DOI:
10.1039/C6RA15040C
(Paper)
RSC Adv., 2016,
6, 69167-69176
Non-precious Mn1.5Co1.5O4–FeNx/C nanocomposite as a synergistic catalyst for oxygen reduction in alkaline media
Received
9th June 2016
, Accepted 13th July 2016
First published on 14th July 2016
Abstract
In this study we show a method of preparing a high performing catalyst by designing functional nano boundaries in a nanocomposite material. A non-precious nanocomposite material composed of spinel Mn1.5Co1.5O4 nano crystals and FeNx-functioned graphene nano platelets (FeNx/C) was synthesized by an ultrasonic process. The crystal structure and elemental composition of the bimetal oxide were determined by X-ray diffraction (XRD) and energy dispersive X-ray spectroscopy (EDS). The surface morphology of the Mn1.5Co1.5O4–FeNx/C nanocomposite was characterized with transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM). The catalytic activity for the oxygen reduction reaction (ORR) was analyzed by an electrochemical method. The enhancement of activity for the ORR at the nanocomposite material is attributed to double synergistic effects from the bimetal particles and the FeNx/C nano sheets. The nanocomposite material is able to catalyze 4-electron oxygen reduction to generate water in alkaline media with a high kinetic rate constant (7.6 × 10−2 cm s−1 at 0.7 V vs. reversible hydrogen electrode, RHE). Finally, the activity and stability of the nanocomposite material were compared with that of 40% Pt supported on active carbon (40% Pt/C), which reaches 95% activity and a comparable stability of 40% Pt/C at 0.7 V (vs. RHE).
1. Introduction
Fuel cells are ideal energy conversion devices that efficiently use fuel with low to zero emission. However, the cost of fuel cell materials and components is unacceptably high for commercial application. For example, platinum catalysts are common core materials in fuel cell devices, which accounts for ∼60% of the total cost of fuel cell stacks.1 Non-precious materials as an alternative for fuel cell catalysts have received much attention. Unpyrolyzed and pyrolyzed transition metal macrocycles,2–10 as a large group of non-precious materials, were extensively investigated for the catalytic oxygen reduction reaction (ORR). It is interesting that the macrocycles after pyrolysis contain only carbon with a micro amount of nitrogen and metal atoms (∼5%), but have better activity and stability than those that have not been pyrolyzed.9,10 Therefore, major efforts have focused on pyrolyzed macrocycles such as nitrogen-doped carbons (Nx/C), as well as metal-nitrogen co-doped carbon (MNx/C). Lately, it has been reported that the Nx/C or MNx/C catalysts may be obtained from non-macrocyclic precursors, such as nitrogen-rich polymers and bio-materials.11–20 Although the Nx/C or MNx/C materials have good initial activity, their activity decays rapidly with time, especially in acidic media. The gradual activity decrease is attributed to continuous loss of catalytic sites in acidic media and harsh fuel cell operating condition. The decrease in catalytic stability is potentially mitigated by increasing the concentration of catalytic sites in the MNx/C materials.20 In addition to Nx/C and MNx/C catalysts, another notable group of non-precious materials investigated for the ORR are transition metal oxides,21–34 which show promising activity and persistent stability in alkaline media. Non-noble metal oxides are earthly-abundant, low cost, and ideal catalyst materials for fuel cells with alkaline electrolyte membranes or anion exchange membranes.35–38 As non-noble metal oxides are poorly electrically conductive, they have to be supported on inert conductive materials such as high surface area carbons. However, the ORR mechanisms for metal oxides are complex. There are 2-electron, mix 2 & 4-electron, and 4-electron reactions, which often depend on the metal oxides' structure, composition, morphology, supporting materials, and dopants. The 2-electron reaction mainly occurs on free metal oxides, carbon supports, and their simple mixtures.21,23,25,26,31,32,34 The 4-electron reaction mainly occurs on the nano-structures of metal oxides coupled with a functional support or a dopant, such as MnO2 intercalated in graphene oxides,23 Co3O4 nano-rod supported on carbon,29 Co3O4 nanocrystals on graphene,30 Co3O4 nano-sheets interleaved with graphene sheets,33 and mesoporous Mn2O3 doped with Pd nanoparticles.32 The mechanism of 4-electron reaction is attributed to a synergistic effect at the nano boundaries between metal atoms and the support. Most recently, more innovative nano materials were synthesized with high catalytic activity and stability. For example, Xia et al.39 synthesized a high performing bifunctional catalyst composed of hollow frameworks of nitrogen-doped carbon nanotubes for oxygen reduction and evolution.
In the present research, we attempt to explore how oxygen is reduced at the nano boundaries between metal oxides and carbon supports. Scheme 1 shows a mixed mechanism containing a 4-electron O2 reduction reaction to produce water (Scheme 1a) and a 2-electron reaction to produce hydrogen peroxide (Scheme 1b) at the boundary between a mono-metal oxide (Mn2O3/C or Co3O4/C) and a traditional carbon support (un-doped).
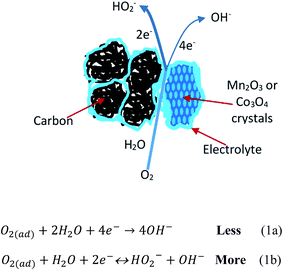 |
| | Scheme 1 Mixed mechanism for ORR at mono-metal oxide and un-doped carbon support. | |
In order to minimize the 2-electron reduction of oxygen, we combined the two metals to form a bimetal oxide, and observed an enhancement of the ORR activity at the boundaries between the bimetal oxide, Mn1.5Co1.5O4, and the carbon support. Scheme 2 shows a mixed mechanism of ORR with a larger fraction of the 4-electron reaction (Scheme 2a) compared to the smaller 2-electron reduction fraction (Scheme 2b).
 |
| | Scheme 2 Mixed mechanism for ORR at bimetal oxide and un-doped carbon support. | |
To further increase the catalytic activity, we used a functional carbon (i.e., a metal-nitrogen co-doped graphene) as a support to replace the traditional carbon. We prepared the nanocomposite material, Mn1.5Co1.5O4, supported on FeNx/C, by ultrasonic processing. Interestingly, we realized a near completed 4-electron oxygen reduction reaction for the nano-composite as a catalyst. Scheme 3 shows a 4-electron reaction mechanism of ORR at the nano boundaries between the bimetal sites and the FeNx/C nano sheets.
 |
| | Scheme 3 A mechanism for ORR at bimetal oxide and FeNx-doped carbon support. | |
The enhancement of catalytic activity is believed to be a synergistic effect of the bimetal atoms (Mn and Co), and a synergistic effect of the bimetal oxide nano crystals and the FeNx/C nano sheets.
2. Experimental section
2.1. Chemicals and materials
Hemin from porcine, cobalt(II) oxalate dihydrate (CoC2O4·2H2O), manganese(II) oxalate dihydrate (MnC2O4·2H2O), and ammonium hydroxide (28% NH3), were purchased from Aldrich. Graphene nano platelets (GNP) with a BET surface area between 600 and 650 m2 g−1 were purchased from Cheap Tubes Inc. Platinum nano catalyst supported on 60% active carbon (40% Pt/C) was purchased from Johnson Matthey. Deionized water was used for all synthesis and experiments.
2.2. Synthesis of Mn2O3, Co3O4 and Mn1.5Co1.5O4
1 g CoC2O4·2H2O and 1 g MnC2O4·2H2O were mixed with 3 mL NH4OH water, ground to a paste and dried at 70 °C. The dry mixture was put in a ceramic boat and heat-treated at 400 °C in the presence of air for two hours. A bimetal oxide with black color was obtained, and determined as Mn1.5Co1.5O4. The Mn2O3 and Co3O4 were synthesized by weighing 2 g MnC2O4·2H2O and 2 g CoC2O4·2H2O, respectively, and following the same procedure as that of making Mn1.5Co1.5O4, except processing and heat-treating them separately. For comparison, a mixture of Mn2O3 and Co3O4 was also prepared by ultrasonic treatment of equal amount of Mn2O3 and Co3O4 in water for 30 minutes and dried at 60 °C.
2.3. Synthesis of FeNx/C
Synthesized according to our previous reports6–8 and briefly described here. 1 g hemin and 1 g GNP were uniformly dispersed in 50 mL N,N-dimethylformamide (DMF). After drying on a hot-plate, the mixture was placed in a ceramic boat and sent to a tubular furnace for heat-treatment for 2 hours under a nitrogen atmosphere at 600 °C. The material after pyrolysis contained 4.9% Fe (w/w), 4.3% N (w/w), 1.5% H (w/w) and 77.8% C (w/w), with N/Fe atomic ratio ∼3.5 by elemental analysis.
2.4. Synthesis of Mn1.5Co1.5O4–FeNx/C nanocomposite
The nanocomposite was synthesized by ultrasonically processing Mn1.5Co1.5O4 and FeNx/C to break the large particles into nano-sized particles. A Branson Sonifier 450 was used with an ultrasound tip source. Here, 32 mg Mn1.5Co1.5O4 and 75 mg FeNx/C were mixed in a small beaker containing 4 mL 1-propanol and 4 mL water. The samples were cooled in an ice bath, and processed at a duty cycle of 40 and output control of 8 for 20 minutes. After ultrasound treatment an ink-like solution was obtained, containing 4 mg mL−1 Mn1.5Co1.5O4 dry sample and 9.4 mg mL−1 FeNx/C. The dried material is referred to a Mn1.5Co1.5O4–FeNx/C nanocomposite. The ink was directly used for electrochemical analysis by adding 5% Nafion solution as binder. As mentioned in our previous report,6 the ultrasound method is a powerful tool for processing nano-materials. Moreover, the FeNx/C is a high surface area nanomaterial with high electric conductivities and relatively good chemical stabilities to accommodate the metal oxides. The Mn2O3/C, Co3O4/C, and Mn1.5Co1.5O4/C samples were prepared by ultrasonically mixing with 70% GNP in chilled in an ice bath.
2.5. Instrumental analysis
X-Ray Diffraction data (XRD) were obtained from a Rigaku Ultima III instrument with Cu Kα radiation (λ = 1.5418 Å) using a Bragg–Brentano configuration. The measurements were conducted with a scan rate of 1.0 degree (2θ) per minute; and each diffraction data point was collected at the interval of 0.02 degree (2θ) for the total spectrum range of 10 to 80 degree (2θ). Transmission electron microscopy (TEM) were obtained with a high-resolution JEOL 2100 FE instrument equipped with an EDAX X-ray detector and a high angle annular dark field (HAADF) detector. Before imaging, a solution was prepared by adding 5 mg of the catalyst into 5 mL of 1-propanol followed by a short ultrasonic bath (Branson 3510) treatment. An aliquot of ink was loaded onto a QUANTIFOIL®-Holey Carbon Films (0.6 μm hole, 1.6 μm space 300 mesh copper grid, Electron Microscopy Science) with a micro-pipette and dried overnight at room temperature. Elemental mapping were obtained by scanning transmission electron microscopy (STEM) based on atomic resolution HAADF imaging. Elemental analysis was obtained by energy dispersive X-ray spectroscopy (EDS).
2.6. Electrochemical characterization
A Pine Bipotentiostat RDE4 was used for electrochemical characterization. A glassy carbon (GC) disk electrode (0.196 cm2) and a ring-disk electrode were used. The ring-disk electrode had a GC disk electrode (0.247 cm2) and a Pt-ring electrode (collection coefficient 0.37). A catalyst sample was first suspended in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/1-propanol solution and cooled in an ice bath. The catalyst ink was prepared by ultrasonic treatment with a Branson Sonifier 450 with a duty cycle of 40 and an output control of 8 for 10 minutes. The final catalyst content was 4 mg mL−1 metal oxides (Mn1.5Co1.5O4, or Mn2O3, or Co3O4). Then, 10 μL of catalyst ink was coated on a GC disk working electrode for RDE experiments; and on the disk electrode of the ring-disk electrode for RRDE experiments, respectively. The ink was dried at 40 °C for over 60 minutes. The final coating on the GC disk electrode contained 95% total catalyst materials and 5% dry Nafion with a metal oxide loading of 0.20 mg cm−2. An ink of 40% Pt/C was prepared using the same procedure as that discussed for the metal oxide samples. The final loading of 40% Pt/C on the GC electrode was 0.20 mg cm−2. The catalyst coated electrode was mounted onto a Pine ASR rotator and used as the working electrode. All voltammetry experiments were performed in argon (Ar) or O2 saturated 0.1 M potassium hydroxide (KOH) solution at room temperature (20 ± 1 °C). A three electrode glass cell was used for electrochemical study containing three compartments separated by porous ceramic layers. A platinum wire counter electrode and a saturated calomel reference electrode (SCE) were used in the cell. The experimental results were reported based on reversible hydrogen electrode (RHE). The difference between SCE and RHE was experimentally determined with an SCE and a fresh-prepared RHE in 0.1 M KOH solution in the same electrochemical cell. The current density reported in this article is based on the geometrical area of the GC disk electrode (mA cm−2), which can be changed to current density based on mass (mA mg−1) by simply multiplying a factor of 5 according to the mass of the catalyst loading.
:1 water/1-propanol solution and cooled in an ice bath. The catalyst ink was prepared by ultrasonic treatment with a Branson Sonifier 450 with a duty cycle of 40 and an output control of 8 for 10 minutes. The final catalyst content was 4 mg mL−1 metal oxides (Mn1.5Co1.5O4, or Mn2O3, or Co3O4). Then, 10 μL of catalyst ink was coated on a GC disk working electrode for RDE experiments; and on the disk electrode of the ring-disk electrode for RRDE experiments, respectively. The ink was dried at 40 °C for over 60 minutes. The final coating on the GC disk electrode contained 95% total catalyst materials and 5% dry Nafion with a metal oxide loading of 0.20 mg cm−2. An ink of 40% Pt/C was prepared using the same procedure as that discussed for the metal oxide samples. The final loading of 40% Pt/C on the GC electrode was 0.20 mg cm−2. The catalyst coated electrode was mounted onto a Pine ASR rotator and used as the working electrode. All voltammetry experiments were performed in argon (Ar) or O2 saturated 0.1 M potassium hydroxide (KOH) solution at room temperature (20 ± 1 °C). A three electrode glass cell was used for electrochemical study containing three compartments separated by porous ceramic layers. A platinum wire counter electrode and a saturated calomel reference electrode (SCE) were used in the cell. The experimental results were reported based on reversible hydrogen electrode (RHE). The difference between SCE and RHE was experimentally determined with an SCE and a fresh-prepared RHE in 0.1 M KOH solution in the same electrochemical cell. The current density reported in this article is based on the geometrical area of the GC disk electrode (mA cm−2), which can be changed to current density based on mass (mA mg−1) by simply multiplying a factor of 5 according to the mass of the catalyst loading.
3. Results and discussion
3.1. XRD results
The composition and morphology of several metal oxides were characterized with XRD, including the mono metal oxides (manganese oxide and cobalt oxide) and the bimetal oxide (manganese–cobalt oxide). As shown in Fig. 1, all of them are highly crystalized. Their structures and compositions were determined with the XRD results, as α-Mn2O3, Co3O4, and (Mn,Co)3O4, respectively. The atomic ratio of Mn to Co in the (Mn,Co)3O4 crystals was further determined with EDS analysis, as ∼1 (see the following EDS results). Therefore, it is determined that the bimetal oxide is Mn1.5Co1.5O4 with a spinel structure. The crystallite size of Mn1.5Co1.5O4 (11.8 nm) is significantly smaller than that of α-Mn2O3 (21.3 nm) or Co3O4 (24.5 nm). As shown in Fig. 1, the XRD pattern of the Mn1.5Co1.5O4–FeNx/C nanocomposite has the main peaks of both Mn1.5Co1.5O4 and FeNx/C. The chemical states of Co and Mn in the nanocomposite have been cleared from the XRD results showing in Fig. 1, as Co3+/Co2+ and Mn3+/Mn2+. The chemical state of nitrogen in the nanocomposite can be determined from X-ray photoelectron spectroscopy (XPS) according to the reports.13,40,41 For Fe-porphyrins pyrolyzed at 600 °C,40,41 the deconvoluted high-resolution XPS spectra at the N 1s region showed three peaks, assigned as pyrrolic, pyridinic and Fe–N4 based nitrogen atoms, with peak positions (binding energy, eV) at 400.3, 398.8 and 398.1, respectively. The chemical state of iron has been determined from the electrochemical results (cyclic voltammograms), as Fe+3/Fe+2.
 |
| | Fig. 1 X-ray diffraction pattern of non-precious metal oxides, FeNx-doped carbon and Mn1.5Co1.5O4–FeNx/C nanocomposite. | |
3.2. TEM and STEM images
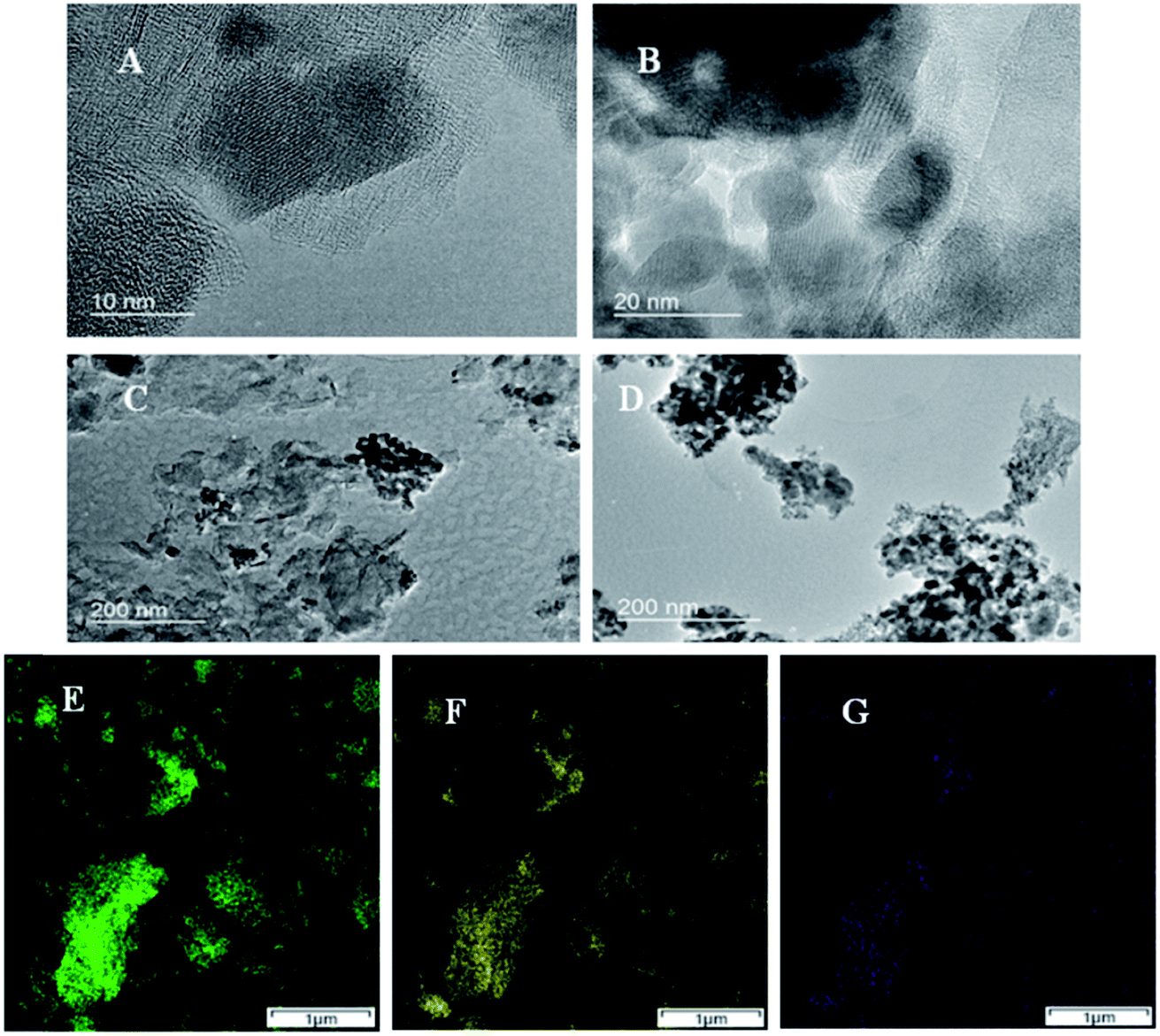
The nano boundaries between these metal oxides and GNP are examined with TEM. Fig. 2A–C shows TEM images of Mn1.5Co1.5O4–FeNx/C. Some small darker and round regions are seen, and identified as Mn1.5Co1.5O4 particles. The positions of GNP can also be recognized from the larger and lighter regions. However, the boundaries between Mn1.5Co1.5O4 particles and GNP are unclear, which implies that the nano boundaries are well formed between the two materials with no gaps. Fig. 2D shows TEM image of unsupported Mn1.5Co1.5O4 with measured particle sizes around 10–20 nm, which is consistent with that of the XRD results. The purity and uniformity of Mn1.5Co1.5O4–FeNx/C are examined by atomic resolution elemental mapping with STEM-EDS imaging. Fig. 2E–G shows elemental mapping of Co, Mn and Fe, respectively. The spatial distributions of Fig. 2E and F are matched very well, which indicates that the Mn1.5Co1.5O4 is a uniform bimetal oxide, instead of a mixture of α-Mn2O3 and Co3O4. The crystals of Mn1.5Co1.5O4, are formed when the Co atoms are replaced by some Mn atoms in the spinel structure of Co3O4. The Fe element is evenly dispersed on the small graphene sheets. The spatial distribution of Fe element (see Fig. 2G) is also approximately matched with that of Mn1.5Co1.5O4, which indicates that the nano boundaries between FeNx/C and Mn1.5Co1.5O4 are well formed. The elemental content of Mn1.5Co1.5O4–FeNx/C and Mn1.5Co1.5O4 is measured with STEM-EDS, and listed in Table 1. Equal Mn and Co atomic concentrations of are seen, which matches the elemental composition of Mn1.5Co1.5O4 and the XRD results.
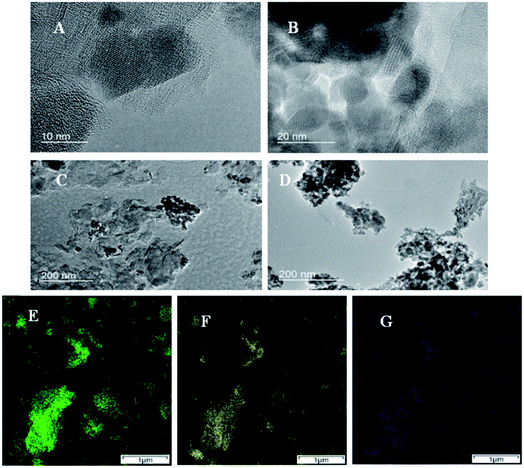 |
| | Fig. 2 TEM images of Mn1.5Co1.5O4–FeNx/C (A–C), and Mn1.5Co1.5O4 (D); and elemental maps of Mn1.5Co1.5O4–FeNx/C obtained from atomic resolution STEM-EDS imaging (E–G) Co Ka1 (E), Mn Ka1 (F), and Fe Ka1 (G). | |
Table 1 Relative metal contents in Mn1.5Co1.5Ox–FeNx/C and Mn1.5Co1.5Ox samples obtained from STEM-EDS analysis (note: the C% content is excluded for clarification)
| Sample |
Mn% |
Co% |
Fe% |
O% |
Total |
| MnCoOx–FeNx/C |
w% |
24.9 |
26.9 |
4.6 |
43.6 |
100 |
| at% |
12.2 |
12.3 |
2.2 |
73.3 |
100 |
| MnCoOx |
w% |
31.1 |
29.3 |
— |
39.6 |
100 |
| at% |
14.9 |
15.1 |
— |
70.0 |
100 |
3.3. Electrocatalytic activity and mechanisms of the ORR at the Mn1.5Co1.5O4–FeNx/C nanocomposite electrode
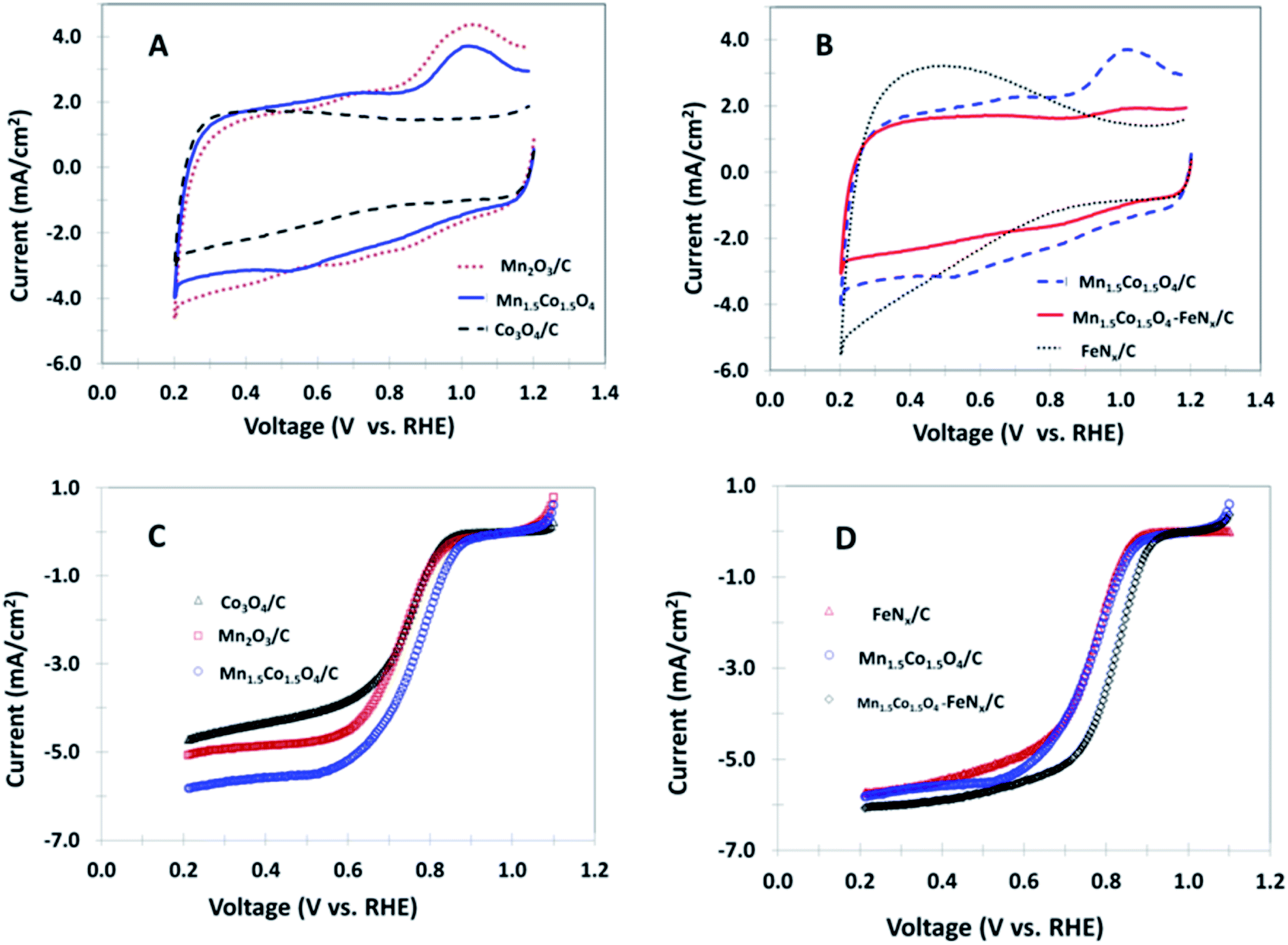
The electrochemical behaviors of various non-precious nanocomposites coated GC electrodes are initially examined by cyclic voltammetry (CV) in 0.1 M KOH in the absence of oxygen, as shown in Fig. 3A and B. An oxidation peak at ∼1.0 V and a reduction peak appearing between 0.5 and 0.7 V are seen for the samples of Mn2O3/C, Mn1.5Co1.5O4/C and Mn1.5Co1.5O4–FeNx/C. It is noticeable that all of the three materials contain Mn element in their composition, indicating redox peaks for Mn ion oxidation and reduction. No peak is observed for the polarization curve obtained from the Co3O4/C nanocomposite. In addition, no peak is observed for the FeNx/C electrode, however, the background current increases when the potential is less than 0.8 V. The rapid increase in background current at the lower potential is likely due to ions adsorption and desorption at the FeNx/C surface. The ORR catalytic activity is detected by bubbling O2 through the electrolyte solution. The catalytic activity is accurately analyzed by the rotating disk electrode method (RDE). Fig. 3C shows polarization curves for the mono-metal oxides, and bimetal oxide coated RDEs in O2 saturated 0.1 M KOH. The ORR limiting current increases in the order of Mn1.5Co1.5O4/C > Mn2O3/C > Co3O4/C. The half wave potential positively shifts from 0.73 V for Mn2O3/C to 0.76 V for Mn1.5Co1.5O4/C, which implies that the Mn1.5Co1.5O4/C has better catalytic activity than that of the mono-metal oxides Co3O4/C and Mn2O3/C investigated in this study. The enhancement of activity at the bimetal oxide is attributed to a synergistic effect of different metal atoms in the bimetal crystal. The Co3O4 shows a spinel structure, with Co2+ ions in tetrahedral interstices and Co3+ ions in the octahedral interstices of the cubic close-packed lattice of oxide anions. The Mn is also able to form a Mn3O4 spinel structure. When Co and Mn form a bimetal oxide, the atoms of Co and Mn are positioned in the same spinel structure, which changes the parameters of their original structure, resulting in smaller crystallite size and particle size as shown by XRD and TEM results. The smaller crystal and particle size of a catalyst are corresponding to the higher surface area, the more active catalytic sites, and the higher catalytic activity for ORR. Therefore, we call this phenomenon as synergistic catalytic effect of bimetal composition. Fig. 3D shows polarization curves of FeNx/C, Mn1.5Co1.5O4/C, and Mn1.5Co1.5O4–FeNx/C coated RDEs in O2 saturated 0.1 M KOH. The highest limiting current and the most positive half wave potential (0.82 V) is observed for the Mn1.5Co1.5O4–FeNx/C nanocomposite-coated RDE.
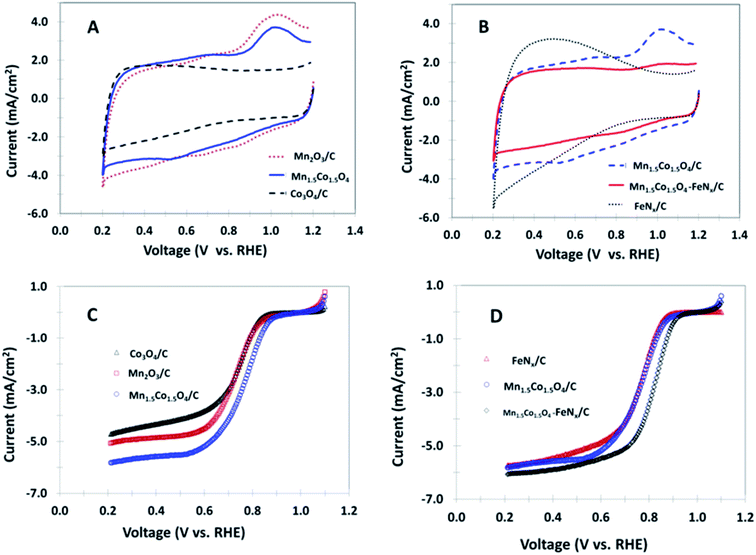 |
| | Fig. 3 Cyclic voltammograms of the different nanocomposites coated GC disk electrodes in argon saturated 0.1 M KOH, 50 mV s−1 (A and B); and polarization curves of the same nanocomposites coated disk electrodes in O2 saturated 0.1 M KOH, 10 mV s−1, rotation rate 1600 rpm (C and D). | |
The enhancement of catalytic activity at the Mn1.5Co1.5O4–FeNx/C nanocomposite is attributed to a secondary synergistic effect of ORR at the nano boundaries, where the oxygen molecules are catalyzed by two types of catalytic sites, or the bimetal atoms, Mn–Co, at the Mn1.5Co1.5O4 site, and the Fe atom at FeNx/C site. The polarization curves of these catalysts coated RDEs are further analyzed with Tafel plots. The half wave potentials and Tafel slopes of ORR at these polarization curves are listed in Table 2. The Mn1.5Co1.5O4–FeNx/C nanocomposite has the smallest Tafel slope (35 mV dec−1), which indicates an advantage of fast electron transfer between the oxygen molecules and the catalyst coated electrode. The number of electron transfer and the yield of intermediate product, H2O2, for ORR are obtained from the RRDE data. Fig. 4A and B shows RRDE polarization curves of ORR at different nanocomposites coated GC disks and Pt-ring electrode in O2 saturated 0.1 M KOH. The current densities in Fig. 4 are somewhat different from that in Fig. 3. This difference often happens when the RDE and RRDE experiments are carried out with different electrodes that have different electrode areas. In the present research we used different electrodes for RDE (0.196 cm2) and RRDE (0.247 cm2) experiments, respectively. Due to the catalyst loading on the RDE and RRDE was exact the same (0.04 mg of metal oxide), which caused the catalyst dispersion and concentration differences (0.2 mg cm−2 for RDE and 0.16 mg cm−2 for RRDE), and the current density a slightly difference. Fortunately, these differences will not affect our comparison of the catalyst performance. The goal of the RDE experiment is used for comparison of the catalyst activity and stability; but the goal of the RRDE experiment is used to compare the H2O2 yielding during the process of ORR. The higher ring current indicates the more H2O2 generated for ORR at the disk electrode and collected at the ring electrode. The magnitude of ring current is Co3O4/C > Mn2O3/C > Mn1.5Co1.5O4/C > FeNx/C > Mn1.5Co1.5O4–FeNx/C. Plots of the number of electron transfer and the yield of H2O2% versus disk potential are shown in Fig. 4C and D, respectively. The yield of H2O2 is potential dependent, and a peak yield appears at ∼0.6 V. The electron transfer number and H2O2% yield at 0.6 V are also summarized in Table 2. The peak yield of H2O2% at the Mn1.5Co1.5O4–FeNx/C nanocomposite is less than 5%. The very small H2O2 production is probably because a small part of electrode is incompletely covered by the catalyst coating. Apparently, the Mn1.5Co1.5O4–FeNx/C nanocomposite mainly catalyzes 4-electron oxygen reduction. The mechanism of ORR at the nanocomposite Mn1.5Co1.5O4–FeNx/C has been explained with Scheme 3, where a nano boundary is composed of Mn1.5Co1.5O4 and FeNx/C. When an oxygen molecule is diffused into a nano boundary, it is catalyzed simultaneously by the bimetal site, Mn–Co, and the FeNx site. If any intermediate product, H2O2, is generated at the Mn–Co surface, it will be continuously reduced at the FeNx site. In addition, the FeNx/C is not only able to catalyze 4-electron O2 reduction to water, but also it is able to catalyze 2-electron H2O2 reduction to water with excellent activity.8,10,20,41,42 The mechanism is shown below,
| | |
2Fe(II)Nx/C + HO−2 + H2O → 3OH− + 2Fe(III)Nx/C
| (1) |
| | |
2Fe(III)Nx/C + 2e− → 2Fe(II)Nx/C
| (2) |
Table 2 Electrochemical parameters of ORR at different nanocomposites coated electrodes in 0.1 M KOH obtained from RDE and RRDE experiments
| Parameters |
Co3O4/C |
Mn2O3/C |
Mn1.5Co1.5O4/C |
FeNx/C |
Mn1.5Co1.5Ox–FeNx/C |
| E1/2 (V) |
0.74 |
0.73 |
0.76 |
0.77 |
0.82 |
| Tafel slope (mV dec−1) |
49 |
63 |
54 |
52 |
35 |
| H2O2% yield at 0.6 V |
57.4 |
21.6 |
16.1 |
7.3 |
4.8 |
| Electron transfer number at 0.6 V |
2.85 |
3.57 |
3.68 |
3.85 |
3.90 |
 |
| | Fig. 4 Polarization curves for the different nanocomposites coated GC disks and Pt-ring electrode in O2 saturated 0.1 M KOH (A and B). Note: the rotation rate is 1600 rpm, scan rate is 10 mV s−1 and the Pt-ring potential was held at 1.2 V vs. RHE with a ring collection coefficient of 0.37. Plots of the number of electrons transferred per O2 molecule (C), and the H2O2% generation as a function of the disk potential (D). | |
At the Mn1.5Co1.5O4–FeNx/C nanocomposite coated electrode, many hetero nano boundaries are evenly dispersed, containing functional groups of Mn1.5Co1.5O4 and FeNx/C, which promote a coordinative catalysis, realizing 4-electron oxygen reduction.
3.4. Catalytic kinetics of ORR at the Mn1.5Co1.5O4–FeNx/C nano composite
The catalytic kinetics of ORR at the Mn1.5Co1.5O4–FeNx/C nanocomposite is investigated by analyzing the variations of the limiting current with rotation rate at the catalyst coated rotating disk electrode. Fig. 5A shows polarization curves of Mn1.5Co1.5O4–FeNx/C coated GC disk electrode in O2 saturated 0.1 M KOH at various rotation rates. The limiting current increases with increasing rotation rate. However, the limiting current is not proportional to the rotation rate at higher rotation rates, which gradually deviates from the values calculated by the Levich equation.7,8 Fig. 5B shows Levich plots at various electrode potentials. The dotted line is the calculated results for 4-electron oxygen reduction by diffusion controlled process. At higher potentials, there are more deviations from that of the calculated dotted line. Fig. 5C shows Kotecky–Levich plots,7,8 all of which are straight lines, and parallel to that of the calculated line (the dotted) for 4-electron oxygen reduction by diffusion controlled process, which indicates that the catalytic process belongs to a process of 4-electron oxygen reduction at the Mn1.5Co1.5O4–FeNx/C nanocomposite coated electrode. The kinetic rate constants are calculated from the intercepts of the Koutecky–Levich plots, and listed in Table 3. At 0.7 V, the kinetic rate constant is 7.6 × 10−2 cm s−1, which is higher than that of silver alloy nano particles (6.8 × 10−2 cm s−1)20 for catalytic ORR in alkaline media.
 |
| | Fig. 5 (A) Polarization curves of Mn1.5Co1.5O4–FeNx/C coated GC disk electrode in O2 saturated 0.1 M KOH at different rotation rates. Scan rate: 10 mV s−1. (B) Levich plots of i versus ω1/2. (C) Koutecky–Levich plots of i−1 versus ω−1/2. | |
Table 3 Kinetic rates constant at Mn1.5Co1.5Ox–FeNx/C nanocomposite coated electrode in O2 saturated 0.1 M KOH
| Catalyst |
Kinetic rate constant (cm s−1) |
| 0.8 V |
0.7 V |
0.6 V |
0.5 V |
| Mn1.5Co1.5O4–FeNx/C |
1.4 × 10−2 |
7.6 × 10−2 |
11.7 × 10−2 |
24.9 × 10−2 |
3.5. Comparison with Pt catalyst
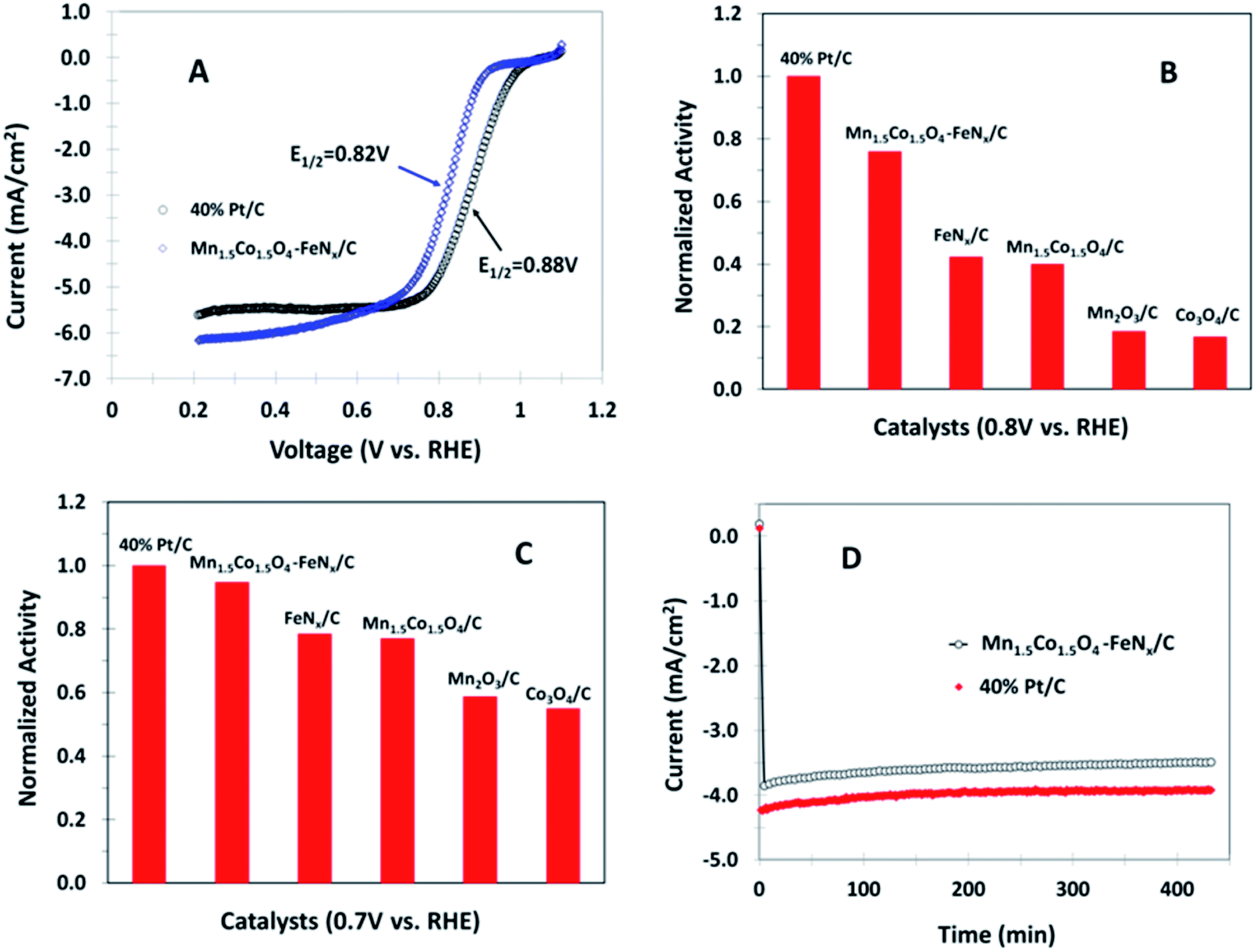
The activity and stability of Mn1.5Co1.5O4–FeNx/C nanocomposite are compared with commercially available Pt catalyst containing 40% Pt and 60% active carbon (40% Pt/C). The catalyst loadings on the electrode were 0.2 mg cm−2 on the electrode for the metal oxide and 40% Pt/C samples, respectively. 5% Nafion was added as a binder in all catalyst coatings. Fig. 6A shows polarization curves of Mn1.5Co1.5O4–FeNx/C and 40% Pt/C coated rotating GC disk electrodes in O2 saturated 0.1 M KOH, respectively. There is 60 mV half wave potential difference between Mn1.5Co1.5O4–FeNx/C and 40% Pt/C. The catalytic activities are compared, as shown in Fig. 6B and C by assuming the activity of 40% Pt/C equal to 1. At 0.7 V the activity of Mn1.5Co1.5O4–FeNx/C reaches 95% that of the 40% Pt/C. The catalytic stability was also examined by chronoamperometric experiment at 0.7 V and 900 rpm rotation rate, during which oxygen gas was bubbled through the electrolyte continuously. Fig. 6D shows chronoamperometric curves of Mn1.5Co1.5O4–FeNx/C and 40% Pt/C coated GC disk electrodes in 0.1 M KOH solution, respectively. The approximately paralleled curves obtained indicates that the catalytic stability of the Mn1.5Co1.5O4–FeNx/C is comparable to that of the 40% Pt/C.
 |
| | Fig. 6 Polarization curves for (A) Mn1.5Co1.5O4–FeNx/C and 40% Pt/C coated GC disk electrodes in O2 saturated 0.1 M KOH with a scan rate of 10 mV s−1 and rotation rate of 1600 rpm. (B and C) Normalized activities at 0.8 and 0.7 V, respectively, where the ORR activity of 40% Pt/C is set equal to 1. (D) Chronoamperometric curves at 0.7 V for Mn1.5Co1.5O4–FeNx/C and 40% Pt/C coated GC electrode in O2 saturated 0.1 M KOH with a rotation rate of 900 rpm. | |
4. Conclusion
In this study we demonstrated a method of making non-precious metal oxide nanocomposite catalysts and achieved double synergistic effects for catalytic oxygen reduction. A precious mono-metal oxide's catalytic activity can be enhanced by incorporating a second non-precious metal element into its crystal structure to generate a nano crystal material with uniform elemental spatial distribution. The addition of the second metal in the crystal structure of a mono-metal oxide results in smaller crystallite size, particle size, and higher active surface area for the bimetal oxide, leading to higher catalytic activity. The catalytic activity of the bimetal oxide can be further enhanced by supporting it on a functional carbon material containing FeNx functional groups. The bimetal oxide, the functional carbon and the electrolyte form nano boundaries, which are the actives sites for catalytic oxygen reduction. The FeNx group is functioned as a mediator to remove H2O2 intermediate generated in the catalytic process of oxygen reduction, and promoting oxygen to be catalyzed through a complete 4-electron reduction. Our work provides a new pathway in design and synthesis of novel non-precious nanocomposite catalysts.
Acknowledgements
The authors would like to thank Dr Wen-An Chiou and Dr Sz-Chian Liou at University of Maryland for assisting TEM and STEM analyses. Thanks to the U.S. Department of the Army and U.S. Army Materiel Command for supporting this work.
References
- J. Marcinkoski, B. D. James, J. A. Kalinoski, W. Podolski, T. Benjamin and J. Kopasz, J. Power Sources, 2011, 196, 5282–5292 CrossRef CAS.
- J. P. Collman, M. Marrocco, P. Denisevich, C. Koval and F. C. Anson, J. Electroanal. Chem., 1979, 101(1), 117 CrossRef CAS.
- J. P. Collman, P. Denisevich, Y. Konai, M. Marrocco, C. Koval and F. C. Anson, J. Am. Chem. Soc., 1980, 102(19), 6027–6036 CrossRef CAS.
- X. M. Hu, D. G. Xia, L. Zhang and J. J. Zhang, J. Power Sources, 2013, 231, 91–96 CrossRef CAS.
- H. D. Sha, X. X. Yuan, L. Li, Z. Ma, Z. F. Ma, L. Zhang and J. J. Zhang, J. Power Sources, 2014, 255, 76–84 CrossRef CAS.
- R. Z. Jiang, D. T. Tran, J. McClure and D. Chu, Electrochem. Commun., 2012, 19, 73–76 CrossRef CAS.
- R. Z. Jiang, D. T. Tran, J. McClure and D. Chu, Electrochim. Acta, 2012, 75, 185–190 CrossRef CAS.
- R. Z. Jiang and D. Chu, J. Power Sources, 2014, 245, 352–361 CrossRef CAS.
- D. Chu and R. Z. Jiang, Solid State Ionics, 2002, 148, 591–599 CrossRef CAS.
- R. Z. Jiang and D. Chu, J. Electrochem. Soc., 2000, 147(12), 4605–4609 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- D. H. Li, C. X. Ly, L. Liu, Y. Z. Xia, X. L. She, S. J. Guo and D. J. Yang, ACS Cent. Sci., 2015, 1, 261–269 CrossRef CAS PubMed.
- J. P. McClure, R. Z. Jiang, D. Chu and P. S. Fedkiw, Carbon, 2014, 79, 457–469 CrossRef CAS.
- D. G. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351(6271), 361–365 CrossRef CAS PubMed.
- J. P. McClure, C. K. Devine, R. Z. Jiang, D. Chu, J. J. Cuomo, G. N. Parsons and P. S. Fedkiw, J. Electrochem. Soc., 2013, 160, F769–F778 CrossRef CAS.
- J. Zhang, S. Wu, X. Chen, M. Pan and S. C. Mu, J. Power Sources, 2014, 271, 522 CrossRef CAS.
- H. Yin, C. Z. Zhang, F. Liu and Y. L. Hou, Adv. Funct. Mater., 2014, 24, 2930–2937 CrossRef CAS.
- J. P. McClure, J. D. Thornton, Z. Jiang, D. Chu, J. J. Cuomo and P. S. Fedkiw, J. Electrochem. Soc., 2012, 159, F733–F742 CrossRef CAS.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- R. Z. Jiang, D. T. Tran, J. McClure and D. Chu, ACS Appl. Mater. Interfaces, 2015, 7, 18530–18539 CAS.
- Y. Y. Ma, R. F. Wang, H. Wang, J. L. Key and S. Ji, J. Power Sources, 2015, 280, 526–532 CrossRef CAS.
- N. I. Andersen, A. Serov and P. Atanassov, Appl. Catal., B, 2015, 163, 623–627 CrossRef CAS.
- H. Y. Park, T. J. Shin, H. I. Joh, J. H. Jang, D. Ahn and S. J. Yoo, Electrochem. Commun., 2014, 41, 35–38 CrossRef CAS.
- T. R. Zhang, F. Y. Cheng, J. Y. X. Hu and J. Chen, Adv. Energy Mater., 2015, 5, 1400654 CrossRef.
- D. J. Davis, T. N. Lambert, J. A. Vigil, M. A. Rodriguez, M. T. Brumbach, E. N. Coker and S. J. Limmer, J. Phys. Chem. C, 2014, 118, 17342–17350 CAS.
- C. Shia, G. L. Zang, Z. Zhang, G. P. Sheng, Y. X. Huang, G. X. Zhao, X. K. Wang and H. Q. Yu, Electrochim. Acta, 2014, 132, 239–243 CrossRef.
- F. Y. Cheng, Y. Su, J. Liang, Z. L. Tao and J. Chen, Chem. Mater., 2010, 22, 898–905 CrossRef CAS.
- Y. G. Wang, L. Cheng, F. Li, H. M. Xiong and Y. Y. Xia, Chem. Mater., 2007, 19, 2095–2101 CrossRef CAS.
- J. B. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333–5339 CAS.
- Y. Y. Liang, Y. G. Li, H. J. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- S. Bag, K. Roy, C. S. Gopinath and C. R. Raj, ACS Appl. Mater. Interfaces, 2014, 6, 2692–2699 CAS.
- H. Q. Dong, Y. Y. Chen, M. Han, S. L. Li, J. Zhang, J. S. Li, Y. Q. Lan, Z. H. Dai and J. C. Bao, J. Mater. Chem. A, 2014, 2, 1272–1276 CAS.
- T. Odedairo, X. C. Yan, J. Ma, Y. L. Jiao, X. D. Yao, A. J. Du and Z. H. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 21373–21380 CAS.
- S. Y. Liu, L. J. Li, H. S. Ahn and A. Manthiram, J. Mater. Chem. A, 2015, 3, 11615–11623 CAS.
- Y. T. Luo, J. H. Guo, Y. H. Liu, Q. Shao, C. S. Wang and D. Chu, J. Membr. Sci., 2012, 423, 209–214 CrossRef.
- Y. T. Luo, J. H. Guo, C. S. Wang and D. Chu, Electrochem. Commun., 2012, 16, 65–68 CrossRef CAS.
- Y. S. Li, T. S. Zhao and X. X. Liang, J. Power Sources, 2009, 187, 387–392 CrossRef CAS.
- E. E. Switzer, T. S. Olson, A. K. Datye, P. Atanassov, M. R. Hibbs, C. Fujimoto and C. Cornelius, Electrochim. Acta, 2010, 55, 3404–3408 CrossRef CAS.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy., 2016, 1, 15006 CrossRef.
- U. I. Kramm, I. Abs-Wurmbach, I. Herrmann-Geppert, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2011, 1(158), B69–B78 CrossRef.
- Q. Wu, M. Nelson, S. Ma, H. Meng, G. F. Cui and P. K. Shen, Carbon, 2011, 49, 3972–3982 CrossRef.
- S. Li, L. Zhang, J. Kim, M. Pan, Z. Shi and J. J. Zhang, Electrochim. Acta, 2010, 55, 7346–7353 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.