
Injectable and self-healing dynamic hydrogel containing bioactive glass nanoparticles as a potential biomaterial for bone regeneration†
Abstract
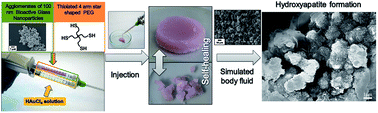
Combination of an Au-based 4-arms thiol terminated poly(ethylene glycol) [Au-(PEGSH)4] dynamic hydrogel exhibiting self-healing ability with 100 nm bioactive glass (BAG) nanoparticles agglomerated in 10 μm clusters, produced via a particulate sol–gel method, resulted in the formation of hydrogel nanocomposites [Au-(PEGSH)4–BAG] with enhanced properties. Au-(PEGSH)4–BAG hydrogel nanocomposites were prepared by injecting simultaneously an aqueous solution of a (PEGSH)4 homopolymer containing different amounts of BAG nanoparticles with an aqueous solution of HAuCl4 using a double barrel syringe. Electron microscopy studies suggested that the clusters of inorganic particles were homogeneously distributed into the polymeric matrix. Rheology studies demonstrated that stiffer hydrogels were obtained after the addition of BAG nanoparticles. The presence of the inorganic colloids appeared to affect slightly the dynamic character of the pristine hydrogel by slowing down the exchange reaction between gold–thiolate (Au–S) and disulfides (SS). Despite slower Au–S/SS exchange, the resulting hydrogel nanocomposites were still exhibiting pH-dependent properties and self-healing abilities, as judged by frequency sweep experiments. In addition, compression tests demonstrated the major drawbacks of each individual material, i.e. brittleness for BAG nanoparticles and weak consistency for Au-(PEGSH)4, were suppressed to result in a material composite with high resistance to stress and relatively large deformation ability. Moreover, the slow diffusion inside the 3D matrix allowed the degradation of the BAG nanoparticles to be delayed as well as the pH to be maintained around physiological values. For the same reason, the incorporation of those BAG nanoparticles into the dynamic hydrogel proved to reduce the cytotoxicity of the organic particles. Finally, in vitro degradation of BAG nanoparticles embedded in the dynamic hydrogel led to the formation of hydroxyapatite. As a result, osteoinductive properties could be anticipated for the Au-(PEGSH)4–BAG hydrogel nanocomposite which allows this new injectable and self-healing dynamic biomaterial to be considered as a scaffold to induce and promote bone self-repair.
 Please wait while we load your content...
Please wait while we load your content...

