. Kim, C. Lee, J. Im, K. Lee, T. Moehl, A. Marchioro, S. Moon, R. Humphry-Baker, J. Yum, J. Moser, M. Grätzel and N. Park, Sci. Rep., 2012, 2, 7 Search PubMed.
- B. Park, B. Philippe, T. Gustafsson, K. Sveinbjörnsson, A. Hagfeldt, E. M. J. Johansson and G. Boschloo, Chem. Mater., 2014, 26, 4466–4471 CrossRef CAS.
- T. J. Savenije, C. S. Ponseca, L. Kunneman, M. Abdellah, K. Zheng, Y. Tian, Q. Zhu, S. E. Canton, I. G. Scheblykin, T. Pullerits, A. Yartsev and V. Sundström, J. Phys. Chem. Lett., 2014, 5, 2189–2194 CrossRef CAS.
- A. Marchioro, J. Teuscher, D. Friedrich, M. Kunst, R. van de Krol, T. Moehl, M. Gratzel and J.-E. Moser, Nat. Photonics, 2014, 8, 250–255 CrossRef CAS.
- C. S. Ponseca, T. J. Savenije, M. Abdellah, K. Zheng, A. Yartsev, T. Pascher, T. Harlang, P. Chabera, T. Pullerits, A. Stepanov, J.-P. Wolf and V. Sundström, J. Am. Chem. Soc., 2014, 136, 5189–5192 CrossRef CAS PubMed.
- L. Wang, C. McCleese, A. Kovalsky, Y. Zhao and C. Burda, J. Am. Chem. Soc., 2014, 136, 12205–12208 CrossRef CAS PubMed.
- I. Dag and E. Lifshitz, J. Phys. Chem., 1996, 100, 8962–8972 CrossRef CAS.
- O. I. Micic, Z. Li, G. Mills, J. C. Sullivan and D. Meisel, J. Phys. Chem., 1987, 91, 6221–6229 CrossRef CAS.
- Y. Nosaka, T. Fukuyama, M. Horiuchi and N. Fujii, Isr. J. Chem., 1993, 33, 71–75 CrossRef CAS.
- B. H. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2708 CrossRef.
- D. I. Kreller and P. V. Kamat, J. Phys. Chem., 1991, 95, 4406–4410 CrossRef CAS.
- A. Wojcik and P. V. Kamat, ACS Nano, 2010, 4, 6697–6706 CrossRef CAS PubMed.
- H. Li, Z. Chen, L. Cheng, Y. Wang, M. Feng and M. Wang, Dalton Trans., 2010, 11000 RSC.
- R. Gottesman, E. Haltzi, L. Gouda, S. Tirosh, Y. Bouhadana and A. Zaban, J. Phys. Chem. Lett., 2014, 5, 2662–2669 CrossRef CAS.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 CAS.
- A. Dualeh, P. Gao, S. I. Seok, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2014 DOI:10.1021/cm502468k.
- J. P. Li, L. H. Li, L. M. Wu and L. Chen, Inorg. Chem., 2009, 48, 1260–1262 CrossRef CAS PubMed.
- J. J. Choi, X. Yang, Z. M. Norman, S. J. L. Billinge and J. S. Owen, Nano Lett., 2013, 14, 127–133 CrossRef PubMed.
- K. W. Tan, D. T. Moore, M. Saliba, H. Sai, L. A. Estroff, T. Hanrath, H. J. Snaith and U. Wiesner, ACS Nano, 2014, 8, 4730–4739 CrossRef CAS PubMed.
- M. Saliba, K. W. Tan, H. Sai, D. T. Moore, T. Scott, W. Zhang, L. A. Estroff, U. Wiesner and H. J. Snaith, J. Phys. Chem. C, 2014, 118, 17171–17177 CAS.
- X. Zhu, H. Su, R. A. Marcus and M. E. Michel-Beyerle, J. Phys. Chem. Lett., 2014, 5, 3061–3065 CrossRef CAS.
- J. Even, L. Pedesseau and C. Katan, J. Phys. Chem. C, 2014, 118, 11566–11572 CAS.
- J.-H. Im, I.-H. Jang, N. Pellet, M. Grätzel and N.-G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee02988g |
| This journal is © The Royal Society of Chemistry 2015 |