
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-doped carbon dots in transesterification reactions for biodiesel synthesis†
Tayline V.
de Medeiros‡
ab,
Alexia
Macina‡
ab,
João P.
de Mesquita
 c and
Rafik
Naccache
*ab
c and
Rafik
Naccache
*ab
aDepartment of Chemistry and Biochemistry and the Centre for NanoScience Research, Concordia University, Montreal, QC H4B 1R6, Canada. E-mail: rafik.naccache@concordia.ca
bQuebec Centre for Advanced Materials, Department of Chemistry and Biochemistry, Concordia University, Montreal, QC H4B 1R6, Canada
cDepartment of Chemistry—Universidade Federal dos Vales do Jequitinhonha e Mucuri, Rodovia MGT 367—Km 583, n° 5000, Alto da Jacuba, CEP 39100-000 Diamantina, MG, Brazil
First published on 22nd September 2023
Abstract
Biodiesel has emerged as a sustainable renewable energy option and a promising substitute for traditional fossil fuel-derived petroleum. However, its current industrial production is financially impractical requiring novel approaches to ensure sustainability and commercial viability. Carbon dots (CDs) have recently been reported as promising heterogeneous catalysts for transesterification of oil to biodiesel yet the role of the surface chemistry remains vaguely understood. Here, we present amine-passivated CDs (N-CDs) as a model in which their surface chemistry, namely the degree of carboxylic acid to amine and amide functionalization, can be controlled by modifying the amine passivating agent. We thoroughly investigated the N-CDs physico-chemical properties and applied them as heterogeneous catalysts to transesterify canola oil to biodiesel. We report biodiesel conversions of ≥97% using 1 wt% catalyst loading at 100 °C for 3 hours even when the catalyst is reused for five reaction cycles. Lastly, we investigate the effects of modifying the carbon dot surface groups and postulate a plausible governing mechanism for the N-CD-catalyzed transesterification of canola oil to biodiesel. Our findings suggest that both carboxylic acids and amines can act as active catalytic sites, and depending on their concentration, two different reaction mechanisms are possible.
Introduction
Driven by a growing worldwide economy and technological advancements in developing countries, global energy demands continue to increase at an alarming speed. With a dominant 80% of current energy consumption stemming from traditional non-renewable fossil fuels such as coal, natural gas and oil,1 there is an urgent need to reduce the world's unsustainable reliance on fossil fuels with clean and renewable forms of energy. This urgent call has spurred financial investments and innovative research into developing and creating renewable forms of energy – ones that are energy-efficient, rapidly regenerated and environmentally friendly while meeting global energy demands.Biodiesel has emerged as a promising alternative form of energy and as a substitute for traditional diesel fuel. It shares similar physico-chemical properties with diesel fuel.2 It offers a myriad of benefits as biodiesel is made from renewable commodities, enabling the efficient usage of waste products and biomass as an energy supply. Moreover, it has high biodegradability and low chemical toxicity and can reduce greenhouse gas and other hazardous emissions by 75–90% compared to diesel fuel combustion.2,3 Biodiesel's chemical composition comprises a mixture of fatty acid methyl or ethyl esters (FAME and FAEE, respectively), formed from the transesterification of triglycerides (e.g. vegetable oils or animal fats) using primary alcohols (e.g. methanol or ethanol), heat and a homogeneous acid/base catalyst.4 The biodiesel transesterification reaction consists of three simultaneous reversible reactions, in which a triglyceride is consecutively converted into methyl or ethyl esters, ending with the formation of glycerol as a byproduct.5
The current commercial production of biodiesel relies on the use of homogeneous base catalysts such as sodium and potassium hydroxide.6,7 While these catalysts are relatively cost-effective and catalytically efficient, achieving high biodiesel conversion yields (<95%) at only 1 wt% catalyst loading (relative to the vegetable oil precursor),8 there lies significant limitations with their current use, ultimately restricting the overall biodiesel supply. These limitations include the fact that: (i) homogenous base catalysts can cause saponification, i.e. the formation of contaminating “soap” (i.e. fatty acid salts),9 leading to catalyst consumption and a decrease in biodiesel yield,10 (ii) the catalyst cannot be reused or regenerated, hindering the process's sustainability,11 and (iii) the current purification of biodiesel is expensive and environmentally unfriendly due to the large amounts of wastewater produced.12
Heterogeneous catalysts have emerged as a potential cost-reducing and efficiency-increasing solution since they can overcome many of the aforementioned challenges. Unlike their homogeneous counterparts, heterogeneous catalysts can be easily recovered after the reaction has been completed since they reside in a different phase relative to the reactants or products.13 Coupled with the ability to regenerate and reuse over several transesterification cycles, heterogeneous catalysts afford an easier and cheaper separation process.14 Several studies have investigated the catalytic efficiency of heterogeneous solid catalysts, including metal oxides such as CaO,15 Ca(OH)2,16 calcium diglyceroxide17 and MgO,18 biochar,19 as well as mesoporous supported materials like silica,20 and have supported the catalysts' efficiency and reusability over several cycles. However, their preparation usually requires tedious synthetic and purification processes, being also prone to metal leaching.10 Furthermore, heterogeneous catalysts typically require more energy-intensive conditions or high catalyst loading since they are not as catalytically efficient as their homogeneous counterparts, limiting their commercial viability both in their production and application.21,22 In light of the above, the search for a novel heterogeneous catalyst for biodiesel production persists. Carbon dots (CDs), a relatively new member of the carbon nanomaterial family, offer an interesting alternative in catalytic applications due to their versatile and highly tuneable surface chemistry, as well as their inexpensive and facile synthesis protocols.
CDs are quasi-spherical amorphous nanoparticles of approximately 10 nm, composed of a mostly sp2 and sp3 hybridized carbon core framework and typically comprise carbon, oxygen and hydrogen.23,24 However, depending on the precursors used during synthesis, heteroatoms such as nitrogen and sulphur can also be incorporated into the surface or carbon framework.25 Their surface chemistry includes a wide range of functional groups such as hydroxyls,26 carboxylic acids,27 amines,28 and amides.29 It has been shown that the degree of surface functionalization can be tuned by modifying precursors and reaction parameters, such as synthesis temperature and precursor concentration.29 In combination with their unique size-specific attributes, and photoluminescent properties, their highly tuneable surface chemistry has enabled the use of CDs in various applications.30–32 Versatile in nature, CDs can be synthesized from a vast number of possible precursors, enabling a cost-effective synthesis since starting reagents include waste materials such as eggshells,33 coffee grounds34 and biomass.27 Due to their robust structure, cost-friendly and sustainable preparation, they have the potential to be used as heterogeneous catalysts in large-scale industrial processes.
Specifically, they have emerged as promising heterogeneous catalysts owing to simple chemical modification protocols to endow their surface with functional groups such as sulfonic acid, carboxylate and/or alkoxide groups. This can translate to engineering specificity for the transesterification reaction by mimicking homogenous acid or base catalysts. Furthermore, their large surface area to volume ratios increase the number of active sites available to participate in the catalytic mechanism. Our previous work was innovative in demonstrating the ability of CDs to catalyze transesterification reactions of canola oil to biodiesel.35 We have demonstrated that carboxylate moieties on the CDs' surface could mimic a basic homogeneous catalytic mechanism. Nonetheless, there is a need to further investigate the role of surface chemistry in dictating the activity of CDs in transesterification reactions. Expanding our fundamental understanding of the surface engineering of CDs is essential to pave the way for systems capable of competing with homogeneous catalysts currently used on an industrial scale.
Organic bases, such as amines, have already been reported to be catalytically efficient in converting triglycerides to biodiesel.36 However, amino-functionalized carbon-based nanomaterials as heterogeneous catalysts remain fairly unexploited, and the catalytic reaction mechanism has yet to be elucidated. Amine-passivated CDs (N-CDs) are well established in the literature, known for their versatility and easy synthesis from citric acid and amine passivating agents. Thus, an in-depth understanding of the governing surface functional groups, necessary to achieve high catalytic efficiencies for transesterification reactions, would be vital in advancing the state of the research and adoption of CDs as heterogeneous catalysts. N-CDs that present amines, amides and carboxylic acids on their surface can be used as a model to investigate the role of functional groups with distinct basicity levels in catalyzing the transesterification reaction. The surface functionalization can be controlled by modifying the type and concentration of the amine passivating agent.
Here, we report the synthesis of amine-passivated carbon dots as catalysts for the transesterification of canola oil. Canola oil was chosen as a model feedstock for this study, as it presents low acidity levels and well-known physico-chemical properties. First, we characterize their physico-chemical properties to glean insights into the effects of the CD surface chemistry on biodiesel yields and better understand the transesterification mechanism. Subsequently, we quantify the conversion of canola oil to biodiesel using a quantitative 1H NMR technique, which is both simple and versatile. We optimize the N-CD-catalyzed transesterification through variation of the reaction parameters, including catalyst loading, methanol content, temperature and reaction time, and demonstrate their reusability over several catalytic cycles. Finally, we postulate a new amine passivated CD-catalyzed mechanism for the transesterification of canola oil to biodiesel.
Experimental section
Materials and reagents
Citric acid (CA), ethylenediamine (ED2), diethylenetriamine (DT3), triethylenetetramine (TT4), tetraethylenepentamine (TP5), pentaethylenehexamine (PH6) and acetone were purchased from Sigma-Aldrich. Sodium hydroxide and methanol were purchased from Fisher Scientific. Food-grade canola oil was purchased from a grocery store. Cellulose dialysis membrane (3.5–5.0 kDa molecular weight cut-off) was purchased from Spectrum Laboratories. All reagents were used without further modification or purification.Synthesis of N-CDs
CDs were synthesized via a one-step microwave-assisted reaction of citric acid and the amine passivating agent (ED2, DT3, TT4, TP5 or PH6) using a CEM Discover SP microwave reactor (Fig. S1†), following a previously reported synthesis with some minor modifications.29 Briefly, 1.5 g (500 mM) of citric acid and 16.0 mL of Milli-Q water were mixed in a glass microwave-transparent reaction vial and stirred until the solution was homogenous. This was followed by adding the amine passivating agent, where the concentration ranged from 125 mM to 750 mM. The reaction vessel was heated to 210 °C for 10 min under constant stirring. Upon completion of the reaction and subsequent cooling to room temperature, the crude CD solutions were dialyzed for 4 days against Milli-Q water, changing the water once a day, allowing for the removal of unreacted precursors and polar impurities. Subsequently, the CDs were further purified using 3 acetone organic washes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v), removing any remaining impurities. Following each organic wash, the sample was vortexed for approximately 30 seconds and centrifuged at 10000 × g for 10 minutes. Subsequently, the supernatant was discarded, and the wash was repeated. After the third acetone wash, the resulting precipitate was dried in an oven at 85 °C for 24 hours.
:10 v/v), removing any remaining impurities. Following each organic wash, the sample was vortexed for approximately 30 seconds and centrifuged at 10000 × g for 10 minutes. Subsequently, the supernatant was discarded, and the wash was repeated. After the third acetone wash, the resulting precipitate was dried in an oven at 85 °C for 24 hours.
Deprotonation of N-CDs
Briefly, 0.1 g of the amine-passivated CDs were dispersed in 10 mL of Milli-Q water and sonicated for 10 minutes. A pH electrode was used to determine the initial pH of the aqueous CD dispersions, and 0.01 M NaOH solution was added dropwise to the samples under constant stirring until the pH of the solution was raised by approximately 2 pH units. The samples were further purified using 3 organic acetone washes, following the same procedure employed during the original purification of the synthesized CDs.N-CD-catalyzed transesterification of canola oil to biodiesel
Approximately 0.5–10 wt% of amine-passivated CDs (wt% relative to canola oil) were added to methanol (0.75–6 mL) (Fig. S2†). The solution was sonicated for 5 minutes until the CDs were homogeneously dispersed. Subsequently, 2.0 mL of both canola oil and the CD dispersion were simultaneously transferred to a 25 mL solvothermal autoclave reactor. The reaction was heated at 150 °C for 3 hours under constant stirring at 600 rpm. Upon completion of the reaction, the reactor was submerged in ice to cool the reaction rapidly. The reaction mixture was then transferred to a vial and heated in an oven at 85 °C for 12 hours to ensure excess methanol evaporation and separation of the glycerol/CDs and biodiesel phases. The biodiesel phase was then centrifuged at 10000 × g for 5 minutes to separate the layer further, and the biodiesel phase was collected for 1H NMR analysis.
Transmission electron microscopy (TEM)
TEM images were acquired using a JEM-2100F (JEOL Ltd) analytical electron microscope under a 5 kV field emission. CDs were dispersed in methanol at a concentration of 10 mg mL−1, and 10 μL of the CD dispersions were pipetted on 300 Mesh Cu (Cu-300 HD) TEM grids coated with a holey/thin carbon film (Grid Tech). The solvent was allowed to evaporate before image acquisition. Image processing and size distribution analysis were carried out using the Fiji imaging software.X-ray powder diffraction (PXRD)
PXRD spectra were acquired using a 2nd Gen D2 Phaser X-ray diffractometer (Bruker AXS). A Cu Kα source at a generator power of 30 kV/10 mA, a scan range from 10 to 80° (2θ) with a step size of 0.11° per 2 s, and a position-sensitive detector opening of 4.79° were used during the analysis. All data were processed using the Bruker DIFFRAC software.Fourier-transform infrared spectroscopy (FT-IR)
FTIR–ATR spectra were acquired using a Thermo Scientific Nicolet iS5 equipped with an iD5 ATR accessory. Dried CD samples were analyzed on a laminate-diamond crystal window using 32 scans at 0.4 cm−1 resolution. The detector was set to a gain of 1, an optical velocity of 0.4747 cm s−1 and an aperture setting of 100. All data were processed using the Thermo Scientific Nicolet Omnic 9 software.Thermogravimetric analysis (TGA)
TGA was performed on dried CD samples in Pt pans using a TGA Q500 analyzer (TA Instruments). Samples were analyzed under an N2 atmosphere at a flow rate of 50 mL min−1 and were heated from 25 to 1000 °C at a heating rate of 1 °C min−1. All data were processed using the Thermal Advantage 5.0 software.Zeta-potential analysis
Zeta-potential measurements were acquired using a Zeta Size (Malvern Instruments – Nano Series). CD samples were dispersed in water at a concentration of 5 mg mL−1 and were analyzed in triplicates of 20 runs each. The average result of the triplicate was used.Elemental analysis
Elemental analysis was carried out using a Vario MICRO cube elemental instrument (Elementar). Approximately 3.0 mg of each sample was weighed and subjected to quantitative high-temperature decomposition under atmospheric oxygen. The gaseous components of the sample were separated and quantified using a direct Temperature Programmed Desorption (direct TPD) column. The samples were analyzed in triplicates, and the results were averaged.Characterization of biodiesel via1H NMR
1H NMR spectra were acquired using a Bruker Fourier Ultrashield™ with an operating frequency of 300 MHz. Biodiesel samples were diluted in CDCl3 before analysis. Biodiesel conversions were determined based on a calibration curve previously obtained by Macina et al.35 The ratio between the integrations of peaks at 3.66 and 2.30 ppm was used to determine the percent conversion by inputting the value into the linear regression equation.Potentiometric titration
The potentiometric titration curves were acquired at 25 °C using a SCHOTT automatic titrator (Titroline 7000) with a combined pH electrode model N6280 (SCHOTT) under a nitrogen atmosphere. In a typical procedure, 5.0 mg of the sample was dispersed in 20 mL HCl solution (0.001 mol L−1) in an electrochemical cell, followed by titration with a CO2-free solution of NaOH (0.003 mol L−1). This aqueous solution was prepared by diluting a stock solution (15 mol L−1) in previously boiled water for 30 minutes. The ionic strengths of the HCl and NaOH solutions were adjusted to 0.100 mol L−1 by the dissolution of sodium chloride to maintain the activity of H+ ions constant throughout the experiment.Surface active site density (SD)
The SD was calculated based on the potentiometric titration results. The functional groups presenting pKa ranging from 2 to 6 were scribed as associated with carboxylic acids and from 7 to 9 to amines. The results were then obtained by dividing the number of active sites of each moiety (per gram of catalyst) by Avogadro's number, as shown in the formula below.| SD = active sites (per g of catalysts) ÷ 6.022 × 10−23 |
Results and discussion
Tailoring the functional groups on the surface of the N-CDs
The functional groups present on the CDs' surface have a major role in dictating the catalytic efficiency of the transesterification reaction. Tailoring the surface functional groups is anticipated to provide key insights into the transesterification reaction's governing catalysis mechanism. Herein, we focused on the role of functional groups such as carboxylates and amines, as these groups are already known for their catalytic efficiency in the transesterification of oils to biodiesel. Diethylenetriamine (DT3) passivated CDs were synthesized using citric acid and a concentration of DT3 ranging from 125 mM to 750 mM (Fig. 1a). It has been previously reported that altering the concentration of DT3 will modify the ratio of carboxylic acids and amines found on the surface of the CDs.29 Furthermore, the N-CDs were synthesized using citric acid and 375 mM of various amine passivating agents, namely ethylenediamine, triethylenetetramine, tetraethylenepentamine, pentaethylenehexamine (ED2, TT4, TP5 and PH6, respectively) since the use of these precursors leads to the formation of N-CDs with different oxygen to nitrogen ratios,37 providing further insights on the governing catalytic mechanism (see the Experimental section for additional information regarding the synthesis of the catalysts). | ||
Fig. 1 (a) Reaction scheme illustrating the reaction between citric acid and DT3 using a microwave reactor to produce N-CDs. (b) Representative TEM electronic image of the 750 mM DT3 CDs displaying quasi-spherical carbon dots, with an average size of 13 ± 3 nm. (c) Representative diffractogram of 750 mM DT3 CDs showing a broad halo spanning 10 to 80° 2θ, confirming the formation of non-crystalline structures. Similar TEM and PXRD results were observed when both amine passivating agents and concentrations were varied. (d) Representative TGA thermogram for 750 mM DT3 CDs displaying a four-step decomposition weight loss profile, ascribed to the evaporation of adsorbed water, decomposition of oxygenated surface groups followed by the nitrogenated groups, and decomposition of the sp2 carbon core, respectively. The residual mass of approximately 12.6% is due to Na+ ions coordinated to the surface of the CDs upon deprotonation with NaOH. (e) FT-IR spectroscopy for the N-CDs exhibits O–H and N–H stretchings, ascribed to the amine and alcohol moieties of the amine passivating agents and citric acid precursors, respectively. The formation of amide groups is observed and is associated with the reaction between the carboxylic and amine moieties from the precursors. Increase in the concentration of DT3 results in spectra that show a stark decrease in carboxylic acid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and a corresponding increase in amide CO stretching. (f) Similar results were observed when using passivating agents containing more amine groups (TT4, TP5 and PH6), in which a strong CO amide stretch is observed, while the acid carbonyls are no longer observed. (g) Potentiometric titration results indicated that the increase in DT3 concentration from 125 to 750 mM leads to a decrease in the percentage of carboxylic acids from 80 to 32%, with a concomitant increase in the amines from 20 to 67%. (h) The variations in the passivation agent did not present major effects on the concentration of carboxylic acids that remained fairly constant. Nonetheless, a major variation in the amine percentage was observed, decreasing from 60% for ED2 to 33% for PH6. O stretching and a corresponding increase in amide CO stretching. (f) Similar results were observed when using passivating agents containing more amine groups (TT4, TP5 and PH6), in which a strong CO amide stretch is observed, while the acid carbonyls are no longer observed. (g) Potentiometric titration results indicated that the increase in DT3 concentration from 125 to 750 mM leads to a decrease in the percentage of carboxylic acids from 80 to 32%, with a concomitant increase in the amines from 20 to 67%. (h) The variations in the passivation agent did not present major effects on the concentration of carboxylic acids that remained fairly constant. Nonetheless, a major variation in the amine percentage was observed, decreasing from 60% for ED2 to 33% for PH6. | ||
Physical properties of N-CDs
The N-CDs morphology and size distribution were investigated by TEM. A representative image for 750 mM DT3 CDs (Fig. 1b) shows quasi-spherical dots with minimal aggregation signs. Statistical analysis revealed a Gaussian size distribution ranging from 6 to 26 nm and an average size of 13 ± 3 nm (Fig. 1b inset). The variation in the concentration of the DT3 precursor does not substantially affect the size of the N-CDs, with average sizes of 14 ± 5 nm, 13 ± 3 nm and 14 ± 5 nm for 375, 250 and 125 mM DT3 CDs, respectively (Fig. S3a–d†). Similar results were also observed when different amine passivating agents were used, with average sizes of 12 ± 3 nm, 13 ± 4 nm, 13 ± 8 nm and 13 ± 4 nm for 375 mM PH6, TP5, TT4 and ED2 CDs, respectively (Fig. S3e–h†).A representative PXRD diffractogram for 750 mM DT3 CDs (Fig. 1c) shows a broad halo and no crystalline reflections spanning 10 to 80° 2θ. The broad hump centred at approximately 25° 2θ has been widely described as characteristic of amorphous carbon.38 PXRD analysis (Fig. S4†) was also carried out on all other N-CDs and confirmed that all synthesized N-CDs were non-crystalline, suggesting that changes in the amine passivating agent had no impact on their microstructures. A small peak centered at 26° 2θ is observed for 375 mM DT3 and TT4, probably due to the presence of graphitic structures in the core of the N-CDs.
TGA was carried out on the N-CDs to determine their thermal stability, noting that high thermal stability is a prerequisite for the CDs catalysts due to the temperatures required for the transesterification reactions, which can reach up to 150 °C. A representative TGA thermogram (Fig. 1d) of 750 mM DT3 CDs shows a four-step decomposition weight loss profile. The initial 12.0% weight loss occurs at 150 °C and is attributed to the evaporation of water adsorbed on the surface of the N-CDs.39 The second weight loss of 13.0% occurring at 310 °C is attributed to the decomposition of oxygenated surface groups, whereas the third weight loss of 29.5% occurring at 465 °C is attributed to the decomposition of nitrogenated surface groups.40,41
The final weight loss (31.9%) occurs at 815 °C and is related to the decomposition of the sp2 carbon core.42 Finally, the 12.6% residual weight remaining at 900 °C is caused by Na+ ions coordinated to the surface of the N-CDs upon deprotonation with NaOH. Differential thermogravimetry curves and TGA thermograms (Fig. S5†) show similar four-step decomposition profiles and weight-loss steps for all N-CDs, confirming their high thermal stability.
Composition and surface properties of N-CDs
The elemental composition of the N-CDs was determined by CHN analysis (Fig. S6†). The percentage of oxygen was determined by subtraction, taking the percentage of sodium obtained by the TGA into account for this calculation. The CHN results indicate that the catalysts primarily comprised carbon, nitrogen, oxygen, and hydrogen due to the precursors used in the synthesis of the N-CDs. The increasing DT3 concentration increases the N:C ratio, while the O:C ratio decreases. Similarly, the N:C ratio augments with the increasing number of amines present in the passivating agent, with a concomitant decrease in the O:C ratio.
Recognizing the importance of the surface groups in the reaction mechanism of heterogeneous catalysts, the surface functional groups of the N-CDs were extensively investigated by FT-IR spectroscopy. All N-CDs (Fig. 1e and f) exhibit O–H and N–H stretchings at ∼3380 cm−1 and ∼3255 cm−1, ascribed to the amine and alcohol moieties of the amine passivating agents and citric acid precursors, respectively.43 The presence of carboxylic acids and amides is confirmed by the bands at ∼1695 cm−1 and ∼1645 cm−1, attributed to carboxylic acid CO and amide CO stretching, respectively.29 Previous studies attribute the amide bond formation to the reaction of carbonyl and amine groups within the precursors.44 The intense band at ∼1540 cm−1 corresponds to CC stretches of the skeletal polycyclic aromatic rings.45,46 Varying the concentration of the DT3 precursors results in spectra that show a stark decrease in carboxylic acid CO stretching and a corresponding increase in amide CO stretching as the concentration of DT3 precursor increases (Fig. 1e); this trend has also been shown in a previous study, which confirms these findings.29 For 750 mM DT3 CDs, the carboxylic CO stretching is no longer observed. CDs passivated with ED2, TT4, TP5, and PH6 agents caused negligible changes to the surface chemistry for the most part when compared to DT3 CDs (Fig. 1f); however, there were notable differences in amide and carboxylic CO stretching, with a strong preference for amide formation. This is observed by the stronger transmittances for amide CO stretching at ∼1645 cm−1 relative to carboxylic acid CO stretching at ∼1695 cm−1. It should be noted that only ED2 and DT3 CDs demonstrated carboxylic acid CO stretching. For PH6, a stretching ascribe to cyclic amides is observed at ∼1705 cm−1.
A more detailed and quantitative analysis of the functional groups present on the surface of the N-CDs was carried out by potentiometric titration studies. Nonlinear regression fitting of the potentiometric titration curves (Fig. S7†) indicates the presence of four main acidic surface groups with pKa values ranging from ∼3–9. It is important to note that the titration was performed in the pH range of 2.7–10.5. As such, groups with pKa values outside this range, such as strong acids or amides, are not accounted for. Despite presenting acidic strength similar to organic functional groups with analogous structures, the assignment of these moieties is challenging since the pKa values are majorly influenced by the substituents present on the α-carbon. It is known that the presence of electron-withdrawing groups increases the acidity, while electron-donating groups have the opposite effect.47 According to the functional groups observed in the FT-IR results and the pKa values for carboxylic acids and aliphatic amines used as precursors, the N-CDs moieties presenting pKa ranging from 2 to 6 were scribed as associated with carboxylic acids and from 7 to 9 to amines. According to the titration results, the increase in DT3 concentration from 125 to 750 mM leads to a decrease in the percentage of carboxylic acids from 80 to 32%, with a concomitant increase in the amines from 20 to 67% (Fig. 1g). These results further corroborate the findings from elemental analysis, indicating that the increase in the passivation agent concentration leads to an increase in the concentration of amines and a decrease in carboxylic acids.
The variations in the passivation agent did not present major effects on the concentration of carboxylic acids that remained fairly constant from 30 to 40%. Nonetheless, a major variation in the amines percentage was observed, decreasing from 60% for ED2 to 33% for PH6 (Fig. 1h). Interestingly, PH6 did not match what was observed from CHN analysis, which suggested a higher C:N ratio when compared to C:O. This finding is associated with the formation of amides, as confirmed by FT-IR results suggesting that with the increase in the number of amines present in the passivating agents, most nitrogenated groups are converted to amides. However, the pKa of the amides is not within the pH range analyzed in the titration experiments. PH6 also presented groups with pKa ~10, most probably associated with cyclic amines, with a more basic character.
The surface charge of the N-CDs was investigated by zeta potential analysis, indicating that all materials present a negative surface charge (Fig. S8†). For the N-CDs synthesized using different concentrations of DT3, a surface charge of −29.6, −26.0, −26.1 and −20.0 mV was observed for 125, 250, 375 and 750 mM of DT3, respectively. When the surface passivating agent was varied, the surface of the materials became more positive, and no discernible trend was observed, with values of −10.3, −5.2, −4.7 and −14.2 mV for 375 mM of ED2, TT4, TP5 and PH6-CDs. These negative values are mostly attributed to the deprotonation of the carboxylic acids upon the post-synthetic treatment with NaOH solution. These findings are further confirmed when comparing the surface charge of the N-CDs before these treatments, in which the surface charge was neutral or slightly negative. No significant changes in the catalysts' morphology and other physico-chemical properties were observed following this step.
Biodiesel quantification following transesterification reaction
In the transesterification of vegetable oils, the reaction yield is characterized by the formation of fatty acid methyl esters (FAME) when using methanol as primary alcohol.4 Conventionally, FAME yields are quantified using gas–liquid chromatography,48 or 1H NMR, which can also accurately determine FAME conversion, circumventing challenges associated with other quantitative techniques. This technique exploits the differences in chemical shifts derived from the triglyceride precursor and the FAME product (Fig. S9†). Furthermore, FT-IR analysis (Fig. S10†) can be used to monitor biodiesel conversion qualitatively.N-CDs-catalyzed transesterification reaction of canola oil
In optimizing the N-CD-catalyzed transesterification of canola oil to produce biodiesel, altering reaction parameters such as catalyst loading, methanol content, temperature and reaction time were investigated.Initially, the catalytic activity of the N-CDs synthesized using 375 mM of the different amine passivating agents was investigated (Fig. 2a). The N-CDs were loaded at a 10 wt% content with oil to methanol ratio of 1:72, and the reaction mixtures were heated at 150 °C for 3 hours. Compared to homogeneous catalysts, heterogeneous catalysts usually require higher oil to methanol molar ratio (ranging from 1:9 to 1:275) as the excess alcohol would drive the reaction to favour the methyl ester formation.3,10 Additionally, in comparison to conventional reaction temperatures of 70 °C, a high reaction temperature was used since heterogeneous catalysts usually require energy-intensive conditions to drive the reaction forward.49
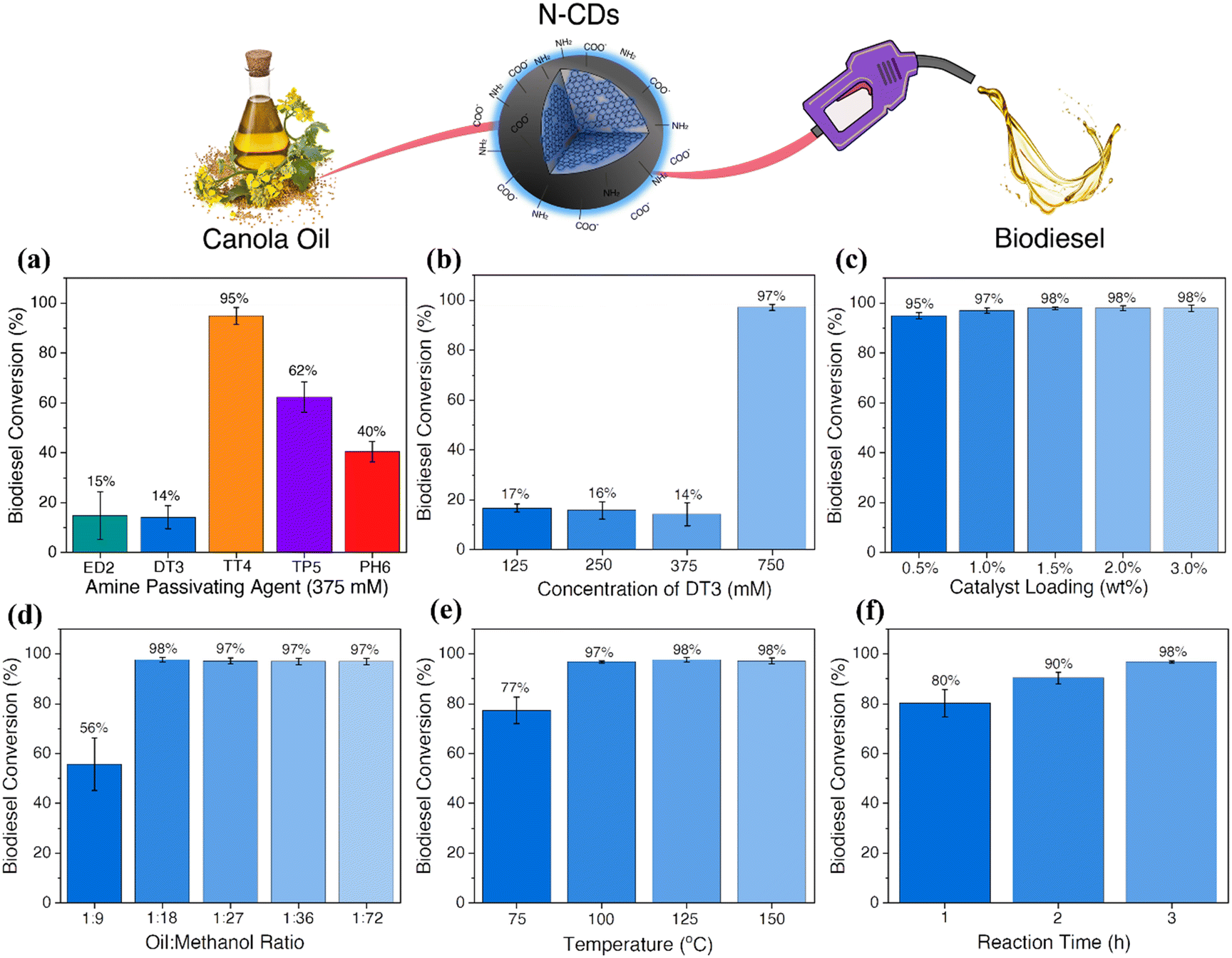 | ||
| Fig. 2 (a) Biodiesel conversion results for the N-CDs synthesized from 375 mM of different passivating agents obtained by 1H NMR. A conversion of 15%, 14%, 95%, 62%, and 40% was achieved when 375 mM of ED2, DT3, TT4, TP5 and PH6 were used. These findings suggest that the N-CDs containing higher concentrations of amines on their surfaces, such as TT4-CDs, are more efficient. In contrast, the presence of high concentrations of carboxylic acids and amines moieties leads to lower yields. (b) Biodiesel conversion results for the N-CDs synthesized using concentrations of DT3 ranging from 125–750 mM. The biodiesel yield obtained by 1H NMR indicated 17%, 16%, 14% and 97% conversion for 125 mM, 250 mM, 375 mM and 750 mM DT3 CDs, respectively. The samples with higher concentrations of carboxylic acids on their surfaces, (125 and 250 mM-DT3 CDs) presented lower conversion yields. (c) Catalyst loading optimization for the 750 DT3-CDs (oil to methanol ratio of 1:72, 150 °C for 3 hours) indicated conversions higher than 97% when 1.0 wt% catalyst loading was used, with a slight decrease to 95% when using 0.5 wt% loadings. The oil to methanol ratio was investigated after determining the optimal catalyst loading to be 1 wt% and keeping all the other parameters constant. (d) The optimal oil to methanol ratio was established as 1:18, with a reaction yield of 98%. The temperature (e) and time (f) were also investigated and optimized to 100 °C and 3 h. | ||
The biodiesel yield obtained by 1H NMR (Fig. S11a†) indicated a conversion of 15%, 14%, 95%, 62% and 40% when 375 mM of ED2, DT3, TT4, TP5 and PH6 were used, respectively. These findings suggest that the N-CDs containing higher concentrations of amines on their surfaces, such as TT4-CDs, are more efficient as catalysts for the transesterification of canola oil to biodiesel. High concentrations of carboxylic acid moieties seem to interfere with the reaction. This might occur due to a competing reaction mechanism or the formation of a hydrogen bond between the amines and carboxylic acids, deactivating these sites. Similarly, as observed for TP5 and PH6-CDs, the decrease in the amine concentration considerably reduces the catalytic efficiency of these materials.
To further confirm these findings and understand if the performance of the catalysts can be improved by changing the concentration of the passivating agent, the sample presenting the lowest conversion rate, DT3-CDs, was investigated (Fig. 2b). The N-CDs, synthesized using different concentrations of DT3, were tested as catalysts for biodiesel production following the same protocol described previously (10 wt% loading; oil to methanol ratio of 1:72, 150 °C for 3 hours). The biodiesel yield obtained by 1H NMR (Fig. S11b†) indicated a conversion of 17%, 16%, 14% and 97% for 125 mM, 250 mM, 375 mM and 750 mM DT3 CDs, respectively. Similar to the results discussed previously, the samples with higher concentrations of carboxylic acids on their surfaces, such as 125 and 250 mM-DT3 CDs, presented lower conversion yields. Despite higher concentrations of amines when compared to carboxylic acids, N-CDs prepared using 375 mM of DT3 do not show substantial biodiesel conversion. Therefore, it is possible to infer that the competitive mechanisms between carboxylic acid and amines are still occurring. For 750 mM DT3-CDs, almost 70% of the surface functional groups are amines, similar to the 375 mM TT4-CDs, which explains the high biodiesel conversion.
The biodiesel reaction parameters were also investigated for the 750 mM DT3-CDs. The first parameter evaluated was the catalyst loading. Heterogeneous base catalysts often require higher catalyst loadings, reaching up to 15 wt%, to have comparable catalytic efficiencies similar to strong bases.21 These experiments were carried out using catalyst loadings ranging from 0.5–3 wt% while all other parameters were kept constant (oil to methanol ratio of 1:72, 150 °C for 3 hours). The biodiesel yield obtained by 1H NMR (Fig. S11c†) indicated conversions >97% when 1.0 wt% catalyst loading was used, with a slight decrease to 95% when using 0.5 wt% loadings. Therefore, the optimal catalyst loading was established to be 1.0 wt%.
An excess of methanol is usually required to drive the transesterification reaction equilibrium towards the formation of biodiesel. However, the oil-to-methanol ratio can be optimized to minimize the volumes of methanol used. Therefore, these experiments were carried out using ratios ranging from 1:9 to 1:72 and 1.0 wt% loading while the temperature and time were kept constant as previously described. When the oil to methanol ratio was 1:9, the biodiesel yield was ∼56%. However, upon increasing this ratio to 1:18, the yield increased to 98%. Therefore, the optimal oil-to-methanol ratio was determined to be 1:18 (Fig. S11d†).
Compared to conventional reaction temperatures of 70 °C (catalyzed by homogeneous alkali bases), a high reaction temperature is required when heterogeneous catalysts are used for transesterification reactions. Therefore, reducing the reaction temperature is extremely important as it also reduces the energy requirements for applying these materials on an industrial scale. For these experiments, the temperature was varied from 75–150 °C, using a 1.0 wt% loading 1:18 oil to methanol ratio for 3 h. Biodiesel conversions higher than 98% were observed when the reaction temperature was higher than 100 °C, while at 75 °C, a conversion of 77% was obtained (Fig. S11e†). Similarly, the reaction time was also evaluated, using 100 °C and keeping all the other parameters constant. The conversion obtained for 1, 2, and 3 h was 80, 90 and 98%, respectively (Fig. S11f†). As such, the optimal reaction time was established to be 3 h. Altogether, the optimal parameters for the transesterification reaction of canola oil to biodiesel catalyzed by the N-CDs were determined as 1.0 wt% catalyst loading, 1:18 oil to methanol ratio, 100 °C for 3 h.
While many acid-functionalized carbon-based nanomaterials have been thoroughly studied in the literature, there is a lack of information regarding the use of amines as active sites in the transesterification reaction. As this work focus on how these surface groups drive the activity of the carbon dots as catalysts for the aforementioned application, a comparative study between the performance of N-CDs and other similar carbon-based materials and homogeneous amines was performed. The results are summarized in Table 1.
| Catalyst | Triglyceride feedstock | Reaction conditions | Biodiesel conversion | Reuse cycles | Ref. |
|---|---|---|---|---|---|
| a Upon reactivation. b As any homogeneous catalyst. | |||||
| Heterogeneous catalysts | |||||
| N-CDs (750 mM DT3) | Canola oil | 1 wt% catalyst loading; 1:18 oil to methanol ratio; 100 °C for 3 h |
≥97% | ≥5 | Present work |
| Glycine-passivated CDs | Canola oil | 1 wt% catalyst loading, 1:9 oil to methanol ratio, 150 °C for 3 h |
≥97% | ≥5 | 50 |
| Amino-functionalized carbon nanotubes | Glyceryl tributyrate | Catalyst loading not specified; 1:12 glyceryl to methanol ratio, 90 °C for 8 h |
≥77% | ≥6a | 51 |
| Homogeneous catalysts | |||||
| Organic amines | Cottonseed oil | 6 wt% catalyst loading, 1:9 oil to methanol ratio, 180 °C for 3 h |
≥94% | 0b | 36 |
| Organic amines | Rapeseed oil | 0.3 wt% catalyst loading, 1:24 oil to methanol ratio, 239 °C (supercritical temperature) for 3 h |
≥80% | 0b | 52 |
Compared to homogeneous catalysts, heterogeneous catalysts usually require higher oil to methanol molar ratio (ranging from 1:9 to 1:275) as the excess alcohol would drive the reaction to favour the methyl ester formation. Villa et al. demonstrated the use of amine-functionalized carbon nanotubes (N-CNTs) as heterogeneous catalysts for the transesterification of triglycerides.51 Their work presented valuable insights correlating the different basicity of the amines to the catalytic activity. The N-CNTs presented conversions ≥77% and remained active for at least six reuse cycles after reactivation. However, the synthesis of carbon nanotubes often requires metallic catalysts and long purification steps. Furthermore, amine grafting is done through a post-synthesis modification, making the process even more laborious. Yao et al. reported using low boiling point organic amines to produce biodiesel. The reactions were carried out at 180 °C for 3 h, with 6 wt% catalyst loading and methanol to oil molar ratio of 1:9. Nonetheless, trace amounts of KOH were required to achieve high conversion yields.36 Tang et al. also reported using amines in supercritical methanol to catalyze the reaction of rapeseed oil to biodiesel. In this case, 0.27 wt% catalyst was used, with a 1:24 0.27, a fixed reaction pressure of 25 MPa amine-passivated carbon nanotubes and homogeneous organic amines and a temperature of 280 °C.52 Both works provided insightful information regarding using organic amines as efficient catalysts for biodiesel reactions. However, the recovery of the catalyst required distillation steps and further purification of the products hindering application on a large scale.
The reusability of heterogeneous catalysts is one of the primary benefits in contrast to their homogeneous counterparts. Therefore, to guarantee the efficiency and sustainability of the process catalyzed by the N-CDs, their reusability was investigated. A slight decrease in biodiesel conversion following the first catalytic cycle was observed. However, the conversion yield seems to plateau at a biodiesel conversion of 95% even after five catalytic cycles.
Mechanism of N-CDs-catalyzed transesterification reaction
Combining the transesterification reaction results and the findings from qualitative and quantitative analysis of the functional groups present on the surface of the N-CD catalysts, we hypothesize that both carboxylates and amines can act as active catalytic sites. Table 2 summarizes the active site density (SD) for all catalysts.| Sample | SDCarboxylates (1019 site/gcatalyst) | SDAmines (1019 site/gcatalyst) | SDTotal (1019 site/gcatalyst) |
|---|---|---|---|
| 125 mM DT3 | 23.3 | 6.1 | 29.4 |
| 250 mM DT3 | 12.4 | 8.1 | 20.5 |
| 375 mM DT3 | 8.2 | 14.2 | 22.4 |
| 750 mM DT3 | 7.5 | 15.8 | 23.3 |
| 375 mM ED2 | 4.0 | 5.8 | 9.8 |
| 375 mM TT4 | 7.4 | 18.2 | 25.6 |
| 375 mM TP5 | 10.4 | 17.9 | 28.3 |
| 375 mM PH6 | 11.3 | 14.6 | 25.8 |
We hypothesize that two mechanisms might occur for the N-CD catalyzed transesterification reaction (Fig. 3). When the concentration of the passivating agent is low, between 125 to 250 mM, the majority of the functional groups present on the surface of the CDs are carboxylates, hence higher SDcarboxylates. In this case, the catalytic mechanism works similarly to one proposed by Macina et al.35 where it was proposed that the methanol attacks the ester, forming a zwitterion intermediate; subsequently, there is a proton transfer from the zwitterion to the functional groups on the surface of the CDs, followed by the traditional base-catalyzed mechanism steps. However, we hypothesize that the conjugate bases of secondary amides acting as weak bases for the N-CDs can also deprotonate the zwitterion.
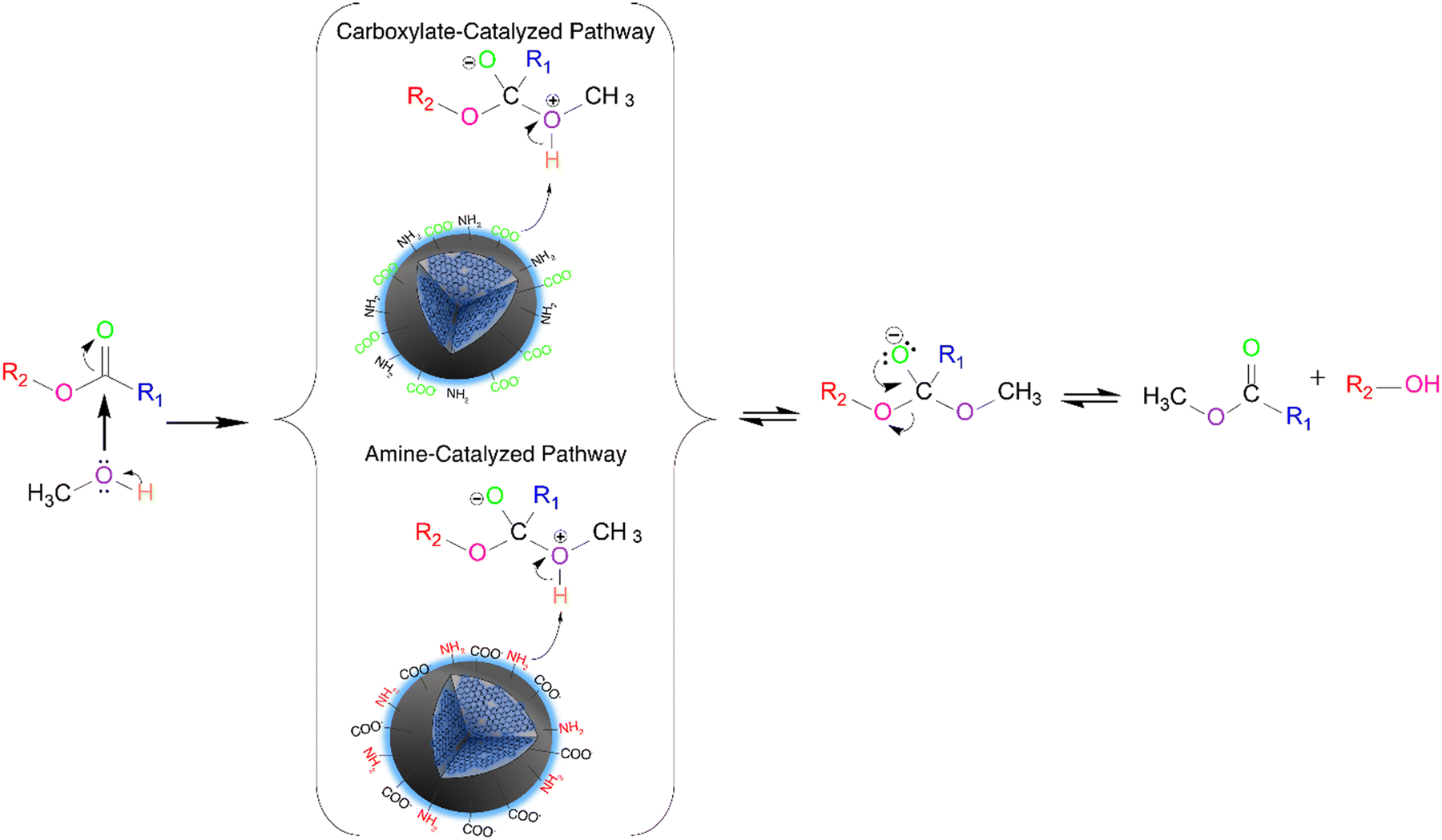 | ||
| Fig. 3 Proposed transesterification reaction mechanism using the N-CDs. Two pathways are possible for the proton abstraction from the tetrahedral intermediate, through the carboxylates or the amine groups. | ||
When the concentration of the passivating agent increases significantly, such as for the 750 mM DT3 CDs, the amines and amides are the main functional groups present on the surface of the catalyst with increased SDamines. In this case, the amines would act as the most active catalytic site, governing the reaction mechanism. It has been previously reported that the transesterification mechanism can occur with weak bases; however, it requires high pressures and reaction temperatures to overcome the energy barrier. In this work, temperatures higher than 100 °C in the hydrothermal reactor (a high-pressure environment) were required to achieve high biodiesel conversions. This supports our claim that weak bases are the key catalytic factors in the CD-catalyzed mechanism.
However, for 375 mM DT3, approximately 40% of the carboxylate moieties have been converted to amides due to the higher concentration of amines from the passivating agent, drastically reducing the availability of these moieties on the surface of the CDs. In this circumstance, both reaction mechanisms catalyzed by the carboxylates and amines can occur through a competitive process that reduces biodiesel conversion. Similar behaviour is observed for the N-CDs synthesized from ED2, PH5 and PH6. For 375 mM TT4, however, the increase in amines on the surface imparts similar properties to the ones observed for increased concentration of passivating agents.
Furthermore, the surface charge of the catalyst also affects its catalytic efficiency. A negative surface charge is required to favor the deprotonation of the methanol during the catalytic mechanism. This is supported by the fact that no biodiesel conversion was achieved when the samples were not subjected to the deprotonation steps (Fig. S11g†).
Conclusions
N-CD heterogeneous catalysts were prepared using different amine passivating agents to tune the number of amine, amide, and carboxylic acid sites on their surface. These materials were thoroughly characterized regarding their physico-chemical properties, confirming the well-established knowledge that changes in the chemical nature of the amine passivating agent and its concentration can indeed be used to modulate their surface chemistry. The catalytic efficiency of amine-passivated carbon dots was investigated for the transesterification of canola oil to biodiesel. We demonstrated a heavy metal-free CD-catalyzed transesterification of canola to biodiesel, achieving biodiesel conversions of ≥97% using 1 wt% catalyst loading at 100 °C and a 3-hour reaction temperature and time, respectively. The biodiesel reaction parameters were optimized to reduce the energy requirements, in line with industry requirements. We highlight their reusability over at least five catalytic cycles, proving their catalytic efficiency and robustness. In addition, we investigated the correlation between the amine, amide and carboxylic surface functional group concentrations and the catalytic efficiency of the N-CDs. Finally, in-depth studies of large-scale reactions, kinetics, and thermodynamics are still required to glean a deeper understanding of this system. Nonetheless, this proof of concept bridges the knowledge gap on the mechanistic aspects of heterogeneous catalysts in biofuel research and can help direct the field toward developing cost-efficient and sustainable catalyst systems.Author contributions
Tayline V. de Medeiros: conceptualization, methodology, validation, writing – original draft, writing – review & editing, visualization. Alexia Macina: conceptualization, methodology, validation, writing – original draft, writing – review & editing, visualization. João Paulo de Mesquita: designed and performed potentiometric titration experiments, writing – review. Rafik Naccache: resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge funding sources for the financial support of this research. RN is grateful to NSERC for funding through the Discovery Grant and Discovery Accelerator Supplement. RN is also grateful for funding from FRQNT through the Établissement de la relève professorale program and to the Quebec Centre for Advanced Materials. TVdM acknowledges the CAPES scholarship program for funding support.References
- M. Höök, S. Davidsson, S. Johansson and X. Tang, Philos. Trans. R. Soc., A, 2014, 372, 1–21 Search PubMed.
- A. Demirbaş, Biodiesel: A Realistic Fuel Alternative for Diesel Engines, Springer, 2008 Search PubMed.
- B. K. Uprety, W. Chaiwong, C. Ewelike and S. K. Rakshit, Energy Convers. Manage., 2016, 115, 191–199 CrossRef CAS.
- D. J. Anneken, S. Both, R. Christoph, G. Fieg, U. Steinberner and A. Westfechtel, Ullmann's Encycl. Ind. Chem., 2012, vol. 14, pp. 73–116 Search PubMed.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405–416 CrossRef CAS PubMed.
- S. L. Dmytryshyn, A. K. Dalai, S. T. Chaudhari, H. K. Mishra and M. J. Reaney, Bioresour. Technol., 2004, 92, 55–64 CrossRef CAS PubMed.
- O. J. Alamu, M. A. Waheed and S. O. Jekayinfa, Fuel, 2008, 87, 1529–1533 CrossRef CAS.
- A. Demirbaş, Energy Convers. Manage., 2003, 44, 2093–2109 CrossRef.
- R. L. Glass, Lipids, 1971, 6, 919–925 CrossRef CAS.
- M. Zabeti, W. M. A. W. Daud and M. K. Aroua, Fuel Process. Technol., 2009, 90, 770–777 CrossRef CAS.
- B. Freedman and S. Saka, J. Am. Oil Chem. Soc., 1984, 61, 1638–1643 CrossRef CAS.
- G. Vicente, M. Martínez and J. Aracil, Bioresour. Technol., 2007, 98, 1724–1733 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- T. F. Dossin, M. F. Reyniers, R. J. Berger and G. B. Marin, Appl. Catal., B, 2006, 67, 136–148 CrossRef CAS.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaría and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317–326 CrossRef CAS.
- A. P. S. Dias, J. Puna, J. Gomes, M. J. N. Correia and J. Bordado, Renewable Energy, 2016, 99, 622–630 CrossRef.
- M. Catarino, S. Martins, A. P. S. Dias, M. F. C. Pereira and J. Gomes, J. Environ. Chem. Eng., 2019, 7, 103099 CrossRef CAS.
- L. Wang and J. Yang, Fuel, 2007, 86, 328–333 CrossRef CAS.
- M. Balajii and S. Niju, Environ. Chem. Lett., 2019, 17, 1447–1469 CrossRef CAS.
- M. Kalantari, M. Yu, Y. Liu, X. Huang and C. Yu, Mater. Chem. Front., 2019, 3, 1816–1822 RSC.
- S. Ramu, N. Lingaiah, B. L. A. P. Devi, R. B. N. Prasad, I. Suryanarayana and P. S. S. Prasad, Appl. Catal., A, 2004, 276, 163–168 CrossRef CAS.
- J. Clohessy and W. Kwapinski, Appl. Sci., 2020, 10, 1–17 Search PubMed.
- T. V. De Medeiros, J. Manioudakis, F. Noun, J.-R. R. Macairan, F. Victoria and R. Naccache, J. Mater. Chem. C, 2019, 7, 7175–7195 RSC.
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef CAS PubMed.
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC.
- Y. Zhang, X. Liu, Y. Fan, X. Guo, L. Zhou, Y. Lv and J. Lin, Nanoscale, 2016, 8, 15281–15287 RSC.
- D. R. da Silva Souza, L. D. Caminhas, J. P. de Mesquita and F. V. Pereira, Mater. Chem. Phys., 2018, 203, 148–155 CrossRef CAS.
- Z. Yi, X. Li, H. Zhang, X. Ji, W. Sun, Y. Yu, Y. Liu, J. Huang, Z. Sarshar and M. Sain, Talanta, 2021, 222, 121663 CrossRef CAS PubMed.
- J. Manioudakis, F. Victoria, C. A. Thompson, L. Brown, M. Movsum, R. Lucifero and R. Naccache, J. Mater. Chem. C, 2019, 7, 853–862 RSC.
- D. Bhattacharya, M. K. Mishra and G. De, J. Phys. Chem. C, 2017, 121, 28106–28116 CrossRef CAS.
- Z. Huang, F. Lin, M. Hu, C. Li, T. Xu, C. Chen and X. Guo, J. Lumin., 2014, 151, 100–105 CrossRef CAS.
- Y. Q. Zhang, D. K. Ma, Y. Zhuang, X. Zhang, W. Chen, L. L. Hong, Q. X. Yan, K. Yu and S. M. Huang, J. Mater. Chem., 2012, 22, 16714–16718 RSC.
- B. N. Jusuf, N. S. Sambudi, I. Isnaeni and S. Samsuri, J. Environ. Chem. Eng., 2018, 6, 7426–7433 CrossRef CAS.
- P. C. Hsu, Z. Y. Shih, C. H. Lee and H. T. Chang, Green Chem., 2012, 14, 917–920 RSC.
- A. Macina, T. V. de Medeiros and R. Naccache, J. Mater. Chem. A, 2019, 7, 23794–23802 RSC.
- J. Yao, L. Ji, P. Sun, L. Zhang and N. Xu, Fuel, 2010, 89, 3871–3875 CrossRef CAS.
- M. Zheng, S. Liu, J. Li, Z. Xie, D. Qu, X. Miao, X. Jing, Z. Sun and H. Fan, J. Mater. Res., 2015, 30, 3386–3393 CrossRef CAS.
- A. B. Siddique, A. K. Pramanick, S. Chatterjee and M. Ray, Sci. Rep., 2018, 8, 1–10 CAS.
- K. M. Tripathi, T. S. Tran, T. T. Tung, D. Losic and T. Kim, J. Nanomater., 2017, 2017, 1–10 CrossRef.
- A. S. Rettenbacher, B. Elliott, J. S. Hudson, A. Amirkhanian and L. Echegoyen, Chem. – Eur. J., 2005, 12, 376–387 CrossRef CAS PubMed.
- T. T. Meiling, R. Schürmann, S. Vogel, K. Ebel, C. Nicolas, A. R. Milosavljević and I. Bald, J. Phys. Chem. C, 2018, 122, 10217–10230 CrossRef CAS.
- A. B. Bourlinos, R. Zbořil, J. Petr, A. Bakandritsos, M. Krysmann and E. P. Giannelis, Chem. Mater., 2012, 24, 6–8 CrossRef CAS.
- L. Cheng, Y. Li, X. Zhai, B. Xu, Z. Cao and W. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 20487–20497 CrossRef CAS PubMed.
- S. Hill and M. C. Galan, Beilstein J. Org. Chem., 2017, 13, 675–693 CrossRef CAS PubMed.
- V. Ţucureanu, A. Matei and A. M. Avram, Crit. Rev. Anal. Chem., 2016, 46, 502–520 CrossRef PubMed.
- D. L. Pavia, G. M. Lampman and G. S. Kriz, Introduction to Spectroscopy, Thomson Learn. Inc., 2001, p. 579 Search PubMed.
- L. A. Alves, A. H. de Castro, F. G. de Mendonça and J. P. de Mesquita, Appl. Surf. Sci., 2016, 370, 486–495 CrossRef CAS.
- P. De Filippis, C. Giavarini, M. Scarsella and M. Sorrentino, J. Am. Oil Chem. Soc., 1995, 72, 1399–1404 CrossRef CAS.
- X. Liu, H. He, Y. Wang and S. Zhu, Catal. Commun., 2007, 8, 1107–1111 CrossRef CAS.
- A. Macina, T. V. de Medeiros and R. Naccache, J. Mater. Chem. A, 2019, 7, 23794–23802 RSC.
- A. Villa, J.-P. Tessonnier, O. Majoulet, D. S. Su and R. Schlögl, Chem. Commun., 2009, 4405 RSC.
- Z. Tang, L. Wang and J. Yang, Eur. J. Lipid Sci. Technol., 2008, 110, 747–753 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lf00060e |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |