
The effect of the carbon surface chemistry and electrolyte pH on the energy storage of supercapacitors
E. G. Calvo,
N. Rey-Raap,
A. Arenillas and
J. A. Menéndez*
Instituto Nacional del Carbón, INCAR-CSIC, Apartado 73, 33080 Oviedo, Spain. E-mail: angelmd@incar.csic.es; Fax: +34 985 29 76 62; Tel: +34 985 11 90 90
First published on 14th July 2014
Abstract
Energy storage in supercapacitors can be enhanced by optimising the electrostatic mechanism and the pseudo-faradaic reactions. In the latter case, it is essential to take into account the surface chemistry of the electrode material and the pH of the electrolyte. In this work, supercapacitors were assembled using three carbon xerogels containing different oxygen surface groups and various aqueous media (Na2SO4, H2SO4 and KOH), in order to evaluate the influence of the surface chemistry on the electrochemical behaviour of the cells. However, it is not only necessary to take into account the chemical nature of the electrodes, but also the combination of this surface chemistry and the pH of the electrolyte, since the performance of the electrode material depends on the type of electrolyte medium used. In the case of a carbon xerogel with quinone-type oxygen groups, it has been demonstrated that there are pseudo-capacitive effects when the pH of the electrolyte is lower than the point of zero charge (pHPZC) of the electrodes, i.e., when the electrode surface is positively charged. However, in the case of an alkaline electrolyte, the carbon samples studied presented similar capacitance values, suggesting that the negative charges make a lower contribution in these carbon-based energy storage systems.
Introduction
In recent years, resorcinol-formaldehyde carbon gels have been used as electrodes for electric double-layer (EDL) capacitors even, in some cases, to the point of displacing activated carbons, i.e. commercial materials widely employed in this field.1–5 The attraction of carbon xerogels as active material for supercapacitors lies in their versatility and excellent properties which include a large surface area, a porous and chemical structure tuneable according to the synthesis conditions, a good electrical conductivity and a recently discovered fast and simple manufacturing process based on microwave heating.6,7Several works reported in the literature have demonstrated the feasibility of this type of carbonaceous material to accumulate energy.1–5 In carbon-based supercapacitors, two energy storage mechanisms can be found: EDL formation by charge separation at the electrode/electrolyte interface (non-faradaic mechanism) and voltage dependent faradaic reactions leading to pseudo-capacitance. It is well known that EDL formation is greatly enhanced by the use of rich micropore electrode materials.8–10 Nevertheless, it is important to take into account the micropore size since this mechanism does not seem to be favoured by ultramicropores (porosity below 0.7 nm), especially, when using hindered electrolytes such as organic media and ionic liquids.9,11–13 Furthermore, a large number of publications have demonstrated the relevance of mesopores, since they facilitate the diffusion of electrolyte ions, thereby increasing the access of the electrolyte to the microporosity.14,15 For this reason carbon xerogels would appear suitable candidates for this application because of their easily tailorable nano-texture which makes it possible to prepare micro-mesoporous carbon gels of specific pore sizes by merely selecting the appropriate conditions for the synthesis process.1,5,6,16–18
Pseudo-capacitive reactions are favoured by the use of electrodes based on transition metal oxides,19–21 conducting polymers22–24 or, as in the case of this study, porous carbons with electroactive species.2,5,8,10,14,15,25–28 Among this last category, the most commonly studied materials are oxygen, nitrogen and phosphorous-containing porous carbons. In the literature, a fairly large number of publications related to the use of carbons with oxygen-containing functional groups can be found.5,8,14,25 Two positive effects are derived from the incorporation of functionalities into the structure of carbonaceous materials. On the one hand, the wettability of the electrode material by the electrolyte solution is improved, the microporosity is more accessible and, therefore, the contribution of capacitance due to electrostatic interactions is increased. On the other hand, fast redox reactions are obtained, i.e. chemical interactions that introduce extra capacitance to the electrochemical cell, which is referred to as pseudo-capacitance. However, it should be borne in mind that sometimes this kind of system does not exhibit a high degree of reversibility since pseudo-capacitance decays with the number of charge–discharge cycles.27–29 Moreover, the electrochemical response of the system is not enhanced by all of the oxygenated groups. Several studies2,30,31 report the positive contribution of reversible oxidation/reduction of hydroquinone/quinone groups but a worse performance by the cells when electrode materials containing acidic oxygen functionalities are used. This does not mean that acidic groups should be discarded in favour of basic groups, because these surface groups behave differently (see Scheme 1) depending on the reaction medium used. For example, carbonyl–quinone type groups produce pseudo-capacitive effects in the presence of acidic aqueous electrolytes (H2SO4, for example), while hardly any influence is observed when an alkaline solution is employed as electrolyte.32 These findings are illustrated in Scheme 1, where it can be seen that the same oxygenated group can behave as donor/acceptor of electrons depending on the reaction media. It is evident that the nature of the surface functionalities of the electrodes is a determinant factor but always depending on the solution used as electrolyte since the same group can give rise to both negative and positive charges.
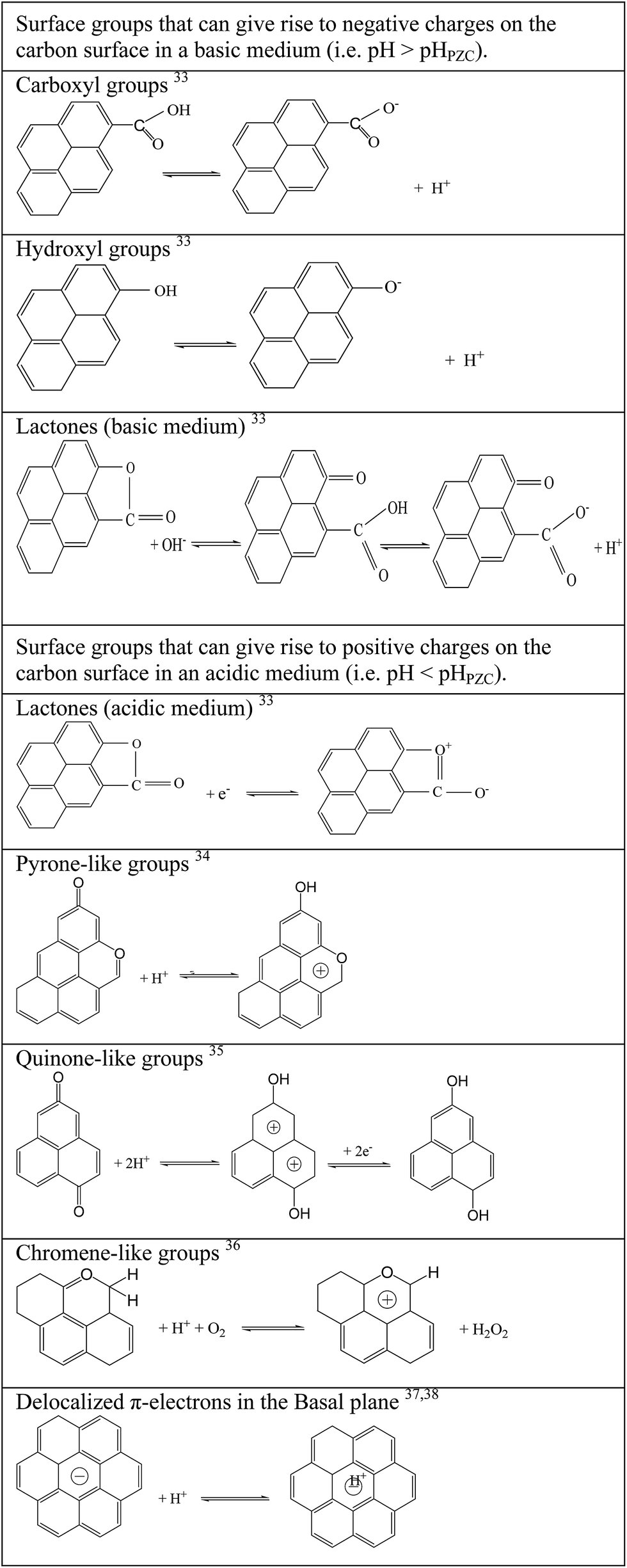 | ||
| Scheme 1 Different forms of surface functionalities depending on the reaction media.33–38 | ||
Thus, the main objective of the present paper is to explore the electrochemical behaviour of cells composed of oxidized carbon xerogels towards various reaction media in order to clarify the relationship between the pseudo-capacitive effects and oxygen functionalities. An attempt will be made to show that redox reactions not only depend on the kind of oxygenated groups, but also on the combination of the point of zero charge (pHPZC), which provides information about the amount and nature of surface groups and the pH of the electrolyte used. In order to achieve these aims, three electrode materials were prepared: an activated xerogel and two oxidized xerogels obtained from two oxidizing agents (O2 and HNO3). Their electrochemical behaviour was tested using three aqueous solutions with different pH (H2SO4, Na2SO4 and KOH).
Experimental section
Materials
Pristine activated carbon xerogel was supplied by Xerolutions Ltd. The sample is an activated carbon xerogel XER-HSA of high purity. This sample was labeled as AX (Activated Xerogel). In order to modify the surface chemistry of the carbon xerogel, two oxidative treatments were applied, gas-phase oxidation in an air atmosphere and wet oxidation using a nitric acid solution. Oxidation with air is a more straightforward process, since it can be performed in a single step and no washing stages are required. Nevertheless, HNO3 (5 M) was also selected as oxidant agent to obtain final materials with diverse oxygen groups and to evaluate their influence on the electrochemical response of the supercapacitors. In the case of air oxidation, the operating procedure was as follows: AX xerogel was introduced into a quartz reactor and was subjected to thermal treatment under an air atmosphere (100 cm3 min−1). The temperature program used was heating rate of 10 °C min−1 and a maximum temperature of 350 °C for 2 h. Oxidative treatment with nitric acid required three steps: (i) mixing of the AX sample with a solution of HNO3 at ambient temperature under stirring for 48 h; (ii) successive washes with deionized water until a pH close to neutrality was achieved and, finally; (iii) a drying step applied overnight at 110 °C to obtain a completely oxidized xerogel free of moisture. The samples subjected to these oxidations with air and nitric acid were labelled AX-O1 and AX-O2, respectively.Analysis of porous and chemical properties
The textural characteristics of the three electrode materials were evaluated from the N2 adsorption–desorption isotherms recorded at 77 K (Micromeritics Tristar 3020). Parameters such as SBET and Vmicro were calculated by applying the Brunauer–Emmett–Teller (BET) and Dubinin–Raduskevich (DR) equations, respectively, to the nitrogen adsorption isotherms. The total pore volume (Vp) was determined from the amount of nitrogen adsorbed at saturation point (p/p° = 0.99).The chemical properties of the samples were analysed by means of three techniques: elemental analysis, determination of the point of zero charge (pHPZC) and temperature-programmed desorption (TPD). The C, N, H and S contents were determined on a LECO-CHNS-932 microanalyzer. This technique involves the combustion of the sample at a temperature close to 1000 °C in an oxygen rich atmosphere. The products of the combustion are CO2, H2O, SO2 and NOx, which is then reduced to N2. These compounds are processed and measured quantitatively by means of a thermal conductivity detector (TC) so the percentage content of each element in the sample is finally obtained. Nevertheless, oxygen was separately measured on a LECO-TF-900 furnace where there is a carbon-rich atmosphere and high temperature (T > 1000 °C). The resulting CO2 was detected and evaluated for its absorption in IR spectrum, directly obtaining the oxygen content of the samples.
The pHPZC of the samples was determined according to the following procedure: approximately 250 mg of sample was introduced into a test tube containing an initial amount of distilled water. After 48 h of stirring at room temperature, the pH of the suspension was measured. Then, a specific volume of water was added in order to modify the mass concentration and, after a stirring period to reach equilibrium, the pH of the solution was measured again. This procedure, consisting in adding various volumes of distilled water and measuring the pH of the different mass concentrations, was repeated until the pH value began to decrease (the pHPZC corresponding to the pH value remaining constant for a certain number of measurements). The third technique used in this study to define the chemical characteristics of the carbon xerogels was temperature-programmed desorption (TPD). TPD experiments were carried out on a Micromeritics AutoChem II analyser coupled to a mass spectrometer (OmniStar Pfeiffer) in order to monitor the amount of CO, CO2 and H2O desorbed during the analysis. The samples were heated at 10 °C min−1 up to a maximum temperature of 1000 °C. The gas used was Argon (50 cm3 min−1).
Electrochemical performance
To evaluate the electrochemical performance of the supercapacitors, disc-shaped electrodes were prepared by mixing powdered carbon xerogel (90 wt%) and a binder (10 wt% of PTFE). The mass of the electrodes was about 6–8 mg and they were about ∼300 μm thick. The procedure used to obtain these pelletized electrodes is described elsewhere.39 The electrochemical measurements were carried out using a two-electrode testing cell (Teflon Swagelok®) and a potentiostat/galvanostat VMP Biologic. The electrolytes employed in this work were 1 M solutions of H2SO4 and Na2SO4 and a 6 M solution of KOH. The capacitance values were calculated from galvanostatic charge–discharge (U = 1.0 V and current densities from 0.2 to 1.0 A g−1) and these were expressed in Farads per mass of active material in one electrode (F g−1). Cyclic voltammetry experiments were also carried out using a voltage window of 1.0 V, a potential range commonly applied for aqueous electrolytes, and a scan rate of 2 mV s−1.Results and discussion
Porous and chemical characterization
The textural and chemical properties of the carbon xerogels used as electrodes are grouped in Table 1. As can be seen, the three xerogels exhibit a highly-developed porosity with SBET values exceeding 2500 m2 g−1 in addition to a considerable amount of narrow mesopores, which is the appropriate pore size for a rapid diffusion of electrolyte ions, especially under fast polarization conditions.14,15 However, it must be emphasized that the porosity decreases slightly both in the case of gas-phase oxidation (air) and liquid-phase treatment (HNO3), probably due to partial pore blockage resulting from the incorporation of oxygenated functionalities (i.e. Vp = 2.25 cm3 g−1 for the AX sample, while the total pore volume was 1.85 and 1.64 cm3 g−1 for the AX-O1 and AX-O2 oxidized xerogels). Similar results have also been observed by other authors.16,40 The variation in porosity only affected the amount of pores, the pore size distribution remaining unchanged, as can be seen from the Fig. 1. The N2 adsorption–desorption isotherms are a combination of type I–IV according to the IUPAC classification corresponding to micro-mesoporous samples in all three cases studied. The isotherms are virtually identical, only in the case of the oxidized xerogels they are shifted towards lower values of nitrogen adsorbed, which results in lower pore volume. The fact that the pore size has not been modify may serve as an advantage for comparing the electrochemical performance of the three materials.| Sample | AX | AX-O1 | AX-O2 |
|---|---|---|---|
| SBET/m2 g−1 | 2876 | 2600 | 2615 |
| VDUB-N2/cm3 g−1 | 0.97 | 0.91 | 0.91 |
| Vp/cm3 g−1 | 2.25 | 1.85 | 1.64 |
| dmicro/nm | 1.0 | 1.0 | 1.0 |
| C/wt% | 97.3 | 89.9 | 88.2 |
| H/wt% | 0.3 | 0.5 | 0.8 |
| N/wt% | 0.1 | 1.5 | 1.1 |
| O/wt% | 2.3 | 8.1 | 9.9 |
| pHPZC | 8.3 | 4.8 | 3.2 |
| pHPZC > pHH2SO4 | ⋙> | ≫ | > |
| pHPZC > pHNa2SO4 | > | ≪ | ⋘ |
| pHPZC < pHKOH | < | ⋘ | ⋘< |
 | ||
| Fig. 1 N2 adsorption–desorption isotherms (A) and pore size distribution (B), obtained by applying DFT method, for the carbon xerogels synthesized. | ||
With respect to the chemical characterization, the AX xerogel mainly consists of C but a slight oxygen content can be appreciated (wt% O = 2.3). This oxygen is mostly of a basic nature, according to the pHPZC value (8.3) and the TPD results recorded in Fig. 2. The oxidized xerogels have higher oxygen content, 8.1 and 9.9 wt% for AX-O1 and AX-O2, respectively. However, although they have similar amounts of oxygen, the chemical nature of these materials differs considerably, both in the quantity of oxygenated groups and in their typology, as is confirmed by the pHPZC values and the CO2/CO desorption profiles. The diverse surface chemistry of the three xerogels used as electrode material may explain their different energy storage capacitances and the differences in their interactions with electrolytes of various pH.
Information about chemistry structure of the carbon xerogels was obtained by analyzing the TPD curves of CO2 and CO (Fig. 2). It can be seen that, by and large, the CO2 and CO-evolving groups have been considerably modified by the two oxidative processes. Furthermore, the three samples studied produce a greater amount of CO-evolving groups, namely, neutral or basic oxygen groups (phenols, carbonyls).41–44 The less stable and more acidic functionalities (carboxylic acids, anhydrides, lactones) incorporated onto the carbon surface are those that produce CO2.41–44 With regard to the CO2 evolution curves, it can be seen that the pristine xerogel shows only one small peak at temperatures below 300 °C, which can be attributed to the release of carboxylic acid groups.41–44 The oxidation of AX xerogel with air (AX-O1) and HNO3 (AX-O2) has produced an increase in the amount of CO2 released, but the type of oxygenated groups created in each case is quite different. AX-O1 displays a peak located at around 580 °C associated with anhydrides and lactone-type groups while AX-O2 exhibits a structure mostly composed of carboxylic acid groups, as can be seen from the evolution of CO2, which shows a maximum at around 280 °C.41–44 This peak has a long tail and CO2 is released up to 700 °C, indicating that other groups, such as anhydrides and lactones are present, although at a lower ratio.41–44 As regards the CO spectrum (Fig. 2, bottom), the AX xerogel has surface groups that evolve up to temperatures higher than 600 °C, the maximum CO desorption peak occurring at around 850 °C, which is clearly assignable to carbonyl–quinone type groups.41–44 According to the literature, phenols evolve between 600–700 °C and carbonyl–quinone groups between 800 and 950 °C.41–44 Therefore, both of the oxidized xerogels used in this study not only have phenolic groups but also carbonyl–quinone type groups, since both peaks display a shoulder, which is prominent in the case of the xerogel oxidized with nitric acid.
 | ||
| Fig. 2 TPD profiles of CO2 (top) and CO (bottom) for the carbon xerogels used as electrode material. | ||
Electrochemical measurements
The first part of the electrochemical characterization was based on using a single electrode material, AX xerogel, and three aqueous electrolytes of different pH. The specific capacitance values obtained with these cells are shown in Table 2 (a cell voltage of 1.0 V and current densities ranging from 0.2 to 1.0 A g−1). If only the trends are considered, it may be significant that a decrease in capacitance occurs in the acidic and neutral medium, while the amount of energy stored remains unchanged, regardless of the current applied with the KOH-based supercapacitor. This can be attributed to the charge storage mechanism of each system. The independence of capacitance from current density is due to the nature of charge storage, which is electrostatic, that is, to the formation of the electric double layer. However, the decline in energy stored with the current evidences the presence of redox phenomena. AX xerogel has a very low amount of oxygen, but most of these oxygenated groups display a nature like quinone–carbonyl groups, that is to say, active groups in acidic media.| Electrolyte | Current density (A g−1) | Capacitance reduction (%) | ||||
|---|---|---|---|---|---|---|
| 0.2 | 0.4 | 0.6 | 0.8 | 1.0 | ||
| H2SO4 | 196 | 186 | 180 | 176 | 173 | 11.7 |
| Na2SO4 | 140 | 135 | 129 | 127 | 124 | 11.4 |
| KOH | 106 | 106 | 105 | 104 | 103 | 2.8 |
From data collected in the Table 1, it can be seen that the pHPZC of the active material is superior to the pHe of both the acid and neutral solutions. In both cases, there is an excess of positive charges on the electrodes surface. These charges are involved in the faradaic reactions with the oxygenated groups on the carbon xerogel surface. It is clear that positive/negative charges have different effects because in the case of basic electrolyte, where there is a predominance of negative charges, there is a total absence of redox reactions, which supports the claim that negative charges could not positively influence the energy storage process via pseudo-capacitive effects, at least, in the case of electrodes that have surface groups of the carbonyl–quinone type.
From the specific capacitance values listed in Table 2, it is apparent that the pseudo-capacitive phenomena occurring in the H2SO4 and Na2SO4 solutions causes an improvement in the charge storage (i.e. 196 and 140 F g−1 in the case of sulfuric acid and sodium sulfate, respectively, while the capacitance barely reaches 100 F g−1 when KOH is used as electrolyte). The tendency reflected by these values is consistent with the hypothesis presented in the previous paragraph since the larger amount of positive loads in the medium, favors redox reactions and, in turn, larger values of specific capacitance are obtained.
Once the electrochemical response of the pristine xerogel versus the three aqueous solutions was evaluated, supercapacitors were assembled with the other two oxidized xerogels and two aqueous media, acid and basic, to detect any further differences in electrochemical behavior. The results from the cyclic voltammetry and charge–discharge tests are reported in Fig. 3 and Table 3, respectively. Fig. 3A includes the curves of the cells composed of three carbon xerogels and the H2SO4 solution as electrolyte. In these cases, because the electrolyte is the same, it is possible to discard the electrolyte conductivity factor and to assign the different responses exclusively to the type of active material used in each supercapacitor. The CV curves associated with the AX and AX-O1 xerogels reveal a good dynamic charge propagation and an excellent wettability, unlike the AX-O2 xerogel which produces more resistive voltammograms with a smaller integrable area. Both the higher resistance and the lower energy capacitance can be attributed to the wettability effect. In the literature, it is possible to find procedures designed to improve the wettability of electrodes in aqueous media by means of functionalization processes. However, not all the surface groups are useful for this purpose.2,8,30,31 There is evidence that the electrochemical response is enhanced with CO-desorbing complexes whereas oxygen groups that give rise to CO2, i.e., mainly acidic functional groups, have a negative effect.2,32 This was also confirmed in the present study because the xerogel oxidized with HNO3, which has the highest percentage of acidic oxygenated groups, led to a worse electrochemical performance in acidic electrolyte. Besides the worse wettability, the lower energy storage exhibited by the AX-O2 xerogel might be related to the loads present on the surface of the electrode. In the case of the electrodes composed of the AX and AX-O1 samples, there is a greater discrepancy between the pHe and the pHPZC value of the material and the surface of the electrodes is positively charged, a phenomenon that occurs to a lesser extent with the AX-O2 xerogel because its pHPZC value is closer to the pH of the sulfuric acid.
 | ||
| Fig. 3 Cyclic voltammograms in an acid (A) and basic (B) medium for the three carbon xerogel-systems evaluated (scan rate: 2 mV s−1). | ||
| Carbon xerogel | Electrolyte | |
|---|---|---|
| H2SO4 (1 M) | KOH (6 M) | |
| AX | 195 | 112 |
| AX-O1 | 175 | 140 |
| AX-O2 | 80 | 110 |
When the voltammograms performed in the potential range of 0–1.0 V with alkaline electrolyte are compared (Fig. 3B) it can be seen that the three carbon xerogels offer a fast current–voltage (i–V) response, i.e., a good rate capacitability. Moreover, the CV curves are more similar to each other than when H2SO4 is used as electrolyte. The integrable area under the AX-O1 curve is the largest, resulting in a specific capacitance value of about 150 F g−1. However, AX-O1 does not have the highest micropore volume, highlighting again the relevance of surface chemistry for this type of electrochemical systems.
If the specific capacitance values listed in Table 3 are examined closely, it can be seen that the amount of energy stored depends on both the electrode material and the electrolyte employed. In an acidic medium, that is, when there is an excess of positive charges on the electrode surface, the C values tend to be higher, suggesting that these loads have a beneficial effect on energy storage capacitance. In the sulphuric acid-based supercapacitors, the difference between the pHe and pHPZC follows the order AX > AX-O1 > AX-O2, coinciding exactly with the trend followed by the values of specific capacitance for these materials. Energy storage therefore is not only influenced by the porosity of the electrode because when both oxidized xerogels are compared, it can be seen that a higher amount of energy is accumulated by the xerogel AX-O1, even though the porous properties of both materials are virtually identical (175 F g−1 as opposed to 80 F g−1 in the case of the AX-O2 xerogel, SBET value for both materials being around 2600 m2 g−1). As mentioned above, the AX-O1 sample has a higher content of oxygenated groups of a basic and neutral nature than AX-O2, as is corroborated by their pHPZC values (4.8 vs. 3.2). In light of this, the difference between the pHPZC and the pH of the H2SO4 is greater in the case of the xerogel oxidized with air which would explain the greater presence of positive loads on the electrode surface. This underlines the importance of the surface chemistry of the electrode material since, when two active materials with exactly the same porosity are used, the amount of energy stored depends on their chemical properties.
However, it is also necessary to take into account the medium used as electrolyte because energy storage varies not only depending on the properties of the electrode materials but also on their chemical characteristics and the electrolyte used. This is supported by the capacitance values obtained with the KOH solution in Table 3. In the case of the pristine xerogel and AX-O1 sample, the capacitance is lower than the energy accumulated in the presence of sulphuric acid, which can be partially attributed to the use of a less conductive electrolyte.43 However, surface chemistry also plays an important role, as can be seen from the fact that the capacitance values of the three systems are closer than in the case of supercapacitors based on H2SO4. In order to explain these results, it is necessary to follow the same line of reasoning as in the previous paragraph. The pH of the KOH solution is greater than the pHPZC of all three carbon xerogels evaluated. The sequence proceeds in this case in the order AX < AX-O1 < AX-O2. That is to say, the greatest variation between pHPZC and pHe corresponds to the oxidized xerogel AX-O2 but in this case, pHpZC < pHe, so the surface of electrode material is negatively charged (see Scheme 1). Therefore, it appears that negative loads do not have such a clear effect as positive loads, since a negatively charged electrode surface does not lead to any improvement in energy storage capacitance. This does not mean that negative loads do not promote pseudocapacitive phenomena and so do not increase the energy accumulated. However, it seems that these phenomena are favoured to a greater extent when there is a positively charged surface and, more specifically, when the active material is composed of carbonyl–quinone-type oxygen groups (such as the pristine xerogel, AX).
Conclusions
In this work, the combination of the surface chemistry of the electrode and the type of aqueous electrolyte was evaluated using activated carbon xerogels of high purity. Samples with diverse oxygen functionalities were obtained and studied in electrolytes of a different nature. The three electrode materials evaluated showed a greater energy storage capacitance in acid media, demonstrating the beneficial effect of positive charges since, when the electrode surface is positively charged, redox phenomena are favored and, hence, the amount of energy stored increases. The effect of negative loads is less evident since with alkaline electrolyte, the electrochemical response of the three supercapacitors is very similar, even when electrodes with a different porosity and chemical properties are used. The main conclusion of this study is that it is essential to take into account the chemical characteristics of the active material but always in combination with the nature of the electrolyte used, because the behavior of the active material may vary a lot. There can be no general rule as to which surface functionality is more beneficial when aqueous electrolytes are used as this depends on the interactions between the active material and the electrolyte.Acknowledgements
The authors gratefully acknowledge the financial support of Ministerio de Economía y Competitividad of Spain MINECO (Ref. MAT-2011-23733 and IPT-2012-0689-420000). N. Rey-Raap also thanks MINECO for her predoctoral research grant.Notes and references
- E. G. Calvo, N. Ferrera-Lorenzo, J. A. Menéndez and A. Arenillas, Microporous Mesoporous Mater., 2013, 168, 206–212 CrossRef CAS PubMed.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS PubMed.
- P. Staiti, A. Arenillas, F. Lufrano and J. A. Menéndez, J. Power Sources, 2012, 214, 137–141 CrossRef CAS PubMed.
- Y.-M. Chang, C.-Y. Wu and P.-W. Wu, J. Power Sources, 2013, 223, 147–154 CrossRef CAS PubMed.
- Z. Zapata-Benabithe, F. Carrasco-Marín and C. Moreno-Castilla, J. Power Sources, 2012, 219, 80–88 CrossRef CAS PubMed.
- E. G. Calvo, E. J. Juárez-Pérez, J. A. Menéndez and A. Arenillas, J. Colloid Interface Sci., 2011, 357, 541–547 CrossRef CAS PubMed.
- E. J. Juárez-Pérez, E. G. Calvo, A. Arenillas and J. A. Menéndez, Carbon, 2010, 48, 3293–3311 CrossRef PubMed.
- E. Frackowiak, Q. Abbas and F. Béguin, J. Energy Chem., 2013, 22, 226–240 CrossRef CAS.
- E. Raymundo-Piñero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2006, 44, 2498–2507 CrossRef PubMed.
- V. V. N. Obreja, Phys. E, 2008, 40, 2596–2605 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 22, 1760–1763 CrossRef PubMed.
- M. M. Jaramillo, A. Mendoza, S. Vaquero, M. Anderson, J. Palma and R. Marcilla, RSC Adv., 2012, 2, 8439–8446 RSC.
- R. Mysyk, E. Raymundo-Piñero and F. Béguin, Electrochem. Commun., 2009, 11, 554–556 CrossRef CAS PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2009, 9, 1774–1785 RSC.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS PubMed.
- J. F. Vivo-Vilches, E. Bailón-García, A. F. Pérez-Cadenas, F. Carrasco-Marín and F. J. Maldonado-Hódar, J. Hazard. Mater., 2013, 263, 533–540 CrossRef CAS PubMed.
- N. Rey-Raap, J. A. Menéndez and A. Arenillas, J. Sol-Gel Sci. Technol., 2014, 69, 488–497 CrossRef CAS PubMed.
- S. Morales-Torres, F. J. Maldonado-Hódar, A. F. Pérez-Cadenas and F. Carrasco-Marín, Microporous Mesoporous Mater., 2012, 153, 24–29 CrossRef CAS PubMed.
- F. Lufrano, P. Staiti, E. G. Calvo, E. J. Juárez-Pérez, J. A. Menéndez and A. Arenillas, Int. J. Electrochem. Sci., 2011, 6, 596–612 CAS.
- J. Cheng, G.-P. Cao and Y.-S. Yang, J. Power Sources, 2006, 159, 734–741 CrossRef CAS PubMed.
- S. Li, L. Qi, L. Lu and H. Wang, RSC Adv., 2012, 2, 3298–3308 RSC.
- D. Salinas-Torres, J. M. Sieben, D. Lozano-Castelló, D. Cazorla-Amorós and E. Morallón, Electrochim. Acta, 2013, 89, 326–333 CrossRef CAS PubMed.
- M. Moniruzzaman, C. Y. Yue and R. K. Jena, RSC Adv., 2014, 4, 5188–5197 RSC.
- R. Ramya, R. Sivasubramanian and M. V. Sangaranaravanan, Electrochim. Acta, 2013, 101, 109–129 CrossRef CAS PubMed.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- Z. Zapata-Benabithe, J. de Vicente, F. Carrasco-Marín and C. Moreno-Castilla, Carbon, 2013, 55, 260–268 CrossRef CAS PubMed.
- M. Zhou, F. Pu, Z. Wang and S. Guan, Carbon, 2014, 68, 185–194 CrossRef CAS PubMed.
- B.-H. Kim, K. S. Yang and H.-G. Woo, Mater. Lett., 2013, 93, 190–193 CrossRef CAS PubMed.
- M. J. Bleda-Martínez, J. A. Maciá-Agulló, D. Lozano-Castelló, E. Morallón, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2005, 43, 2677–2684 CrossRef PubMed.
- C.-T. Hsieh and H. Teng, Carbon, 2002, 40, 667–674 CrossRef CAS.
- K. Okajima, K. Ohta and M. Sudoh, Electrochim. Acta, 2005, 50, 2227–2231 CrossRef CAS PubMed.
- V. Ruiz, C. Blanco, E. Raymundo-Piñero, V. Khomenko, F. Béguin and R. Santamaría, Electrochim. Acta, 2007, 52, 4969–4973 CrossRef CAS PubMed.
- C. A. León y León and L. R. Radovic, in Chemistry and Physics of Carbon, ed. P.A. Thrower, 1994, vol. 24, pp. 213–310 Search PubMed.
- D. Suárez, J. A. Menéndez, E. Fuente and M. A. Montes-Morán, Langmuir, 1999, 15, 3897–3904 CrossRef.
- M. A. Montes-Moran, D. Suárez, J. A. Menéndez and E. Fuente, in Novel Carbon Adsorbents, ed. J. M. D. Tascon, 2012, Elsevier Ltd, pp. 173–203 Search PubMed.
- V. A. Garten and D. E. Weiss, Aust. J. Chem., 1957, 10, 309–328 CrossRef CAS.
- C. A. León, J. M. Solar, V. Calemma and L. R. Radovic, Carbon, 1992, 30, 797–811 CrossRef.
- M. A. Montes-Morán, J. A. Menéndez, E. Fuente and D. Suárez, J. Phys. Chem. B, 1998, 102, 5995 CrossRef.
- E. G. Calvo, F. Lufrano, P. Staiti, A. Brigandì, J. A. Menéndez and A. Arenillas, J. Power Sources, 2013, 241, 776–782 CrossRef CAS PubMed.
- B. Gryzb, C. Hildenbrand, S. Berthon-Fabry, D. Bégin, N. Job, R. Rigacci and P. Achard, Carbon, 2010, 48, 2297–2307 CrossRef PubMed.
- C. Moreno-Castilla, F. Carrasco-Marín, F. J. Maldonado-Hódar and J. Rivera-Utrilla, Carbon, 1998, 36, 145–151 CrossRef CAS.
- J. F. Vivo-Vilches, E. Bailón-García, A. F. Pérez-Cadenas, F. Carrasco-Marín and F. J. Maldonado-Hódar, Carbon, 2014, 68, 520–530 CrossRef CAS PubMed.
- J. L. Figueiredo and M. F. R. Pereira, Catal. Today, 2010, 150, 2–7 CrossRef CAS PubMed.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |