Zeolite and mesoporous silica nanomaterials: greener syntheses, environmental applications and biological toxicity
Sean E.
Lehman
and
Sarah C.
Larsen
*
Department of Chemistry, University of Iowa, Iowa City, IA 52242, USA. E-mail: sarah-larsen@uiowa.edu
First published on 17th April 2014
Abstract
Zeolites and mesoporous silica nanoparticles are silicate or aluminosilicate nanomaterials with well-defined pore networks. Zeolites are widely used in industry for applications such as catalysis, separations and gas adsorption. Mesoporous silica nanomaterials have not been as extensively applied relative to zeolites due to the cost and reduced thermal stability, but are being intensively investigated for potential environmental and biomedical applications. In this article, zeolite and mesoporous silica nanomaterials are reviewed with emphasis on connections to the environment. Specifically, the topics of greener syntheses, environmental applications and biological toxicity will be addressed. Both of these materials are typically synthesized with a template, which is then removed to produce the pore volume. Synthetic strategies for the “greening” of the syntheses of zeolites and mesoporous silica will be discussed. Environmental applications including the adsorption of environmental contaminants and environmental catalysis will also be presented. Finally, the toxicity of zeolite and mesoporous silica nanomaterials will be considered.
Nano impactZeolites and mesoporous silica nanoparticles are porous nanomaterials that have two dimensions on the nanoscale, the pore size and the particle or crystal size. In this article, the environmental implications and applications relevant to these porous nanomaterials are reviewed. Critical issues with respect to greener synthetic strategies designed to mitigate the environmental impacts of the synthesis will be presented. Future and developing environmental applications for zeolite and mesoporous silica nanomaterials including the adsorption of environmental contaminants and environmental catalysis will be discussed. As zeolites and mesoporous silica nanomaterials grow in their applications, increased exposures will result, leading to heightened concerns about the toxicity. The current status of the biological toxicity in the context of recent studies will be reviewed. |
1. Introduction
In this article, zeolite and mesoporous silica nanomaterials will be reviewed with respect to greener synthesis strategies, environmental applications and general toxicity considerations. Zeolites are crystalline aluminosilicates (or silicates) with regular arrangements of micropores, high surface areas, and exchangeable cations.1 Ordered mesoporous silica materials are amorphous silica materials with a regular arrangement of mesopores that results in very high surface areas. Zeolites and mesoporous silica are both readily modifiable using well-known silanol organofunctionalization methods. Representative zeolite and mesoporous silica structures are shown in Fig. 1.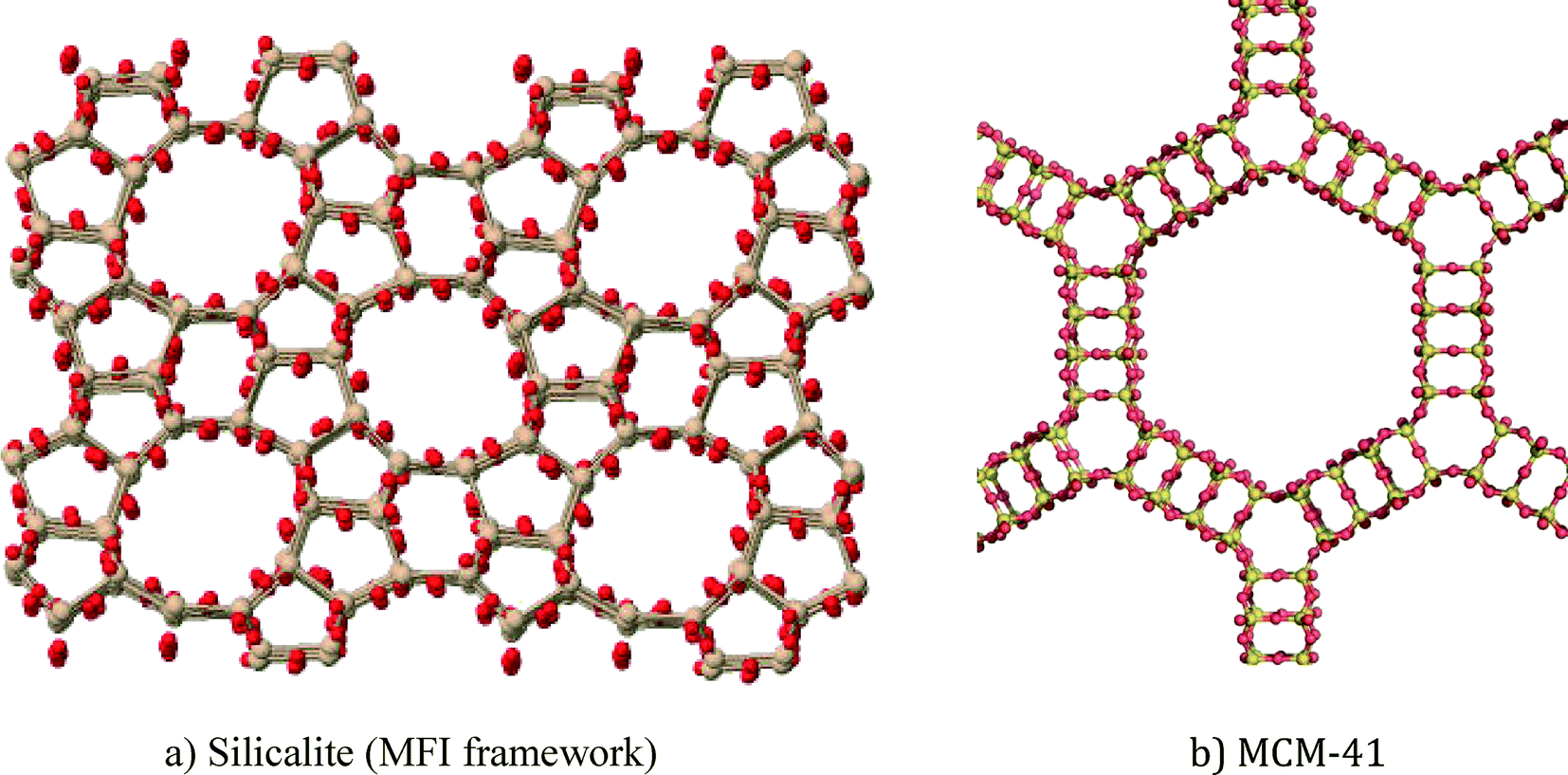 | ||
| Fig. 1 Representative structures of porous nanomaterials: a) ZSM-5 zeolite and b) MCM-41 mesoporous silica. | ||
In recent years, there has been a great deal of interest in zeolites and mesoporous silica nanomaterials in which the particle or crystal size of these porous materials is less than approximately 100 nm.2 Thus, zeolites and mesoporous silica nanomaterials have two dimensions on the nanoscale – the particle size and the pore size. There have been several review articles focused on the synthesis and applications of nanoscale zeolites3,4 and mesoporous silica nanoparticles.2,5,6
Recently there has been growing interest in the green synthesis of porous nanomaterials such as zeolites and mesoporous silica. The principles of green chemistry were introduced in the early 1990's and articulate the objective of reducing (and/or eliminating) negative environmental impacts by decreasing waste or increasing efficiency of chemical processes or syntheses.7,8 For example, the porosity in zeolites and mesoporous silica nanomaterials is typically produced using a structure-directing organic template or surfactant that is subsequently removed via calcination or solvent extraction leading to potential negative environmental impacts, such as high energy use and the release of toxic by-products. Greener synthetic routes to zeolites include recycling methods, low-cost or degradable templates, template-free synthesis methods, microwave synthesis, and solvent-free or ionic liquid-mediated syntheses.9–11
The environmental applications of zeolites and mesoporous silica nanomaterials include environmental catalysis,12–14 renewable energy applications, such as biomass conversion15,16 and adsorption of environmental contaminants.17 One of the concerns with the use of zeolite or mesoporous silica adsorbents is the increased cost of these materials compared to more common adsorbents, such as activated carbon. A major factor in the cost is the use of a template, which is ultimately removed to afford the void space. Therefore, the development and implementation of greener synthetic methods for preparing these materials may help address a critical issue in the commercialization of zeolite and mesoporous silica for adsorption applications.
Interest in the toxicity of zeolite and mesoporous silica nanomaterials stems from the concern that as zeolite and mesoporous silica nanomaterials are more widely used in industry and in commercial products, environmental and human exposures will increase dramatically. Crystalline silica dusts have been extensively studied and it is well-established that prolonged exposure leads to silicosis, lung cancer and autoimmune diseases in humans.18,19 Recently, there have been a number of studies of the biological toxicity of zeolite and mesoporous silica nanomaterials and the key results will be presented.
In this review article, three major areas with respect to zeolite and mesoporous silica nanomaterials and the environment will be reviewed. The first area is the synthesis of zeolite and mesoporous silica nanomaterials using greener synthetic routes. The second area is the largely unrealized commercial potential of these porous nanomaterials for environmental applications and the third area is biological toxicity.
2. Overview of greener synthesis of zeolite and mesoporous silica nanomaterials
The synthesis of zeolites and mesoporous silica nanomaterials are distinctly different but do share some common characteristics. Zeolites are commonly synthesized under hydrothermal conditions in an autoclave using an organic template or structure directing agent, commonly a tetraalkylammonium cation, such as tetrapropylammonium cation (TPA+) (Fig. 2A). The zeolite structure forms around the organic template, which is subsequently removed by a high temperature calcination process that ultimately leads to the desired porosity. The key to producing nanoscale zeolites is careful control of the zeolite nucleation process.2,3 Many methods for the synthesis of mesoporous silica are based on the Stöber method which was introduced in the 1960's for preparation of colloidal solutions of nonporous silica.20 The Stöber method was modified by the incorporation of a surfactant into the synthesis which lead to the formation of ordered pores. The size of the pores and the pore structure were varied by judicious choice of surfactant and/or modifying the concentration of surfactant, respectively. The particle size was also varied by adjusting reactant concentrations.2,21 Similar to zeolites, the porosity is a result of removal of the surfactant either by extraction or high temperature calcination (Fig. 2B).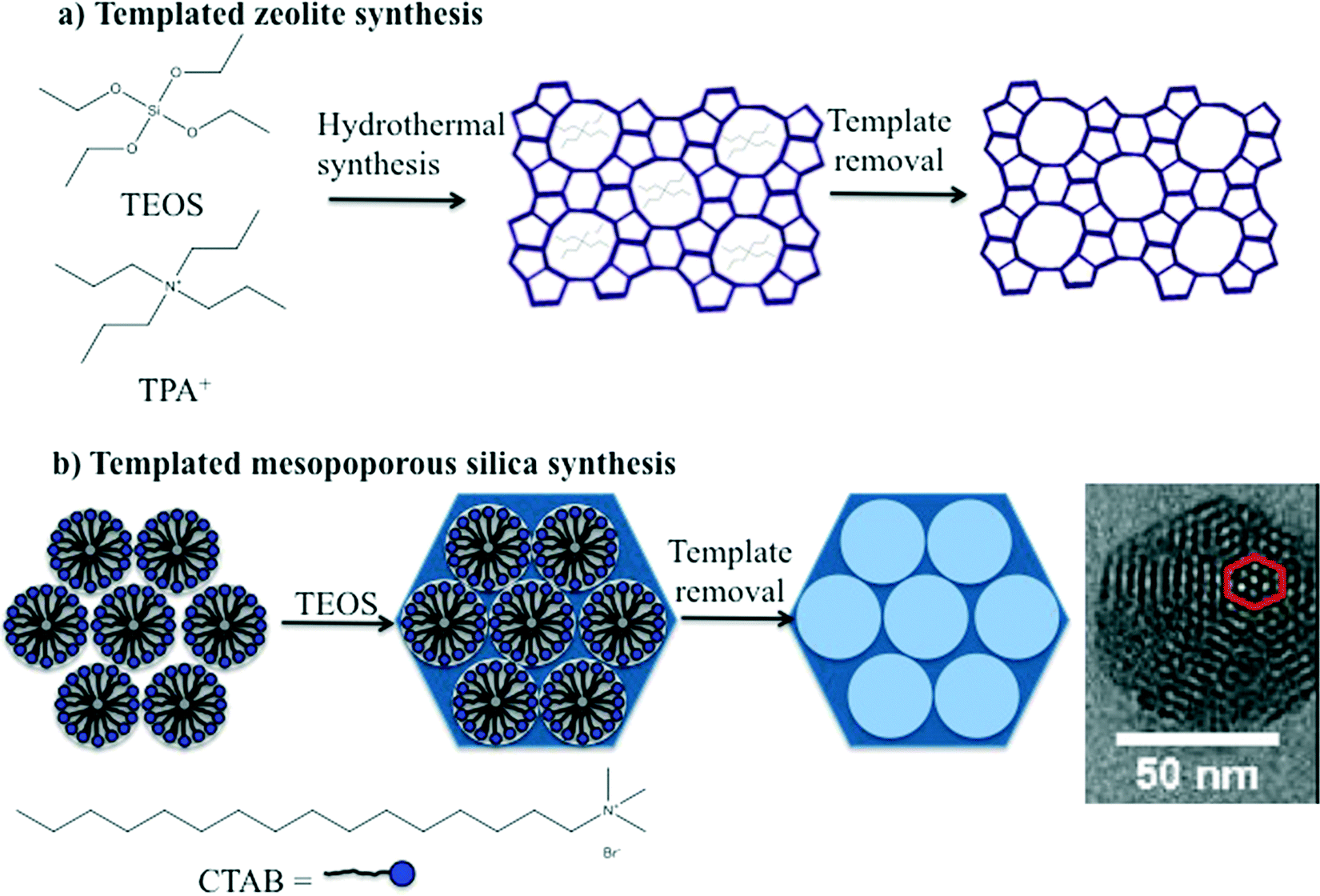 | ||
| Fig. 2 Schematic illustration of the templated synthesis of a) a representative zeolite, ZSM-5 and b) a representative form of mesoporous silica, such as MCM-41. | ||
The syntheses of zeolites and mesoporous silica are inherently challenging from a green chemistry perspective because of the reliance on organic templates or surfactants that are ultimately removed by calcination at high temperature to combust the template, or extraction using an organic solvent. The financial and energy costs associated with template removal are high as well as the environmental cost associated with the formation of greenhouse gases during calcination of the organic template and the toxicity of the amphiphilic surfactants, such as cetyltrimethylammonium bromide (CTAB).9,22,23 Synthetic strategies (Fig. 3) will be described here that have been developed to improve the zeolite and mesoporous silica syntheses with respect to green chemistry criteria. The green strategies can be organized into three main categories: solvent, template, and heating. One obvious focus is on synthetic methods that do not require a template or use a recyclable, inexpensive or renewable template. Other strategies for greener syntheses of zeolites and mesoporous silica nanomaterials involve replacing the solvent, developing solvent-free methods and using microwave heating.
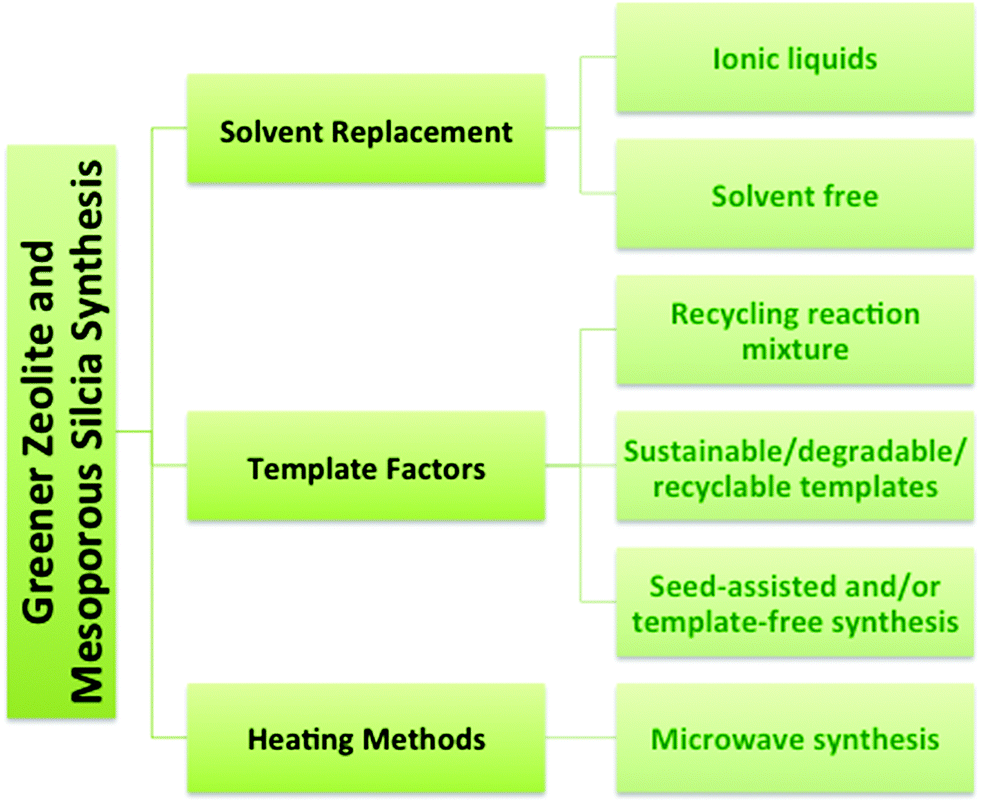 | ||
| Fig. 3 Strategies for the greener synthesis of zeolites and mesoporous silica. | ||
2.1 Template recycling in zeolite nanocrystal synthesis
From the green chemistry perspective, the synthesis of zeolites is deficient in several areas.9 First, as mentioned above, the use of an organic template is problematic because it must be removed to afford the void pore space. Removal is typically accomplished by high temperature calcination, which adds to the energy costs, decomposes the template, and results in the formation of air pollutants, such as NOx and CO2. Second, the syntheses are usually conducted under hydrothermal conditions of elevated temperature and pressure. Third, the syntheses often require long times which decreases the overall efficiency and increases the energy costs of the process.Several strategies have been implemented to make the zeolite synthesis greener and more sustainable. These strategies include: 1) the replacement of the organic template with a recyclable template, removal of the template altogether from the synthesis or recycling of unused template in low yield syntheses; 2) replacing the solvent with ionic liquids resulting in lower pressure and temperature synthesis conditions24–26 or solvent-free methods27 and 3) using microwave heating to save energy and time in the syntheses.28
A common strategy used to synthesize nanoscale zeolites is to increase nucleation and slow down the crystal growth. Then, the synthesis is quenched before the zeolite crystals grow to large sizes. The challenge of this approach is that the product yield is very low with large amounts of template and reactants remaining in the solution after the nanocrystalline zeolites are removed by centrifugation. In order to avoid wasting the unreacted template and precursors, a recycling strategy can be implemented in which the zeolite nanocrystals are removed from the synthesis solution by centrifugation and then the synthesis solution is reheated to produce more zeolite nanocrystals.29–31 The recycling strategy is depicted schematically in Fig. 4.
 | ||
| Fig. 4 Recycling process to increase yield of nanoscale zeolite synthesis. | ||
The recycling method has been applied to the synthesis of nanoscale zeolites such as silicalite (purely siliceous form of ZSM-5), NaY and recently, Ge-substituted NaY.29,32 The synthetic method involves an organic template, such as tetrapropylammonium hydroxide (TPAOH) or tetramethylammonium hydroxide (TMAOH) for silicalite and zeolite Y, respectively. Significantly higher product yields (up to ten-fold) were achieved for nanocrystalline silicalite-1 and Y zeolites by recycling the clear synthesis solutions after periodically removing zeolite nanocrystals and reheating the supernatant.29 The crystal sizes and Si/Al ratio of zeolites synthesized using the recycling method show very good batch-to-batch consistency. By application of the recycling method, high quality, uniform, nanocrystalline zeolites, such as silicalite-1 and NaY, can be synthesized rapidly and more efficiently. The recycling process is environmentally beneficial because the amount of waste produced is minimized by the reuse of the organic template and reactants. Recently, it was shown that the recycling synthesis method could also be applied to the synthesis of nanoscale zeolites with heteroatom substitution of germanium into the framework.32
Another approach that has been developed by Lee and coworkers involves a cleavable organic template that can be used as a structure-directing agent in the synthesis of ZSM-5.33 After the ZSM-5 structure forms, the template is cleaved so that the two organic molecules are small enough to be removed from the zeolite.33 After removal of the fragments from the ZSM-5, they can then be reassembled and reused as structure-directing agents.
2.2 Organic template-free synthesis of zeolite nanocrystals
Prior results in the literature have shown that template-free syntheses and seed-assisted template-free synthesis of ZSM-534–36 and βeta35,37,38 can be effectively used to prepare crystalline materials. The first example of the seed-assisted template-free synthesis was reported by Xie and coworkers who demonstrated that this approach could be used in the synthesis of beta zeolites.37 EMT-type zeolite, which is synthesized with a costly crown ether template, was recently prepared on the nanoscale without a template.39 The key components to the synthesis were the low temperature crystallization and careful variation of the precursor chemical composition and crystallization time. This template-free strategy has been extended to other nanosized zeolites, such as faujasite and sodalite zeolites. The template-free synthesis of nanoscale zeolites typically leads to significantly higher yields relative to conventional synthesis using organic templates.36 To summarize, the advantages of organic template-free methods are to decrease the environmental impact and cost of the zeolite synthesis process while increasing the yield.Seed-assisted template-free synthesis is also used to prepare zeolite nanocrystals. The use of seed crystals allows better control of the crystal size and phase and can also lead to increased crystallization rates. Majano and coworkers studied the seed-induced crystallization of ZSM-5 and found that the seed content and the crystallization temperature were critical to the formation of ZSM-5 crystals.35 They also determined that the ZSM-5 product was highly aggregated most likely due to the aggregation of the seeds used. Prior work in the literature has shown that template-free methods are effective for preparing high alumina content zeolite βeta.35,37
2.3 Other practices for the greening of the synthesis of zeolite nanocrystals
Other methods for improving the synthesis of zeolite nanocrystals in accordance with the principles of green chemistry include changing or eliminating the solvent and microwave heating. The use of green solvents, such as ionic liquids, in the preparation of zeolites is a relatively new area of research and some success has been achieved as discussed below.22,24–26,40 Another route to greener synthesis of zeolites is a solvent-free approach in which the mixture of raw solid materials are heated in an autoclave.27,41 The synthesis of hierarchical mesoporous aluminophosphates with grinding and heating in the absence of solvent has also been reported.42 Finally, the use of microwaves in zeolite synthesis as a rapid, homogeneous method of heating will be discussed.28Due to their desirable properties as solvents relative to traditional organic solvents, ionic liquids have been explored as solvents for the ionothermal synthesis of zeolites.26 Ionic liquids are a relatively new class of organic salts that exist as liquids below ~100 °C. Some common ionic liquids are 1-butyl-3-methylimidazolium chloride (BMIMCl) and 1-allyl-3-methylimidazolium chloride (AMIMCl). There has been a great deal of interest in applications of ionic liquids as green solvents due to their chemical and thermal stability, very low vapor pressure and relative inflammability especially when compared to traditional organic solvents. With respect to zeolite synthesis, the advantages are that the ionothermal synthesis can be performed at ambient pressure and relatively low temperature due to the low vapor pressures of ionic liquids. In an ionothermal synthesis, the ionic liquid, defined for this application as a salt that melts below the zeolite synthesis temperature, acts as both a solvent and a template. Examples in the literature include the use of ionic liquids in the synthesis of aluminophosphates.43,44 Synthesis of siliceous zeolites with ionic liquids requires the use of OH− as the counterion in the ionic liquid, BMIM, in order to promote the dissolution of the silica source.25
In a further greening of the synthesis, Ren and coworkers reported a solvent-free approach to the synthesis of silicalite and aluminosilicate zeolites, such as zeolites Y and βeta.27 The solvent-free route involves mixing and grinding the solid precursors prior to heating in an autoclave. The yield of zeolites in this approach is >90% eliminating the need for recycling of reactants from alkaline solvent mixtures.
Microwaves can be used to heat zeolite reaction mixtures both in traditional solvent systems and in ionic liquids.28,45,46 The advantages are the increased efficiency in heating time, the corresponding decrease in energy required and the uniform crystals. With microwave irradiation, the synthesis time of zeolites is typically reduced by an order of magnitude thus lowering the energy costs associated with long periods of heating in an oven for the typical hydrothermal process in a standard autoclave.9,28,36,45 Since radiative processes heat water very effectively, the transfer of energy occurs from water to the zeolite synthesis gel components at a faster rate than conventional heating via conduction/convection in an oven. Therefore, microwave-assisted zeolite synthesis generally occurs more rapidly and at lower temperatures relative to conventional heating. In addition, the zeolite product is often more uniform in size and shape relative to zeolites synthesized using conventional heating methods.28,36 While microwave-assisted synthesis of zeolites is generally regarded as a promising and green synthetic method, the enhancement of synthesis rate is still not well-understood.28
The syntheses of different zeolites by microwave heating have been reported but there are far fewer examples in the literature of template-free methods utilizing microwaves. Recently, Ng and coworkers demonstrated that nanosized EMT zeolite could be synthesized in the absence of the expensive crown ether template using microwave radiation.39 ZSM-5 membranes have been synthesized in a seed-assisted approach using microwave radiation.47 In general, it was found that the presence of seed crystals increased the crystallization rate due to increased nucleation.9 Future improvements in zeolite synthesis with respect to green chemistry criteria will most certainly involve combining one or more of these greener synthetic strategies, such as the combination of template-free or solvent-free methods with microwave heating. Some of these studies have been reported but more research is needed to develop viable greener zeolite syntheses.
2.4 Green synthetic strategies for synthesis of MSNs
The synthesis of colloidal suspensions of nonporous spherical silica nanoparticles by the Stöber method is well-established and forms the basis for many methods for the synthesis of mesoporous silica nanoparticles.2 The Stöber method involves the hydrolysis and condensation of tetralkylorthosilicates (Si–(OR)4) in the presence of ammonia and alcohol based solvents.20 The particle size of Stöber silica particles is controlled by systematically varying the amount of silicate, ammonia concentration and identity of the alcohol solvent. To prepare mesoporous silica materials, a surfactant, such as CTAB, is added to a typical Stöber synthesis protocol. In recent work, Haynes and coworkers reported the preparation of mesoporous silica nanoparticles ranging in size from approximately 25–200 nm by systematic variation of the ammonia and silica concentrations and the synthesis temperature.21,48 Others have controlled the particle size of mesoporous silica by varying the pH of the reaction mixture and the synthesis time, by adding swelling agents or quenching agents to stop the particle growth.2Mesoporous silica materials are generally regarded as high cost materials due to the use of expensive surfactants that are removed in the final stages of preparation by calcination or extraction. One strategy for a greener synthesis of mesoporous silica nanoparticles involves using surfactants that can be recovered or recycled which improves both the cost of the materials as well as the environmental impact.23 Another strategy is to use surfactants synthesized from renewable raw materials or from biological systems, but the results have been mixed in terms of producing high quality mesoporous materials. Two recent reviews have discussed the ecodesign of mesoporous silica materials where ecodesign is defined as an approach to product design such that the environmental effects of the product during its entire life cycle are included in the design process.22,23
In the first strategy, one example of the recycling of surfactants is provided by Wang and coworkers who prepared mesoporous silica with a worm-like pore structure using citric acid as the template.49 The citric acid was then removed from the mesoporous silica product by washing with water and drying to recover the citric acid which could then be reused. The use of citric acid provides a lower cost, greener synthetic route to the production of worm-like mesoporous silica. A second example in the literature involves the use of the triblock copolymer, F127, which is water soluble. In this case, F127 was used as a template in the synthesis of mesoporous silica and was then extracted from the mesoporous silica using water at 80 °C.49 However, the recycling of the extracted F127 was not reported.
In the second strategy of using surfactants based on renewable resources, there are many examples in the literature and these have been recently reviewed.22,23 In this article, only two examples will be briefly described. Canlas and Pinnavaia demonstrated that the standard alkyl amine surfactants could be replaced by oleyl amine surfactants that are available from plants.50 The resulting mesoporous silica products had worm-like or lamellar pore structures. In a second example, Wei-Dong and coworkers used an amino acid, glycine, as the basis for amphoteric surfactants.51 An industrial grade polyglycoside was utilized as a structure-directing agent to produce mesoporous silica with a worm-like pore structure with pore diameters of ~3.6 nm. The reader should refer to more extensive review articles for additional information on this strategy.22,23
3. Environmental applications of zeolite and mesoporous silica nanomaterials
Zeolite and mesoporous silica nanoparticles have many desirable properties that are useful in applications ranging from catalysis to adsorption to drug delivery. The porosity of these materials results in very high surface areas and shape selectivity in catalytic reactions and separations. The advantages of these porous nanomaterials for applications are the high surface area, accessible adsorption sites and fast mass transport, as well as the high concentration of functional groups on the surface and the generally good thermal stability.52–54 The silanol groups on the surface are easily modifiable and can be used to vary the surface properties, such as the hydrophobicity/hydrophilicity, surface charge and/or biocompatibility. Ion exchange, which is the basis for the water-softening properties of zeolites, can also be used to incorporate transition metals into zeolites for catalysis. The environmental applications of zeolites and mesoporous silica nanomaterials include environmental catalysis12–14 in areas such as emission control and biorenewable energy production via biomass conversion15,16 and adsorption for air and water pollution remediation.173.1 Environmental catalysis
Zeolites are well-known catalysts used in industrial applications such as petroleum refining and chemical production. In recent years, there has been growth in the development of environmental catalysts designed to address environmental issues, such as reduction of automobile emissions, greener methods for production of chemicals and cleaner and more renewable energy sources.55 Zeolite catalysts have been used in environmental applications, such as the production of lead-free octane enhancers in petroleum and as ion-exchangers in powdered laundry detergents to remove calcium and magnesium ions and soften the water.56 Zeolites have the potential to be used as catalysts in many other environmental applications.In general, applications for zeolites as environmental catalysts can be grouped into three major categories:
i. Catalytic emission abatement of nitrogen oxides and volatile organic compounds (VOCs)12,14 produced by high temperature combustion processes.
ii. Environmentally benign synthesis and manufacturing of chemicals using the principles of green chemistry.57,58
iii. Renewable and clean energy production.
In the first area related to the reduction of nitrogen oxides and VOCs, the applications of zeolites in the decomposition and selective catalytic reduction of nitrogen oxides has been an area of intense interest for the past 25 years or so.12,14 Transition metal-exchanged zeolites exhibit promising catalytic activity for emission abatement applications that depends on the identity of the zeolite host and of the exchanged transition metal. Much research has focused on optimizing the transition metal-exchanged zeolites for direct NOx decomposition and/or selective catalytic reduction (SCR) of NOx.55
The environmentally benign synthesis and manufacturing of chemicals is a broad area that will not be discussed in detail here. Zeolites are utilized as catalysts in various chemical processes and, in many cases, are improving the processes with respect to environmental considerations. Zeolite catalysts can be used as solid acids to replace liquid acids such as HF, HCl or H2SO4 in chemical processes and to decrease waste and energy consumption by providing more efficient catalytic routes. Biomass conversion is an area where solid acid zeolite catalysts may contribute to renewable energy production in the future.
The conversion of cellulosic biomass to chemicals is currently an area of intense research and development across the globe.59–63 Lignocellulosic biomass is abundant and consists of cellulose, hemicellulose and lignin. Cellulose consists of glucose units linked by β-glycosidic bonds within a rigid crystalline polymer structure. An important step in the conversion of biomass into useful chemicals or fuels is the hydrolysis of cellulose into glucose and other monosaccharides. Typically, this is accomplished using liquid acids as catalysts but separation of product from the acids is a disadvantage of this process. Alternatively, solid acid catalysts are easily separated from the products and the catalyst can typically be reused.59,61,62,64 One of the challenges is that cellulose is not soluble in traditional solvents and therefore a great deal of solid acid catalyst must be used to maximize the catalyst/reactant contact.
Zeolite catalysts have a great deal of unrealized potential as solid acid catalysts for the conversion of biomass to fuels and chemicals and this represents a critical area of research.15,16 Currently, research into the use of zeolite catalysts in biomass conversion processes has focused on acidic zeolites, such as H-ZSM-5 and H-βeta, which can transform biomass pyrolysis oil into hydrocarbons,65–67 and cellulose into glucose,68–70 and on iron- or tin-substituted zeolite catalysts which are utilized for the production of olefins from ethanol and isomerization of glucose to fructose, respectively.71–74 Under mild hydrothermal conditions, acid zeolites H–Y, H-βeta and H-ZSM-5 were evaluated for cellulose hydrolysis.68 The higher the Si/Al, and therefore the more hydrophobic the zeolite, the higher the conversion and the better the selectivity for glucose. The hydrolysis of cellulose to glucose and hydroxymethylfurfural (HMF) on H-βeta and H-mordenite has also been demonstrated.69 Research is in progress to further develop zeolite catalysts for biomass conversion.
3.2 Adsorption of environmental contaminants
Zeolites and mesoporous silica are both considered excellent adsorbents for environmental contaminants due to their high surface areas and readily modifiable surface properties. Surface modification of zeolite and mesoporous silica nanomaterials enhances their adsorption properties by providing a better surface for ionic (electrostatic) interaction or by providing binding sites for chelation of heavy metals. Two methods are employed in surface functionalization of zeolites and mesoporous silica with an organosilane. In the first method shown in Fig. 5A, called co-condensation, the organosilane, aminopropyltriethoxysilane (APTES) is added with the silicon source (often tetraethylorthosilicate, TEOS) so that functionalization occurs simultaneously with formation of the framework. The second method (shown in Fig. 5B) is postsynthesis grafting in which the calcined porous material is modified by heating an organosilane reactant and the porous material in an aprotic solvent, such as toluene. Due to the larger pore size and surface area, mesoporous silica materials exhibit greater adsorption capacities relative to zeolites. In the rest of this section, the focus will be on the development of mesoporous silica adsorbents.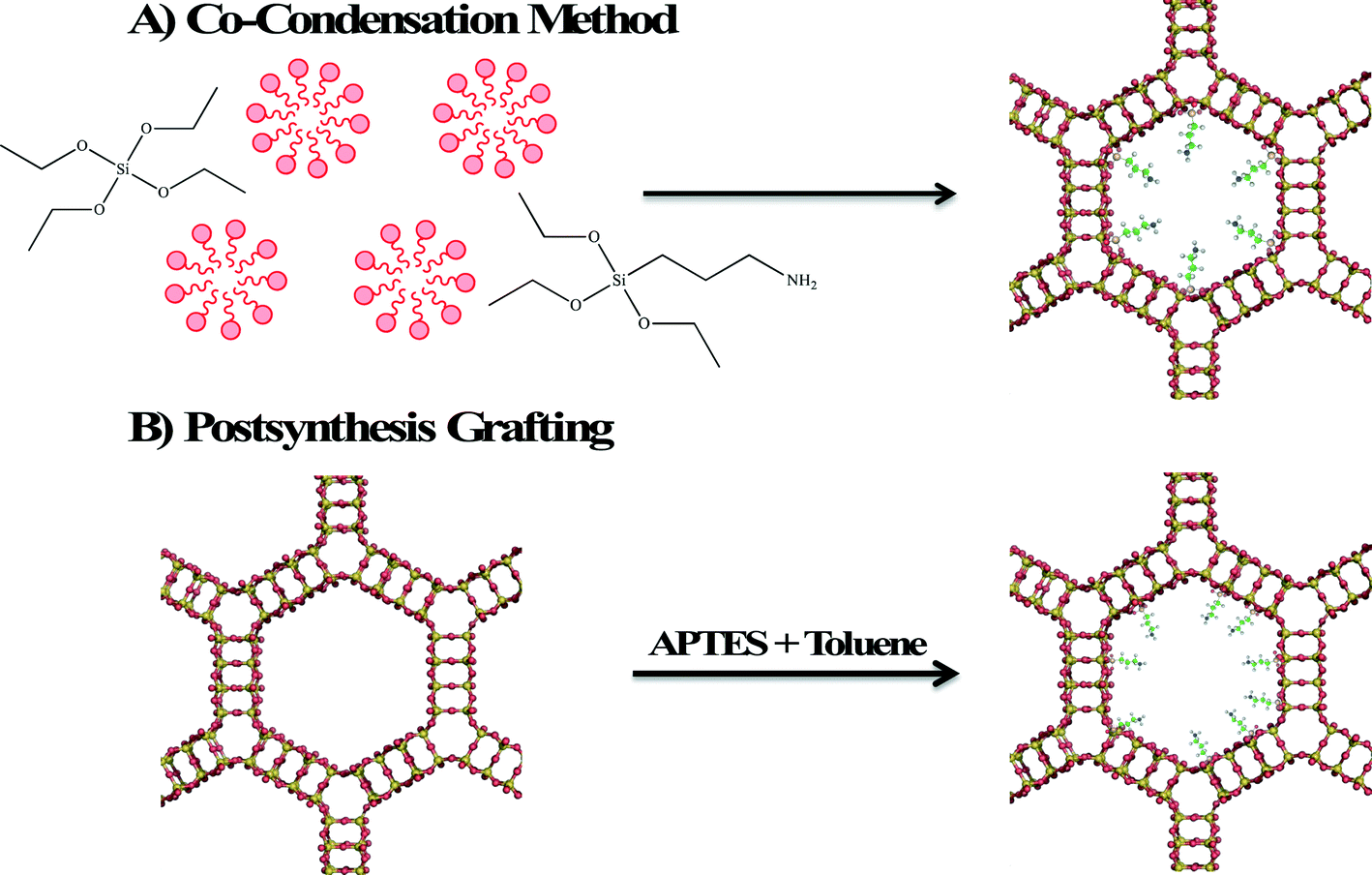 | ||
| Fig. 5 Schematic diagram illustrating the functionalization of MCM-41 by A) co-condensation and B) postsynthesis grafting. | ||
The efficacy of using mesoporous silica materials for removal of a broad range of inorganic or organic pollutants was recently reviewed.54 The literature precedent shows different functional groups, such as amine or thiol groups or more complex groups such as sugars, can be grafted on the surface of microporous or mesoporous materials to promote adsorption processes.75–86 Functionalized MCM-41 has been extensively used in the adsorption of heavy metals such as As, Hg, Cr, Cd, Pb and Fe.75,77,78,83,87–89 Typically an electrostatic or metal binding process drives the adsorption process. Fryxell and coworkers demonstrated the sequestration of radioactive water contaminants, such as U, La, Nd, Eu, and Am, on self-assembled monolayers on mesoporous supports (SAMMS).90
Recently, composite materials, such as magnetic mesoporous silica, have been explored as adsorbents for environmental contaminants.87,91,92 The magnetic mesoporous silica has an iron oxide core typically consisting of ~5–7 nm iron oxide nanoparticles and a mesoporous silica shell as shown in Fig. 6. By combining the magnetic behavior of iron oxide nanoparticles with mesoporous silica materials, the properties of both of these materials will be coupled providing increased adsorption capacity and effective magnetic separation. Several groups have reported the synthesis and characterization of magnetic iron oxide nanoparticles with mesoporous silica shells.87,93–96 In work by Yeung and coworkers, magnetic mesoporous silica was evaluated for the adsorption of Cr(VI) and Cu(II).87,91,94 Similarly, Zhang and coworkers used iron oxide/silica core shell particles to adsorb the heavy metal ions, chromium and arsenic, from aqueous solution.95 Magnetic zeolite materials have been investigated for applications in the adsorption Cr(VI) and Hg.97–99 The advantage of using iron oxide nanoparticles as a core for mesoporous silica is to enable the separation of the magnetic solid adsorbents from polluted water using a permanent magnet.87,88,91,95 Although not discussed here, magnetic mesoporous silica composites are also being widely investigated for use in biomedical applications, such as imaging.100,101
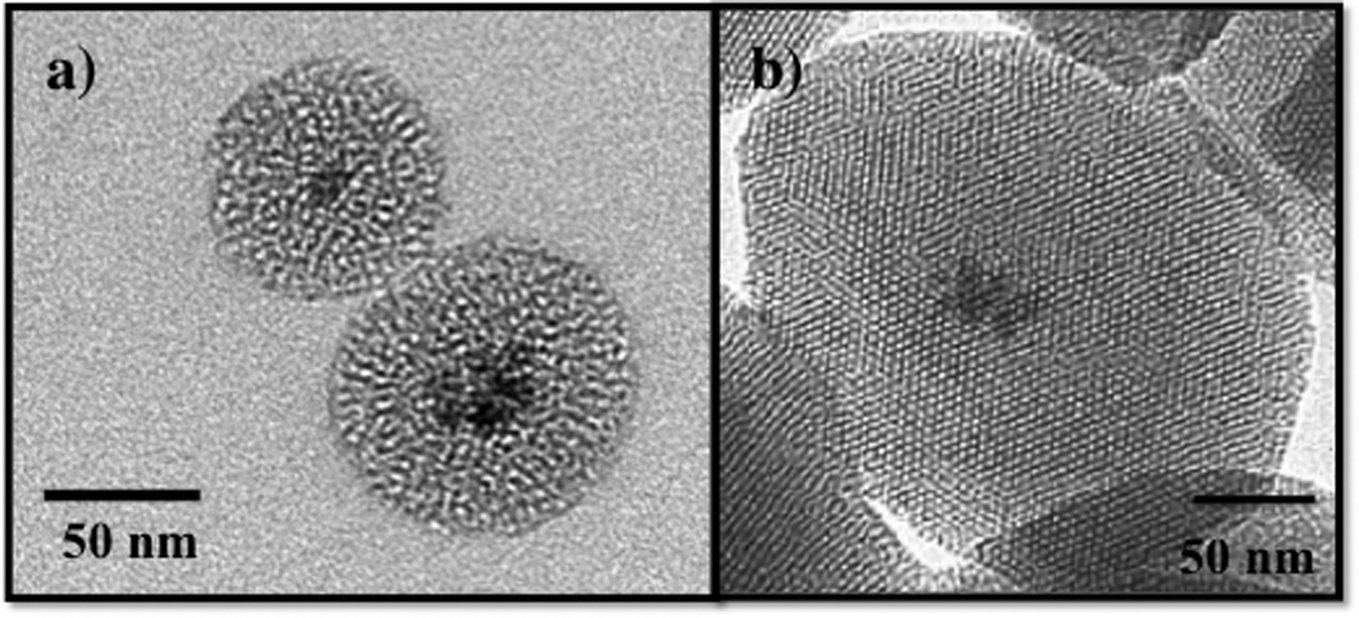 | ||
| Fig. 6 TEM images of a) wormhole mesoporous silica with an iron oxide nanoparticle core and b) MCM-41 with an iron oxide nanoparticle core. | ||
4. Biological toxicity of zeolite and mesoporous silica nanoparticles
Silicon dioxide (SiO2) or silica, the second most commonly found mineral in the Earth's crust, is most commonly found in the form of crystalline quartz, which possesses tetrahedral coordination around each silicon atom. The abundance of naturally occurring silicon dioxide, in combination with the growing diversification and implementation of engineered nanomaterials, has generated a great need for understanding silica toxicity in environmental and biological systems. The intrinsic pulmonary toxicity of a collection of six silicate minerals commonly known as asbestos has been established for quite some time. These materials raised the original concern of toxicological effects of silicas and more recent work has focused on the growing body of nanoscale silicate materials, including the porous forms, zeolites and mesoporous silicas.102Ingestion of silica appears to generate a much less significant toxicological response than inhaled silicas.103 Inhalation of these particles can lead to local cellular inflammation, cytokine responses, silicosis and increased rates of lung cancer. Even with recent advances in the field, the precise mechanism of toxicity is poorly understood. An overarching theme that has emerged is that different forms, sizes and surface chemistries all play a critical role in determining toxic response. Current work is now focused on direct, systematic studies to determine the functional relationship between these critical variables.
Amorphous silica, without an ordered crystal structure, does not appear to engender the same toxic response as the crystalline silicas. Comparison between studies of these two different materials, including non- and meso-porous nanomaterials has revealed that toxicity is complex and arises directly from the previously mentioned physicochemical properties. Another critical aspect of these toxicity studies is that different cell lines (in vitro) have a differential response to these materials. The precise underlying reasons and mechanisms remain elusive at the present time.
There are comparatively few in vivo studies of nanoparticle toxicity for silica-based species. Kohane and coworkers examined the biocompatibility of unfunctionalized micro- and mesoporous silica nanoparticles using in vitro and in vivo injection.104 Yu and coworkers in a similar fashion demonstrated that intravenous injection of particles resulted in in vivo toxicity dictated by surface chemistry and porosity.105 Sayes et al. studied crystalline and amorphous silica micron-sized particles and found little correlation between in vitro and in vivo pulmonary toxicity. They found that crystalline silica microparticles produced sustained inflammatory response and toxicity compared to amorphous silica microparticles which resulted in reversible and transient inflammation.106 Overall, results by Sayes and others indicate that silica toxicity is more complex in vivo than it appears to be from the multitude of in vitro studies.106,107 Another clear finding is that the toxicity observed is highly dependent on the cell types used in the study. This implies that in vitro studies must be examined carefully in terms of the methodology to obtain meaningful information about toxicity. Furthermore, another conclusion of these studies is that the ultimate toxicant may be reactive oxygen species (ROS) generated by reactions at the particle surface. Clearly, studies which link the ultimate toxicant to surface chemistry and physicochemical properties are required to understand biological toxicity in a holistic and meaningful way. In the following two sections, relevant aspects of mesoporous silica and zeolite toxicity will be discussed in the broader context of what is known about silica toxicity as discussed above.
4.1 Toxicity of mesoporous silica nanoparticles
Mesoporous silica has seen great strides in recent years as a possible drug delivery vehicle due to increased cellular uptake of nanoscale materials.108 However, an associated concern that has risen concomitantly is the concern of physiological toxicity. One of the primary themes that has emerged is that physical structure plays a decisive role in mesoporous silica toxicity with specific emphasis placed on the external surface area.109 Crystallinity of silica also plays a key role in toxicity. When compared to crystalline materials such as quartz, mesoporous silica, with its amorphous structure exhibits greatly attenuated toxicity against in vitro cell lines.110In vitro studies involving various cell lines demonstrate a very low (below detection limit) amount of silica hydrolysis. Recent work in nanotoxicity has revealed a profound effect of the release of chemically modified species from nanoparticles.105 Not surprisingly, this matches up well with the low levels of toxicity of mesoporous silica, even in comparison with other amorphous silicas, such as fumed silica.111Haynes and co-workers further clarified the immune response of mesoporous silica using a highly innovative amperometric assay to visualize release events of mast cell granules following uptake of silica and titania (TiO2).109 The outcome of this work demonstrated that even though mesoporous silica exhibits increased cellular uptake, lower toxicity seems to result from less reactive surfaces for particle–cell interactions.109 Other work has focused on hemolytic activity of mesoporous silica and confocal imaging of in situ particles to assess distribution in human liver cells.21,112 This is important in terms of the viability as a drug delivery vehicle as previous work in this area has demonstrated that mesoporous silica can be shunted from the systemic circulation, accumulating in the liver and spleen.113 Any positive effects of a drug delivery-type application would be negated while negative effects of bioaccumulation in these organs would naturally increase.
Silica toxicity is directly correlated to the surface silanol functionality (Si–OH). These moieties participate in hydrogen-bonding interactions and have well-documented acid–base properties arising from the acidity of the silanol proton (pKa ≈ 3.0). At physiological pH (≈7.4), silanol groups are uniformly deprotonated across the surface as silicate anions (Si–O−). This uniformly negative charge at the surface can interact with positively-charged tetraalkylammonium (N–R4)+ phospholipids in cellular membranes. These interactions can entropically disrupt the membranes leading to cellular lysis and expulsion of intracellular fluid and contents into the surrounding tissue. Work from just the last few years has elucidated the correlation between surface silanol density and toxicity. This was used to explain the decreased toxicity of MCM-41 type silica in comparison to other similar mesoporous silica materials, like SBA-15.114 Due to the different ways the silicon is hydrolyzed and ultimately condenses around the surfactant constructs, the intrinsic amounts of free silanols on the surfaces of MCM-41 and SBA-15 are quite different. This difference leads to differences in observed toxicity, even between these different forms of mesoporous silica. The toxicity attributed to surface silanols can be directly mitigated by functionalization of the surface, but the overall surface charge must also be considered. Mesoporous silicas have lower surface silanol densities, and thus provoke a lower toxic response than nonporous silicas.21 Zhang et al. used Raman spectroscopy to identify three-membered siloxane rings in fumed silica samples that are not observed in mesoporous silica.111 Homolytic cleavage of these strained siloxane rings can ultimately give rise to different types of radicals including hydroxyl radicals via the proposed scheme shown in Fig. 7.111,115
 | ||
| Fig. 7 Proposed reaction of three-membered strained siloxane rings to form hydroxyl radicals. Reprinted with permission from ref. 111. Copyright 2012 American Chemical Society. | ||
As expected, in vivo studies are much less prevalent, even in the recent literature. One surprising finding is that even though mesoporous silicas show very little tissue toxicity, they do appear to invoke a systemic response. Hudson and associates revealed that death of mice subjects exposed to mesoporous silica was possibly due to thrombosis, the formation of blood clots within the circulatory system.104 This is not apparent from the multitude of in vitro studies. This is further enhanced by work carried out by Yu et al. which demonstrated that mesoporous silica accumulated in lung tissue to a greater degree than nonporous silica of a very similar size.116 One caveat of this is that previously established work has shown lower accumulation by phagocytic mechanisms, implying that truly nanoscale materials may have decreased clearance in vivo.117
A major theme that has emerged is that toxicity is associated with formation of reactive oxygen species (ROS), such as ˙OH radicals. ROS have been linked to oxidative stress in vivo and studies suggest that this may be a paradigm for nanoparticle toxicity. The method by which ROS causes damage leading to programmed cell death (apoptosis) or membrane disruption (necrosis) is not new. These destructive processes are the end product of cascade events triggered and proliferated by ROS. The formation of ROS in silica has been attributed to Fenton-like chemistry from the presence of trace iron. In vivo, the cellular concentration of free iron is kept very low due to tight binding to the transferrin protein. In fact, transferrin binds Fe3+ with overall formation constant β = 1.1 × 1031 M−2, but this low level of iron may still result in generation of toxic ROS species.118 Work by Ahmad and coworkers reveals that nanoparticle-derived ROS can overcome the natural cellular defense mechanism, glutathione-mediated redox chemistry.119 Classically, species like hydroxyl radicals can proliferate in free-radical reactions leading to oxidation of lipids, DNA damage, and production of even more damaging species such as highly reactive organic peroxynitrite (R–ONOO−).115 The reactions that produce such species have been previously described.120 Recent developments detect ROS in amorphous silica, in the absence of iron, and ROS production has been linked to highly strained three-membered siloxane rings which can undergo cleavage leading to radical generation at the particle surface.111
Free radicals have been implicated as the possible ultimate toxicant in these sorts of complex systems. Reactions at the surface of the silica nanoparticles may generate dangerous ROS which can cause cellular havoc in biological systems. The mechanism of damage is well-described and can lead to cell death, programmed or otherwise. Work on nonporous silicas seems to suggest that the production of these species arises from a Fenton-like chemistry associated with trace amounts of heavy metal ions.121 This was believed to have been the case with mesoporous silica, but has since been disproved. Instead, what appears to be the primary mechanism by which these ROS species are formed from mesoporous silica is from differential chemistry ultimately stemming from the surface silanols.111
The ultimate question that arises is how this apparent toxicity can be mitigated. The overarching answer seems to be surface differentiation. Strategies vary from functionalization by alkylamine or other organic functional moieties to gross PEG-ylation by larger polymeric molecules. The literature has established a marked decrease in toxicity with amine functionalization of the surface.122 This seems to be a convergent effect between the addition of the functional moiety with an overall change in effective surface charge in aqueous media. Yet some new work in the field has demonstrated that the linkage is perhaps not as direct as once thought.123 Polymeric functionalization of the surface is clearly complicated by any adjoining functionalities. In this case, a PEG–quaternary ammonium (R–N–Me3)+ cation (whose charge is pH-independent) showed a significantly different response from a PEG–tertiary amine (R–N–R2).123 One interpretation of this is that the surface chemistry plays perhaps the most critical role in determining toxic response. Reducing the free silanols, whether through surface functionalization or physical modification of the surface, are simply different means to the same end. The chemical nature of the surface ultimately controls interactions, reactions and biological fate.
4.2 Toxicity of zeolite nanoparticles
Compared to mesoporous silica, significantly less work has been done on zeolite nanoparticles. Recent work has revealed that a protein corona accumulates on nanocrystalline zeolites.124 This work also demonstrated the low cytotoxicity of the evaluated disc-shaped nanoscale zeolite L at low concentrations. Yet, at high concentrations a toxic response was observed, which was linked to positively charged functionalities on the surface. Similar to silica studies, the zeolites were internalized by the HeLa cancerous cell line. An important caveat of this work is the intentionally anisotropic structure of these materials. In terms of chemical formulation, it seems that for at least silicalite (no Al in framework) there is very little toxic response against multiple cell types assessed through various standard assays.111 This was primarily attributed to the lack of strained siloxane rings on the surface as detected by Raman vibrational spectroscopy.Work similar to that carried out with mesoporous silica nanoparticles reveals a similar Fenton-like chemistry that may generate ROS in situ, even with very low levels of heavy metal ions. For zeolites, this is not surprising due to their intrinsic ion exchange properties. As synthetic methods multiply in the recent literature, the concern has moved towards nanoscale materials. Micron-sized zeolites seem to be effectively non-toxic and environmentally safe, but nanoscale zeolites have not yet been evaluated fully to assess their risk in the biological or environmental domains.
In 2011, Kihara and coworkers demonstrated that in contrast to the purely siliceous zeolite, silicalite, aluminum-containing nanoscale zeolites show a distinct dose–response relationship of toxicity. The different morphologies also seem to play a key role in determining toxicity. Another odd effect revealed that contradicts work carried out on mesoporous silica is that the surface charge differences seem to play little role in determining toxic effects. Finally, recent work showed a definitive preference for cellular necrosis rather than programmed cell death via apoptotic mechanisms for the assayed conditions.125
A study by Petushkov and coworkers reinforced previously reported themes in in vivo toxicity.126 First, the observed toxicity was cell line-dependent in in vitro assays. This was attributed to possible different mechanisms of cellular uptake. Cytotoxicity was also confounded by surface functionalization on each cell line, and must be considered when drawing gross conclusions about these types of materials. The mechanism of toxicity was found to be different based on the material involved. In particular, apoptosis (as measured using the caspase activity assay) was observed in carboxyl-functionalized silicalite, yet thiol-functionalized silicalite of the same size was cytotoxic by a necrotic pathology instead.126 Annexin V–propidium iodide (V–PI) is a staining method used to provide visual evidence of the type of cell death occurring in a system. Annexin V is a 35.8 kDa protein that has a strong natural affinity for phosphatidylserine and that translocates from the inner to the outer surface of the cell membrane after apoptosis begins. If annexin V–PI enters the cells due to the presence of a damaged membrane, then the DNA is stained red. Cells with undamaged membranes are not stained red. Results of annexin V–PI staining experiments previously published are shown in Fig. 8 and support the conclusion that thiol-functionalized silicalite exhibited toxicity by necrosis.126
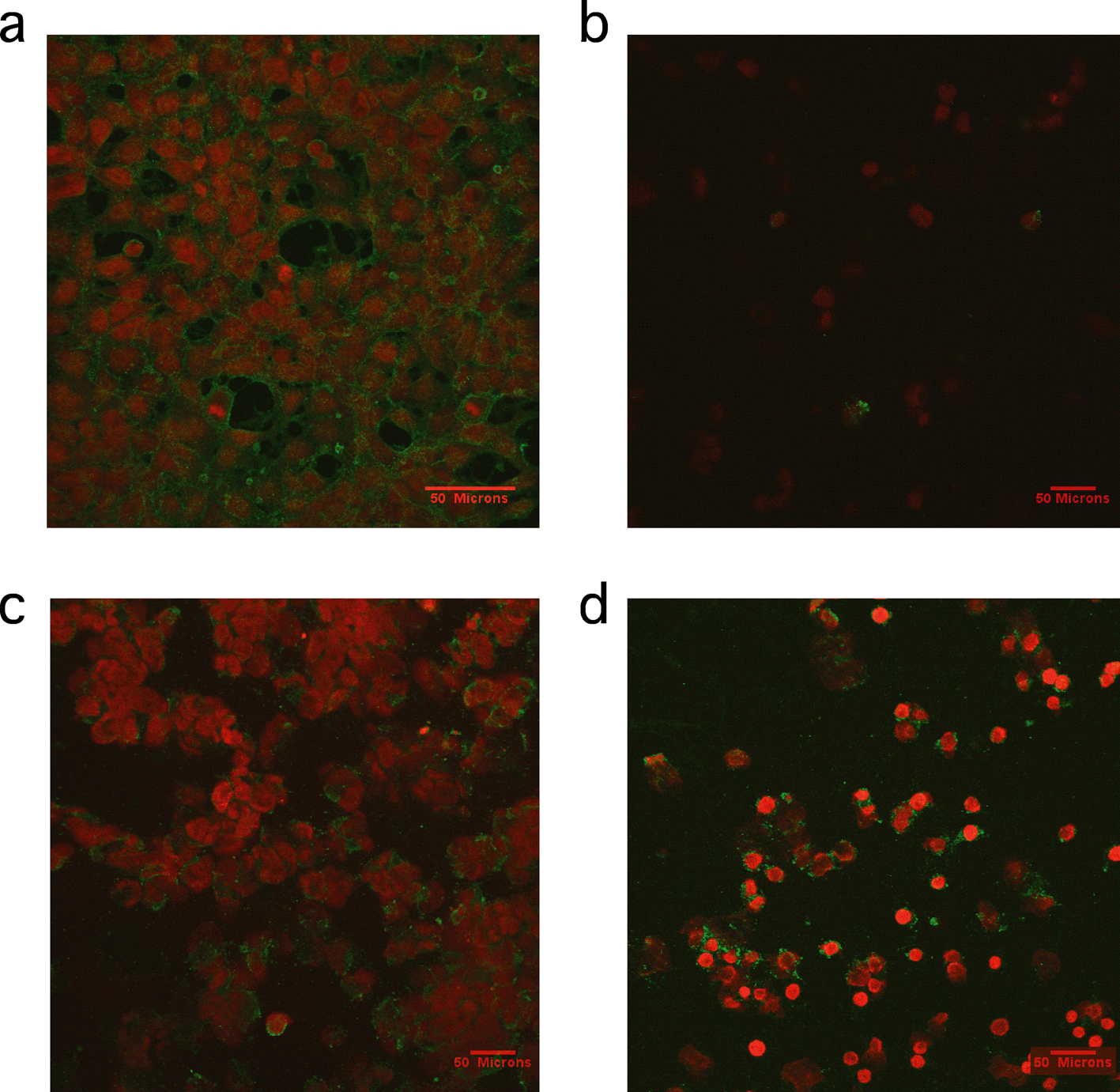 | ||
| Fig. 8 Representative confocal microscopy images of cells treated with annexin V–PI staining. a) Positive control of cells treated with 1 μM staurosporine for 2 h b) HEK293 cells incubated with 30 nm silicalite nanoparticles functionalized with thiol groups for 4 h, c) HEK293 cells incubated with 500 nm silicalite nanoparticles functionalized with carboxy groups for 4 h and d) RAW264.7 macrophages incubated with 500 nm silicalite nanoparticles functionalized with carboxy groups for 4 h. Reprinted with permission from ref. 126. Copyright 2009, American Chemical Society. | ||
Additionally, this same work demonstrated a contradiction for zeolites in that the unfunctionalized, calcined material showed less cytoxicity, with amine-functionalized being most toxic among the materials tested. This is the opposite trend from that observed in mesoporous silica materials. This further provides clear evidence for the complex, intrinsically multidimensional nature of toxic interactions between nanomaterials/nanoparticles and biological systems.
5. Conclusions and outlook
Zeolites and mesoporous silica are important porous nanomaterials due to their varied physicochemical properties that open up a wide range of potential applications. In this review, the environmental applications and implications of zeolite and mesoporous silica nanomaterials were described. First, green methods for the synthesis of zeolite and mesoporous silica were covered. The environmental applications presented here included environmental catalysis which is most applicable to zeolites and the adsorption of environmental contaminants which, while relevant for both zeolites and mesoporous silica, was focused more on the mesoporous silica materials due to the higher surface area. Finally, due to concerns that increased use of these materials translates to increased exposures, toxicity studies of zeolites and mesoporous silica materials were reviewed.Acknowledgements
Paul Mueller and Shani Egodawatte are acknowledged for experimental and editing assistance with TEM figures. This material is based upon work partially supported by the National Science Foundation under grant CHE-0847974 and the Center for Health Effects of Environmental Contamination at the University of Iowa.References
- D. W. Breck, Zeolite Molecular Sieves: Structure, Chemistry, and Use, Wiley-Interscience, New York, 1974 Search PubMed.
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef CAS PubMed.
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494–2513 CrossRef CAS.
- S. C. Larsen, J. Phys. Chem. C, 2007, 111, 18464–18474 CAS.
- Z. X. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- B. G. Trewyn, I. I. Slowing, S. Giri, H. T. Chen and V. S. Y. Lin, Acc. Chem. Res., 2007, 40, 846–853 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- X. Meng and F.-S. Xiao, Chem. Rev., 2013, 114, 1521–1543 CrossRef PubMed.
- W. Song, V. H. Grassian and S. C. Larsen, Chem. Commun., 2005, 2951–2953 RSC.
- O. Larlus, S. Mintova and T. Bein, Microporous Mesoporous Mater., 2006, 96, 405–412 CrossRef CAS PubMed.
- J. N. Armor, Appl. Catal., B, 1992, 1, 221–256 CrossRef CAS.
- M. Iwamoto, in Zeolites and Related Microporous Materials: State of the Art 1994, ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Holderich, 1994, vol. 84, pp. 1395–1410 Search PubMed.
- S. Roy, M. S. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283–2297 CrossRef CAS PubMed.
- C. Perego and A. Bosetti, Microporous Mesoporous Mater., 2011, 144, 28–39 CrossRef CAS PubMed.
- E. Taarning, C. M. Osmundsen, X. B. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Energy Environ. Sci., 2011, 4, 793–804 CAS.
- A. Walcarius and L. Mercier, J. Mater. Chem., 2010, 20, 4478–4511 RSC.
- D. Napierska, L. C. J. Thomassen, D. Lison, J. A. Martens and P. H. Hoet, Part. Fibre Toxicol., 2010, 7 Search PubMed.
- B. Fubini, Ann. Occup. Hyg., 1998, 42, 521–530 CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- Y. S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979–3986 CrossRef CAS.
- N. Baccile, F. Babonneau, B. Thomas and T. Coradin, J. Mater. Chem., 2009, 19, 8537–8559 RSC.
- C. Gerardin, J. Reboul, M. Bonne and B. Lebeau, Chem. Soc. Rev., 2013, 42, 4217–4255 RSC.
- Z. Ma, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef CAS PubMed.
- P. S. Wheatley, P. K. Allan, S. J. Teat, S. E. Ashbrook and R. E. Morris, Chem. Sci., 2010, 1, 483–487 RSC.
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS PubMed.
- L. Ren, Q. Wu, C. Yang, L. Zhu, C. Li, P. Zhang, H. Zhang, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 15173–15176 CrossRef CAS PubMed.
- G. A. Tompsett, W. C. Conner and K. S. Yngvesson, ChemPhysChem, 2006, 7, 296–319 CrossRef CAS PubMed.
- W. Song, V. H. Grassian and S. C. Larsen, Chem. Commun., 2005, 2951–2953 RSC.
- K. B. Myli, S. C. Larsen and V. H. Grassian, Catal. Lett., 1997, 48, 199–202 CrossRef CAS.
- O. Larlus, S. Mintova and T. Bein, Microporous Mesoporous Mater., 2006, 96, 405–412 CrossRef CAS PubMed.
- P. S. Mueller and S. C. Larsen, Microporous Mesoporous Mater., 2013, 180, 229–234 CrossRef CAS PubMed.
- H. Lee, S. I. Zones and M. E. Davis, Nature, 2003, 425, 385–388 CrossRef CAS PubMed.
- G. Majano, A. Darwiche, S. Mintova and V. Valtchev, Ind. Eng. Chem. Res., 2009, 48, 7084–7091 CrossRef CAS.
- G. Majano, L. Delmotte, V. Valtchev and S. Mintova, Chem. Mater., 2009, 21, 4184–4191 CrossRef CAS.
- S. Mintova, J.-P. Gilson and V. Valtchev, Nanoscale, 2013, 5, 6693–6703 RSC.
- B. Xie, J. Song, L. Ren, Y. Ji, J. Li and F.-S. Xiao, Chem. Mater., 2008, 20, 4533–4535 CrossRef CAS.
- H. Y. Zhang, B. Xie, X. J. Meng, U. Muller, B. Yilmaz, M. Feyen, S. Maurer, H. Gies, T. Tatsumi, X. H. Bao, W. P. Zhang, D. De Vos and F. S. Xiao, Microporous Mesoporous Mater., 2013, 180, 123–129 CrossRef CAS PubMed.
- E.-P. Ng, D. Chateigner, T. Bein, V. Valtchev and S. Mintova, Science, 2012, 335, 70–73 CrossRef CAS PubMed.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS PubMed.
- G. T. Neumann and J. C. Hicks, Cryst. Growth Des., 2013, 13, 1535–1542 CAS.
- P. Zhang, L. Wang, L. Ren, L. Zhu, Q. Sun, J. Zhang, X. Meng and F.-S. Xiao, J. Mater. Chem., 2011, 21, 12026–12033 RSC.
- E. R. Parnham, P. S. Wheatley and R. E. Morris, Chem. Commun., 2006, 380–382 RSC.
- H. Ma, Z. Tian, R. Xu, B. Wang, Y. Wei, L. Wang, Y. Xu, W. Zhang and L. Lin, J. Am. Chem. Soc., 2008, 130, 8120–8121 CrossRef CAS PubMed.
- C. S. Cundy, Collect. Czech. Chem. Commun., 1998, 63, 1699–1723 CrossRef CAS.
- R. E. Morris, Angew. Chem., Int. Ed., 2008, 47, 442–444 CrossRef CAS PubMed.
- Z. Tang, S.-J. Kim, X. Gu and J. Dong, Microporous Mesoporous Mater., 2009, 118, 224–231 CrossRef CAS PubMed.
- Y.-S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132, 4834–4842 CrossRef CAS PubMed.
- Y. Wang, S. Huang, S. Kang, C. Zhang and X. Li, Mater. Chem. Phys., 2012, 132, 1053–1059 CrossRef CAS PubMed.
- C. P. Canlas and T. J. Pinnavaia, RSC Adv., 2012, 2, 7449–7455 RSC.
- X. Wei-Dong, Y. Yu-Xiang, C. Jing, W. Zhaolun and L. Xiang-Nong, J. Am. Ceram. Soc., 2008, 91, 1517–1521 CrossRef.
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef CAS PubMed.
- R. Ciriminna, A. Fidalgo, V. Pandarus, F. Béland, L. M. Ilharco and M. Pagliaro, Chem. Rev., 2013, 113, 6592–6620 CrossRef CAS PubMed.
- A. Walcarius and L. Mercier, J. Mater. Chem., 2010, 20, 4478–4511 RSC.
- S. C. Larsen, in Environmental Catalysis, ed. V. H. Grassian, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- B. K. Marcus and W. E. Cormier, Chem. Eng. Prog., 1999, 47–53 CAS.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3–13 CrossRef CAS.
- R. A. Sheldon, J. Chem. Technol. Biotechnol., 1997, 68, 381–388 CrossRef CAS.
- E. Gürbüz, J. Q. Bond, J. A. Dumesic and Y. Román-Leshkov, in The Role of Catalysis for the Sustainable Production of Bio-fuels and Bio-chemicals, ed. T. Kostas, L. Angelos and M. Stöcker, Elsevier, Amsterdam, 2013, pp. 261–288 Search PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, ACS Catal., 2012, 2, 1487–1499 CrossRef CAS.
- Y.-B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2610–2618 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Prieto and J. Bilbao, Energy Fuels, 2004, 18, 1640–1647 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, B. Valle and J. Bilbao, J. Chem. Technol. Biotechnol., 2005, 80, 1244–1251 CrossRef CAS.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- P. Lanzafame, D. M. Temi, S. Perathoner, A. N. Spadaro and G. Centi, Catal. Today, 2012, 179, 178–184 CrossRef CAS PubMed.
- L. Zhou, M. Shi, Q. Cai, L. Wu, X. Hu, X. Yang, C. Chen and J. Xu, Microporous Mesoporous Mater., 2013, 169, 54–59 CrossRef CAS PubMed.
- M. Inaba, K. Murata, M. Saito and I. Takahara, Green Chem., 2007, 9, 638–646 RSC.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CAS.
- J. Aguado, J. M. Arsuaga, A. Arencibia, M. Lindo and V. Gascón, J. Hazard. Mater., 2009, 163, 213–221 CrossRef CAS PubMed.
- L. Wang, T. Qi and Y. Zhang, Colloids Surf., A, 2006, 275, 73–78 CrossRef CAS PubMed.
- T. Kang, Y. Park and J. Yi, Ind. Eng. Chem. Res., 2004, 43, 1478–1484 CrossRef CAS.
- E. Da'na and A. Sayari, Desalination, 2012, 285, 62–67 CrossRef CAS PubMed.
- A. Vinu, K. Z. Hossain and K. Ariga, J. Nanosci. Nanotechnol., 2005, 5, 347–371 CrossRef CAS PubMed.
- S. Huh, J. W. Wiench, J. C. Yoo, M. Pruski and V. S. Y. Lin, Chem. Mater., 2003, 15, 4247–4256 CrossRef CAS.
- A. S. M. Chong and X. S. Zhao, J. Phys. Chem. B, 2003, 107, 12650–12657 CrossRef CAS.
- I. Diaz, C. Marquez-Alvarez, F. Mohino, J. Perez-Pariente and E. Sastre, J. Catal., 2000, 193, 283–294 CrossRef CAS.
- L. Mercier and T. J. Pinnavaia, Environ. Sci. Technol., 1998, 32, 2749–2754 CrossRef CAS.
- L. Mercier and T. J. Pinnavaia, Adv. Mater., 1997, 9, 500–503 CrossRef CAS.
- W. Yantasee, R. D. Rutledge, W. Chouyyok, V. Sukwarotwat, G. Orr, C. L. Warner, M. G. Warner, G. E. Fryxell, R. J. Wiacek, C. Timchalk and R. S. Addleman, ACS Appl. Mater. Interfaces, 2010, 2, 2749–2758 CAS.
- V. Valtchev, G. Majano, S. Mintova and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 263–290 RSC.
- X. Chen, K. F. Lam, Q. Zhang, B. Pan, M. Arruebo and K. L. Yeung, J. Phys. Chem. C, 2009, 113, 9804–9813 CAS.
- G. L. Li, Z. S. Zhao, J. Y. Liu and G. B. Jiang, J. Hazard. Mater., 2011, 192, 277–283 CAS.
- S. J. Wu, F. T. Li, R. Xu, S. H. Wei and G. T. Li, J. Nanopart. Res., 2010, 12, 2111–2124 CrossRef CAS.
- G. E. Fryxell, Y. Lin, S. Fiskum, J. C. Birnbaum, H. Wu, K. Kemner and S. Kelly, Environ. Sci. Technol., 2005, 39, 1324–1331 CrossRef CAS.
- X. Q. Chen, K. F. Lam and K. L. Yeung, Chem. Eng. J., 2011, 172, 728–734 CrossRef CAS PubMed.
- J. Liu, S. Z. Qiao, Q. H. Hu and G. Q. Lu, Small, 2011, 7, 425–443 CrossRef CAS PubMed.
- Y.-S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979–3986 CrossRef CAS.
- M. Arruebo, W. Y. Ho, K. F. Lam, X. G. Chen, J. Arbiol, J. Santamaria and K. L. Yeung, Chem. Mater., 2008, 20, 486–493 CrossRef CAS.
- F. Zhang, J. Lan, Z. S. Zhao, Y. Yang, R. Q. Tan and W. J. Song, J. Colloid Interface Sci., 2012, 387, 205–212 CrossRef CAS PubMed.
- Y.-S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979–3986 CrossRef CAS.
- M. Yin, Z. Li, Z. Liu, X. Yang and J. Ren, ACS Appl. Mater. Interfaces, 2011, 4, 431–437 Search PubMed.
- K. Barquist and S. C. Larsen, Microporous Mesoporous Mater., 2010, 130, 197–202 CrossRef CAS PubMed.
- L. C. A. Oliveira, D. I. Petkowicz, A. Smaniotto and S. B. C. Pergher, Water Res., 2004, 38, 3699–3704 CrossRef CAS PubMed.
- K. R. Hurley, Y.-S. Lin, J. Zhang, S. M. Egger and C. L. Haynes, Chem. Mater., 2013, 25, 1968–1978 CrossRef CAS PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- A. Petushkov, N. Ndiege, A. K. Salem and S. C. Larsen, in Advances in Molecular Toxicology, ed. J. C. Fishbein, 2010, vol. 4, pp. 223–266 Search PubMed.
- W. Lin, Y.-W. Huang, X.-D. Z. Zhou and Y. Ma, Toxicol. Appl. Pharmacol., 2006, 217, 252–259 CrossRef CAS PubMed.
- S. P. Hudson, R. F. Padera, R. Langer and D. S. Kohane, Biomaterials, 2008, 29, 4045–4055 CrossRef CAS PubMed.
- Y. Yu, Y. Li, W. Wang, M. Jin, Z. Du, Y. Li, J. Duan, Y. Yu and Z. Sun, PLoS One, 2013, 8 Search PubMed.
- C. M. Sayes, K. L. Reed and D. B. Warheit, Toxicol. Sci., 2007, 97, 163–180 CrossRef CAS PubMed.
- D. Liu, B. Lin, W. Shao, Z. Zhu, T. Ji and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 2131–2136 CAS.
- Z. M. Tao, B. B. Toms, J. Goodisman and T. Asefa, Chem. Res. Toxicol., 2009, 22, 1869–1880 CrossRef CAS PubMed.
- M. A. Maurer-Jones, Y.-S. Lin and C. L. Haynes, ACS Nano, 2010, 4, 3363–3373 CrossRef CAS PubMed.
- D. Napierska, L. C. Thomassen, D. Lison, J. A. Martens and P. H. Hoet, Part. Fibre Toxicol., 2010, 7, 1–39 CrossRef PubMed.
- H. Y. Zhang, D. R. Dunphy, X. M. Jiang, H. Meng, B. B. Sun, D. Tarn, M. Xue, X. Wang, S. J. Lin, Z. X. Ji, R. B. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef CAS PubMed.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- S.-H. Wu, Y.-S. Lin, Y. Hung, Y.-H. Chou, Y.-H. Hsu, C. Chang and C.-Y. Mou, ChemBioChem, 2008, 9, 53–57 CrossRef CAS PubMed.
- M. T. A. Samri, A. V. Biradar, A. R. Alsuwaidi, G. Balhaj, S. Al-Hammadi, S. Shehab, S. Al-Salam, S. Tariq, T. Pramathan, S. Benedict, T. Asefa and A.-K. Souid, Int. J. Nanomed., 2012, 7, 3111–3121 Search PubMed.
- J. S. Valentine, D. L. Wertz, T. J. Lyons, L.-L. Liou, J. J. Goto and E. B. Gralla, Curr. Opin. Chem. Biol., 1998, 2, 253–262 CrossRef CAS.
- T. Yu, D. Hubbard, A. Ray and H. Ghandehari, J. Controlled Release, 2012, 163, 46–54 CrossRef CAS PubMed.
- S. D. Li, Y. C. Chen, M. J. Hackett and L. Huang, Mol. Ther., 2008, 16, 163–169 CrossRef CAS PubMed.
- P. Aisen, A. Leibman and J. Zweier, J. Biol. Chem., 1978, 253, 1930–1937 CAS.
- J. Ahmad, M. Ahamed, M. J. Akhtar, S. A. Alrokayan, M. A. Siddigui, J. Musarrat and A.-K. Al-Khedhairy, Toxicol. Appl. Pharmacol., 2012, 259, 160–168 CrossRef CAS PubMed.
- C. Murray, E. L. Derro, T. D. Sechler and M. I. Lester, Acc. Chem. Res., 2009, 42, 419–427 CrossRef CAS PubMed.
- M. Ghiazza, M. Polimeni, I. Fenoglio, E. Gazzano, D. Ghigo and B. Fubini, Chem. Res. Toxicol., 2010, 23, 620–629 CrossRef CAS PubMed.
- A. Yildirim, E. Ozgur and M. Bayindir, J. Mater. Chem. B, 2013, 1, 1909–1920 RSC.
- J. L. Townson, Y.-S. Lin, J. O. Agola, E. C. Carnes, H. S. Leong, J. D. Lewis, C. L. Haynes and C. J. Brinker, J. Am. Chem. Soc., 2013, 135, 16030–16033 CrossRef CAS PubMed.
- Z. Li, J. Huve, C. Krampe, G. Luppi, M. Tsotsalas, J. Klingauf, L. De Cola and K. Riehemann, Small, 2013, 9, 1809–1820 CrossRef CAS PubMed.
- T. Kihara, Y. Zhang, Y. Hu, Q. Mao, Y. Tang and J. Miyake, J. Biosci. Bioeng., 2011, 111, 725–730 CrossRef CAS PubMed.
- A. Petushkov, J. Intra, J. B. Graham, S. C. Larsen and A. K. Salem, Chem. Res. Toxicol., 2009, 22, 1359–1368 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |