Graphene–inorganic nanocomposites
Song
Bai
and
Xiaoping
Shen
*
School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang, China 212013. E-mail: xiaopingshen@163.com
First published on 28th November 2011
Abstract
Graphene (GN) has received intense interest in fields such as physics, chemistry, biology and materials science due to its exceptional electrical, mechanical, thermal and optical properties as well as its unique two-dimensional (2D) structure and large surface area. Recently, GN–inorganic nanocomposites have been opened up an exciting new field in the science and technology of GN. From the viewpoint of chemistry and materials, this account presents an overview of the synthesis and application of GN–inorganic nanocomposites. The challenges and perspective of these emerging nanocomposites are also discussed.
 Song Bai | Song Bai is a Master degree candidate under the supervision of Prof. Xiaoping Shen at School of Chemistry and Chemical Engineering, Jiangsu University, China. His current research concentrates on the preparation and application of graphene-based materials. |
 Xiaoping Shen | Xiaoping Shen is a Professor at Jiangsu University, China and a Director of the Department of Chemistry. His current research interests focus on synthesis and property of nanostructural materials including metal oxides, metal sulfides, graphene-based composites and coordination polymers. He received his PhD degree in inorganic chemistry from Nanjing University, China (2005). From 2008 to 2009 he worked as a visiting scholar at University of Wollongong, Australia. He is author or co-author of more than 80 original papers in peer-reviewed international journals and 10 patents. |
1. Introduction
Graphene (GN), a two-dimensional material (2D), composed of layers of carbon atoms packed into a honeycomb network, has become a sparkling rising star on the horizon of material science in the last several years.1,2 Even though GN is the mother of all graphitic forms, including 0D fullerene, 1D carbon nanotubes (CNT) and 3D graphite, which have been intensively studied for decades, not too much attention was paid to GN before.3 Until in 2004, Geim and Novoselov at Manchester University first isolated single-layer sheets of GN from bulk graphite, which were awarded the 2010 Nobel Prize in Physics for their groundbreaking experiments regarding GN.4–6Long-range π-conjugation in GN yields remarkable and unique properties, such as high values of its Young’s modulus (∼1.0 TPa),7 large theoretical specific surface area (2630 m2 g−1),8 excellent thermal conductivity (∼5000 W m−1 K−1),9 high mobility of charge carriers (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1),10 and optical transmittance (∼97.7%).11 These excellent properties support GN as an ideal building blocks in nanocomposites. Nanocomposites are multiphase materials, in which one phase (dispersed phase) in the nanosize regime is dispersed in a second phase (matrix/continuous phase), resulting in a combination of the individual properties of the component materials. It is well known that a new type of nanocomposite—CNT–inorganic nanocomposites—have attracted significant research attention in the last decade, due to the combination of the intrinsic properties of CNTs and inorganic materials, thus their exceptional performance in various applications.12,13 In comparison with CNTs, GN exhibits some similar behaviors, but some vastly distinct properties, such as quantum Hall effect,6,14,15 and ambipolar electric field effect.4,16 More important is the tubular geometry of CNTs while GN has a planar structure. All of these make the emergence of novel nanocomposites—GN–inorganic nanocomposites—viz. the hybrids of GN with inorganic nanomaterials. Over the past few years, GN–inorganic nanocomposites have been intensively developed, and found to exhibit a range of unique and useful properties, which are attracting more and more attention from researchers.
000 cm2 V−1 s−1),10 and optical transmittance (∼97.7%).11 These excellent properties support GN as an ideal building blocks in nanocomposites. Nanocomposites are multiphase materials, in which one phase (dispersed phase) in the nanosize regime is dispersed in a second phase (matrix/continuous phase), resulting in a combination of the individual properties of the component materials. It is well known that a new type of nanocomposite—CNT–inorganic nanocomposites—have attracted significant research attention in the last decade, due to the combination of the intrinsic properties of CNTs and inorganic materials, thus their exceptional performance in various applications.12,13 In comparison with CNTs, GN exhibits some similar behaviors, but some vastly distinct properties, such as quantum Hall effect,6,14,15 and ambipolar electric field effect.4,16 More important is the tubular geometry of CNTs while GN has a planar structure. All of these make the emergence of novel nanocomposites—GN–inorganic nanocomposites—viz. the hybrids of GN with inorganic nanomaterials. Over the past few years, GN–inorganic nanocomposites have been intensively developed, and found to exhibit a range of unique and useful properties, which are attracting more and more attention from researchers.
In this review, we focus on synthesis and potential applications of GN–inorganic nanocomposites. Starting with an attempt to introduce GN as building blocks for nanocomposites (section 2), we discuss the preparation of GN for nanocomposite synthesis. This will be followed by the classification of GN-based nanocomposites. In the second part, we provide a comprehensive summary of various techniques in synthesizing GN–inorganic nanocomposites, which are grouped into two main categories: in situ synthesis (section 3.1) and ex situ approaches (section 3.2). Finally, we demonstrate the superb advantages of GN–inorganic nanocomposites for a wide range of applications (section 4) and discuss the remaining challenges and future prospects (section 5).
2. Graphene in nanocomposite materials
2.1. Why choose graphene?
Not surprisingly, the main motivating factor for probing the research of the GN-based nanocomposites is the desire to combine the favorable properties of GN with other constituent nanomaterials (the second components).17,18 The benefits of GN in constructing inorganic nanocomposites include the following:(1) Planar structure. It is usually difficult to achieve a good decoration of nanomaterials on the CNTs, when the size of them is in the same range as or larger than the diameters of the CNT. Whereas, GN have a unique basal plane structure, which make it possible for GN to load microspheres with diameter size even bigger than several hundred nanometres.19 Furthermore, the 2D structure makes it possible for GN-based nanocomposites to be synthesized by new synthesis methods which could not be used in CNT-based nanocomposites synthesis, such as the thermal decomposition of GN precursor-intercalated inorganic materials.20,21
(2) High surface area. Compared with CNTs, GN, with higher surface area, improves interfacial contact with the other components. The large surface of GN could prevent the aggregation of secondary components, so that some unique properties in the nanoscale could be preserved.22 On the other hand, most of the extraordinary properties of GN nanosheets are only associated with individual sheets. However, the high surface area make GN tend to form irreversible agglomerates.23 While the second components could act as spacers between GN nanosheets to minimize the agglomeration.24
(3) Electrical and optical properties. Most of the research on GN focuses on the electrical properties. As a zero band gap semiconductor, GN displays a remarkably high electron mobility under ambient conditions, with reported values up to 15000 cm2 V−1 s−1.5 Moreover, the observed mobilities are nearly independent of temperature, suggesting that an ultrahigh mobility could be realized in GN at room temperature.25 By minimizing impurity scattering, mobilities of GN in excess of 200000 cm2 V−1 s−1 were achieved.10 This is of importance as nanocomposites with GN as the electron carrier may perform better in applications that involve charge transfer processes, such as sensors, supercapacitors, and electrocatalysis. Moreover, considering the high optical transparency of GN, GN-based nanocomposites could be fabricated into transparent conductive films,26,27 which show promise for application in solar cells, advanced electronics, etc.
(4) Mechanical properties. It was reported that defect-free monolayer GN has a Young's modulus of 1.0 TPa and a fracture strength of 130 GPa.7 Despite some structural distortion, the measured elastic modulus of freely suspended GN monolayers is still as high as 0.25 TPa.28 The advantages of GN in mechanical properties make it facile to fabricate and process GN-based nanocomposites into devices with various application. Furthermore, it was also reported that the mechanical stability of GN nanosheets could be improved by dispersing inorganic nanomaterials on them.29
(5) Thermal properties. In a noncontact measurement using the confocal micro-Raman spectroscopy, a thermal conductivity value of about 5000 W m−1 K−1 was obtained for a suspended single-layer GN.9 The high thermal conductivity make GN-based nanocomposites with excellent thermal stability, which could be important in some electronic devices or catalytic reactions with heat release, such as fuel cells and lithium-ion batteries (LIBs).
(6) Low cost and simple procedure of production. GN can readily form stable colloids dispersion in various solvents,30 which make it possible to process GN-based nanocomposites directly using solution processing techniques, while the chemical functionlization in advance is necessary for CNT, due to their poor dispersion.31 In addition, the solution processing methods start with graphite as the raw material, resulting in the production cost of GN-based materials in large quantities much lower than that of CNT.32
2.2. Preparation of graphene for use in nanocomposite materials
| Synthesis technique | Description | Product | Advantages | Disadvantages |
|---|---|---|---|---|
| micromechanical exfoliation | exfoliation from bulk graphite using Scotch tape | GN particles | simple process, few defects | small area, low yield, |
| CVD | surface segregation of carbon or decomposition of hydrocarbons. | GN films | large area | low yield, poor scalability |
| epitaxial growth | high temperature evaporation of Si on SiC wafer surfaces | GN films | few defects | costly method, small area, low yield |
| longitudinal “unzipping” of CNTs | Ar plasma etching or edge oxidation | GN nanoribbons | high yield, controllable widths, smooth edges | poor scalability |
| organic synthsis | stepwise organic reactions to extend polycyclic aromatic hydrocarbons | GN quantum dots (QDs) | few defects, easy scalability | small area, costly method |
| colloidal suspension | exfoliation suspension of graphite or graphite derivatives | CMG/GO/RGO nanosheets | high yield, large area, easy scalability | significant numbers of defects |
Among the six methods, the colloidal suspension method from graphite or graphite derivatives (method 6) stands out as the primary strategy that can not only yield large amounts of chemically modified graphene (CMG), but also be suited to chemical functionalization and used for a wide range of applications.49 Among the synthesized CMG, reduced graphene oxide (RGO) is the most common product, which is often obtained through graphite oxide exfoliation-chemical reduction route.50Graphite oxide, formerly called graphitic oxide or graphitic acid, is obtained by treating graphite with strong oxidizers. Nowadays, most GN-based nanocomposites are fabricated with graphite oxide as the starting material. This is much similar to the acid-treated CNT, which is often used in CNT-based nanocomposites synthesis. In fact, most of “GN” in GN-based nanomaterials is CMG, or to be exact RGO.
Methods for the oxidation of graphite to graphite oxide include: Brodie,51 Staudenmaier,52 Hummers method,53 and some of these methods with minor modifications.54,55 A comparison of these methods can be seen in a related review.56 The oxidation of graphite can destroy the sp2 hybridized structure with a increase of layer distance.57 The precise structure of graphite oxide is still under debate today, but it is sure that graphite oxide is strongly hydrophilic,58 which can be exfoliated to colloidal suspensions of graphene oxide (GO) sheets in water and organic solvents by simple sonication due to the oxygen-containing functional groups on both basal planes and the edges of GO.59,60 A thorough discussion on graphite oxide and GO can be found in a recent review.61
Besides reducing agents, some other methods have also been used to reduce GO. Thermally-mediated reduction including oil-bath,84,85 hydrothermal,86 solvothermal,87 and microwave-assisted heating approaches,88 have been implemented to reduce GO at high temperatures. With no hazardous reductants used and only simple equipment, the thermally-mediated methods are believed to be perfect for mass production of RGO. Another clean method showing promise for the reduction of GO relies on the electrochemical removal of the oxygen functional groups.89,90 This reduction process can be monitored and controlled by its reduction peaks and the redox current peaks.91 However, this method mostly yields solid RGO films on the surface of electrode, which is less suitable for processing of GN colloid dispersions. Furthermore, other methods, such as photo-irradiation,92 bacterial respiration,93 laser,94 and plasmas95 were also used to reduce GO. Liu and Sun et al.96 also propose an evaluation criterion for the reduction method of GO with several factors, including dispersibility, reduction degree, defect repair degree, and electrical conductivity. The production of RGO from graphite oxide is a hotspot in the field of GN-based materials, and a recent review was dedicated to this topic.97
Covalent functionalization is achieved through the chemical bonding of the functional molecules with the basal planes and edges of GN nanosheets. However, the functionalization of defect-free GN is not so easily accomplished, and most reported covalent chemical modifications of GN occurred with GO or RGO (GO/RGO) at reactive sites of oxygen-containing functional groups. Actually, GO itself can be also regarded as covalent functionalized GN with oxygen functional groups. At present, introduction of amines to conjugate with oxygen functional groups is a common approach to covalent functionalizaion of GN,104,105 which have been investigated for various applications.106,107 In contrast, GN can also be non-covalently functionalized viahydrogen bonding,108,109 π–π stacking,110,111 electrostatic interactions,108,112 and van der Waals interactions.108,113 The main advantage of the noncovalent functionalization of GN is that functional groups could be introduced into GN without affecting its structure and electronic network, so that the novel properties of GN are retained.
2.3. Classification of graphene-based nanocomposites
In 2006, Ruoff’s group reported the first GN-based nanocomposite, a GN–polystyrene nanocomposite,114 which has attracted tremendous attention and was followed by further development a broad of new class of GN-based nanocomposites. So far, there has not been an authoritative classification of GN-based nanocomposites. Herein, we classified the GN-based nanocomposites according to two criteria: the second component of the nanocomposites and the architecture of the nanocomposites.Based on the different kinds of inorganic components, GN–inorganic nanocomposites could be further classified into: GN–metal nanocomposites, GN–carbon nanocomposites, GN–metal compound nanocomposites, and GN–nonmetal nanocomposites. In GN–metal nanocomposites reported in literature, the majority of the second components are noble metals, such as Au,121–124Ag,125–127Pt,126,128–130 and Pd131,132. GN can not only reduce the consumption of noble metal, but also establish significant electronic interaction with them.133 In addition, other metal nanomaterials such as Cu,126,134,135Sn,126,136Co,137 bimetals,138 and alloys126 were also used for fabricating inorganic composites of GN.
The combination of GN with other carbon building blocks (CNT,139–141fullerene,142carbon black (CB),143carbon sphere,144 and carbon nanofibers145) offers fascinating prospects for the design of new carbon materials, especially the composites of GN with CNT. Both GN and CNT show remarkable electrical, thermal, mechanical and structural properties. The assembly of them into nanocomposites produces carbon materials with large specific surfaces, high electrical conductivities, and unique mechanical properties.
The integration of metal compound nanomaterials with GN forms GN–metal compound nanocomposites. The reported metal compounds include: oxides,146–151hydroxides,152,153sulfides,154,155selenides,156,157nitride,158inorganic salt,159 clay,160etc. Among these metal compounds, semiconductor and magnetic nanomaterials were intensively studied. It has been demonstrated that there exists charge-transfer as well as electronic and magnetic interactions between GN and the attached semiconducting oxide or magnetic nanomaterials.161GN as an electron transport channel could bring enhanced properties and improved performance of metal compound nanomaterials in various applications.
Non-metallic materials such as S,162,163Si,164,165SiO2,166Si3N4,167SiOC,168 CN,169,170 and C3N4171,172 have also been reported for preparing their corresponding nanocomposites with GN. Some of the GN–nonmetal nanocomposites were synthesized to develop metal-free catalysts to substitute metal catalysts. For example, a GN–C3N4 nanocomposite was reported to be a high-performance catalyst to activate molecular oxygen for selective oxidation of secondary C–H bonds of saturated alkanes with good conversion and high selectivity to the corresponding ketones.171 The classification of GN-based nanocomposites by the second component was listed in Table 2.
| GN-based nanocomposites | Second component | Ref. | |
|---|---|---|---|
| GN–polymer nanocomposites | Polymer | 114–120 | |
| GN–inorganic nanocomposites | GN–metal nanocomposites | Metal | 121–138 |
| GN–carbon nanocomposites | Carbon building blocks | 139–145 | |
| GN–metal compound nanocomposites | Metal compound | 146–160 | |
| GN–nonmetal nanocomposites | Nonmetal | 162–172 | |
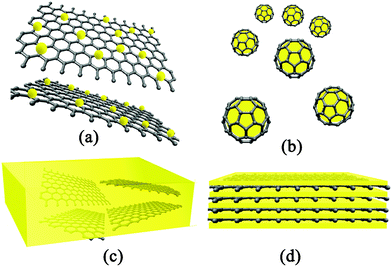 | ||
| Fig. 1 Schematic illustration of architectures of GN-based nanocomposites: (a) GN-supported nanocomposites; (b) GN-encapsulated nanocomposites; (c) GN-incorporated nanocomposites and (d) GN-based multilayered nanocomposites. | ||
GN-encapsulated nanocomposites are fabricated through enwrapping the second components with GN nanosheets. The nanostructures of the second component could be NPs,191 hollow particles,192nanotubes,193etc. In this structure, GN nanosheets functioned as protection layers, which could more effectively prevent the aggregation of the second components in comparison with GN-supported nanocomposites. Meanwhile, the contact surface between GN and the second components is much larger, and thus this structure could be more stable to avoid the exfoliation of the second components from the GN. This structure has been fabricated for high-performance lithium storage electrode materials with the reason that GN could compensate for the volume change of the inner active material during the Li+ insertion and extraction as well as improve the electrical conductivity.
In GN-incorporated nanocomposites, GN nanosheets play the role of nanofillers and distribute in the matrix of second components. The second matrix could be bulk materials,194 or made-up of nanomaterials.195 In this structure, usually, the second components were polymers. However, some inorganic compounds, particularly, the ceramic materials were also embedded with GN nanosheets to form GN-incorporated inorganic nanocomposites. The large surface area and high electrical conductivity of GN functionalize the inorganic materials with interesting properties and valuable applications. For example, by incorporation of individual GN sheets into a silica matrix, GN–ceramic composite thin films were fabricated as transparent conductors.196 GN-incorporated alumina ceramic composites were prepared by spark plasma sintering, which show far higher electrical conductivities than CNT–alumina ceramic composites with the same conductive phase content.197,198 Corral et al.167 also reported that incorporating a small amount of GN nanosheets could greatly increase the toughness of silicon nitride ceramics.
GN-based multilayered nanocomposites are formed by stacking GN nanosheets with the second components alternately. This structure could maximize the interfacial area, and is propitious to charge generation, transfer, and separation, thus has potential applications in energy storage. The second components could be NPs. As an example, Yang et al.199 reported a multilayered structure of a GN–NiO composite. The tightly fixed NiO NPs and planar GN form the skeleton of such structures, which acts as an ideal buffer to accommodate volume changes of the NiO, and thus has a better resilience and structural stability in the electrochemical charge–discharge process. However, in most cases, the second components were in the form of nanosheets. Jin et al.200 reported the multilayered composite of GO sheets with Co-Al layered double hydroxide (LDH) nanosheets for application as a pseudocapacitor. Their results demonstrated that the composite exhibited a high specific capacitance. This is due to the single atomic layered structure, in which all Co atoms occupy the surface of sheets and thus have an opportunity to contribute to redox reaction. In addition, the face-to-face assembly of GO and Co-Al LDH nanosheets optimizes their contact area, which is advantageous to efficient electron transport. Table 3 summarizes the classification of GN-based nanocomposites by the architectures of the composites.
| GN-based nanocomposites | Dispersed phase | Matrix/continuous phase | Ref. | |||
|---|---|---|---|---|---|---|
| GN-supported nanocomposites | NPs, Nanorods, | |||||
| Nanosheets, Nanoplates, | GN nanosheets | 173–190 | ||||
| Nanowires, Nanotubes, etc. | ||||||
| GN-encapsulated nanocomposites | NPs, Nanotubes, etc. | GN nanosheets | 191–193 | |||
| GN-incorporated nanocomposites | GN nanosheets | Bulk materials or Buildup of NPs | 194–198 | |||
| GN-based multilayered nanocomposites | Alternating layers of GN nanosheets with the second components (NPs or nanosheets) | 199, 200 | ||||
3. Synthesis of graphene–inorganic nanocomposites
Nowadays, a number of strategies have been put forward for the fabrication of GN–inorganic nanocomposites. Nonetheless, they fall into two basic classes. One main approach involves the formation of nanocrystallites in the present of pristine or functionalized GN nanosheets, and then the nanocrystallites directly grow into nanomaterials such as NPs, nanowires, nanorods and nanofilms on the surface of the GN nanosheets, which belong to in situ techniques; while the second key approach involves the prior synthesis of nanomaterials in the desired dimensions and morphology, then modified and subsequently connected to the surface of functionalized GN nanosheets, which is named as ex situ techniques here.3.1. In situ growth on a graphene surface
Among the two approaches, in situgrowth is more widely used in the synthesis of GN–inorganic nanocomposites. The main advantage of this route is that the protecting surfactant or extra linker molecules could be avoided, which may imply a tedious experimental procedure, and also influence the functions of the nanocomposites. Another advantage of the in situ approach is that a variety of chemical and physical synthesis techniques could be used, including solution deposition methods, direct decomposition of precursors, hydrothermal/solvothermal techniques, gas-phase deposition, sol–gel processing, template method, and so on.3.1.1.1. Synthesis of graphene–metal nanocomposites by reduction of mixed solution. The simultaneous reduction of metal precursors and GO in a mixed solution is a common way to prepare GN–metal nanocomposites. During the synthesis process, NaBH4 is a frequently-used reducing agent. For example, RGO–Ag nanocomposites have been prepared by reducing AgNO3 with NaBH4 in a GO suspension127,202,203. Pt204 and Sn136 NPs were grown on RGO nanosheets through a similar procedure with H2PtCl4 and SnCl2 as the metal precursors, respectively. Furthermore, this method was also used to synthesize RGO–alloy nanocomposites.205–207Ethylene glycol (EG) is another important reducing agent for producing RGO–metal nanocomposites. Xu and Wang et al.208 developed a general approach for producing RGO–metal nanocomposites. They dispersed GO and a metal precursor (K2PtCl4, K2PdCl4, or HAuCl4·3H2O) in a water–EG mixed solvent. Then, both GO and the metal precursor were reduced by EG, forming the corresponding nanocomposites. Through this EG co-reduction route, RGO nanosheets supported Pt and Pt-based alloy NPs were also fabricated.209–211 Recently, Yu et al.201 demonstrated an efficient one-step approach to prepare RGO–Ag nanocomposites with formaldehyde as the reducing agent under mild conditions. In this case, formaldehyde is highly effective in reducing both Ag+ and GO within several minutes at 60 °C.
However, the co-reduction route also has challenges to obtain uniformly dispersed metal NPs on GN with high-dispersibility. On the one hand, before the reduction of GO, randomly distributed oxygen-containing groups on the its surface could result in non-homogenous distribution of metal NPs. On the other hand, reduction of GO could result in poor dispersibility due to π–π stacking interactions. An important and effective improvement is to attach capping agents on GO/RGO nanosheets, which could evenly bind in situ reduced metal NPs, control the size and shape of the NPs, and also make obtained composites well-dispersed after reduction of GO. For example, sodium citrate was introduced to in situ reduce Au NPs onto the surface of RGO nanosheets.212 In this case, the hydrophilic property of sodium citrate binding on Au NPs make the composites well-dispersed in water. Chen et al.213 adhered a perylene thiol derivative on the basal plane of GO and then reduced GO into well-dispersed RGO nanosheets, which serve as an excellent stabilizer in solution for in situ nucleation and orderly growth of Au nanodots (NDs) viathiol–Au bonding. Transmission electron microscopy (TEM) images indicate that the small Au NDs are uniformly decorated on RGO surface with a narrow size distribution (Fig. 2).
 | ||
| Fig. 2 TEM images of Au NDs uniformly decorated on RGO sheets. Reprinted with permission from ref. 213. Copyright 2011, Royal Society of Chemistry. | ||
3.1.1.2. Precipitation of graphene–metal compound nanocomposites from a mixed solution. A variety of GO–metal oxide nanocomposites have been synthesized by precipitating the mixed solution of metal salts and GO, which would then be reduced to RGO–metal oxide nanocomposites. For example, in situdecomposition of cobalt nitrate and Cu(OAc)2 in the dispersion of GO give GO–Co3O4 and GO–CuO nanocomposites, respectively.214,215Precipitation of Fe3+/Fe2+ ions and GO in an alkali solution produce GO–Fe3O4 nanocomposites, which could be reduced to RGO–Fe3O4 nanocomposites by hydrazine.216–218RGO–TiO2,219 and RGO–SnO2220 nanocomposites were also prepared by mixing GO with corresponding metal salts in solutions, followed by the reduction of GO. The advantage of this method is that the abundant oxygen-containing groups distributed on the GO nanosheets ensure the high dispersion of NPs during the whole fabrication process. However, the two-step procedure could be avoided, with the growth of metal oxide NPs and reduction of GO synchronistically. For instance, a one-pot method has been reported to synthesize a RGO–TiO2 composite using TiCl3 as both a precursor of TiO2 and a reducing agent of GO.221,222SnO2 NPs could be in situ grown on RGO nanosheets based on a redox reaction between GO and Sn2+, in which Sn2+ ions were oxidized and hydrolyzed to form SnO2 NPs, while GO were synchronously reduced to RGO nanosheets.222–224
Also, surfactants have been used in directing the assembly of oxide nanomaterials on GN. Liu and coworkers developed a unique process for the construction of ordered CMG–metal oxide nanocomposites.225 In their synthetic system, anionic surfactants were used to assist the dispersion of CMG nanosheets in the hydrophobic domains of the surfactant micelles and self-assembly of the oppositely charged metal cations on the CMG surface. After converting the metal cations to oxides, ordered nanostructures of CMG–NiO, CMG–SnO2 and CMG–MnO2 nanocomposites were formed. Müllen and coworkers chose cetyltrimethyl ammonium bromide (CTAB) as cationic surfactants to electrostatically adsorb onto the surface of highly negatively charged GO, which not only effectively solved the incompatibility and aggregation problems between GO and inorganic materials, but also directed the formation of mesoporous silica through the hydrolysis of TEOS around the surface of GO sheets.226 The constructed GO-based mesoporous silica sheets would then be reduced to RGO–silica sheets via thermal treatment at high temperature.
Precipitation by a one-pot reaction could be also used to prepare GN–chalcogenide nanocomposites. Apart from metal salts and GO in the mixed solution, S/Se sources were also needed. For instance, RGO–sulfide nanocomposites were prepared from the mixed aqueous solution of metal ions (Cd2+, Zn2+) and GO with H2S as both of sulfur source and reducing agent.227 Pan and coworkers reported the in situ synthesis of a RGO–CdSe nanocomposite by adding RGO directly into the reaction solution during the process of synthesizing CdSe NPs.157 Li et al.228 synthesized CdS QDs on an electrophoretically deposited GN layer using a sequential chemical bath deposition (S–CBD) from Cd2+ and S2− aqueous solutions, and repeating the electrophoretic deposition and S–CBD gives a multilayered GN–CdS nanocomposite (Fig. 3).
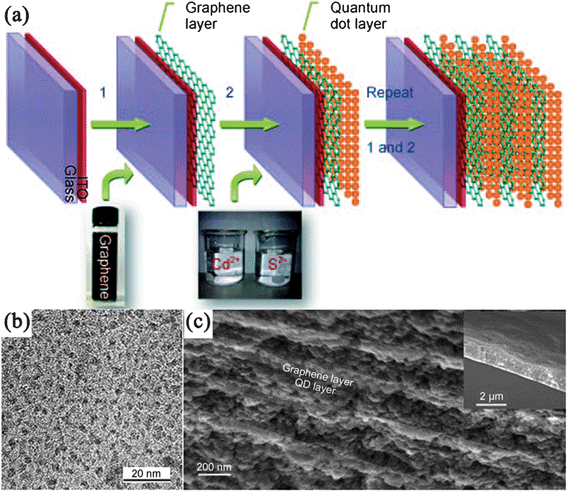 | ||
| Fig. 3 (a) Fabrication of multilayered GN–CdS QDs nanocomposite. (b) TEM images of CdS QDs on GN nanosheets, (c) Cross-sectional scanning electron microscopy (SEM) image of a (GN–CdS QDs)10 sample. The inset shows its thickness. Reprinted with permission from ref. 228. Copyright 2010, John Wiley & Sons, Inc. | ||
3.1.1.3. Electrochemical deposition and electroless deposition. As mentioned above, the electrochemical approach is simple, fast and green, which does not result in contamination of the synthesized materials. Recently, electrochemical deposition methods have been developed to fabricate GN–inorganic nanocomposites, which are typically carried out via two steps, namely, GN nanosheets being first assembled on the electrodes, shortly thereafter, the GN-coated electrodes being immersed in electrolyte solution containing metallic precursor to perform the electrochemical synthesis. Nowadays, noble metals such as Au229–232, Pt,233,234 and bimetallic Pt–Au235 with high purity have been formed quickly on GN nanosheets through an electrochemical reduction of the corresponding metal salts under an applied potential. For example, Xia et al.234 proposed three strategies to electrochemically deposit Pt NPs on RGO nanosheets. The first route is the electrochemical reduction of GO at −1.5 V, and electrodeposition of Pt NPs at −0.25 V. In the second route, Pt NPs were deposited on GO first and then GO was reduced. While in the third approach, electrochemical reduction of Pt and GO was simultaneously performed. It is found that only with the third approach, highly dispersed Pt NPs with small sizes could be formed on RGO nanosheets.
Although research currently concentrates on the electrodeposition of metal NPs on GN nanosheets, there have also been several reports on the electrodeposition of metal oxides. For instance, Zhang's group236 have successfully electrochemically deposited ZnO nanorods, as well as p-type and n-type Cu2O films on RGO electrodes with polyethylene terephthalate (PET) as the substrate. In the deposition of ZnO, the electrochemical reactions occurred with the reduction of O2 and precipitation of ZnO at a potential of −1.9 V in an oxygen-saturated Zn2+ aqueous solution electrolyte, while the electrochemical deposition of Cu2O includes the reduction of Cu2+ ions and the formation of Cu2O, in which the pH of the electrolyte affects the type of the obtained Cu2O semiconductor. They also demonstrated the electrochemical deposition of Cl-doped n-type Cu2O on RGO electrodes.237 Tang et al.238 fabricated RGO–MnO2 composite by in situ anodic electrodeposition of γ-MnO2 nanoflowers on the RGO electrode. The fabrication process includes making a RGO suspension into paper by vacuum filtering, after which the RGO paper is cut into pieces of designated dimensions for anodic MnO2 electrodeposition as the electrode.
Similar to electrochemical deposition, electroless deposition is another clean method to deposit metal NPs, which take advantage of the redox potential differences between substrate and the metal ions without any reducing agent used. The galvanic displacement between metal cations and negatively charged GO/RGO could in situ reduce the metal ions into metal NPs on the GO/RGO nanosheets. For example, Zhang et al.239 reported that Ag NPs formed spontaneously on RGO nanosheets when RGO substrate was immersed in an aqueous solution of AgNO3 under N2 protection at 75 °C. It was proposed that negatively charged RGO acted as the electron-donating source to reduce Ag+ to Ag NPs, with the reason that the reduction potential of the RGO is +0.38 V vs.SHE (standard hydrogen electrode), much lower than that of Ag+ (+0.73 V vs.SHE). With the galvanic displacement mechanism, noble metal nanoparticles such as Au124,240 and Pd241 NPs were also deposited on GO/RGO nanosheets driven by the difference between the reduction potential of AuCl42− (+1.002 V vs.SHE), PdCl42− (+0.83 V vs.SCE, saturated calomel electrode) and RGO (+0.38 V vs.SHE)/GO (+0.48 V vs.SCE). However, with this method, RGO can't reduce the metal ion with reduction potential lower than +0.38 V, such as Cu2+. Wei et al.242 fabricated various RGO–metal (Ag, Au, Pd, Pt and Cu) composites with an improved electroless deposition method. Instead of using negatively charged RGO, they made use of the redox potential differences between substrates (Cu or Zn foil) of RGO and the metal ions with RGO as an electron transport carrier. The reduction potentials of Cu or Zn were much lower than that of RGO, thus it could be performed to reduce metal ion which can’t be reduced by RGO.
3.1.1.4. Photo-assisted reduction. Photochemical reduction is another “green” process, which can provide a uniform reducing environment in solution and no additional reagents are introduced. Semiconductors with large-band-gaps are photocatalytically active under UV-visible light irradiation, which could be used to reduce GO through a photogenerated electron transfer process, thus RGO–semiconductor nanocomposites can be obtained.243 Kamat et al.244 reported the photo-assisted preparation of a RGO–TiO2 composite with the UV irradiation of GO–TiO2 dispersion in an inert atmosphere using ethanol as a hole scavenger for the TiO2 photocatalyst. So far, several semiconductors have been reported to blend with RGO nanosheets through a photo-assisted reduction process, such as UV active TiO2245,246 and ZnO247,248 as well as visible light driven WO3 and BiVO4.249
Besides the photocatalytic reduction of GO in the synthesis of GN–semiconductor nanocomposites, GN-supported metal NPs could also be produced via the photo-irradiation reduction of metal salts. One-pot synthesis of Au NDs on RGO nanosheets was performed by the photochemical reduction of HAuCl4 in the presence of octadecanethiol (ODT) molecules (Fig. 4a).250 Interestingly, the Au NDs assembled in situ into particle chains along the <100> directions of the RGO lattice, directed by the thiol groups of the self-assembled ODT molecules (Fig. 4b, c). Later, Kamat et al.251 demonstrated that the RGO nanosheets could store photogenerated electrons from UV-irradiated TiO2, and then shuttle the electrons to reduce Ag+ into Ag NPs at a location distinct from the TiO2 anchored site on RGO nanosheets. More recently, Choi et al.252 deposited Au, Ag and Pd NPs on RGO nanosheets using a phosphotungstate as a homogeneous photocatalyst under UV irradiation. Photoreduced phosphotungstates as well as electrons stored in RGO directly reduced the metal ions.
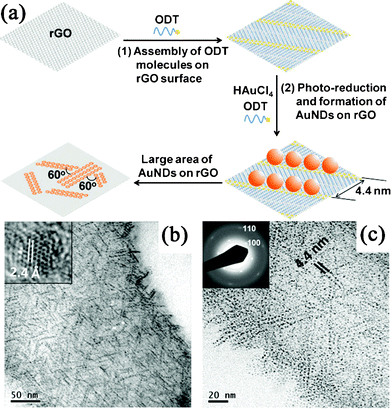 | ||
| Fig. 4 (a) Schematic illustration of the in situ synthesis and assembly of Au NDs on RGO. (b) TEM and (c) magnified TEM images of ODT-capped Au NDs synthesized in situ on RGO surface. Reprinted with permission from ref. 250. Copyright 2010, John Wiley & Sons, Inc. | ||
3.1.1.5. Microwave-assisted deposition. The main advantage of microwave irradiation (MWI) over other conventional methods is heating the reaction mixture uniformly and rapidly. As a consequence, a variety of nanomaterials including metals, alloy, and semiconductors with very small particle size and narrow size distribution could be obtained. The MWI process allows the simultaneous reduction of GO and metal ions, resulting in the fast formation of GN–metal nanocomposites. El-Shall and co-workers253 reported a microwave-assisted synthesis of various RGO–metal (Au, Ag, Cu, Pd, CuPd) nanocomposites in aqueous and organic media, respectively. In their experiments, GO and a variety of metal salts were reduced simultaneously with reductants such as hydrazine hydrate or reducing solvent such as oleylamine during the MWI process. Besides, RGO–Pt nanocomposites were synthesized by employing a microwave-assisted polyol process with EG as both dispersing and reductive agent for both metal ions and GO.254,255 Berry et al.256 also demonstrated that without any reducing agents, bare-surfaced Au NPs could also be produced in situ on GO sheets by a microwave reduction process.
Microwave-assisted methods have also been used in the synthesis of RGO–metal compound nanocomposites. A facile MWI method for the synthesis of RGO–CdSe nanocomposites were developed by EI-Shall and co-workers, which allows the simultaneous reduction of GO and the nucleation and growth of CdSe nanocrystallites.257 In this system, dimethyl sulfoxide (DMSO) was used as both the reducing agent of GO and solvent for the reaction between the Cd and Se precursors, resulting in the formation of hexagonal and cubic CdSe NPs on RGO nanosheets. Furthermore, metal compounds, such as ZnS,258Fe3O4,259Co3O4,260SnO2,261 and ZnO262 were reported to grow on RGO nanosheets with MWI methods. Lin and coworkers also reported a solvent-free method to prepare GN-supported metal (Ag, Au, Co, Ni, Pd, Pt) and metal oxide (Fe3O4, MnO, TiO2) NPs in high yields through direct solid-state Joule heating of GN and various organic metal salts within short duration of MWI.263
3.1.1.6. Sonication-assisted deposition. Similar to microwave-induced heating, sonication of a liquid also results in rapid heating, although the mechanism is fundamentally different. The sonication of a liquid results in sonic cavitation (the growth and violent collapse of microbubbles) that creates localized “hot spots” with effective temperatures of 5000 K and lifetimes on the order of a few nanoseconds or less. As such, the chemical reactions largely take place inside the bubbles. Nowadays, sonication has been used in exfoliation of graphite into GN nanosheets and cut them into GN nanoribbons.264 Also, sonochemical synthesis has become a new method to synthesize GN–inorganic nanocomposites. Vinodgopal et al.265 reported the ultrasound-induced reduction of GO and HAuCl4 in 2% aqueous solution of poly(ethylene glycol) at an ultrasonic frequency of 211 kHz to prepare RGO–Au nanocomposites. The proposed mechanism is as follows: highly reactive H·• and OH·• radicals are generated within the bubbles by the homolysis of water molecules due to the high-temperature conditions in aqueous solutions. The oxidizing radicals are scavenged by adding alcohols, thus providing a reducing condition. Finally, RGO–Au nanocomposites could be produced through simultaneous and sequential sonolytic reduction of GO and Au3+, respectively. Besides, sonochemical strategy has been successfully implemented to prepare RGO–Rh and RGO–TiO2 composites.266,267 Shi and Zhu et al.268,269 also used the sonoelectrochemical technique to fabricate RGO–Pd and RGO–PdPt alloy nanocomposites, in which an ultrasound emitter also acts as a cathode.
:1) were assembled on negatively charged GO nanosheets.276 Subsequent thermal treatments resulted in the formation of the RGO–NiCo2O4 composites.
So far, decomposition of metal complexes has also been used to prepare GN–inorganic nanocomposites, such as the utilization of metal acetylacetonate in synthesis of GN–metal oxide composites,277–279 and metal carbonyl complex in fabrication of GN–metal nanocomposites.280,281 In this case, functional groups of the metal complex can connect to the surface of GN through covalent or noncovalent interaction, which could ensure a homogenous dispersion of the NPs on GN. For instance, Müllen and coworkers reported the dispersion of cobalt phthalocyanine onto GN through π–π stacking interactions, which could then turn into RGO–Co and RGO–Co3O4 nanocomposites after simple pyrolysis and oxidation processes, respectively.282 Gotoh et al.283,284 investigated the preparation of RGO–metal/metal oxide nanocomposites by heat treatment of ion-exchanged graphite oxide, which include cations of amine complexes ([M(NH3)n]x+, M = Pt, Ru, Pd, Cu, Co, Ni), bipyridyl complex ([Au(bipy)Cl2]+), and imidazole complex ([Ag(imH)2]+). In obtained materials, it is found that all noble metals existed on RGO sheets were metal NPs, whereas Cu and Co existed as metal oxides, and Ni with a partly oxidized surface.
GN-supported metal oxide and hydroxide NPs synthesized through hydrothermal/solvothermal process mainly include ZnO,286,287TiO2,288–291Fe3O4,19,292–295SnO2,296,297Co3O4,298Bi2O3,299Fe2O3,300CoO,300MnOOH,301 and Co(OH)2.302 Also, hydrothermal synthesis can also be employed to grow vertically aligned metal oxide nanowires on GN films. Kim et al.303 reported a nanocomposite consisting of ZnO nanowires hydrothermally grown on RGO substrates. Firstly, a ZnO seed layer was obtained by spin casting of ethanol solution of zinc acetate on RGO surface for several times, and then dried at 90 °C. In the second step, ZnO nanowires were grown by suspending the seeded RGO substrates in an aqueous solution of zinc nitrate hexahydrate and hexamethylenetetramine at 90 °C for 3 h. The two-step method for the synthesis of GN-supported ZnO nanowires could also be found in other literature.304,305
Wang and Dai et al.179,306 also developed a two-step method for growing Ni, Fe, and Co hydroxide and oxide nanocrystals with well-defined morphologies on GN surfaces. In the first step, they uniformly nucleated dense Ni(OH)2 NPs onto GN by hydrolysis of Ni(CH3COO)2 in a N,N-dimethylformamide (DMF)/water mixture at 80 °C. In the second step, they hydrothermally treated the product of the first step at 180 °C and the dense Ni(OH)2 NPs coated on GN nanosheets diffused and recrystallized into single-crystalline hexagonal Ni(OH)2 nanoplates. However, this phenomenon was not observed from the GO–Ni(OH)2 nanocomposite with the reason that Ni(OH)2 NPs precoated on GO were pinned by the high concentration functional groups and defects on GO without recrystallization into well-defined morphologies (Fig. 5). These results confirm that GN with various degrees of oxidation can be used as a novel substrate for the growth of nanocrystals into various sizes and morphologies. The two-step hydrothermal method has also been developed to grow Mn3O4,307TiO2,308 and CuO309nanocrystals on the surface of GN nanosheets.
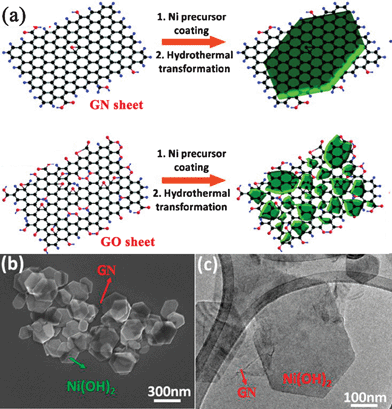 | ||
| Fig. 5 (a) Schematic illustration of two-step Ni(OH)2 nanocrystals hydrothermal growth on (top) GN and (bottom) GO sheets. (b) SEM and (c) TEM images of hexagonal Ni(OH)2 nanoplates formed on top of GN sheets. Reprinted with permission from ref. 306. Copyright 2010, American Chemical Society. | ||
Currently, various chalcogenide QDs, such as CdS,310–312 ZnS,311,313 Cu2S,297MoS2,314Sn3S4,155 and CdTe315 have been successfully immobilized on GN through hydrothermal/solvothermal processes. Besides GO and metal salts, sources of S or Te were also needed. In the fabrication of RGO–sulfide nanocomposites, sulfur sources often acted as reducing agents of GO.310,313,314 For instance, Cao et al.310 developed a solvothermal process to synthesize RGO–CdS composite directly from GO and Cd(CH3COO)2 in DMSO at 180 °C. In the one-pot reaction, DMSO not only serves as a solvent and a source of sulfur, but also produces H2S to reduce GO simultaneously.
3.1.4.1. Physical vapor deposition . PVD is a general term used to describe a variety of purely physical methods to deposit solid material by condensation of a vaporized form of the material onto various surfaces, which involves vacuum deposition and sputtering deposition. The advantage of PVD in GN-based inorganic nanocomposites synthesis is that it can directly deposit films on the basal planes of defect-free pristine GN without any functionlization, which can preserve the intrinsic properties of GN. Sun et al.316 evaporated the Au film onto the surface of micromechanical exfoliated n-layer (n: 1∼4) GN in a vacuum thermal evaporator at a deposition rate of 1.0 Å s−1 under a vacuum of ∼10−4 Pa, followed by heat treatment at 1260 °C. It was found that the morphologies of Au film on n-layer GN are dependent on the layer number, the lower GN layer number, the smaller average size of Au NPs, and the higher density of NPs. They also thermally deposited Ag films onto n-layer GN and found that the morphologies of Ag NPs on n-layer GN are also dependent on the layer number.317 This phenomenon was also observed and further researched by Johnson and co-workers.318 They proposed a theoretical model predicting a particle size distribution characterized by a mean diameter D that follows a D ∝ n1/3 scaling law where n is the number (1∼9) of layers in the GN film. This method could be applied in checking the different layer numbers of GN and growing NPs with controlled dimensions. Besides, an ultra-high vacuum (UHV) system was often used for the deposition of metal nanoclusters on moiré-patterned GN supported on transition metals, such as Ir, Pt, W, Re on GN/Ir(111);319,320Pt, Pd, Rh, Co and Au on GN/Ru(0001).321,322 Moreover, magnetron sputtering has been used to deposit Pt on the CVD-growth's GN nanoflakes.323 The thickness of Pt nanoclusters, ranging from 2.1 to 85 nm, could be simply controlled by tuning the sputtering time. The deposition of high dielectric constant Al2O3 by radio frequency magnetron sputtering on multilayer epitaxial GN grown by graphitization of SiC was also reported.324
3.1.4.2. Chemical vapor deposition . CVD is a chemical process used to produce high-purity, high-performance solid materials on a substrate surface. In a typical CVD process, one or more gaseous precursors flow over a substrate surface, which react or decompose to produce the desired material on it. In comparison with PVD, the advantages of CVD include high deposition rate, controllable composition and inexpensive equipment. CVD has been used to synthesize GN-supported semiconductors, such as CdSe NPs,325ZnO nanorods,326 and ZnS nanowires.181 It is interesting that through CVD, nanocrystals with various morphologies can grow on the surface of GN. A representative example is that ZnO nanostructures were successfully grown on GN with zinc powder as a precursor carried by Ar flow at different temperature, and followed by oxygen flow in a tube furnace.326 It was observed that ordered crystalline ZnO with high density is preferably vertically grown along the <0001> direction. Different nanorods and nanoneedles were obtained when the growth temperatures were 450 °C, 550 °C and 650 °C, respectively.
The more important aspect in CVD synthesis of GN–inorganic composites is the vertical growth of CNTs on GN nanosheets.327–329 Different from the CVD synthesis of GN-semiconductor composites, catalysts were indispensable in the formation process of GN–CNT composites. Zhao et al.328 prepared the GO/RGO sheets pillared with CNTs using CVD method with acetonitrile as the carbon source and Ni NPs as the catalyst. The Ni NPs were deposited on GO/RGO platelets, and CNTs were then grown on them through a tip growth model with Ni catalysts residing at the top of them. Both the amount and length of CNTs could be adjusted by the amount of Ni catalyst and CVD times, respectively (Fig. 6). With the same growth mechanism, Fan et al.329 prepared a RGO–CNT composite with Co as a catalyst and CO as the carbon source. They also employed Co NPs as a catalyst to deposit carbon nanofibers on the RGO sheets stemming from the pyrolysis of GO during the CVD process.145
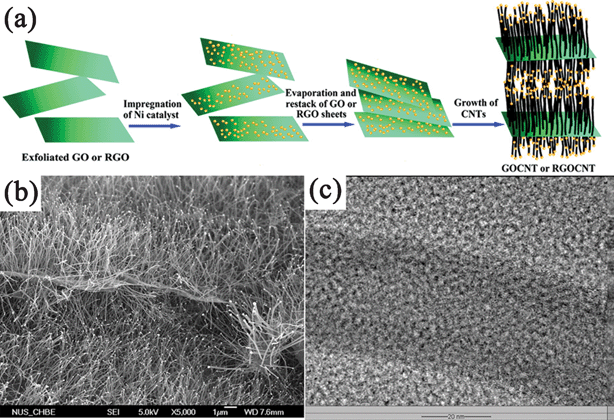 | ||
| Fig. 6 (a) Scheme illustrating the experimental steps of pillaring GO/RGO platelets with CNTs. (b) Field emission SEM (FESEM) and (c) TEM images of the CNT-pillared GO nanosheets. Reprinted with permission from ref. 328, Copyright 2010, American Chemical Society. | ||
3.1.4.3. Atomic layer deposition . ALD is a thin-film growth technique with atomically precise control over thickness and uniformity. ALD is different from the CVD method with the distinctive feature of alternate and cyclic supply of each gaseous precursors with two sequential half-reactions, which makes film growth self-limiting based on surface reactions. The work of Dai et al. provides an excellent example of ALD in coating GN with metal oxide films.330 Through alternating pulses of trimethylaluminum and water as precursors at ∼100 °C, the authors deposited Al2O3 NPs on pristine and 3,4,9,10-perylene tetracarboxylic acid (PTCA)-coated GN respectively. They found Al2O3 cannot directly be deposited on pristine GN due to the lack of dangling bonds and functional groups, and only preferentially grow actively on edges and defect sites, which could be used as a simple and effective probe to defects in GN. Uniform ultrathin Al2O3 deposition was achieved on PTCA-coated GN because of their densely packed functional groups (Fig. 7). Besides Al2O3, other dielectrics such as HfO2 films grown on GN by ALD were also reported,331,332 revealing that the ALD method is promising to be used for depositing ultrathin high-κ dielectrics for future GN electronics.
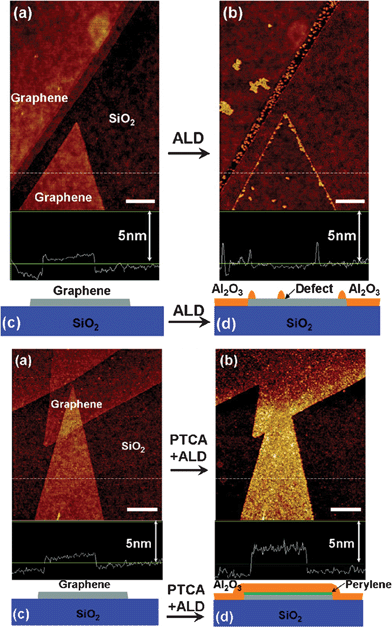 | ||
| Fig. 7 ALD of Al2O3 on (Top) pristine GN and (Bottom) PCTA-coated GN. (a) Atomic force microscopy (AFM) image of GN on SiO2 before ALD. (b) AFM image of the same area as (a) after Al2O3 ALD deposition. (c) and (d) Schematics of GN on SiO2 before and after ALD. Reprinted with permission from ref. 330. Copyright 2008, American Chemical Society. | ||
The ALD strategy exhibits many unique advantages in fabricating GN–metal oxide nanocomposites, which can tune and control the deposited metal oxide on both morphologies and structural phases. Sun et al.333 demonstrated the coating of SnO2 on RGO sheets with SnCl4 and H2O as the ALD precursors. Both amorphous and crystalline SnO2 NPs have been uniformly grown on RGO sheets by adjusting the growth temperature to 200 and 400 °C, respectively. The change of structural phases was attributed to the temperature dependent surface reactions. By adjusting their cycling numbers, the as-deposited SnO2 could present NPs or nanofilms on RGO. They also deposited TiO2 on RGO nanosheets by ALD with titanium isopropoxide and water as precursors.334 It was found that a lower temperature (150 °C) contributed to amorphous TiO2, while a higher temperature (250 °C) produced crystalline anatase TiO2.
Through template approaches, regular distribution of arrays on GN sheets was also realized, which is difficult to achieve through other methods. Kim et al.340 created a nanopatterned iron catalyst array on the GO film by means of self-assembled block-copolymer nanoporous templates and then highly aligned CNTs were grown from the catalyst by plasma-enhanced CVD (PE-CVD). In this growth method, GO films were reduced during the high-temperature PE-CVD process, and the nanoporous templates enabled the precise adjustments of the particle size of the catalyst, and thus tuning the CNT diameter. Hong et al.341 synthesized a CdSe QDs array on the basal plane of GN using a mesoporous silica thin film as a template: (a) GN nanosheets were synthesized by CVD of methane on thin Ni layers formed on SiO2/Si substrates; (b) a mesoporous silica film was formed on top of the GN by spin-coating, aging and calcination; (c) CdSe was electrochemically deposited onto a GN surface through the pores of the mesoporous silica film; (d) silica template and Ni layer underneath the GN was removed (Fig. 8). Other nanoarrays such as ZnO nanorods,342 and NiO nanocapacitors343 have also been fabricated on GN following the template procedures.
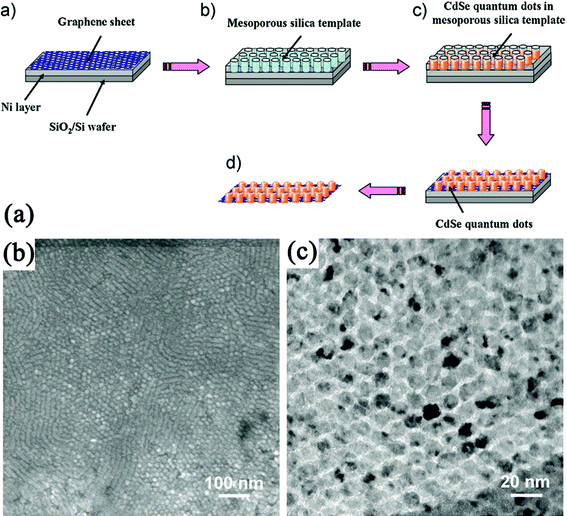 | ||
| Fig. 8 (a) Procedure to synthesize a CdSe QDs array on the basal plane of a GN using a mesoporous silica thin film as a template. (b) TEM and (c) High-resolution TEM (HRTEM) images of a CdSe quantum-dot array grown on the GN. Reprinted with permission from ref. 341. Copyright 2010, John Wiley & Sons, Inc. | ||
3.2. Ex situ approach: assembly on graphene surface
In the ex situ approach, inorganic components were synthesized in advance and then attached to the surface of GNvia linking agents that utilize covalent or noncovalent interactions (van der Waals interactions, hydrogen bonding, π–π stacking, or electrostatic interactions). In this approach, either the inorganic components or GN (or both) require modification with functional groups. The type of functionalization and thus, the strength of interaction determine the loading of the inorganic nanomaterials on the GN surface. This self-assembly based method is excellent at overcoming any incompatibilities between nanomaterials syntheses and the formation of nanocomposites. Compared to in situgrowth, the better distribution, size, and feeding amount control of the second components on GN could be obtained by the self-assembled methods.344 For example, Bovine serum albumin (BSA), a complex amphiphilic protein were reported to absorb on the basal plane of GN for the assembly of NPs.345 In this system, BSA could not only attach to GO, but also reduce GO to RGO at a suitable pH value and reaction temperature. By simply mixing BSA–GO/RGO with pre-synthesized Au, Pt, Ag or Pd NPs, corresponding GO/RGO-metal nanocomposites were prepared (Fig. 9). The density of metal NPs on BSA–GO could be easily controlled by changing the concentrations of BSA during the assembly process. This well-controlled self-assembly method also produces coassembly of presynthesized NPs with distinctively different sizes, compositions, shapes, and properties on the same GO/RGO nanosheets.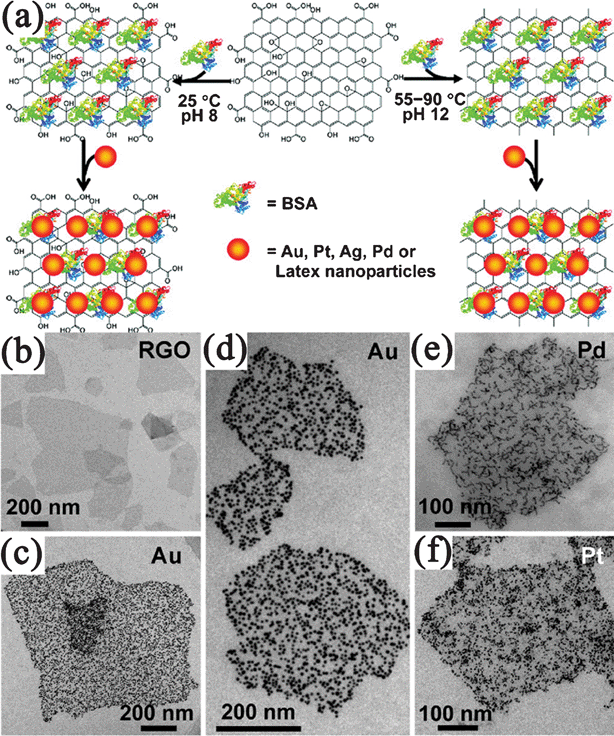 | ||
| Fig. 9 (a) Illustration of assembly of noble metal NPs on GO/RGO nanosheets with the assistance of BSA. TEM images of (b) BSA–RGO nanosheets and BSA–RGO supported (c,d) Au, (e) Pd, and (f) Pt NPs. Reprinted with permission from ref. 345. Copyright 2010, American Chemical Society. | ||
In addition, covalent interactions were also used in synthesis of GN–carbon nanocomposites. For example, the covalent linkage between the aminated CNTs and the acid chloride-activated GO formed GO–CNT composites.351 Chen et al. reported two methods for the covalent linkage of C60 with GO sheets. One was to adopt a mild coupling reaction between hydroxyl groups of fullerenol and carboxylic groups of GO,352 while another was to form amide carbonyl groups between pyrrolidine fullerene and carboxylic group of GO via a coupling reaction.353,354 Wang et al.355 reported the covalent bonding of fullerenes onto GO by the chemical reaction through the Fisher esterification between the hydroxyl groups on GO and the carboxyl groups on (1,2-methanofullerene C60)-61-carboxylic acid.
3.2.2.1. π–π stacking. Noncovalent functionlization of GN with aromatic organic compounds through π–π stacking has been mentioned earlier (Section 2.1.3.). Herein, we discuss the attachment of inorganic nanomaterials on the surface of GN through this functionlization process. Generally, these aromatic compounds are terminated with thiol (mercaptan), amine, or acid groups, which can connect to the inorganic NPs and enable their attachment to GNvia π–π stacking. For example, pyridine, with an aromatic structure, has provided π–π stacking interactions for anchoring Au356 and CdSe357 NPs on the basal planes of GO/RGO sheets. DNA molecules containing both purine and pyrimidine bases could be also used to mediate the fabrication of GN nanocomposites via π–π stacking interactions. Both thiolated DNAs and pyrene-labeled DNAs have been used to stablize Au NPs on GN.358,359 Following a similar strategy, Huang et al.360 fabricated DNA-conjugated Au and Ag NPs on GO nanosheets, respectively. Zhan et al.361 reported the decoration of RGO with CdS QDs by using benzyl mercaptan (BM) as the interlinker. During the synthesis process, the mercapto substituent of BM binds to the CdS QDs during their nucleation and growth process, and then the phenyl comes into contact with the RGO sheets via the π–π stacking interaction.
It is also worthy of note that GO behaves like an amphiphilic molecule containing hydrophobic aromatic regions on the basal plane and hydrophilic oxygen groups on the edges, which can adsorb the pristine CNT through the π–π interaction with the hydrophobic basal plane, and assist to stabilize the dispersion of CNT in aqueous media due to the hydrophilic edges. This means that the GO–CNT composite films could be readily formed by vacuum filtration of the aqueous dispersion without any additional organic solvents or pre-functionalisation involved.362,363
3.2.2.2. Electrostatic interactions. Electrostatic interactions have also been utilized to modify GN with various inorganic NPs. GO/RGO are negatively charged as a result of the ionization of the oxygen functional groups on them, which could be used to assemble with positively charged inorganic NPs through electrostatic interactions. For instance, RGO–Fe3O4 nanocomposites have been formed by mixing the positively charged Fe3O4 NPs and negatively charged RGO nanosheets.364 Chen et al.365 noncovalently decorated GO sheets with oppositely charged aerosol Ag nanocrystals synthesized from an arc plasma source using an electrostatic force directed assembly technique. A novel strategy for the fabrication of RGO-encapsulated oxide (silica, Co3O4) NPs was developed by coassembly between negatively charged GO and positively charged oxide NPs with three steps: oxides were firstly modified by surface grafting of aminopropyltrimethoxysilane (APS) to render the oxide surface positively charged; then the modified oxide NPs were assembled with negatively charged GO by electrostatic interactions; finally, the resulting composites were chemically reduced with hydrazine (Fig. 10).191,366 However, the negative charge of RGO is often too weak to assemble some NPs directly. So the functionlization of GN is also needed, such as the use of 1-pyrene butyric acid functionalized GN to anchor positively charged Au NPs.367
 | ||
| Fig. 10 (a) Schematic illustration of fabrication of RGO-encapsulated oxide NPs. (b,c) Typical SEM, and (d) TEM images of RGO-encapsulated silica spheres. Reprinted with permission from ref. 366. Copyright 2010, John Wiley & Sons, Inc. | ||
On the contrary, when the NPs are negatively charged, it is necessary to alter the surface of GO/RGO to positively charged. The deposition of a cationic polyelectrolyte is a common route. For example, RGO nanosheets could accept negatively charged Ag NPs after noncovalent functionalized with polyelectrolyte PQ11.368 Cationic polyelectrolyte poly(diallyldimethyl ammonium chloride) (PDDA) coated GN nanosheets were served as platforms for citrate-capped Au NPs and thioglycolic acid modified CdSe QDs, respectively.369,370 Wang et al.371 self-assembled negatively charged mixed Pt and Au NPs onto positively charged PDDA-functionalized RGO nanosheets as effective electrocatalysts, while dispersed MnO2 nanosheets were dispersed on PDDA-functionalized RGO nanosheets as electrode materials for pseudocapacitors.372 Besides, the self-assembly of GN with CNT can also be accomplished with this approach. Cationic PEI modified RGO was used for assembly with negatively charged acid-oxidized CNT.373 On the other hand, GN–CNT nanocomposites were prepared by a combination of positively charged aminated CNTs with negatively charged GO.374
3.2.2.3. Layer-by-layer self-assembly. LBL self-assembly is a widely used method for fabricating GN–inorganic nanocomposite films. The obtained films are typically formed by alternating layers of GN nanosheets with the second components. There are many advantages of the LBL assembly technique, such as simplicity and thickness controllability in the nanoscale. Moreover, the blending of two components into composites and the fabrication of composites into films were carried out simultaneously. Other film fabrication techniques, such as vacuum assisted self-assembly were also used in fabricating GN–inorganic nanocomposite multilayer films, but the composites usually need to be prepared in advance.375
The LBL self-assembly route to fabricate films of GN–inorganic composites were mainly performed through the electrostatic interaction of alternating layers with opposite charge. For example, positively charged imidazolium salt-based ionic liquid-functionalized GN were LBL self-assembled with negatively charged citrate-stabilized Pt NPs into 3D composite films.376 Loh et al.377 assembled negatively charged GO and negatively charged titania nanosheets into multilayer films with cationic PEI as linkers. Exposure of the films to UV irradiation allows both the reduction of GO and the photocatalytic removal of the PEI moiety, and multilayered composite films consisting of alternating RGO and titania nanosheets were obtained. RGO and CdSe NPs multilayers were fabricated by negatively charged pyrene-grafted poly(acrylic acid)-RGO nanosheets with positively charged CdSe NPs.378 Also, with the electrostatic LBL self-assembly, thin films of (RGO/CNT)n,26 and (GN/carbon nanospheres)n379 composites were also fabricated.
Positively charged polyelectrolytes of PDDA were often used as building blocks to fabricate 3D multilayer architectures by the electrostatic LBL technique. Wang et al.380 employed the negatively charged species of poly(sodium 4-styrenesulfonate) (PSS) modified RGO, negatively charged MnO2 sheets, and positive charged PDDA as building blocks to fabricate 3D multilayer architectures (PDDA/PSS-RGO/PDDA/MnO2)10. Yu et al.381 fabricated (PDDA/GO/PDDA/TiO)20 composite films on the glass substrate through alternative LBL self-assembly with GO, titania nanosheets, and PDDA. Besides, hydrogen-bonding LBL assembly was also used to fabricate GN–inorganic composite films. By utilizing the hydrogen bonding interaction of poly(vinyl alcohol) (PVA) with both GO and LDH, followed with a reduction process, Liu et al. fabricated electrically conductive (LDH/PVA/RGO/PVA)50 multilayer films.382
4. Applications of graphene–inorganic nanocomposites
As discussed above, GN can be combined with various inorganic nanomaterials through different architecture types and synthesis techniques. The GN–inorganic nanocomposites are expected to not only preserve the favorable properties of GN and the second components, but also greatly enhance the intrinsic properties due to the synergetic effect between them. In this section, we presents the improved performance of GN–inorganic nanocomposites in catalysis, energy storage and conversion, sensors, and other applications.4.1. Catalysis
Carbon materials have been widely used as the support for immobilizing inorganic catalysts, mainly due to their large specific surface area, excellent electrical and thermal conductivity, low price, high chemical inertness, easy modification and loading. Like other carbon-based catalysts, GN–inorganic nanocomposites have been developed as active catalysts in chemical, electrochemical as well as photochemical reactions.000 h−1 as well as very low Pd leaching (< 1 ppm (parts-per-million)) in the Suzuki–Miyaura coupling reaction. Moreover, these novel heterogeneous catalysts were readily available and easy to handle as they are stable in air. Also, another important coupling reaction, the Heck reaction, with GN–Pd composites as catalysts was also reported.384,385 Wang et al.385 immobilized Pd NPs on GO and assembled the composite into a 3D macroscopic porous structures. This catalyst exhibited excellent catalytic activity and selectivity for the Heck reaction. Both the selectivity and conversion were measured to be 100% when K2CO3 was used as the base in the reaction of iodobenzene and methyl acrylate. These values are much higher than the results obtained when the Pd catalyst is loaded onto other supports. The ideal supporting material also make GN–Au nanocomposites active catalysts for Suzuki reactions, though Au NPs alone were generally poor in catalyzing these reactions.174,386
The most widely used electrocatalysts with GN as the supporting material were Pt-based NPs, such as Pt,130,204,209,233,234,254,324,388–392Pt-based alloy (PtRu,210,393–396PtCo,205,397PtCr,397PtNi,207PtAu,398 and PtPd269), bimetal (Pt–Pd,399 Pt–Au235), and mixed Pt and Au nanocatalysts.371 For example, Honma et al.130 reported the enhanced activity and improved tolerance of a GN–Pt electrocatalyst for MOR compared to the CB–Pt catalyst. Liu et al.388 demonstrated GN–Pt nanocomposites exhibited a higher electrochemical surface area and oxygen reduction activity with improved stability as compared with the Pt catalysts supported with other carbon materials (CNT and Vulcan XC-72 carbon). They also synthesized indium tin oxide (ITO) NPs directly on functionalized GN sheets, and then deposited Pt NPs, forming a unique triple-junction structure.400 The GN–ITO–Pt nanocomposite was investigated as an electrocatalyst for oxygen reduction for potential application in polymer electrolyte membrane fuel cell, which showed enhanced performance not only better than the widely used Pt electrocatalysts supported with other carbon materials, but also better than Pt supported on GN sheets.
Considering the high cost and CO poisoning of platinum, many other metals and metal oxides NPs have also been combined with GN nanosheets to construct highly active non-platinum electrocatalysts. RGO–Pd nanocomposites have been developed as alternatives in electrocatalytic oxidation of ethanol, methanol or reduction of oxygen.241,401–403 Meanwhile, RGO–Pd nanocomposites also exhibit excellent electrocatalytic activity and stability towards formic acid oxidation.241,338,404 Niu et al.405 deposited Au–ionic liquid on PTCA functionalized GN. It is found that the composite showed good electrocatalytic behavior toward ORR. Dai et al.406 reported a composite consisting of Co3O4 NPs grown on RGO nanosheets (Fig. 11a) as a high-performance catalyst for ORR. Although Co3O4 or RGO alone has little catalytic activity, their composite exhibits an unexpected, surprisingly high activity that is further enhanced by nitrogen doping of the GN nanosheets (Fig. 11b, c, d). The N-doped RGO–Co3O4 (N-RGO–Co3O4) composite exhibits similar catalytic activity but superior stability to a fresh commercial C–Pt catalyst in alkaline solutions (Fig. 11e, f). The unusual catalytic activity arises from synergetic chemical coupling effects between Co3O4 and GN. They also grew MoS2 NPs on RGO nanosheets, which exhibited superior electrocatalytic activity in the hydrogen evolution reaction relative to other MoS2 catalysts.407 In other cases, nanocomposites such as RGO–MnO2408 and RGO–SnO2396 were also reported for electrocatalytic applications. Besides, possessing the advantages of low costs and environmental friendliness, GN-based metal-free electrocatalysts were also developed. A typical example of this is a RGO–CN nanocomposite, which exhibits an excellent electrocatalytic performance for ORR, including high electrocatalytic activity, long-term durability, and high selectivity, all of which are superior to those observed for CN sheets without GN as well as for commercially available C–Pt catalysts.169,170
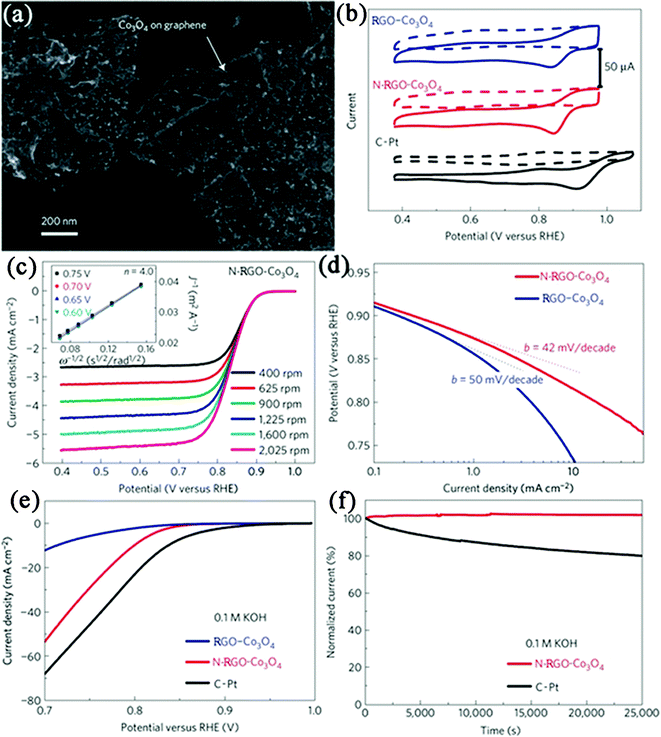 | ||
| Fig. 11 (a) SEM image of N-RGO–Co3O4 composite deposited on a silicon substrate from a suspension in solution. (b) Cyclic voltammetry curves of RGO–Co3O4, N–RGO–Co3O4 and C–Pt on glassy carbon electrodes in O2-saturated (solid line) or Ar-saturated 0.1 M KOH (dash line). (c) Rotating-disk voltammograms of N-RGO–Co3O4 composite in O2-saturated 0.1 M KOH with a sweep rate of 5 mV s−1 at the different rotation rates indicated. (d) Tafel plots of RGO–Co3O4 and N-RGO–Co3O4 composites derived by the mass-transport correction of corresponding rotating-disk electrode data. (e) Oxygen reduction polarization curves of RGO–Co3O4, N-RGO–Co3O4 and C–Pt catalyst dispersed on carbon fibre paper in O2-saturated 0.1 M KOH electrolytes. (f) Chronoamperometric responses of N–RGO–Co3O4 and C–Pt on carbon fibre paper electrodes kept at 0.70 V versus reversible hydrogen electrode in O2-saturated 0.1 M KOH electrolytes. Reprinted with permission from ref. 406. Copyright 2011, Nature Publishing Group. | ||
TiO2 has been considered as one of the best photocatalytic materials, due to its efficient photoactivity, photo- and chemical stability, nontoxicity, and low production cost. To date, a large number of reports have focused on the photocatalytic application of GN–TiO2 nanocomposites in removal of pollutants.21,222,288,308,409–415 The GN–TiO2 photocatalysts were reported to exhibit a significant enhancement in the photodegradation activity, compared with the bare TiO2 and CNT–TiO2.409,410,414 However, the detailed research of Xu et al. towards the gas-phase degradation of benzene strongly manifested that RGO–TiO2 nanocomposite was in essence the same as other carbon-based TiO2 composites on enhancement of photocatalytic activity of TiO2, although GN had unique structural and electrical properties.411 Besides decomposition of organic compounds, RGO–TiO2 composites have also been reported as photocatalysts for the evolution of hydrogen under UV-vis illumination.335,416
Despite of the advantages above, TiO2 is mainly excited by high energy UV irradiation, which greatly limits its practical applications because of the low content of UV light in the solar spectrum. So efforts have been made to exploit visible-light responsive photocatalysts with GN as the support. For instance, RGO–Au,417RGO–Bi2WO6,418 as well as 3D CNT-pillared RGO328 nanocomposites have been found to display excellent performance in the photocatalytic degradation of dyes under visible light. Also, photocatalytic production of H2 from water splitting under visible-light irradiation has been achieved by GN-supported photocatalysts. For instance, a remarkable 10-fold enhancement in photoelectrochemical water splitting reaction was observed on RGO-incorporated BiVO4 nanocomposite compared with pure BiVO4 under visible illumination.419 This improvement was mainly attributed to the presence of GN, which serves as an electron collector and transporter to efficiently lengthen the lifetime of the photogenerated charge carriers. With the same charge recombination mechanism, a high efficiency of the photocatalytic H2 production was achieved using RGO–CdS nanocomposite as visible-light-driven photocatalysts.312 The composite reaches a high H2-production rate of 1.12 mmol h−1 (about 4.87 times higher than that of pure CdS NPs) at RGO content of 1.0 wt % under visible-light irradiation. Xu et al.420 also reported the N-doped RGO–CdS composite shows higher photocatalytic activity in the photocatalytic H2 production, compared with RGO–CdS, and GO–CdS composites. This finding demonstrates that N-doped GN is a more promising candidate for the development of high-performance photocatalysts. As a metal-free photocatalyst in visible-light catalytic hydrogen production, RGO–C3N4 (∼1.0 wt% RGO) also shows a H2-production rate of 451 μmol h−1 g−1.172 Besides, significantly promoted visible-light photocatalytic performance has also been observed in GN-supported ZnO,421,422SnO2,222Bi2MoO6,423InNbO4,424ZnFe2O4,425 and GN-encapsulated Ag/AgX(X = Br, Cl)426 composites, etc.
4.2. Energy storage and conversion
The increasing demand for energy and growing concerns about air pollution and global warming have stimulated considerable effort to the development of energy storage/conversion devices with high power and energy densities. GN-based materials have been considered as one of the promising alternatives as electrode materials in energy-related devices, because of their superior electrical and thermal conductivities, high surface area, good chemical stability, excellent mechanical strength and broad electrochemical window.427,428 Recently, GN–inorganic nanocomposites have been demonstrated for applications in energy storage/conversion devices, such as supercapacitors, LIBs, solar cells, and fuel cells.Among these compounds, MnO2 is one of the most promising electrode materials for supercapacitor applications, due to its low-cost, being environment-friendly and high specific capacitance. Fan et al.430 reported a rapid and facile method to prepare GN–MnO2 composites as novel electrode materials by MWI (Fig. 12a). The obtained GN–MnO2 (78 wt% MnO2) nanocomposite shows a specific capacitance of 310 F g−1 at a scan rate of 2 mV s−1 (Fig. 12b), which is higher than other kinds of carbon-MnO2 composites. After 15000 cycles, the composite only shows 4.6% capacity loss, indicating a long cycle life (Fig. 12c). The excellent electrochemical performances are believed to be due to the unique microstructures of the composite, which not only improve the diffusion rate and reduce the diffusion length of the cations within the MnO2 NPs, but also enhance the conductivity with GN as conductive channels (Fig. 12a). Besides metal oxides and hydroxides, other carbon materials (such as CNT,329,373,446,447CB,143 and carbon spheres144) can also enhance the electrochemical performances of GN for supercapacitor applications. The incorporation of carbon materials into GN layers not only inhibits the agglomeration of GN nanosheets, but also provides highly conductive channels for electron transport and form well-defined nanopores for fast ion diffusion. The electrochemical performance of supercapacitors with various GN–inorganic composites as electrode materials is shown in Table 4.
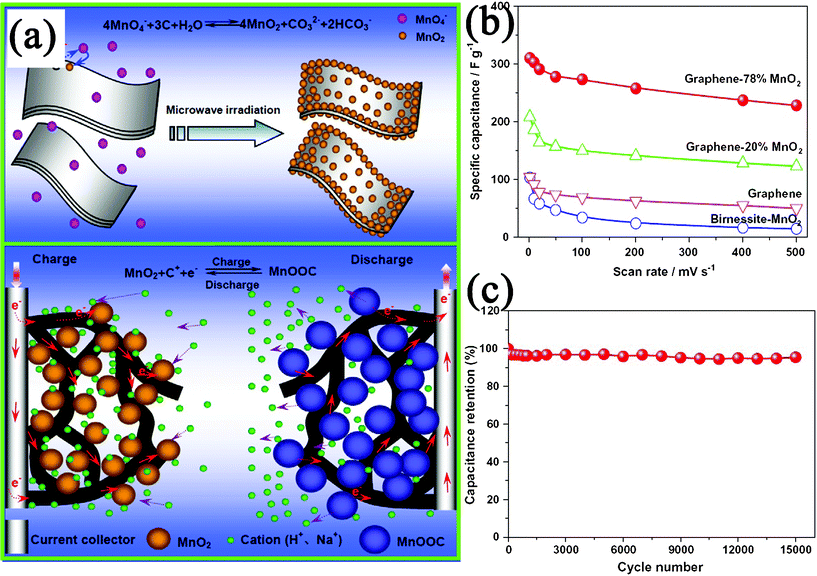 | ||
| Fig. 12 (a) Schematic illustration for the synthesis and electrochemical performance of a GN–MnO2 nanocomposite. (b) Specific capacitance of GN–MnO2 as well as GN and pure MnO2 at different scan rates in 1 M Na2SO4 solution. (c) Variation of the specific capacitance of GN-78% MnO2 electrode as a function of cycle number measured at 500 mV s−1 in 1 M Na2SO4 aqueous solution. Reprinted with permission from ref. 430. Copyright 2010, Elsevier. | ||
| GN–inorganic nanocomposites | Specific capacitance (F g−1) | Current density (A g−1) | Electrolyte | Ref. |
|---|---|---|---|---|
| GN-supported MnO2 nanowalls | 122 | — | 1 M Na2SO4 | 184 |
| GN-supported MnO2 nanoflowers | 328 | — | 1 M KCl | 238 |
| GN-supported MnO2 nanosheets | 188 | 4.0 | 1 M Na2SO4 | 372 |
| GN–MnO2 multilayer films | 263 | 0.283 | 0.1 M Na2SO4 | 380 |
| GN-supported MnO2 NPs | 310 | — | 1 M Na2SO4 | 430 |
| GN-supported MnO2 nanowires | 24.5 | 0.5 | 1 M Na2SO4 | 432 |
| GN-supported MnO2 NPs | 113.5 | 1.2 | 1 M Na2SO4 | 433 |
| GN–MnO2 textiles | 315 | 2.2 | 0.5 M Na2SO4 | 434 |
| GN-supported Mn3O4 NPs | 175 | — | 1 M Na2SO4 | 435 |
| 256 | — | 6 M KOH | ||
| GN-supported Co3O4 NPs | 243.2 | 10 (mA cm−2) | 6 M KOH | 436 |
| GN–Co3O4 nanoscrolls | 163.8 | 1 | 6 M KOH | 437 |
| GN-supported SnO2 NPs | 43.4 | — | 1 M· H2SO4 | 223 |
| GN-supported ZnO NPs | 61.7 | — | 1 M KCl | 438 |
| GN-incorporated ZnO composite | 308 | 1 | 1 M· Na2SO4 | 440 |
| GN-supported Bi2O3 NPs | 255 | 1 | 6 M KOH | 299 |
| GN-supported RuO2 NPs | ∼570 | 0.1 | 1M· H2SO4 | 272 |
| GN-supported Fe3O4 NPs | 480 | 5 | 1 M KOH | 295 |
| GN–NiO multilayer membranes | 150∼220 | 0.1 | 30% KOH | 199 |
| GN-supported porous NiO | 400 | 2 | 1 M KOH | 442 |
| GN-supported CeO2 NPs | 208 | 1 | 3 M KOH | 443 |
| GN-supported Cu2O NPs | 24.0 | 0.2 | Saturated KCl | 444 |
| GN-supported β-Ni(OH)2 nanoplates | ∼1335 | 2.8 | 1 M KOH | 179 |
| GN-supported α-Ni(OH)2 NPs | 1215 | — | 6 M KOH | 445 |
| GN-supported Co(OH)2 NPs | 972.5 | 0.5 | 6 M KOH | 152 |
| GN-supported NiCo2O4 NPs | 835 | 1 | 6 M KOH | 276 |
| GN-supported Ni–Al LDH nanosheets | 781.5 | 10 (mA cm−2) | 6 M KOH | 153 |
| GN–Co–Al LDH multilayered composite | 1031 | 20 | 1 M KOH | 200 |
| GN-supported CNT | 290.4 | 0.5 | 1 M KCl | 447 |
| GN-supported CB | 175 | 5 | 6 M KOH | 143 |
| GN-supported carbon spheres | 39.4 | 0.1 | 6 M KOH | 144 |
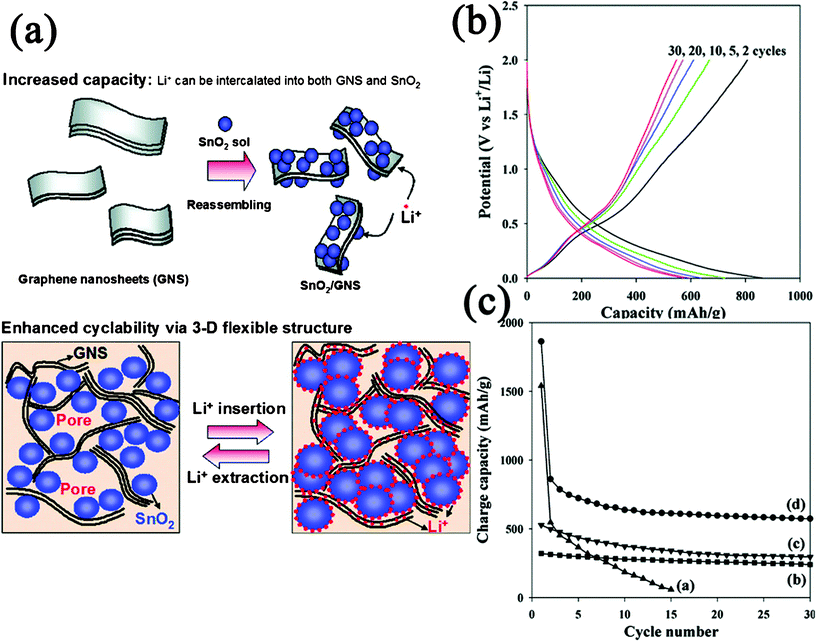 | ||
| Fig. 13 (a) Schematic illustration for the synthesis and structure change of GN–SnO2 nanocomposite during Li+ insertion and extraction. (b) Charge–discharge profile for GN–SnO2 nanocomposite. (c) Cyclic performances for (a) bare SnO2 NPs, (b) graphite, (c) GN, and (d) GN–SnO2 nanocomposite. Reprinted with permission from ref. 450. Copyright 2009, American Chemical Society. | ||
On the basis of this concept, a wide range of nanomaterials of oxides, hydroxides and chalcogenides, such as SnO2,220,224,296,451–455Fe3O4,259,273,293,441,456,457Co3O4,183,270,271,298,366,458CuO,309,459TiO2,158,192,415,460,461Mn3O4,307CoO,183,300Fe2O3,300,462,463NiO,178CeO2,464Li4Ti5O12,337,465Co(OH)2,302Ni(OH)2,466MoS2,314,467 and SnSe180 have been anchored on GN nanosheets for application as anode materials for LIBs. Besides, GN-supported Si,164,165,468Sn-based136,206 NPs, GN–Sn multilayered nanostructure,469 and GN-incorporated ceramic composite168 have also been developed as anode materials for LIBs. All these composites showed large reversible capacities, excellent cycling performances and high coulombic efficiencies. Furthermore, Honma and coworkers reported that the incorporation of CNT and C60 could further increase the specific capacity of GN as anode materials.470 The enhanced performance of the composites were attributed to the improvement of lithium accommodation in the increased layered spaces between GN through intercalating CNT or C60. Fan et al. also reported a RGO–carbon nanofiber composite for high-performance anode materials for LIBs.145 The performance of LIBs with GN–inorganic composites as anode materials are summarized in Table 5.
| GN–inorganic nanocomposites | Reversible capacity (mAh h−1) | Current density (mA g−1) | Cycles | Ref. |
|---|---|---|---|---|
| GN-supported SnO2 NPs | 840 | 67 | ∼30 | 224 |
| GN-supported SnO2 nanorods | 838 | 50 | ∼20 | 296 |
| GN-supported SnO2 NPs | 765 | 55 | ∼100 | 451 |
| GN-supported echinoid-like SnO2 NPs | 634 | 100 | ∼50 | 454 |
| GN-supported SnO2 nanosheets | 518 | 160 | ∼50 | 455 |
| GN-encapsulated Fe3O4NPs | 1026 | 35 | ∼30 | 273 |
| 580 | 700 | ∼100 | ||
| GN-encapsulated Fe3O4 NPs | 650 | 100 | ∼100 | 293 |
| GN-supported Fe3O4 NPs | 1048 | 100 | ∼90 | 456 |
| GN-supported Co3O4 nanowalls | 673 | 180 | ∼100 | 183 |
| GN-supported Co3O4 nanosheets | 931 | 4450 | ∼30 | 270 |
| GN-supported Co3O4 NPs | 935 | 50 | ∼30 | 271 |
| GN-supported Co3O4 NPs | 941 | 200 | ∼60 | 298 |
| GN-encapsulated Co3O4 composite | 1000 | 74 | ∼130 | 366 |
| GN-supported CuO NPs | 583.5 | 67 | ∼50 | 309 |
| GN-encapsulated CuO nanomicroflowers | 600 | 65 | ∼100 | 459 |
| GN-encapsulated TiO2 hollow NPs | ∼90 | 1700 | ∼180 | 192 |
| GN-supported mesoporous anatase TiO2 nanospheres | 97 | 8400 | ∼100 | 415 |
| GN-supported anatase TiO2 nanosheets | 161 | 170 | ∼120 | 461 |
| GN-supported Mn3O4 NPs | 730 | 400 | ∼40 | 307 |
| GN-supported CoO nanowalls | 732 | 150 | ∼100 | 183 |
| GN-supported CoO NPs | 600 | 215 | ∼90 | 300 |
| GN-supported Fe2O3 NPs | 881 | 302 | ∼90 | |
| GN-supported NiO nanosheets | 1031 | 71.8 | ∼40 | 178 |
| GN-supported CeO2 NPs | 605 | 50 | ∼100 | 464 |
| GN-supported Co(OH)2 NPs | 910 | 200 | ∼30 | 302 |
| GN-incorporated β-Ni(OH)2 composite | 507 | 200 | ∼30 | 466 |
| GN-supported MoS2 nanosheets | 1290 | 100 | ∼50 | 314 |
| GN-supported MoS2 nanosheets | ∼1100 | 100 | ∼100 | 467 |
| GN-supported SnSe nanoplates | 470∼640 | 40 | ∼30 | 180 |
| GN-supported Si NPs | 1168 | 100 | ∼30 | 164 |
| GN-supported Si NPs | 708 | 50 | ∼100 | 165 |
| GN-supported Sn NPs | 508 | 55 | ∼100 | 136 |
| GN-Sn multilayered nanostructure | 679 | 50 | ∼30 | 469 |
| GN-incorporated SiOC composite | 357 | 40 | ∼30 | 168 |
| GN-supported C60 | 600 | 50 | ∼20 | 470 |
| GN-supported CNT | 480 | 50 | ∼20 | |
| GN-supported carbon nanofibers | 667 | 0.12 (mA cm−2) | ∼20 | 145 |
In another case, GN nanosheets were also used as supporting materials to enhance the capacity delivery and cycle performances of cathode materials. LiFePO4 has been a promising cathode-active material for LIBs because of its low-cost, low toxicity and high stability. However, its poor electronic conductivity and lithium-ion diffusivity limit its practical application. GN–LiFePO4 composites have shown improved specific capacity and rate capability with the reason that GN can facilitate electron transport effectively and thus improve the kinetics and rate performance of LiFePO4.471–473 Besides, LiMn1-xFexPO4 nanorods,175Li3V2(PO4)3 NPs,336LiNi1/3Co1/3Mn1/3O2 NPs,474 and V2O5·nH2O xerogel nanoribbons475 were also grown on GN nanosheets as improved cathode materials for LIBs.
Another example of the successful application of GN–inorganic nanocomposites in solar cells is the development of QDSSCs.485–487 QDs such as CdS and CdSe with large extinction coefficient and upon light absorption could harvest light energy and transfer excited electrons to the conduction band of TiO2 or ZnO film. Similar to DSSCs, capturing photoinduced electrons as quickly as they are generated and efficiently transporting them in the metal oxide film is one of the most difficult challenges in QDSSCs system. The introduction of GN was reported to suppress electron recombination and back-transport reaction, enhance the electron transport as well as increase the QDs adsorption, thus improve the performance of QDSSCs. For example, Deng et al.485 demonstrated the use of a GN–ZnO nanorod composite as a photoanode, leading to 54.7% improvement of power conversion efficiency in CdSe QDSSCs compared to a quantum dot sensitized ZnO nanorods without the RGO layer. Pan et al.486 reported CdS QDSSCs, based on a GN-incorporated TiO2 film photoanode, which demonstrated a maximum power conversion efficiency of 1.44%, 56% higher than that without RGO.
4.3. Sensing
The rapid development of GN provides new opportunities for the progress of analytical science. Nowadays, GN nanosheets with large active area and high conductivity have been introduced into analytical chemistry and implanted novel functions into analytical sensing systems.427,488–490 In this section, we present the application of GN–inorganic nanocomposites in constructing high-performance gas sensors and biosensors.Besides, GN–inorganic nanocomposites were also developed to fabricate gas sensors based on cataluminescence (CTL), which refer to the kind of chemiluminescence (CL) that is emitted during the catalytic oxidization of organic vapors on the surface of sensing material in an atmosphere containing oxygen. GN–supported metal oxide NPs with good distribution can provide greater versatility in carrying out adsorption, selective catalytic and sensing processes. Their porosity and large specific surface area are attractive advantages for CTL sensing materials. For instance, RGO–Al2O3 composite was reported to display high CL sensitivity and high selectivity to ethanol gas.496 The CTL sensor based on a RGO–SnO2 composite demonstrated a linear range 1.34∼266.67 μg mL−1 and detection limit of 0.3 μg mL−1 in propanal sensing.497
Most of the reported biosensors were based on an eletrochemical strategy with techniques such as amperometric, voltammetric and electrochemiluminescence (ECL) methods. Occasionally, GN-based inorganic composites were also used to detect biomolecules based on field effect transistors (FETs) signal conversion strategies.191,509,510 As an example, a specific protein detection FET biosensor was fabricated using thermally-reduced graphene oxide (TRGO) sheets decorated with Au NP–antibody conjugates.509 In this sensing strategy, the Au NP–antibody conjugates were assembled onto the surface of TRGO sheets by electrostatic assembly. Then a blocking buffer was used to prevent possible nonspecific binding events. When the target proteins (IgGs) were introduced, the protein binding events induced significant changes in the electrical characteristics of the device, which could be studied by FET and direct current measurements. The schematic of the TRGO–Au NP FET device and the FET sensor fabrication process are shown in Fig. 14 a and b, respectively. The performance of the novel biosensor is among the best carbon nanomaterial-based protein sensors, which exhibited a low detection limit of ∼13 pM.
 | ||
| Fig. 14 (a) Schematic of the FET device. (b) Schematic illustration of the FET biosensor fabrication process. Reprinted with permission from ref. 509. Copyright 2010, John Wiley & Sons, Inc. | ||
Besides, GN–inorganic nanocomposites have initially been used as a advanced carrier to construct immunosensors,370,519–521 which are important analytical tools based on the property of highly specific molecular recognition of antigen by antibodies. For instance, Zhu et al.370 fabricated an advanced electrogenerated chemiluminescence immunosensor by using PDDA-protected GN–CdSe composites. After two successive steps of amplification via the conjugation of PDDA and Au NPs, the immunosensor has an extremely sensitive response to human IgG in the linear range 0.02∼2000 pg mL−1 with a detection limit of 0.005 pg mL−1. Table 6 summarizes the biosensors fabricated with GN–inorganic nanocomposites. It is also worthy of note that GN–inorganic nanocomposites have also been fabricated as sensing platforms for detection of pollutants and dangerous substances.499,522
| Inorganic component | Analyte | Detection techniques | Detection limits | Ref. |
|---|---|---|---|---|
| Ag | H2O2 | Amperometric | 2.8 μμM | 368 |
| Au | H2O2 | Amperometric | 0.44 μμM | 369 |
| Pt | H2O2 | Amperometric | 80 nM | 499 |
| Au | glucose | Amperometric | 35 μμM | 212 |
| Pd | glucose | Amperometric | 1 μμM | 502 |
| Pt-Au | glucose | Amperometric | 1 μμM | 504 |
| Pt | DA | Amperometric | 0.03 μμM | 506 |
| AA | 0.15 μμM | |||
| UA | 0.05 μμM | |||
| Au | UA | Amperometric | 0.2 μμM | 367 |
| Pt | cholesterol | Amperometric | 0.2 μμM | 508 |
| Au | protein | FETs | ∼13 pM | 509 |
| Au | DNA | FETs | — | 510 |
| Au | organophosphate pesticide | Amperometric | 0.1 pM | 511 |
| Au | L-histidine | Voltammetric | 0.1 pM | 512 |
| Au | NADH | Amperometric | 1.2 μμM | 513 |
| Fe3O4 | H2O2 | Amperometric | 0.6 μμM | 292 |
| MnO2 | H2O2 | Amperometric | 0.8 μμM | 515 |
| TiO2 | DA | Voltammetric | 2 μμM | 289 |
| ZrO2 | organophosphorus agents | Voltammetric | 0.1 ng mL−1 | 516 |
| CdS | H2O2 | ECL | 1.7 μμM | 517 |
| CdSe | thrombin | Photocurrent | 4.5 × ×10−13 M | 378 |
| SiO2 | cancer biomarker | FETs | 1 pM | 191 |
| CdSe | human IgG | ECL | 0.005 pg mL−1 | 370 |
| Au | human IgG | Voltammetric | 70 ng mL−1 | 519 |
| Au–Ag | alpha-fetoprotein | Amperometric | 0.5 pg mL−1 | 521 |
4.4. Other applications
Also, works have been carried out to study the enhanced Raman signal of adsorbed molecules on GN–metal composites. For instance, Shen et al.527 deposited Au films on single-layer GN as SERS substrates for the characterization of rhodamine molecules with low concentration. While Au films effectively enhance both the Raman and photoluminescence (PL) signals of the molecules, single-layer GN effectively quenches the PL signals from the Au film and molecules. Tang et al.528 investigated the Raman signal of 4-aminothiophenol (4-ATP) with GN–Ag composite as SERS substrates. The Raman intensity of 4-ATP increased with increasing weight ratio of Ag in the composite. Based on the enhanced Raman mechanism, the GN–metal composites have been used as efficient SERS substrates to detect molecules.529,530 The RGO–Ag and RGO–Au composites fabricated by Zhang et al. exhibited strong adsorption and dramatic Raman enhancement to aromatic molecules.529 Subsequently, the SERS substrates were used to detect the adsorbed aromatic molecules with a low detection limit at nM level. Wang et al.530 self-assembled Ag NPs with PDDA-functionalized GO as SERS substrates for the detection of folic acid. The modified GO with a positive potential exhibited strong enrichment of negatively charged folic acid due to the electrostatic interaction, and the self-assembled Ag NPs greatly enhanced the SERS spectra of folic acid with the strongest peak located at 1595 cm−1. The intensity of the peak increased with the increase of the concentration of folic acid. The SERS spectra of the folic acid showed that the minimum detected concentration of folic acid in serum was as low as 9 nM with a linear response range from 9 to 180 nM (Fig. 15).
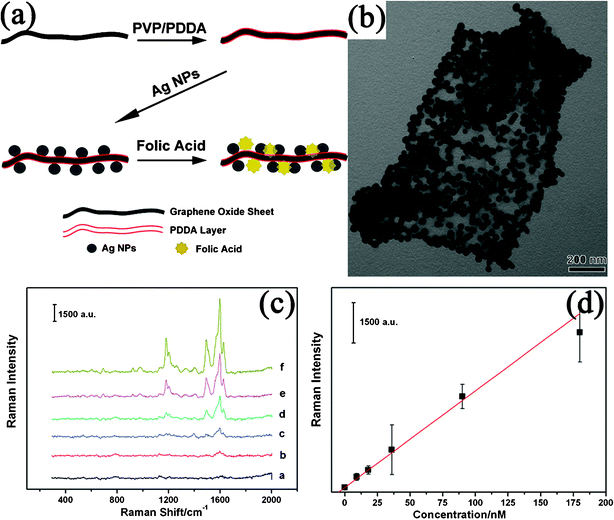 | ||
| Fig. 15 (a) Scheme illustrated of the fabricated of PDDA–GO-supported Ag NPs and the procedure of SERS detection using PDDA–GO–Ag as substrates; (b) TEM image of PDDA–GO-supported Ag NPs; (c) SERS spectra of different concentrations of folic acid with PDDA–GO–Ag as SERS substrates in diluted serum: (a) blank, (b) 9 nM, (c) 18 nM, (d) 36 nM, (e) 90 nM, and (f) 180 nM; and (d) SERS dilution series of folic acid in diluted serum based on the peak located at 1595 cm−1. Reprinted with permission from ref. 530. Copyright 2011, American Chemical Society. | ||
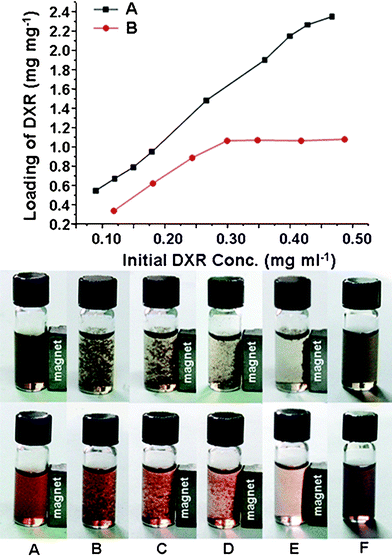 | ||
| Fig. 16 (Top) Loading capacity of DXR on GO (A) and GO–Fe3O4 composite (B) in different initial DXR concentrations. (Bottom) Photographic images of the behaviors of GO–Fe3O4 composite (upper) and GO–Fe3O4 composite loaded with DXR (lower) in the magnetic field under different conditions: neutral conditions (A), acidic conditions (pH 2–3) (B–E) and basic conditions (pH 8–9) (F). Reprinted with permission from ref. 216. Copyright 2009, Royal Society of Chemistry. | ||
Besides GN-supported magnetic NPs as adsorbents as well as GN-supported photocatalyst NPs as reductants262,414 (Section 4.1.3.), contaminants and heavy metal ion removal was also reported with other GN-supported NPs as adsorbents, such as ZnO,421Cu2O,444 ferric hydroxide,534MnO2 and Ag.535 Apart from anincrease of the adsorption capacity, the introduction of GN could also improve the chemical stability and mechanical strength of the adsorbents, which is important in separation technology. Also, GN–Ag composites were reported to exhibit excellent antibacterial activity towards bacteria such as Escherichia coli, Pseudomonous aeruginosa, etc.202,536–539Ag has been known as an antibacterial agent for centuries. Studies have shown that the toxicity of Ag NPs is size dependent and the smaller NPs exhibit higher antibacterial activity. GN can effectively stabilize highly monodispersed Ag NPs. Meanwhile, GN could adsorb the bacteria, and keep Ag NPs well-dispersed in solutions. Also GN (GO) nanosheets display antibacterial activity themselves. All of these contributed to the superior antibacterial activity of the nanocomposites.
5. Conclusion, challenges and perspective
In summary, GN nanosheets have been integrated with various inorganic components (including metal, nano-carbon, metal compounds, and nonmetal materials) to form nanocomposites. GN nanosheets could act as a substrate for supporting the inorganic nanomaterials, form shells to enwrap the inorganic nanostructures, play a role of fillers to disperse in inorganic matrices, and also stack with inorganic nanosheets alternately to form multilayer films. Diverse approaches and techniques, such as solution deposition methods, directly decomposition of precursors, hydrothermal/solvothermal techniques, gas-phase deposition, sol–gel processing, template method, covalent grafting and noncovalent interactions have been developed for fabricating GN–inorganic nanocomposites. GN–inorganic nanocomposites have become one of the most important GN-based materials.540 The nanocomposites exhibit excellent properties and improved functionalities due to the synergetic effects between GN and the inorganic components, thus have been widely used in the fields of catalysis, energy, analytical, biotechnology and pollutants removal applications. Especially, the catalysis application is of significance in respect that it is the basement in developing many other applications of GN–inorganic nanocomposites, such as electrocatalysis in developing electrochemical sensors and fuel cells, as well as photocatalysis in hydrogen energy, solar cells, and pollutants removal applications (Fig. 17). It is expected that the development of clean and renewable energy resources to improve the quality of environment is one of the most promising directions in the progress of GN–inorganic nanocomposites. | ||
| Fig. 17 Relationship between the various applications of GN–inorganic nanocomposites. | ||
With the rapid development of GN–inorganic nanocomposites, some new kinds of inorganic materials are being blended with GN, and many new applications are arising. For instance, GN-supported polyoxometalate,541–544 and Prussian blue545–547 NPs have been synthesized for application in hydrogen storage, electronics, electrochemical catalysis and sensing. However, further effort for GN–inorganic nanocomposites is still needed to realize their practical applications. First, as an essential component of the nanocomposites, high-quality GN was required, which greatly affect the reinforcement of GN in the novel properties and potential applications of the nanocomposites. So far, it is still difficult to synthesize GN with controllable sizes, shapes, layers, compositions and defects in a low-cost, high-yield, and environmentally friendly manner, although we have witnessed a promising step forward in this direction. The second challenge involves the control of the size, morphology, crystallinity, phase composition, and distribution of the inorganic components on GN nanosheets. The properties and functions of the nanocomposites depend strongly on micro structures of the second components. However, the exact mechanism of how the synthesis variable precisely correlates with inorganic nanostructures stabilized on GN is rarely known. Therefore, the assembly behaviors of GN sheets with inorganic building blocks are required to be investigated more clearly for synthesizing nanocomposites with uniform morphology and distribution in the nanoscale. The third one is the improvement of the adhesion between GN and inorganic components, which is especially important for applications that involve charge transfer processeses. A poor interfacial contact will destroy the synergetic effects, and thus diminish the performance of the nanocomposites. So various surface functionalization methods, synthesis techniques, and novel architectures should be developed to make sure a large interfacial area between GN and inorganic components. Fourthly, a successful design and application of GN–inorganic nanocomposites also requires improvements in methodology to ensure better understanding of the components interaction mechanism and structure–property relationship in the nanocomposites. This needs to be studied systematically from both theoretical and experimental aspects, which will be a key to guide the future work of researchers and promote the development of GN–inorganic nanocomposites. Finally, further development and successful implementation of GN–inorganic nanocomposites require the multidisciplinary efforts from physics, chemistry, biology and materials science.
Acknowledgements
The authors are grateful for financial support from the Natural Science Foundation of Jiangsu Province (No. BK2009196) and the National Natural Science Foundation of China (No. 51072071).References
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S.Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308–1308 CrossRef CAS.
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS.
- X. H. Peng, J. Y. Chen, J. A. Misewich and S. S. Wong, Chem. Soc. Rev., 2009, 38, 1076–1098 RSC.
- Y. B. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS.
- K. S. Novoselov, E. McCann, S. V. Morozov, V. I. Fal''ko, M. I. Katsnelson, U. Zeitler, D. Jiang, F. Schedin and A. K. Geim, Nat. Phys., 2006, 2, 177–180 CrossRef.
- S. Latil and L. Henrard, Phys. Rev. Lett., 2006, 97, 036803 CrossRef.
- E. T. Thostenson, Z. F. Ren and T. W. Chou, Compos. Sci. Technol., 2001, 61, 1899–1912 CrossRef CAS.
- H. Qian, E. S. Greenhalgh, M. S. P. Shaffe and A. Bismarck, J. Mater. Chem., 2010, 20, 4751–4762 RSC.
- K. F. Zhou, Y. H. Zhu, X. L. Yang and C. Z. Li, New J. Chem., 2010, 34, 2950–2955 RSC.
- C. Nethravathi, M. Rajamathi, N. Ravishankar, L. Basit and C. Felser, Carbon, 2010, 48, 4343–4350 CrossRef CAS.
- B. J. Jiang, C. G. Tian, W. Zhou, J. Q. Wang, Y. Xie, Q. J. Pan, Z. Y. Ren, Y. Z. Dong, D. Fu, J. L. Han and H. G. Fu, Chem.–Eur. J., 2011, 17, 8379–8387 CrossRef CAS.
- Z. Jin, D. Nackashi, W. Lu, C. Kittrell and J. M. Tour, Chem. Mater., 2010, 22, 5695–5699 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- Y. C. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792–6797 CrossRef CAS.
- S. V. Morozov, K. S. Novoselov, M. I. Katsnelson, F. Schedin, D. C. Elias, J. A. Jaszczak and A. K. Geim, Phys. Rev. Lett., 2008, 100, 016602 CrossRef CAS.
- T. K. Hong, D. W. Lee, H. J. Choi, H. S. Shin and B. S. Kim, ACS Nano, 2010, 4, 3861–3868 CrossRef CAS.
- V. C. Tung, L. M. Chen, M. J. Allen, J. K. Wassei, K. Nelson, R. B. Kaner and Y. Yang, Nano Lett., 2009, 9, 1949–1955 CrossRef CAS.
- C. Gomez-Navarro, M. Burghard and K. Kern, Nano Lett., 2008, 8, 2045–2049 CrossRef CAS.
- K. J. Jeon and Z. Lee, Chem. Commun., 2011, 47, 3610–3612 RSC.
- S. Park, J. H. An, I. W. Jung, R. D. Piner, S. J. An, X. S. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS.
- N. Karousis, N. Tagmatarchis and D. Tasis, Chem. Rev., 2010, 110, 5366–5397 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun''ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov, A. K. Geim, Proc. Natl. Acad. Sci.USA, 2005, 102, 10451–10453 Search PubMed.
- A. Reina, X. T. Jia, J. Ho, D. Nezich, H. B. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 30–35 CrossRef CAS.
- C. Mattevi, H. Kima and M. Chhowalla, J. Mater. Chem., 2011, 21, 3324–3334 RSC.
- W. A. de Heer, C. Berger, X. S. Wu, P. N. First, E. H. Conrad, X. B. Li, T. B. Li, M. Sprinkle, J. Hass, M. L. Sadowski, M. Potemski and G. Martinez, Solid State Commun., 2007, 143, 92–100 CrossRef CAS.
- P. W. Sutter, J. I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406–411 CrossRef CAS.
- S. Shivaraman, R. A. Barton, X. Yu, J. Alden, L. Herman, M. V. S. Chandrashekhar, J. Park, P. L. McEuen, J. M. Parpia, H. G. Craighead and M. G. Spencer, Nano Lett., 2009, 9, 3100–3105 CrossRef CAS.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS.
- L. Y. Jiao, L. Zhang, X. R. Wang, G. Diankov and H. J. Dai, Nature, 2009, 458, 877–880 CrossRef CAS.
- L. Y. Jiao, X. R. Wang, G. Diankov, H. L. Wang and H. J. Dai, Nat. Nanotechnol., 2010, 5, 321–325 CrossRef CAS.
- X. Yan, X. Cui and L. S. Li, J. Am. Chem. Soc., 2010, 132, 5944–5945 CrossRef CAS.
- X. Yan and L. S. Li, J. Mater. Chem., 2011, 21, 3295–3300 RSC.
- S. Stankovich, R. D. Piner, X. Q. Chen, N. Q. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- K. A. Worsley, P. Ramesh, S. K. Mandal, S. Niyogi, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2007, 445, 51–56 CrossRef CAS.
- S. Park, J. H. An, R. D. Piner, I. Jung, D. X. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- D. C. Wei and Y. Q. Liu, Adv. Mater., 2010, 22, 3225–3241 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS.
- B. C. Brodie, Philos. Trans. R. Soc.London, 1859, 149, 249–259 Search PubMed.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481–1487 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud''homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS.
- O. C. Compton, D. A. Dikin, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2010, 22, 892–896 CrossRef CAS.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner, G. G. Wallace and G. Gordon, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- J. F. Shen, Y. Z. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. X. Ye, Chem. Mater., 2009, 21, 3514–3520 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- G. X. Wang, J. Yang, J. Park, X. L. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CrossRef CAS.
- G. X. Wang, X. P. Shen, B. Wang, J. Yao and J. Park, Carbon, 2009, 47, 1359–1364 CrossRef CAS.
- X. B. Fan, W. C. Peng, Y. Li, X. Y. Li, S. L. Wang, G. L. Zhang and F. B. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- W. F. Chen, L. F. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885–19890 CrossRef CAS.
- T. N. Zhou, F. Chen, K. Liu, H. Deng, Q. Zhang, J. W. Feng and Q. A. Fu, Nanotechnology, 2011, 22, 045704 CrossRef.
- X. P. Shen, L. Jiang, Z. Y. Ji, J. L. Wu, H. Zhou and G. X. Zhu, J. Colloid Interface Sci., 2010, 354, 493–497 Search PubMed.
- J. F. Che, L. Y. Shen and Y. H. Xiao, J. Mater. Chem., 2010, 20, 1722–1727 RSC.
- M. J. Fernandez-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. M. D. Tascon, J. Phys. Chem. C, 2010, 114, 6426–6432 CrossRef CAS.
- J. Gao, F. Liu, Y. L. Liu, N. Ma, Z. Q. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- C. Z. Zhu, S. J. Guo, Y. X. Fang and S. J. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS.
- D. R. Dreyer, S. Murali, Y. Zhu, R. S. Ruoff and C. W. Bielawski, J. Mater. Chem., 2011, 21, 3443–3447 RSC.
- S. F. Pei, J. P. Zhao, J. H. Du, W. C. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- Z. J. Fan, K. Wang, T. Wei, J. Yan, L. P. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS.
- Z. J. Fan, K. Wang, J. Yan, T. Wei, L. J. Zhi, J. Feng, Y. M. Ren, L. P. Song and F. Wei, ACS Nano, 2011, 5, 191–198 CrossRef CAS.
- Z. Zhang, H. H. Cheng, C. Y. Xing, M. Y. Guo, F. G. Xu, X. D. Wang, H. Gruber, B. L. Zhang and J. L. Tang, Nano Res., 2011, 4, 599–611 CrossRef CAS.
- Y. Wang, Z. X. Shi and J. Yin, ACS Appl. Mater. Interfaces, 2011, 3, 1127–1133 CrossRef CAS.
- F. Yang, Y. Q. Liu, L. A. Gao and J. Sun, J. Phys. Chem. C, 2010, 114, 22085–22091 CrossRef CAS.
- Y. W. Zhu, M. D. Stoller, W. W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS.
- W. F. Chen and L. F. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- Y. Zhou, Q. L. Bao, L. A. L. Tang, Y. L. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Chem. Mater., 2009, 21, 5004–5006 CrossRef CAS.
- W. F. Chen, L. F. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- M. Zhou, Y. L. Wang, Y. M. Zhai, J. F. Zhai, W. Ren, F. A. Wang and S. J. Dong, Chem.–Eur. J., 2009, 15, 6116–6120 CrossRef CAS.
- S. J. An, Y. W. Zhu, S. H. Lee, M. D. Stoller, T. Emilsson, S. Park, A. Velamakanni, J. H. An and R. S. Ruoff, J. Phys. Chem. Lett., 2010, 1, 1259–1263 Search PubMed.
- Y. Y. Shao, J. Wang, M. Engelhard, C. M. Wang and Y. H. Lin, J. Mater. Chem., 2010, 20, 743–748 RSC.
- Y. H. Ding, P. Zhang, Q. Zhuo, H. M. Ren, Z. M. Yang and Y. Jiang, Nanotechnology, 2011, 22, 215601 CrossRef CAS.
- E. C. Salas, Z. Z. Sun, A. Luttge and J. M. Tour, ACS Nano, 2010, 4, 4852–4856 CrossRef CAS.
- L. Huang, Y. Liu, L. C. Ji, Y. Q. Xie, T. Wang and W. Z. Shi, Carbon, 2011, 49, 2431–2436 Search PubMed.
- M. Baraket, S. G. Walton, Z. Wei, E. H. Lock, J. T. Robinson and P. Sheehan, Carbon, 2010, 48, 3382–3390 Search PubMed.
- D. C. Guo, G. X. Zhang, J. F. Liu and X. M. Sun, J. Phys. Chem. C, 2011, 115, 11327–11335 CAS.
- J. I. Paredes, S. Villar-Rodil, M. J. Fernandez-Merino, L. Guardia, A. Martinez-Alonso and J. M. D. Tascon, J. Mater. Chem., 2011, 21, 298–306 RSC.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720–7721 CrossRef CAS.
- Y. X. Xu, H. Bai, G. W. Lu, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS.
- Z. F. Liu, Q. Liu, Y. Huang, Y. F. Ma, S. G. Yin, X. Y. Zhang, W. Sun and Y. S. Chen, Adv. Mater., 2008, 20, 3924–3930 CrossRef CAS.
- X. L. Li, X. R. Wang, L. Zhang, S. W. Lee and H. J. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS.
- H. F. Yang, C. S. Shan, F. H. Li, D. X. Han, Q. X. Zhang and L. Niu, Chem. Commun., 2009, 3880–3882 RSC.
- M. C. Hsiao, S. H. Liao, M. Y. Yen, P. I. Liu, N. W. Pu, C. A. Wang and C. C. M. Ma, ACS Appl. Mater. Interfaces, 2010, 2, 3092–3099 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS.
- Y. F. Xu, Z. B. Liu, X. L. Zhang, Y. Wang, J. G. Tian, Y. Huang, Y. F. Ma, X. Y. Zhang and Y. S. Chen, Adv. Mater., 2009, 21, 1275–1279 CrossRef CAS.
- A. J. Patil, J. L. Vickery, T. B. Scott and S. Mann, Adv. Mater., 2009, 21, 3159–3164 CrossRef CAS.
- X. Y. Yang, X. Y. Zhang, Z. F. Liu, Y. F. Ma, Y. Huang and Y. S. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS.
- W. Lv, M. Guo, M. H. Liang, F. M. Jin, L. Cui, L. J. Zhi and Q. H. Yang, J. Mater. Chem., 2010, 20, 6668–6673 RSC.
- J. Geng and H. T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 CrossRef CAS.
- Y. Y. Liang, D. Q. Wu, X. L. Feng and K. Mullen, Adv. Mater., 2009, 21, 1679–1683 CrossRef CAS.
- S. Z. Zu and B. H. Han, J. Phys. Chem. C, 2009, 113, 13651–13657 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- J. J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. F. Ma, T. Y. Guo and Y. S. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. H. Song, Z. Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef CAS.
- T. Kuilla, S. Bhadra, D. H. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, Polymer, 2011, 52, 5–25 CrossRef CAS.
- R. Verdejo, M. M. Bernal, L. J. Romasanta and M. A. Lopez-Manchado, J. Mater. Chem., 2011, 21, 3301–3310 RSC.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263–5266 CrossRef CAS.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Gracio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- X. Huang, Z. S. Li, Y. Z. Huang, S. X. Wu, X. Z. Zhou, S. Z. Li, C. L. Gan, F. Boey, C. A. Mirkin and H. Zhang, Nat. Commun., 2011, 2, 292 Search PubMed.
- B. S. Kong, J. X. Geng and H. T. Jung, Chem. Commun., 2009, 2174–2176 RSC.
- R. Pasricha, S. Gupta and A. K. Srivastava, Small, 2009, 5, 2253–2259 CrossRef CAS.
- S. Bai, X. P. Shen, G. X. Zhu, Z. Xu and Y. J. Liu, Carbon, 2011, 49, 4563–4570 Search PubMed.
- J. Li and C. Y. Liu, Eur. J. Inorg. Chem., 2010, 1244–1248 CrossRef CAS.
- S. Zhang, Y. Y. Shao, H. G. Liao, M. H. Engelhard, G. P. Yin and Y. H. Lin, ACS Nano, 2011, 5, 1785–1791 CrossRef CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CrossRef CAS.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Muelhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS.
- N. Li, Z. Y. Wang, K. K. Zhao, Z. J. Shi, S. K. Xu and Z. N. Gu, J. Nanosci. Nanotechnol., 2010, 10, 6748–6751 Search PubMed.
- K. S. Subrahmanyam, A. K. Manna, S. K. Pati and C. N. R. Rao, Chem. Phys. Lett., 2010, 497, 70–75 CrossRef CAS.
- N. A. Luechinger, E. K. Athanassiou and W. J. Stark, Nanotechnology, 2008, 19, 445201 CrossRef.
- X. B. Bin, J. Z. Chen, H. Cao, L. T. Chen and J. Z. Yuan, J. Phys. Chem. Solids, 2009, 70, 1–7 CrossRef CAS.
- G. X. Wang, B. Wang, X. L. Wang, J. Park, S. X. Dou, H. Ahn and K. Kim, J. Mater. Chem., 2009, 19, 8378–8384 RSC.
- J. H. Warner, M. H. Ruemmeli, A. Bachmatiuk, M. Wilson and B. Buechner, ACS Nano, 2010, 4, 470–476 CrossRef CAS.
- J. Bian, X. W. Wei, L. Wang and Z. P. Guan, Chinese Phys. Lett., 2011, 22, 57–60 Search PubMed.
- J. K. Wassei, K. C. Cha, V. C. Tung, Y. Yang and R. B. Kaner, J. Mater. Chem., 2011, 21, 3391–3396 RSC.
- S. Das, R. Seelaboyina, V. Verma, I. Lahiri, J. Y. Hwang, R. Banerjee and W. B. Choi, J. Mater. Chem., 2011, 21, 7289–7295 RSC.
- X. C. Dong, B. Li, A. Wei, X. H. Cao, M. B. Chan-Park, H. Zhang, L. J. Li, W. Huang and P. Chen, Carbon, 2011, 49, 2944–2949 CrossRef CAS.
- D. S. Yu, K. Park, M. Durstock and L. M. Dai, J. Phys. Chem. Lett., 2011, 2, 1113–1118 Search PubMed.
- J. Yan, T. Wei, B. Shao, F. Q. Ma, Z. J. Fan, M. L. Zhang, C. Zheng, Y. C. Shang, W. Z. Qian and F. Wei, Carbon, 2010, 48, 1731–1737 CrossRef CAS.
- Z. B. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 RSC.
- Z. J. Fan, J. Yan, T. Wei, G. Q. Ning, L. J. Zhi, J. C. Liu, D. X. Cao, G. L. Wang and F. Wei, ACS Nano, 2011, 5, 2787–2794 CrossRef CAS.
- T. T. Baby and R. Sundara, J. Phys. Chem. C, 2011, 115, 8527–8533 CrossRef CAS.
- P. Wang, Y. M. Zhai, D. J. Wang and S. J. Dong, Nanoscale, 2011, 3, 1640–1645 RSC.
- H. Y. Koo, H. J. Lee, H. A.Go, Y. B. Lee, T. S. Bae, J. K. Kim and W. S. Choi, Chem.–Eur. J., 2011, 17, 1214–1219 CrossRef CAS.
- C. Xu, X. Wang, L. C. Yang and Y. P. Wu, J. Solid State Chem., 2009, 182, 2486–2490 CrossRef CAS.
- C. Chen, W. M. Cai, M. C. Long, B. X. Zhou, Y. H. Wu, D. Y. Wu and Y. J. Feng, ACS Nano, 2010, 4, 6425–6432 CrossRef CAS.
- H. K. He and C. Gao, ACS Appl. Mater. Interfaces, 2010, 2, 3201–3210 CrossRef CAS.
- S. Chen, J. W. Zhu and X. Wang, J. Phys. Chem. C, 2010, 114, 11829–11834 CrossRef CAS.
- Z. Gao, J. Wang, Z. S. Li, W. L. Yang, B. Wang, M. J. Hou, Y. He, Q. Liu, T. Mann, P. P. Yang, M. L. Zhang and L. H. Liu, Chem. Mater., 2011, 23, 3509–3516 CrossRef CAS.
- J. L. Wu, S. Bai, X. P. Shen and L. Jiang, Appl. Surf. Sci., 2010, 257, 747–751 CrossRef CAS.
- C. Z. Yuan, L. R. Hou, L. Yang, C. G. Fan, D. K. Li, J. M. Li, L. F. Shen, F. Zhang and X. G. Zhang, Mater. Lett., 2011, 65, 374–377 Search PubMed.
- Y. Wang, H. B. Yao, X. H. Wang and S. H. Yu, J. Mater. Chem., 2011, 21, 562–566 RSC.
- Y. Lin, K. Zhang, W. F. Chen, Y. D. Liu, Z. G. Geng, J. Zeng, N. Pan, L. F. Yan, X. P. Wang and J. G. Hou, ACS Nano, 2010, 4, 3033–3038 CrossRef CAS.
- Y. C. Qiu, K. Y. Yan, S. H. Yang, L. M. Jin, H. Deng and W. S. Li, ACS Nano, 2010, 4, 6515–6526 CrossRef CAS.
- S. J. Kim, S. H. Ku, S. Y. Lim, J. H. Kim and C. B. Park, Adv. Mater., 2011, 23, 2009–2014 Search PubMed.
- C. Nethravathi, B. Viswanath, C. Shivakumara, N. Mahadevaiah and M. Rajamathi, Carbon, 2008, 46, 1773–1781 CrossRef.
- B. Das, B. Choudhury, A. Gomathi, A. K. Manna, S. K. Pati and C. N. R. Rao, ChemPhysChem, 2011, 12, 937–943 Search PubMed.
- Y. L. Cao, X. L. Li, I. A. Aksay, J. Lemmon, Z. M. Nie, Z. G. Yang and J. Liu, Phys. Chem. Chem. Phys., 2011, 13, 7660–7665 RSC.
- H. L. Wang, Y. Yang, Y. Y. Liang, J. T. Robinson, Y. G. Li, A. Jackson, Y. Cui and H. J. Dai, Nano Lett., 2011, 11, 2644–2647 Search PubMed.
- S. L. Chou, J. Z. Wang, M. Choucair, H. K. Liu, J. A. Stride and S. X. Dou, Electrochem. Commun., 2010, 12, 303–306 CrossRef CAS.
- J. Z. Wang, C. Zhong, S. L. Chou and H. K. Liu, Electrochem. Commun., 2010, 12, 1467–1470 CrossRef CAS.
- L. Kou and C. Gao, Nanoscale, 2011, 3, 519–528 RSC.
- L. S. Walker, V. R. Marotto, M. A. Rafiee, N. Koratkar and E. L. Corral, ACS Nano, 2011, 5, 3182–3190 Search PubMed.
- F. Ji, Y. L. Li, J. M. Feng, D. Su, Y. Y. Wen, Y. Feng and F. Hou, J. Mater. Chem., 2009, 19, 9063–9067 RSC.
- S. B. Yang, X. L. Feng, X. C. Wang and K. Mullen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS.
- Y. Q. Sun, C. Li, Y. X. Li, H. Bai, Z. Y. Yao and G. Q. Shi, Chem. Commun., 2010, 46, 4740–4742 RSC.
- X. H. Li, J. S. Chen, X. C. Wang, J. H. Sun and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074–8077 CrossRef CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS.
- R. Kumar, D. Varandani, B. R. Mehta, V. N. Singh, Z. H. Wen, X. L. Feng and K. Mullen, Nanotechnology, 2011, 22, 275719 CrossRef.
- N. Zhang, H. X. Qiu, Y. Liu, W. Wang, Y. Li, X. D. Wang and J. P. Gao, J. Mater. Chem., 2011, 21, 11080–11083 RSC.
- H. L. Wang, Y. Yang, Y. Y. Liang, L. F. Cui, H. S. Casalongue, Y. G. Li, G. S. Hong, Y. Cui and H. J. Dai, Angew. Chem., Int. Ed., 2011, 50, 7364–7368 CrossRef CAS.
- H. X. Chang, Z. H. Sun, K. Y. F. Ho and X. M. Tao, Nanoscale, 2011, 3, 258–264 RSC.
- H. J. Li, G. Zhu, Z. H. Liu, Z. P. Yang and Z. L. Wang, Carbon, 2010, 48, 4391–4396 CrossRef CAS.
- Y. Q. Zhou and Y. Wang, Nanoscale, 2011, 3, 2615–2620 RSC.
- H. L. Wang, H. S. Casalongue, Y. Y. Liang and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS.
- J. Choi, J. Jin, I. G. Jung, J. M. Kim, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 5241–5243 RSC.
- S. Kim, H. Choi, M. Jung, S. Y. Choi and S. Ju, Nanotechnology, 2010, 21, 425203 Search PubMed.
- B. Kumar, K. Y. Lee, H. K. Park, S. J. Chae, Y. H. Lee and S. W. Kee, ACS Nano, 2011, 5, 4197–4204 CrossRef CAS.
- J. X. Zhu, Y. K. Sharma, Z. Y. Zeng, X. J. Zhang, M. Srinivasan, S. Mhaisalkar, H. Zhang, H. H. Hng and Q. Y. Yan, J. Phys. Chem. C, 2011, 115, 8400–8406 CrossRef CAS.
- C. Z. Zhu, S. J. Guo, Y. X. Fang, L. Han, E. K. Wang and S. J. Dong, Nano Res., 2011, 4, 648–657 CrossRef CAS.
- V. Jousseaume, J. Cuzzocrea, N. Bernier and V. T. Renard, Appl. Phys. Lett., 2011, 98, 123103 CrossRef.
- R. Lv, T. Cui, M. Jun, Q. Zhang, A. Cao, D. S. Su, Z. Zhang, S. Yoon, J. Miyawaki, I. Mochida and F. Kang, Adv. Funct. Mater., 2011, 21, 999–1006 CrossRef CAS.
- Y. J. Kim, J. H. Lee and G. C. Yi, Appl. Phys. Lett., 2009, 95, 213101 CrossRef.
- C. Y. Li, Z. Li, H. W. Zhu, K. L. Wang, J. Q. Wei, X. A. Li, P. Z. Sun, H. Zhang and D. H. Wu, J. Phys. Chem. C, 2010, 114, 14008–14012 CrossRef CAS.
- J. J. Xu, K. Wang, S. Z. Zu, B. H. Han and Z. X. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS.
- H. T. Hu, X. B. Wang, J. C. Wang, L. Wan, F. M. Liu, H. Zheng, R. Chen and H. C. Xu, Chem. Phys. Lett., 2010, 484, 247–253 CrossRef CAS.
- S. Myung, A. Solanki, C. Kim, J. Park, K. S. Kim and K. B. Lee, Adv. Mater., 2011, 23, 2221–2225 CrossRef CAS.
- J. S. Chen, Z. Y. Wang, X. C. Dong, P. Chen and X. W. Lou, Nanoscale, 2011, 3, 2158–2161 RSC.
- X. Liu, Y. S. Hu, J. O. Müller, R. Schlogl, J. Maier and D. S. Su, ChemSusChem, 2010, 3, 261–265 CrossRef CAS.
- L. Jiang, X. P. Shen, J. L. Wu and K. C. Shen, J. Appl. Polym. Sci., 2010, 118, 275–279 CAS.
- Y. B. Tang, C. S. Lee, J. Xu, Z. T. Liu, Z. H. Chen, Z. B. He, Y. L. Cao, G. D. Yuan, H. S. Song, L. M. Chen, L. B. Luo, H. M. Cheng, W. J. Zhang, I. Bello and S. T. Lee, ACS Nano, 2010, 4, 3482–3488 CrossRef CAS.
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung, G. H. B. Dommett, G. Evmenenko, S. E. Wu, S. F. Chen, C. P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888–1892 CrossRef CAS.
- Z. J. Fan, K. Wang, J. Yan and T. Wei, Mater. Res. Bull., 2011, 46, 315–318 CrossRef CAS.
- Y. C. Fan, L. J. Wang, J. L. Li, J. Q. Li, S. K. Sun, F. Chen, L. D. Chen and W. Jiang, Carbon, 2010, 48, 1743–1749 CrossRef CAS.
- W. Lv, F. Sun, D. M. Tang, H. T. Fang, C. Liu, Q. H. Yang and H. M. Cheng, J. Mater. Chem., 2011, 21, 9014–9019 RSC.
- L. Wang, D. Wang, X. Y. Dong, Z. J. Zhang, X. F. Pei, X. J. Chen, B. Chen and J. Jin, Chem. Commun., 2011, 47, 3556–3558 RSC.
- X. Z. Tang, Z. W. Cao, H. B. Zhang, J. Liu and Z. Z. Yu, Chem. Commun., 2011, 47, 3084–3086 RSC.
- M. R. Das, R. K. Sarma, R. Saikia, V. S. Kale, M. V. Shelke and P. Sengupta, Colloids Surf., B, 2011, 83, 16–22 CrossRef CAS.
- H. W. Tien, Y. L. Huang, S. Y. Yang, J. Y. Wang and C. C. M. Ma, Carbon, 2011, 49, 1550–1560 CrossRef CAS.
- Y. M. Li, L. H. Tang and J. H. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- O. L. Yue, K. Zhang, X. M. Chen, L. Wang, J. S. Zhao, J. F. Liu and J. B. Jia, Chem. Commun., 2010, 46, 3369–3371 RSC.
- S. Q. Chen, P. Chen, M. H. Wu, D. Y. Pan and Y. Wang, Electrochem. Commun., 2010, 12, 1302–1306 CrossRef CAS.
- K. Zhang, Q. L. Yue, G. F. Chen, Y. L. Zhai, L. Wang, H. S. Wang, J. S. Zhao, J. F. Liu, J. B. Jia and H. B. Li, J. Phys. Chem. C, 2011, 115, 379–389 CrossRef CAS.
- C. Xu, X. Wang and J. W. Zhu, J. Phys. Chem. C, 2008, 112, 19841–19845 CrossRef CAS.
- Y. J. Li, W. Gao, L. J. Ci, C. M. Wang and P. M. Ajayan, Carbon, 2010, 48, 1124–1130 CrossRef CAS.
- L. F. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. F. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- C. V. Rao, A. L. M. Reddy, Y. Ishikawa and P. M. Ajayan, Carbon, 2011, 49, 931–936 CrossRef CAS.
- Y. Chen, Y. Li, D. Sun, D. B. Tian, J. R. Zhang and J. J. Zhu, J. Mater. Chem., 2011, 21, 7604–7611 RSC.
- X. Yang, M. S. Xu, W. M. Qiu, X. Q. Chen, M. Deng, J. L. Zhang, H. Iwai, E. Watanabe and H. Z. Chen, J. Mater. Chem., 2011, 21, 8096–8103 RSC.
- C. Xu, X. Wang, J. W. Zhu, X. J. Yang and L. Lu, J. Mater. Chem., 2008, 18, 5625–5629 RSC.
- J. W. Zhu, G. Y. Zeng, F. D. Nie, X. M. Xu, S. Chen, Q. F. Han and X. Wang, Nanoscale, 2010, 2, 988–994 RSC.
- X. Y. Yang, X. Y. Zhang, Y. F. Ma, Y. Huang, Y. S. Wang and Y. S. Chen, J. Mater. Chem., 2009, 19, 2710–2714 RSC.
- X. Y. Zhang, X. Y. Yang, Y. F. Ma, Y. Huang and Y. S. Chen, J. Nanosci. Nanotechnol., 2010, 10, 2984–2987 CrossRef CAS.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS.
- T. N. Lambert, C. A. Chavez, B. Hernandez-Sanchez, P. Lu, N. S. Bell, A. Ambrosini, T. Friedman, T. J. Boyle, D. R. Wheeler and D. L. Huber, J. Phys. Chem. C, 2009, 113, 19812–19823 CrossRef CAS.
- L. S. Zhang, L. Y. Jiang, H. J. Yan, W. D. Wang, W. Wang, W. G. Song, Y. G. Guo and L. J. Wan, J. Mater. Chem., 2010, 20, 5462–5467 RSC.
- C. Z. Zhu, S. J. Guo, P. Wang, L. Xing, Y. X. Fang, Y. M. Zhai and S. J. Dong, Chem. Commun., 2010, 46, 7148–7150 RSC.
- J. T. Zhang, Z. G. Xiong and X. S. Zhao, J. Mater. Chem., 2011, 21, 3634–3640 RSC.
- F. H. Li, J. F. Song, H. F. Yang, S. Y. Gan, Q. X. Zhang, D. X. Han, A. Ivaska and L. Niu, Nanotechnology, 2009, 20, 455602 CrossRef.
- X. Y. Wang, X. F. Zhou, K. Yao, J. G. Zhang and Z. P. Liu, Carbon, 2011, 49, 133–139 CrossRef CAS.
- D. H. Wang, R. Kou, D. Choi, Z. G. Yang, Z. M. Nie, J. Li, L. V. Saraf, D. H. Hu, J. G. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587–1595 CrossRef CAS.
- S. B. Yang, X. L. Feng, L. Wang, K. Tang, J. Maier and K. Mullen, Angew. Chem. Int. Ed., 2010, 49, 4795–4799 CAS.
- C. Nethravathi, T. Nisha, N. Ravishankar, C. Shivakumara and M. Rajamathi, Carbon, 2009, 47, 2054–2059 CrossRef CAS.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 CAS.
- M. Du, T. Yang and K. Jiao, J. Mater. Chem., 2010, 20, 9253–9260 RSC.
- C. P. Fu, Y. F. Kuang, Z. Y. Huang, X. A. Wang, N. N. Du, J. H. Chen and H. H. Zhou, Chem. Phys. Lett., 2010, 499, 250–253 CrossRef CAS.
- Y. J. Hu, J. Jin, P. Wu, H. Zhang and C. X. Cai, Electrochim. Acta, 2010, 56, 491–500 CrossRef CAS.
- C. B. Liu, K. Wang, S. L. Luo, Y. H. Tang and L. Y. Chen, Small, 2011, 7, 1203–1206 CrossRef CAS.
- S. Liu, J. Q. Wang, J. Zeng, J. F. Ou, Z. P. Li, X. H. Liu and S. R. Yang, J. Power Sources, 2010, 195, 4628–4633 CrossRef CAS.
- Y. G. Zhou, J. J. Chen, F. B. Wang, Z. H. Sheng and X. H. Xia, Chem. Commun., 2010, 46, 5951–5953 RSC.
- Y. J. Hu, H. Zhang, P. Wu, H. Zhang, B. Zhou and C. X. Cai, Phys. Chem. Chem. Phys., 2011, 13, 4083–4094 RSC.
- S. X. Wu, Z. Y. Yin, Q. Y. He, X. A. Huang, X. Z. Zhou and H. Zhang, J. Phys. Chem. C, 2010, 114, 11816–11821 CrossRef CAS.
- S. X. Wu, Z. Y. Yin, Q. Y. He, G. Lu, X. Z. Zhou and H. Zhang, J. Mater. Chem., 2011, 21, 3467–3470 RSC.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L. C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS.
- X. Z. Zhou, X. Huang, X. Y. Qi, S. X. Wu, C. Xue, F. Y. C. Boey, Q. Y. Yan, P. Chen and H. Zhang, J. Phys. Chem. C, 2009, 113, 10842–10846 CrossRef CAS.
- Y. K. Kim, H. K. Na, Y. W. Lee, H. Jang, S. W. Han and D. H. Min, Chem. Commun., 2010, 46, 3185–3187 RSC.
- X. M. Chen, G. H. Wu, J. M. Chen, X. Chen, Z. X. Xie and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS.
- X. W. Liu, J. J. Mao, P. d. Liu and X. W. Wei, Carbon, 2011, 49, 477–483 Search PubMed.
- P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 520–527 Search PubMed.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- B. Li, X. T. Zhang, X. H. Li, L. Wang, R. Y. Han, B. B. Liu, W. T. Zheng, X. L. Li and Y. C. Liu, Chem. Commun., 2010, 46, 3499–3501 RSC.
- T. N. Lambert, C. A. Chavez, N. S. Bell, C. M. Washburn, D. R. Wheeler and M. T. Brumbache, Nanoscale, 2011, 3, 188–191 RSC.
- G. Williams and P. V. Kamat, Langmuir, 2009, 25, 13869–13873 CrossRef CAS.
- O. Akhavan, Carbon, 2011, 49, 11–18 CrossRef CAS.
- Y. H. Ng, A. Iwase, N. J. Bell, A. Kudo and R. Amal, Catal. Today, 2011, 164, 353–357 CrossRef CAS.
- X. Huang, X. Z. Zhou, S. X. Wu, Y. Y. Wei, X. Y. Qi, J. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 513–516 CrossRef CAS.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577–583 CrossRef CAS.
- G. H. Moon, Y. Park, W. Kim and W. Choi, Carbon, 2011, 49, 3454–3462 Search PubMed.
- H. M. A. Hassan, V. Abdelsayed, A. E. R. S. Khder, K. M. AbouZeid, J. Terner, M. S. El-Shall, S. I. Al-Resayes and A. A. El-Azhary, J. Mater. Chem., 2009, 19, 3832–3837 RSC.
- S. Sharma, A. Ganguly, P. Papakonstantinou, X. P. Miao, M. X. Li, J. L. Hutchison, M. Delichatsios and S. Ukleja, J. Phys. Chem. C, 2010, 114, 19459–19466 CrossRef CAS.
- P. Kundu, C. Nethravathi, P. A. Deshpande, M. Rajamathi, G. Madras and N. Ravishankar, Chem. Mater., 2011, 23, 2772–2780 CrossRef CAS.
- K. Jasuja, J. Linn, S. Melton and V. Berry, J. Phys. Chem. Lett., 2010, 1, 1853–1860 Search PubMed.
- A. F. Zedan, S. Sappal, S. Moussa and M. S. El-Shall, J. Phys. Chem. C, 2010, 114, 19920–19927 CrossRef CAS.
- H. T. Hua, X. B. Wang, F. M. Liu, J. C. Wang and C. H. Xu, Synth. Met., 2011, 161, 404–410 CrossRef CAS.
- M. Zhang, D. N. Lei, X. M. Yin, L. B. Chen, Q. H. Li, Y. G. Wang and T. H. Wang, J. Mater. Chem., 2010, 20, 5538–5543 RSC.
- J. Yan, T. Wei, W. M. Qiao, B. Shao, Q. K. Zhao, L. J. Zhang and Z. J. Fan, Electrochim. Acta, 2010, 55, 6973–6978 CrossRef CAS.
- M. Zhang, D. Lei, Z. F. Du, X. M. Yin, L. B. Chen, Q. H. Li, Y. G. Wang and T. H. Wang, J. Mater. Chem., 2011, 21, 1673–1676 RSC.
- X. J. Liu, L. K. Pan, T. Lv, T. Lu, G. Zhu, Z. Sun and C. Q. Sun, Catal. Sci. Technol., 2011, 1, 1189–1193 RSC.
- Y. Lin, D. W. Baggett, J. W. Kim, E. J. Siochi and J. W. Connell, ACS Appl. Mater. Interfaces, 2011, 3, 1652–1664 CrossRef CAS.
- Z. S. Wu, W. C. Ren, L. B. Gao, B. L. Liu, J. P. Zhao and H. M. Cheng, Nano Res., 2010, 3, 16–22 CrossRef CAS.
- K. Vinodgopal, B. Neppolian, I. V. Lightcap, F. Grieser, M. Ashokkumar and P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 1987–1993 Search PubMed.
- S. Chandra, S. Bag, R. Bhar and P. Pramanik, J. Nanopart. Res., 2011, 13, 2769–2777 Search PubMed.
- J. J. Guo, S. M. Zhu, Z. X. Chen, Y. Li, Z. Y. Yong, Q. L. Liu, J. B. Li, C. L. Feng and D. Zhang, Ultrason. Sonochem., 2011, 18, 1082–1090 CrossRef CAS.
- J. J. Shi and J. J. Zhu, Electrochim. Acta, 2011, 56, 6008–6013 CrossRef CAS.
- J. J. Shi, G. H. Yang and J. J. Zhu, J. Mater. Chem., 2011, 21, 7343–7349 RSC.
- S. Q. Chen and Y. Wang, J. Mater. Chem., 2010, 20, 9735–9739 RSC.
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- G. M. Zhou, D. W. Wang, F. Li, L. L. Zhang, N. Li, Z. S. Wu, L. Wen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- Z. Y. Ji, J. L. Wu, X. P. Shen, H. Zhou and H. T. Xi, J. Mater. Sci., 2011, 46, 1190–1195 CrossRef CAS.
- K. H. Chang, Y. F. Lee, C. C. Hu, C. I. Chang, C. L. Liu and Y. L. Yang, Chem. Commun., 2010, 46, 7957–7959 RSC.
- H. W. Wang, Z. A. Hu, Y. Q. Chang, Y. L. Chen, H. Y. Wu, Z. Y. Zhang and Y. Y. Yang, J. Mater. Chem., 2011, 21, 10504–10511 RSC.
- V. K. Singh, M. K. Patra, M. Manoth, G. S. Gowd, S. R. Vadera and N. Kumar, New Carbon Mater., 2009, 24, 147–152 Search PubMed.
- J. F. Shen, Y. Z. Hu, M. Shi, N. Li, H. W. Ma and M. X. Ye, J. Phys. Chem. C, 2010, 114, 1498–1503 CrossRef CAS.
- H. P. Cong, J. J. He, Y. Lu and S. H. Yu, Small, 2010, 6, 169–173 CrossRef CAS.
- D. Marquardt, C. Vollmer, R. Thomann, P. Steurer, R. Mülhaupt, E. Redel and C. Janiak, Carbon, 2011, 49, 1326–1332 CrossRef CAS.
- Q. Su, Y. Y. Liang, X. L. Feng and K. Mullen, Chem. Commun., 2010, 46, 8279–8281 RSC.
- S. B. Yang, G. L. Cui, S. P. Pang, Q. Cao, U. Kolb, X. L. Feng, J. Maier and K. Mullen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS.
- K. Gotoh, K. Kawabata, E. Fujii, K. Morishige, T. Kinumoto, Y. Miyazaki and H. Ishida, Carbon, 2009, 47, 2120–2124 CrossRef CAS.
- K. Gotoh, T. Kinumoto, E. Fujii, A. Yamamoto, H. Hashimoto, T. Ohkubo, A. Itadani, Y. Kuroda and H. Ishida, Carbon, 2011, 49, 1118–1125 CrossRef CAS.
- J. F. Shen, M. Shi, B. Yan, H. W. Ma, N. Li and M. X. Ye, J. Mater. Chem., 2011, 21, 7795–7801 RSC.
- W. B. Zou, J. W. Zhu, Y. X. Sun and X. Wang, Mater. Chem. Phys., 2011, 125, 617–620 CrossRef CAS.
- J. L. Wu, X. P. Shen, L. Jiang, K. Wang and K. M. Chen, Appl. Surf. Sci., 2010, 256, 2826–2830 CrossRef CAS.
- K. F. Zhou, Y. H. Zhu, X. L. Yang, X. Jiang and C. Z. Li, New J. Chem., 2011, 35, 353–359 RSC.
- Y. Fan, H. T. Lu, J. H. Liu, C. P. Yang, Q. S. Jing, Y. X. Zhang, X. K. Yang and K. J. Huang, Colloids Surf., B, 2011, 83, 78–82 CrossRef CAS.
- J. F. Shen, M. Shi, B. Yan, H. W. Ma, N. Li and M. X. Ye, Nano Res., 2011, 4, 795–806 CrossRef CAS.
- J. F. Shen, B. Yan, M. Shi, H. W. Ma, N. Li and M. X. Ye, J. Mater. Chem., 2011, 21, 3415–3421 RSC.
- K. F. Zhou, Y. H. Zhu, X. L. Yang and C. Z. Li, Electroanalysis, 2011, 23, 862–869 CrossRef CAS.
- J. Z. Wang, C. Zhong, D. Wexler, N. H. Idris, Z. X. Wang, L. Q. Chen and H. K. Liu, Chem.–Eur. J., 2011, 17, 661–667 CrossRef CAS.
- X. P. Shen, J. L. Wu, S. Bai and H. Zhou, J. Alloys Compd., 2010, 506, 136–140 CrossRef CAS.
- W. H. Shi, J. X. Zhu, D. H. Sim, Y. Y. Tay, Z. Y. Lu, X. J. Zhang, Y. Sharma, M. Srinivasan, H. Zhang, H. H. Hng and Q. Y. Yan, J. Mater. Chem., 2011, 21, 3422–3427 RSC.
- X. D. Huang, X. F. Zhou, L. Zhou, K. Qian, Y. H. Wang, Z. P. Liu and C. Z. Yu, ChemPhysChem, 2011, 12, 278–281 CrossRef CAS.
- G. B. Jung, Y. Myung, Y. J. Cho, Y. J. Sohn, D. M. Jang, H. S. Kim, C. W. Lee, J. Park, I. Maeng, J. H. Son and C. Kang, J. Phys. Chem. C, 2010, 114, 11258–11265 Search PubMed.
- B. J. Li, H. Q. Cao, J. Shao, G. Q. Li, M. Z. Qu and G. Yin, Inorg. Chem., 2011, 50, 1628–1632 CrossRef CAS.
- H. W. Wang, Z. A. Hu, Y. Q. Chang, Y. L. Chen, Z. Q. Lei, Z. Y. Zhang and Y. Y. Yang, Electrochim. Acta, 2010, 55, 8974–8980 CrossRef CAS.
- J. X. Zhu, T. Zhu, X. Z. Zhou, Y. Y. Zhang, X. W. Lou, X. D. Chen, H. Zhang, H. H. Hng and Q. Y. Yan, Nanoscale, 2011, 3, 1084–1089 RSC.
- S. Chen, J. W. Zhu, H. A. Huang, G. Y. Zeng, F. D. Nie and X. Wang, J. Solid State Chem., 2010, 183, 2552–2557 Search PubMed.
- Y. S. He, D. W. Bai, X. W. Yang, J. Chen, X. Z. Liao and Z. F. Ma, Electrochem. Commun., 2010, 12, 570–573 CrossRef CAS.
- J. O. Hwang, D. H. Lee, J. Y. Kim, T. H. Han, B. H. Kim, M. Park, K. No and S. O. Kim, J. Mater. Chem., 2011, 21, 3432–3437 RSC.
- D. Choi, M. Y. Choi, W. M. Choi, H. J. Shin, H. K. Park, J. S. Seo, J. Park, S. M. Yoon, S. J. Chae, Y. H. Lee, S. W. Kim, J. Y. Choi, S. Y. Lee and J. M. Kim, Adv. Mater., 2010, 22, 2187–2192 CrossRef CAS.
- T. V. Cuong, V. H. Pham, J. S. Chung, E. W. Shin, D. H. Yoo, S. H. Hahn, J. S. Huh, G. H. Rue, E. J. Kim, S. H. Hur and P. A. Kohl, Mater. Lett., 2010, 64, 2479–2482 CrossRef CAS.
- H. L. Wang, J. T. Robinson, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS.
- H. L. Wang, L. F. Cui, Y. A. Yang, H. S. Casalongue, J. T. Robinson, Y. Y. Liang, Y. Cui and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS.
- Y. Y. Liang, H. L. Wang, H. S. Casalongue, Z. Chen and H. J. Dai, Nano Res., 2010, 3, 701–705 CrossRef CAS.
- Y. J. Mai, X. L. Wang, J. Y. Xiang, Y. Q. Qiao, D. Zhang, C. D. Gu and J. P. Tu, Electrochim. Acta, 2011, 56, 2306–2311 CrossRef CAS.
- A. N. Cao, Z. Liu, S. S. Chu, M. H. Wu, Z. M. Ye, Z. W. Cai, Y. L. Chang, S. F. Wang, Q. H. Gong and Y. F. Liu, Adv. Mater., 2010, 22, 103–106 CrossRef CAS.
- P. Wang, T. F. Jiang, C. Z. Zhu, Y. M. Zhai, D. J. Wang and S. J. Dong, Nano Res., 2010, 3, 794–799 CrossRef CAS.
- Q. Li, B. D. Guo, J. G. Yu, J. R. Ran, B. H. Zhang, H. J. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS.
- L. P. Xue, C. F. Shen, M. B. Zheng, H. L. Lu, N. W. Li, G. B. Ji, L. J. Pan and J. M. Cao, Mater. Lett., 2011, 65, 198–200 Search PubMed.
- K. Chang and W. X. Chen, Chem. Commun., 2011, 47, 4252–4254 RSC.
- Z. S. Lu, C. X. Guo, H. B. Yang, Y. Qiao, J. Guo and C. M. Li, J. Colloid Interface Sci., 2011, 353, 588–592 CrossRef CAS.
- H. Q. Zhou, C. Y. Qiu, Z. Liu, H. C. Yang, L. J. Hu, J. Liu, H. F. Yang, C. Z. Gu and L. F. Sun, J. Am. Chem. Soc., 2010, 132, 944–946 CrossRef CAS.
- H. Q. Zhou, C. Y. Qiu, F. Yu, H. C. Yang, M. J. Chen, L. J. Hu and L. F. Sun, J. Phys. Chem. C, 2011, 115, 11348–11354 CAS.
- Z. T. Luo, L. A. Somers, Y. P. Dan, T. Ly, N. J. Kybert, E. J. Mele and A. T. C. Johnson, Nano Lett., 2010, 10, 777–781 CrossRef CAS.
- A. T. N''Diaye, S. Bleikamp, P. J. Feibelman and T. Michely, Phys. Rev. Lett., 2006, 97, 215501 CrossRef.
- A. T. N''Diaye, T. Gerber, C. Busse, J. Myslivecek, J. Coraux and T. Michely, New J. Phys., 2009, 11, 103045 CrossRef.
- Y. Pan, M. Gao, L. Huang, F. Liu and H. J. Gao, Appl. Phys. Lett., 2009, 95, 093106 CrossRef.
- Z. H. Zhou, F. Gao and D. W. Goodman, Surf. Sci., 2010, 604, L31–L38 CrossRef CAS.
- N. G. Shang, P. Papakonstantinou, P. Wang, S. Ravi and P. Silva, J. Phys. Chem. C, 2010, 114, 15837–15841 CrossRef CAS.
- M. H. Maneshian, F. L. Kuo, K. Mahdak, J. Hwang, R. Banerjee and N. D. Shepherd, Nanotechnology, 2011, 22, 205703 Search PubMed.
- K. H. Yu, G. H. Lu, S. Mao, K. H. Chen, H. Kim, Z. H. Wen and J. H. Chen, ACS Appl. Mater. Interfaces, 2011, 3, 2703–2709 Search PubMed.
- J. A. Lin, M. Penchev, G. P. Wang, R. K. Paul, J. B. Zhong, X. Y. Jing, M. Ozkan and C. S. Ozkan, Small, 2010, 6, 2448–2452 CrossRef CAS.
- D. Kondo, S. Sato and Y. Awano, Appl. Phys. Express, 2008, 1, 074003 Search PubMed.
- L. L. Zhang, Z. G. Xiong and X. S. Zhao, ACS Nano, 2010, 4, 7030–7036 CrossRef CAS.
- Z. J. Fan, J. Yan, L. J. Zhi, Q. Zhang, T. Wei, J. Feng, M. L. Zhang, W. Z. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS.
- X. R. Wang, S. M. Tabakman and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 8152–8153 CrossRef CAS.
- H. Alles, J. Aarik, A. Aidla, A. Fay, J. Kozlova, A. Niilisk, M. Paers, M. Raehn, M. Wiesner, P. Hakonen and V. Sammelselg, Cent. Eur. J. Phys., 2011, 9, 319–324 Search PubMed.
- K. Zou, X. Hong, D. Keefer and J. Zhu, Phys. Rev. Lett., 2010, 105, 126601 Search PubMed.
- X. B. Meng, D. S. Geng, J. A. Liu, M. N. Banis, Y. Zhang, R. Y. Li and X. L. Sun, J. Phys. Chem. C, 2010, 114, 18330–18337 CrossRef CAS.
- X. B. Meng, D. S. Geng, J. A. Liu, R. Y. Li and X. L. Sun, Nanotechnology, 2011, 22, 165602 Search PubMed.
- X. Y. Zhang, H. P. Li, X. L. Cui and Y. H. Lin, J. Mater. Chem., 2010, 20, 2801–2806 RSC.
- H. D. Liu, P. Gao, J. H. Fang and G. Yang, Chem. Commun., 2011, 47, 9110–9112 RSC.
- H. F. Xiang, B. B. Tian, P. C. Lian, Z. Li and H. H. Wang, J. Alloys Compd., 2011, 509, 7205–7209 Search PubMed.
- H. Zhao, J. Yang, L. Wang, C. G. Tian, B. J. Jiang and H. G. Fu, Chem. Commun., 2011, 47, 2014–2016 RSC.
- Z. M. Wang, W. D. Wang, N. Coombs, N. Soheilnia and G. A. Ozin, ACS Nano, 2010, 4, 7437–7450 Search PubMed.
- D. H. Lee, J. E. Kim, T. H. Han, J. W. Hwang, S. Jeon, S. Y. Choi, S. H. Hong, W. J. Lee, R. S. Ruoff and S. O. Kim, Adv. Mater., 2010, 22, 1247–1252 CrossRef CAS.
- Y. T. Kim, J. H. Han, B. H. Hong and Y. U. Kwon, Adv. Mater., 2010, 22, 515–518 CrossRef CAS.
- J. M. Lee, Y. B. Pyun, J. Yi, J. W. Choung and W. I. Park, J. Phys. Chem. C, 2009, 113, 19134–19138 CrossRef CAS.
- J. Y. Son, Y. H. Shin, H. Kim and H. M. Jang, ACS Nano, 2010, 4, 2655–2658 CrossRef CAS.
- F. A. He, J. T. Fan, F. Song, L. M. Zhang and H. L. W. Chan, Nanoscale, 2011, 3, 1182–1188 RSC.
- J. B. Liu, S. H. Fu, B. Yuan, Y. L. Li and Z. X. Deng, J. Am. Chem. Soc., 2010, 132, 7279–7281 CrossRef CAS.
- F. A. He, J. T. Fan, D. Ma, L. M. Zhang, C. Leung and H. L. Chan, Carbon, 2010, 48, 3139–3144 CrossRef CAS.
- Y. Zhang, B. A. Chen, L. M. Zhang, J. Huang, F. H. Chen, Z. P. Yang, J. L. Yao and Z. J. Zhang, Nanoscale, 2011, 3, 1446–1450 RSC.
- T. A. Pham, B. C. Choi and Y. T. Jeong, Nanotechnology, 2010, 21, 465603 CrossRef.
- A. P. Tuan, B. C. Choi, K. T. Lim and Y. T. Jeong, Appl. Surf. Sci., 2011, 257, 3350–3357 Search PubMed.
- T. A. Pham, N. A. Kumar and Y. T. Jeong, Synth. Met., 2010, 160, 2028–2036 Search PubMed.
- Y. K. Kim and D. H. Min, Carbon, 2010, 48, 4283–4288 CrossRef CAS.
- X. Y. Zhang, Z. B. Liu, Y. Huang, X. J. Wan, J. G. Tian, Y. F. Ma and Y. S. Chen, J. Nanosci. Nanotechnol., 2009, 9, 5752–5756 CrossRef CAS.
- X. Y. Zhang, Y. Huang, Y. Wang, Y. F. Ma, Z. F. Liu and Y. S. Chen, Carbon, 2009, 47, 334–337 CrossRef CAS.
- Z. B. Liu, Y. F. Xu, X. Y. Zhang, X. L. Zhang, Y. S. Chen and J. G. Tian, J. Phys. Chem. B, 2009, 113, 9681–9686 CrossRef CAS.
- Y. Zhang, L. Q. Ren, S. R. Wang, A. Marathe, J. Chaudhuri and G. G. Li, J. Mater. Chem., 2011, 21, 5386–5391 RSC.
- J. Huang, L. M. Zhang, B. A. Chen, N. Ji, F. H. Chen, Y. Zhang and Z. J. Zhang, Nanoscale, 2010, 2, 2733–2738 RSC.
- X. M. Geng, L. Niu, Z. Y. Xing, R. S. Song, G. T. Liu, M. T. Sun, G. S. Cheng, H. J. Zhong, Z. H. Liu, Z. J. Zhang, L. F. Sun, H. X. Xu, L. Lu and L. W. Liu, Adv. Mater., 2010, 22, 638–642 CrossRef CAS.
- J. B. Liu, Y. L. Li, Y. M. Li, J. H. Li and Z. X. Deng, J. Mater. Chem., 2010, 20, 900–906 RSC.
- F. Liu, J. Y. Choi and T. S. Seo, Chem. Commun., 2010, 46, 2844–2846 RSC.
- Y. Wang, S. J. Zhen, Y. Zhang, Y. F. Li and C. Z. Huang, J. Phys. Chem. C, 2011, 115, 12815–12821 CrossRef CAS.
- M. Feng, R. Q. Sun, H. B. Zhan and Y. Chen, Nanotechnology, 2010, 21, 075601 CrossRef.
- L. Qiu, X. W. Yang, X. L. Gou, W. R. Yang, Z. F. Ma, G. G. Wallace and D. Li, Chem.–Eur. J., 2010, 16, 10653–10658 CrossRef CAS.
- C. Zhang, L. L. Ren, X. Y. Wang and T. X. Liu, J. Phys. Chem. C, 2010, 114, 11435–11440 CrossRef CAS.
- G. X. Zhu, Y. J. Liu, Z. Xu, T. A. Jiang, C. Zhang, X. Li and G. Qi, ChemPhysChem, 2010, 11, 2432–2437 CrossRef CAS.
- G. H. Lu, S. Mao, S. Park, R. S. Ruoff and J. H. Chen, Nano Res., 2009, 2, 192–200 CrossRef CAS.
- S. B. Yang, X. L. Feng, S. Ivanovici and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS.
- W. J. Hong, H. Bai, Y. X. Xu, Z. Y. Yao, Z. Z. Gu and G. Q. Shi, J. Phys. Chem. C, 2010, 114, 1822–1826 CrossRef CAS.
- S. Liu, J. Q. Tian, L. Wang, H. L. Li, Y. W. Zhang and X. P. Sun, Macromolecules, 2010, 43, 10078–10083 CrossRef CAS.
- Y. X. Fang, S. J. Guo, C. Z. Zhu, Y. M. Zhai and E. K. Wang, Langmuir, 2010, 26, 11277–11282 CrossRef CAS.
- L. L. Li, K. P. Liu, G. H. Yang, C. M. Wang, J. R. Zhang and J. J. Zhu, Adv. Funct. Mater., 2011, 21, 869–878 CrossRef CAS.
- S. Y. Wang, X. Wang and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 6883–6891 RSC.
- J. T. Zhang, J. W. Jiang and X. S. Zhao, J. Phys. Chem. C, 2011, 115, 6448–6454 CrossRef CAS.
- D. S. Yu and L. M. Dai, J. Phys. Chem. Lett., 2010, 1, 467–470 Search PubMed.
- Y. K. Kim and D. H. Min, Langmuir, 2009, 25, 11302–11306 CrossRef CAS.
- J. L. Xiang and L. T. Drazl, ACS Appl. Mater. Interfaces, 2011, 3, 1325–1332 CrossRef CAS.
- C. Z. Zhu, S. J. Guo, Y. M. Zhai and S. J. Dong, Langmuir, 2010, 26, 7614–7618 CrossRef CAS.
- K. K. Manga, Y. Zhou, Y. L. Yan and K. P. Loh, Adv. Funtc. Mater., 2009, 19–3643 Search PubMed.
- X. R. Zhang, S. G. Li, X. Jin and S. S. Zhang, Chem. Commun., 2011, 47, 4929–4931 RSC.
- M. Alazemi, I. Dutta, F. Wang, R. H. Blunk and A. P. Angelopoulos, Carbon, 2010, 48, 4063–4073 Search PubMed.
- Z. P. Li, J. Q. Wang, X. H. Liu, S. Liu, J. F. Ou and S. R. Yang, J. Mater. Chem., 2011, 21, 3397–3403 RSC.
- H. B. Yao, L. H. Wu, C. H. Cui, H. Y. Fang and S. H. Yu, J. Mater. Chem., 2010, 20, 5190–5195 RSC.
- D. Chen, X. Y. Wang, T. X. Liu, X. D. Wang and J. Li, ACS Appl. Mater. Interfaces, 2010, 2, 2005–2011 CrossRef CAS.
- Y. Li, X. B. Fan, J. J. Qi, J. Y. Ji, S. L. Wang, G. L. Zhang and F. B. Zhang, Nano Res., 2010, 3, 429–437 CrossRef CAS.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS.
- Z. H. Tang, S. L. Shen, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 4603–4607 CrossRef CAS.
- Y. Li, X. B. Fan, J. J. Qi, J. Y. Ji, S. L. Wang, G. L. Zhang and F. B. Zhang, Mater. Res. Bull., 2010, 45, 1413–1418 CrossRef CAS.
- L. H. Tang, Y. Wang, Y. M. Li, H. B. Feng, J. Lu and J. H. Li, Adv. Funct. Mater., 2009, 19, 2782–2789 CrossRef CAS.
- R. Kou, Y. Y. Shao, D. H. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. M. Wang, Y. H. Lin, Y. Wang, I. A. Aksay and J. Liu, Electrochem. Commun., 2009, 11, 954–957 CrossRef CAS.
- S. M. Choi, M. H. Seo, H. J. Kim and W. B. Kim, Carbon, 2011, 49, 904–909 CrossRef CAS.
- Y. C. Xin, J. G. Liu, W. M. Liu, J. Gao, Y. Xie, Y. Yin and Z. G. Zou, J. Power Sources, 2011, 196, 1012–1018 CrossRef.
- E. Yoo, T. Okada, T. Akita, M. Kohyama, I. Honma and J. Nakamura, J. Power Sources, 2011, 196, 110–115 CrossRef CAS.
- Y. Y. Shao, S. Zhang, C. M. Wang, Z. M. Nie, J. Liu, Y. Wang and Y. H. Lin, J. Power Sources, 2010, 195, 4600–4605 CrossRef CAS.
- H. Y. Li, X. H. Zhang, H. L. Pang, C. T. Huang and J. H. Chen, J. Solid State Electrochem., 2010, 14, 2267–2274 CrossRef CAS.
- C. Nethravathi, E. A. Anumol, M. Rajamathi and N. Ravishankar, Nanoscale, 2011, 3, 569–571 RSC.
- S. Bong, Y. R. Kim, I. Kim, S. Woo, S. Uhm, J. Lee and H. Kim, Electrochem. Commun., 2010, 12, 129–131 CrossRef CAS.
- S. Y. Wang, S. P. Jiang and X. Wang, Electrochim. Acta, 2011, 56, 3338–3344 CrossRef CAS.
- C. V. Rao, A. L. M. Reddy, Y. Ishikawa and P. M. Ajayan, Carbon, 2011, 49, 931–936 CrossRef CAS.
- S. Zhang, Y. Y. Shao, H. G. Liao, J. Liu, I. A. Aksay, G. P. Yin and Y. H. Lin, Chem. Mater., 2011, 23, 1079–1081 CrossRef CAS.
- S. J. Guo, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS.
- R. Y. Y. Shao, D. H. Mei, Z. M. Nie, D. H. Wang, C. M. Wang, V. V. Viswanathan, S. Park, I. A. Aksay, Y. H. Lin, Y. Wang and J. Liu, J. Am. Chem. Soc., 2011, 133, 2541–2547 CrossRef CAS.
- Y. C. Zhao, L. Zhan, J. N. Tian, S. L. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967–1972 CrossRef CAS.
- M. H. Seo, S. M. Choi, H. J. Kim and W. B. Kim, Electrochem. Commun., 2011, 13, 182–185 CrossRef CAS.
- R. N. Singh and R. Awasthi, Catal. Sci. Technol., 2011, 1, 778–783 RSC.
- J. Yang, C. G. Tian, L. Wang and H. G. Fu, J. Mater. Chem., 2011, 21, 3384–3390 RSC.
- F. H. Li, H. F. Yang, C. S. Shan, Q. X. Zhang, D. X. Han, A. Ivaska and L. Niu, J. Mater. Chem., 2009, 19, 4022–4025 RSC.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 Search PubMed.
- Y. G. Li, H. L. Wang, L. M. Xie, Y. Y. Liang, G. S. Hong and H. J. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS.
- Y. Qian, S. B. Lu and F. L. Gao, Mater. Lett., 2011, 65, 56–58 Search PubMed.
- H. Zhang, X. J. Lv, Y. M. Li, Y. Wang and J. H. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS.
- J. C. Liu, H. W. Bai, Y. J. Wang, Z. Y. Liu, X. W. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175–4181 CrossRef CAS.
- Y. H. Zhang, Z. R. Tang, X. Z. Fu and Y. J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS.
- H. J. Zhang, P. P. Xu, G. D. Du, Z. W. Chen, K. Oh, D. Y. Pan and Z. Jiao, Nano Res., 2011, 4, 274–283 CrossRef CAS.
- J. Du, X. Y. Lai, N. L. Yang, J. Zhai, D. Kisailus, F. B. Su, D. Wang and L. Jiang, ACS Nano, 2011, 5, 590–596 CrossRef CAS.
- G. D. Jiang, Z. F. Lin, C. Chen, L. H. Zhu, Q. Chang, N. Wang, W. Wei and H. Q. Tang, Carbon, 2011, 49, 2693–2701 CrossRef CAS.
- N. Li, G. Liu, C. Zhen, F. Li, L. L. Zhang and H. M. Cheng, Adv. Funct. Mater., 2011, 21, 1717–1722 CrossRef CAS.
- W. Q. Fan, Q. H. Lai, Q. H. Zhang and Y. Wang, J. Phys. Chem. C, 2011, 115, 10694–10701 CrossRef CAS.
- Z. G. Xiong, L. L. Zhang, J. Z. Ma and X. S. Zhao, Chem. Commun., 2010, 46, 6099–6101 RSC.
- E. P. Gao, W. Z. Wang, M. Shang and J. H. Xu, Phys. Chem. Chem. Phys., 2011, 13, 2887–2893 RSC.
- N. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 Search PubMed.
- L. Jia, D. H. Wang, Y. X. Huang, A. W. Xu and H. Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CAS.
- B. J. Li and H. Q. Cao, J. Mater. Chem., 2011, 21, 3346–3349 RSC.
- T. G. Xu, L. W. Zhang, H. Y. Cheng and Y. F. Zhu, Appl. Catal., B, 2011, 101, 382–387 CrossRef CAS.
- F. Zhou, R. Shi and Y. F. Zhu, J. Mol. Catal. A: Chem., 2011, 340, 77–82 CrossRef CAS.
- X. F. Zhang, X. Quan, S. Chen and H. T. Yu, Appl. Catal., B, 2011, 105, 237–242 CrossRef CAS.
- Y. S. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210–7218 CrossRef CAS.
- M. S. Zhu, P. L. Chen and M. H. Liu, ACS Nano, 2011, 5, 4529–4536 CrossRef CAS.
- M. Pumera, Energy Environ. Sci., 2011, 4, 668–674 RSC.
- S. J. Guo and S. J. Dong, Chem. Soc. Rev., 2011, 40, 2644–2672 RSC.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983–5992 RSC.
- J. Yan, Z. J. Fan, T. Wei, W. Z. Qian, M. L. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- S. Chen, J. W. Zhu, X. D. Wu, Q. F. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS.
- Z. S. Wu, W. C. Ren, D. W. Wang, F. Li, B. L. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS.
- Z. J. Fan, J. Yan, T. Wei, L. J. Zhi, G. Q. Ning, T. Y. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- G. Yu, L. Hu, M. Vosgueritchian, H. Wang, X. Xie, J. R. McDonough, X. Cui, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 2905–2911 CrossRef CAS.
- B. Wang, J. Park, C. Y. Wang, H. Ahn and G. X. Wang, Electrochim. Acta, 2010, 55, 6812–6817 CrossRef CAS.
- J. Yan, T. Wei, W. M. Qiao, B. Shao, Q. K. Zhao, L. J. Zhang and Z. J. Fan, Electrochim. Acta, 2010, 55, 6973–6978 CrossRef CAS.
- W. W. Zhou, J. P. Liu, T. Chen, K. S. Tan, X. T. Jia, Z. Q. Luo, C. X. Cong, H. P. Yang, C. M. Li and T. Yu, Phys. Chem. Chem. Phys., 2011, 13, 14462–14465 RSC.
- T. Lu, Y. P. Zhang, H. B. Li, L. K. Pan, Y. L. Li and Z. Sun, Electrochim. Acta, 2010, 55, 4170–4173 CrossRef CAS.
- Y. P. Zhang, H. B. Li, L. K. Pan, T. Lu and Z. Sun, J. Electroanal. Chem., 2009, 634, 68–71 CrossRef CAS.
- Y. L. Chen, Z. A. Hu, Y. Q. Chang, H. W. Wang, Z. Y. Zhang and Y. Y. Yang, J. Phys. Chem. C, 2011, 115, 2563–2571 CrossRef CAS.
- B. J. Li, H. Q. Cao, J. Shao, M. Z. Qu and J. H. Warner, J. Mater. Chem., 2011, 21, 5069–5075 RSC.
- X. H. Xia, J. P. Tu, Y. J. Mai, R. Chen, X. L. Wang, C. D. Gu and X. B. Zhao, Chem.–Eur. J., 2011, 17, 10898–10905 Search PubMed.
- Y. Wang, C. X. Guo, J. H. Luo, T. Chen, H. B. Yang and C. M. Li, Dalton Trans., 2011, 40, 6388–6391 RSC.
- B. J. Li, H. Q. Cao, G. Yin, Y. X. Lu and J. F. Yin, J. Mater. Chem., 2011, 21, 10645–10648 RSC.
- J. W. Lee, T. Ahn, D. Soundararajan, J. M. Ko and J. D. Kim, Chem. Commun., 2011, 47, 6305–6307 RSC.
- S. Y. Yang, K. H. Chang, H. W. Tien, Y. F. Lee, S. M. Li, Y. S. Wang, J. Y. Wang, C. C. M. Ma and C. C. Hu, J. Mater. Chem., 2011, 21, 2374–2380 RSC.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L. C. Qin, Phys. Chem. Chem. Phys., 2011, 13, 17615–17624 RSC.
- G. X. Wang, X. P. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- M. H. Liang and L. J. Zhi, J. Mater. Chem., 2009, 19, 5871–5878 RSC.
- S. M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72–75 CrossRef CAS.
- J. Yao, X. P. Shen, B. Wang, H. K. Liu and G. X. Wang, Electrochem. Commun., 2009, 11, 1849–1852 CrossRef CAS.
- Y. M. Li, X. J. Lv, J. Lu and J. H. Li, J. Phys. Chem. C, 2010, 114, 21770–21774 CrossRef CAS.
- Z. Y. Wang, H. Zhang, N. Li, Z. J. Shi, Z. N. Gu and G. P. Cao, Nano Res., 2010, 3, 748–756 CrossRef CAS.
- H. Kim, S. W. Kim, Y. U. Park, H. Gwon, D. H. Seo, Y. Kim and K. Kang, Nano Res., 2010, 3, 748–756 CrossRef CAS.
- S. J. Ding, D. Luan, F. Y. C. Beoy, J. S. Chen and X. W. Lou, Chem. Commun., 2011, 47, 7155–7157 RSC.
- P. C. Lian, X. F. Zhu, H. F. Xiang, Z. Li, E. S. Yang and H. H. Wang, Electrochim. Acta, 2010, 56, 834–840 CrossRef CAS.
- L. W. Ji, Z. K. Tan, T. R. Kuykendall, S. Aloni, S. D. Sun, E. Lin, V. Battaglia and Y. G. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 7170–7177 RSC.
- H. Kim, D. H. Seo, S. W. Kim, J. Kim and K. Kang, Carbon, 2011, 49, 326–332 CrossRef CAS.
- B. Wang, X. L. Wu, C. Y. Shu, Y. G. Guo and C. R. Wang, J. Mater. Chem., 2010, 20, 10661–10664 RSC.
- D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907–914 CrossRef CAS.
- S. J. Ding, J. S. Cheng, D. Y. Luan, F. Y. C. Boey, S. Madhavi and X. W. Lou, Chem. Commun., 2011, 47, 5780–5782 RSC.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS.
- G. Wang, T. Liu, Y. J. Luo, X. Tong, L. J. Wan, Y. Zhao, H. Wang, Z. Y. Ren and J. B. Bai, J. Alloys Compd., 2011, 509, L216–L220 CrossRef CAS.
- G. Wang, J. T. Bai, Y. H. Wang, Z. Y. Ren and J. B. Bai, Scr. Mater., 2011, 65, 339–342 Search PubMed.
- L. F. Shen, C. Z. Yuan, H. J. Luo, X. G. Zhang, S. D. Yang and X. J. Lu, Nanoscale, 2011, 3, 572–574 RSC.
- B. J. Li, H. Q. Cao, J. Shao, H. Zheng, Y. X. Lu, J. F. Yin and M. Z. Qu, Chem. Commun., 2011, 47, 3159–3161 RSC.
- K. Chang and W. X. Cheng, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS.
- J. K. Lee, K. B. Smith, C. M. Hayner and H. H. Kung, Chem. Commun., 2010, 46, 2025–2027 RSC.
- L. W. Ji, Z. K. Tan, T. Kuykendall, E. J. An, Y. B. Fu, V. Battaglia and Y. G. Zhang, Energy Environ. Sci., 2011, 4, 3611–3616 RSC.
- E. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
- Y. Ding, Y. Jiang, F. Xu, J. Yin, H. Ren, Q. Zhuo, Z. Long and P. Zhang, Electrochem. Commun., 2010, 12, 10–13 CrossRef CAS.
- D. W. Choi, D. H. Wang, V. V. Viswanathan, I. T. Bae, W. Wang, Z. M. Nie, J. G. Zhang, G. L. Graff, J. Liu, Z. G. Yang and T. Duong, Electrochem. Commun., 2010, 12, 378–381 CrossRef CAS.
- X. F. Zhou, F. Wang, Y. M. Zhu and Z. P. Liu, J. Mater. Chem., 2011, 21, 3353–3358 RSC.
- C. V. Rao, A. L. M. Reddy, Y. Ishikawa and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2011, 3, 2966–2972 Search PubMed.
- G. D. Du, K. H. Seng, Z. P. Guo, J. Liu, W. X. Li, D. Z. Jia, C. Cook, Z. W. Liu and H. K. Liu, RSC Adv., 2011, 1, 690–697 RSC.
- Y. H. Hu, H. Wang and B. Hu, ChemSusChem, 2010, 3, 782–796 CrossRef CAS.
- N. L. Yang, J. Zhai, D. Wang, Y. S. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS.
- S. R. Sun, L. Gao and Y. Q. Liu, Appl. Phys. Lett., 2010, 96, 083113 CrossRef.
- M. Y. Yen, M. C. Hsiao, S. H. Liao, P. I. Liu, H. M. Tsai, C. C. M. Ma, N. W. Pu and M. D. Ger, Carbon, 2011, 49, 3597–3606 CrossRef CAS.
- S. R. Kim, M. K. Parvez and M. Chhowalla, Chem. Phys. Lett., 2009, 483, 124–127 CrossRef CAS.
- H. B. Yang, G. H. Guai, C. X. Guo, Q. L. Song, S. P. Jiang, Y. L. Wang, W. Zhang and C. M. Li, J. Phys. Chem. C, 2011, 115, 12209–12215 CrossRef CAS.
- S. S. Li, Y. H. Luo, W. Lv, W. J. Yu, S. D. Wu, P. X. Hou, Q. H. Yang, Q. B. Meng, C. Liu and H. M. Cheng, Adv. Energy Mater., 2011, 1, 486–490 Search PubMed.
- H. Choi, H. Kim, S. Hwang, M. Kang, D. W. Jung and M. Jeon, Scr. Mater., 2011, 64, 601–604 CrossRef CAS.
- H. Choi, H. Kim, S. Hwang, W. Choi and M. Jeon, Sol. Energy Mater. Sol. Cells, 2011, 95, 323–325 CrossRef CAS.
- J. Chen, C. Li, G. Eda, Y. Zhang, W. Lei, M. Chhowalla, W. I. Milne and W. Q. Deng, Chem. Commun., 2011, 47, 6084–6086 RSC.
- G. Zhu, T. Xu, T. A. Lv, L. K. Pan, Q. F. Zhao and Z. Sun, J. Electroanal. Chem., 2011, 650, 248–251 CrossRef CAS.
- S. R. Sun, L. Gao, Y. Q. Liu and J. Sun, Appl. Phys. Lett., 2011, 98, 093112 CrossRef.
- Y. Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. H. Lin, Electroanal., 2010, 22, 1027–1036 CAS.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, TrAC, Trends Anal. Chem., 2010, 29, 954–965 CrossRef CAS.
- D. Chen, L. H. Tang and J. H. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 RSC.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- J. L. Johnson, A. Behnam, S. J. Pearton and A. Ural, Adv. Mater., 2010, 22, 4877–4880 CrossRef CAS.
- U. Lange, T. Hirsch, V. M. Mirsky and O. S. Wolfbeis, Electrochim. Acta, 2011, 56, 3707–3712 Search PubMed.
- M. Shafiei, P. G. Spizzirri, R. Arsat, J. Yu, J. du Plessis, S. Dubin, R. B. Kaner, K. Kalantar-Zadeh and W. Wlodarski, J. Phys. Chem. C, 2010, 114, 13796–13801 CAS.
- A. Kaniyoor, R. I. Jafri, T. Arockiadoss and S. Ramaprabhu, Nanoscale, 2009, 1, 382–386 RSC.
- Z. X. Jiang, J. J. Wang, L. H. Meng, Y. D. Huang and L. Liu, Chem. Commun., 2011, 47, 6350–6352 RSC.
- H. J. Song, L. C. Zhang, C. L. He, Y. Qu, Y. F. Tian and Y. Lv, J. Mater. Chem., 2011, 21, 5972–5977 RSC.
- H. Y. Jeong, D. S. Lee, H. K. Choi, D. H. Lee, J. E. Kim, J. Y. Lee, W. J. Lee, S. O. Kim and S. Y. Choi, Appl. Phys. Lett., 2010, 96, 213105 CrossRef.
- S. J. Guo, D. Wen, Y. M. Zhai, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 3959–3968 CrossRef CAS.
- K. F. Zhou, Y. H. Zhu, X. L. Yang, J. Luo, C. Z. Li and S. R. Luan, Electrochim. Acta, 2010, 55, 3055–3060 CrossRef CAS.
- K. J. Huang, D. J. Niu, X. Liu, Z. W. Wu, Y. Fan, Y. F. Chang and Y. Y. Wu, Electrochim. Acta, 2011, 56, 2947–2953 Search PubMed.
- L. M. Lu, H. B. Li, F. L. Qu, X. B. Zhang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2011, 26, 3500–3504 CrossRef CAS.
- Q. Zeng, J. S. Cheng, X. F. Liu, H. T. Bai and J. H. Jiang, Biosens. Bioelectron., 2011, 26, 3456–3463 CrossRef CAS.
- T. T. Baby, S. S. J. Aravind, T. Arockiadoss, R. B. Rakhi and S. Ramaprabhu, Sens. Actuators, B, 2010, 145, 71–77 CrossRef.
- C. S. Shan, H. F. Yang, D. X. Han, Q. X. Zhang, A. Ivaska and L. Niu, Biosens. Bioelectron., 2010, 25, 1070–1074 CrossRef CAS.
- C. L. Sun, H. H. Lee, J. M. Yang and C. C. Wu, Biosens. Bioelectron., 2011, 26, 3450–3455 CrossRef CAS.
- F. H. Li, J. Chai, H. F. Yang, D. X. Han and L. Niu, Talanta, 2010, 81, 1063–1068 CrossRef CAS.
- R. S. Dey and C. R. Raj, J. Phys. Chem. C, 2010, 114, 21427–21433 CrossRef CAS.
- S. Mao, G. H. Lu, K. H. Y. Z. Bo and J. H. Chen, Adv. Mater., 2010, 22, 3521–3526 CrossRef CAS.
- X. C. Dong, W. Huang and P. Chen, Nanoscale Res. Lett., 2011, 6, 60 Search PubMed.
- Y. Wang, S. Zhang, D. Du, Y. Y. Shao, Z. H. Li, J. Wang, M. H. Engelhard, J. H. Li and Y. H. Lin, J. Mater. Chem., 2011, 21, 5319–5325 RSC.
- J. F. Liang, Z. B. Chen, L. Guo and L. D. Li, Chem. Commun., 2011, 47, 5476–5478 RSC.
- H. C. Chang, X. J. Wu, C. Y. Wu, Y. Chen, H. Jiang and X. M. Wang, Analyst, 2011, 136, 2735–2740 RSC.
- Y. P. He, Q. L. Shen, J. B. Zheng, M. Z. Wang and B. Liu, Electrochim. Acta, 2011, 56, 2471–2476 Search PubMed.
- L. M. Li, Z. F. Du, S. A. Liu, Q. Y. Liu, Q. Y. Hao, Y. G. Wang, Q. H. Li and T. H. Wang, Talanta, 2010, 82, 1637–1641 CrossRef CAS.
- D. Du, J. Liu, X. Y. Zhang, X. L. Cui and Y. H. Li, J. Mater. Chem., 2011, 21, 8032–8037 RSC.
- K. Wang, Q. Liu, X. Y. Wu, Q. M. Guan and H. N. Li, Talanta, 2010, 82, 372–376 CrossRef CAS.
- K. Wang, Q. Liu, Q. M. Guan, J. Wu, H. N. Li and J. J. Yan, Biosens. Bioelectron., 2011, 26, 2252–2257 CrossRef CAS.
- G. F. Wang, H. Huang, G. Zhang, X. J. Zhang, B. Fang and L. Wang, Anal. Methods, 2010, 2, 1692–1697 RSC.
- M. H. Yang, A. Javadi and S. Q. Gong, Sens. Actuators, B, 2011, 155, 357–360 CrossRef.
- B. L. Su, D. P. Tang, Q. F. Li, J. Tang and G. N. Chen, Anal. Chim. Acta, 2011, 692, 116–124 Search PubMed.
- J. M. Gong, T. Zhou, D. D. Song and L. Z. Zhang, Sens. Actuators, B, 2010, 150, 491–497 CrossRef.
- C. Xu and X. Wang, Small, 2009, 5, 2212–2217 CrossRef CAS.
- J. Lee, S. Shim, B. Kim and H. S. Shin, Chem. –Eur. J., 2011, 17, 2381–2387 CAS.
- X. Q. Fu, F. L. Bei, X. Wang, S. O'’Brien and J. R. Lombardi, Nanoscale, 2010, 2, 1461–1466 RSC.
- K. Jasuja and V. Berry, ACS Nano, 2009, 3, 2358–2366 CrossRef CAS.
- Y. Y. Wang, Z. H. Ni, H. L. Hu, Y. F. Hao, C. P. Wong, T. Yu, J. T. L. Thong and Z. X. Shen, Appl. Phys. Lett., 2010, 97, 163111 CrossRef.
- Z. Zhang, F. G. Xu, W. S. Yang, M. Y. Guo, X. D. Wang, B. L. Zhang and J. L. Tang, Chem. Commun., 2011, 47, 6440–6442 RSC.
- G. Lu, H. Li, C. Liusman, Z. Y. Yin, S. X. Wu and H. Zhang, Chem. Sci., 2011, 2, 1817–1821 RSC.
- R. Wen, Y. X. Fang and E. K. Wang, ACS Nano, 2011, 5, 6425–6433 CrossRef CAS.
- H. M. Sun, L. Y. Cao and L. H. Lu, Nano Res., 2011, 4, 550–562 CrossRef CAS.
- N. W. Li, M. B. Zheng, X. F. Chang, G. B. Ji, H. L. Lu, L. P. Xue, L. J. Pan and J. M. Cao, J. Solid State Chem., 2011, 184, 953–958 Search PubMed.
- H. Jabeen, V. Chandra, S. Jung, J. W. Lee, K. S. Kim and S. B. Kim, Nanoscale, 2011, 3, 3583–3585 RSC.
- K. Zhang, V. Dwivedi, C. Y. Chi and J. S. Wu, J. Hazard. Mater., 2010, 182, 162–168 CrossRef CAS.
- T. S. Sreeprasad, S. M. Maliyekkal, K. P. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS.
- W. P. Xu, L. C. Zhang, J. P. Li, Y. Lu, H. H. Li, Y. N. Ma, W. D. Wang and S. H. Yu, J. Mater. Chem., 2011, 21, 4593–4597 RSC.
- J. Z. Ma, J. T. Zhang, Z. G. Xiong, Y. Yong and X. S. Zhao, J. Mater. Chem., 2011, 21, 3350–3352 RSC.
- L. Liu, J. C. Liu, Y. J. Wang, X. L. Yan and D. D. Sun, New J. Chem., 2011, 35, 1418–1423 RSC.
- J. F. Shen, M. Shi, N. Li, B. Yan, H. W. Ma, Y. Z. Hu and M. X. Ye, Nano Res., 2010, 3, 339–349 CrossRef.
- H. Bai, C. Li and G. Q. Shi, Adv. Mater., 2011, 23, 1088–1114 CrossRef.
- D. Zhou and B. H. Han, Adv. Funct. Mater., 2010, 20, 2717–2722 CrossRef CAS.
- H. L. Li, S. P. Pang, S. Wu, X. L. Feng, K. Mullen and C. Bubeck, J. Am. Chem. Soc., 2011, 133, 9423–9429 CrossRef CAS.
- H. Li, S. Pang, X. Feng, K. Mullen and C. Bubeck, Chem. Commun., 2010, 46, 6243–6245 RSC.
- L. Y. Cao, H. M. Sun, J. Li and L. H. Lu, Anal. Methods, 2011, 3, 1587–1594 RSC.
- L. Y. Cao, Y. L. Liu, B. H. Zhang and L. H. Lu, ACS Appl. Mater. Interfaces, 2010, 2, 2339–2346 CrossRef CAS.
- E. Jin, X. F. Lu, L. L. Cui, D. M. Chao and C. Wang, Electrochim. Acta, 2010, 55, 7230–7234 CrossRef CAS.
- Y. Zhang, X. M. Sun, L. Z. Zhu, H. B. Shen and N. Q. Jia, Electrochim. Acta, 2011, 56, 1239–1245 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |