
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantiopure polythiophene nanoparticles. Chirality dependence of cellular uptake, intracellular distribution and antimicrobial activity†
Ilaria Elena Palamà *a,
Francesca Di Maria*ab,
Mattia Zangolibc,
Stefania D'Amonea,
Giovanni Manfredid,
Jonathan Barsottid,
Guglielmo Lanzanide,
Luca Ortolanif,
Elisabetta Salatellig,
Giuseppe Gigliah and
Giovanna Barbarellabc
*a,
Francesca Di Maria*ab,
Mattia Zangolibc,
Stefania D'Amonea,
Giovanni Manfredid,
Jonathan Barsottid,
Guglielmo Lanzanide,
Luca Ortolanif,
Elisabetta Salatellig,
Giuseppe Gigliah and
Giovanna Barbarellabc
aCNR NANOTEC, c/o Campus Ecotekne, Via Monteroni, 73100 Lecce, Italy. E-mail: ilaria.palama@nanotec.cnr.it
bCNR-ISOF, Via P. Gobetti 101, 40129 Bologna, Italy. E-mail: francesca.dimaria@isof.cnr.it
cMediteknology srl, Via P. Gobetti 101, Bologna, Italy
dPolitecnico di Milano, Dept. of Physics, Via Pascoli 70/3, I-20133 Milano, Italy
eCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, 40136, Via Pascoli 70/3, I-20133 Milano, Italy
fCNR-IMM, Via P. Gobetti 101, 40129 Bologna, Italy
gDept. of Industrial Chemistry Toso Montanari, University of Bologna, 40136 Bologna, Italy
hDept. of Mathematics and Physics Ennio De Giorgi, University of Salento, 73100 Lecce, Italy
First published on 25th July 2019
Abstract
The use of intrinsic chiral molecules opens the door to bio-imaging specific tools and to the development of target-therapy. In this work the synthesis and characterization of polythiophenes with alkyl side chains containing one R or S chiral carbon is reported. Enantiopure chiral nanoparticles (R or S NPs) were prepared from the polymers by a reprecipitation method. UV-vis, photoluminescence and circular dichroism spectroscopy of the NPs are described. In vitro analysis and metabolic assays show that both R and S NPs are efficiently taken-up by fibroblast cells without signs of toxicity. SDS-PAGE experiments show that formation of hard protein ‘corona’ enhances the chirality difference between nanoparticles. Co-localization experiments demonstrate that the cells are able to discriminate between the enantiomeric R and S nanoparticles. Finally, experiments carried out on Gram negative and Gram positive bacteria show that the enantiomeric NPs display different antibacterial activity.
1. Introduction
Chiral nanostructures – namely nano-objects non-superimposable to their mirror image – are currently attracting increasing attention in materials science, nanoscience and nanotechnology. Applications in chiral catalysis, chiral sensing and chiroptical devices have been reported.1–6 Chiral innovative materials are being explored in biology and medicine,7–10 as most molecular and supramolecular biological compounds are chiral. The chirality of biomolecules influences physiological events and through chiral recognition and stereoselective non-bonding interactions the enantiomorphs of chiral nanostructures may interact differently with chiral biomolecules, depending on the sign of chirality. Hydrogen bonding, electrostatic forces, dipole–dipole, steric and hydrophobic interactions, are the main weak forces driving the behaviour of chiral nanostructures towards biomolecules. In the presence of live cells, chiral nanostructures may give rise to different interactions between the chiral surface and the surrounding chiral proteins. In this way, they behave as tools capable to control the type and the amount of biomolecules interacting with the cells. Up to now most of the studies demonstrating selective interactions of enantiomorphs with live cells have concerned inorganic nanoparticles coated with organic chiral compounds. Functional porous nanometer-scale materials coated with chiral polymers display chirality-dependent cellular uptake11 while for CdSe/ZnS quantum dots (QDs) capped with L- and D-cysteine in living Ehrlich ascite carcinoma cells the uptake of L-Cys QDs is almost twice that of D-Cys QDs.12 Selective behaviour is also displayed by chiral gold nanoparticles coated with poly(acryloyl-L-valine)-b-poly(2-hydroxyethyl methylacrylate) (L)-PAV-b-PHEMA or its D counterpart in A549 and HepG2 cancer cells.13 It has been found that magnetic nanoparticles decorated with L-phosphotyrosine (L-pY) are innocuous to cells while D-pY-decorated ones selectively inhibit cancer cells.14 Recently, graphene quantum dots functionalized with chiral biomolecules have been reported as new efficient selective antimicrobial nanoagents.15Systematic investigations on the behaviour of purely organic chiral nanoparticles of opposite chirality in the presence of live cells are still lacking, despite the fact that several chiral nanoparticles based on conjugated polymers and oligomers have already been described.16–19 The interest in organic nanoparticles comes from the fact that they are generally not toxic to living cells, are fluorescent and may be used as probes for live cell imaging.20
We have recently reported that poly(3-hexylthiophene) nanoparticles easily enter within the cytoplasm of HEK-293 cells, leaving the viability and the physiological equilibrium unaltered.21 We have also demonstrated that nanoparticles, prepared from appropriately functionalized thiophene polymers, can act as phototransducers able to absorb visible light and convert it into an electrical signal through cell membrane polarization.22,23 We report here the synthesis, characterization and behaviour in the presence of live cells of enantiopure thiophene based nanoparticles prepared from chiral R and S thiophene polymers. Thiophene oligomers and polymers can be made chiral either by scaffold chirality or by the presence of chiral centers on the side chains of linear polymers.24–28 The NPs described here were obtained by linear thiophene polymers containing enantiopure R and S carbon centres in the side chains. The objective of our work was to check whether chiral selectivity can be observed on NPs uptake and intracellular distribution in live cells, such as NIH-3T3 fibroblasts, as well as on antimicrobial activity of the chiral nanoparticles.
2. Experimental section
Materials: all chemicals were used as received without further purification. The detailed synthesis of polymers and intermediates (Scheme 1) is reported in ESI.† All tissue culture media and chemical reagents were purchased from Sigma-Aldrich. Cell line and bacteria stock were purchased from American Tissue Type Collection (ATTC). Lyso-tracker, ER-tracker and Mito-Tracker were purchased from Life Technology. Optical characterization and circular dichroism (CD): absorption of the polymer solutions and nanoparticles dispersions was measured in quartz cuvettes (Suprasil® QS) using a PerkinElmer Lambda 1050 spectrophotometer (3 nm resolution, 2 nm sampling). Photoluminescence was acquired using a Jobin Yvon spectrofluorimeter exciting at 480 nm (5 nm excitation slit, 5 nm emission slit, 3 nm sampling). CD spectra were collected using a spectropolarimeter JASCO J-715 under ambient conditions. Protein corona analysis of chiral NPs: for the protein corona analyses SDS-PAGE (Sodium Dodecyl Sulphate PolyAcrylamide Gel Electrophoresis) was employed. R and S NPs (0.05 mg mL−1) were incubated at 37 °C in a shaker for 1 hour in complete culture medium with 10% FBS and subsequently NPs were first centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min in order to eliminate hard corona and then resuspended in PBS 1×. Washing process was performed three times prior to SDS-PAGE analysis. Eluted hard corona proteins from all samples were mixed with SDS-sample buffer and boiled at 100 °C for 10 min. The samples were subjected to 4–12% SDS-PAGE for 90 min at 120 V. Coomassie blue staining was used for detection of protein bands. Proteins concentration on NPs was determined by using Bradford reagent (BCA, Sigma Aldrich) at 590 nm. Representative results of three independent experiments are reported. NPs cellular uptake and intracellular localization: mouse fibroblast cells (NIH-3T3) were cultured in DMEM supplemented with 10% fetal bovine serum at 37 °C in 5% CO2 in a humidified incubator. Quantification of cellular uptake: for cellular uptake efficiency (%), NIH-3T3 fibroblasts (105 cells) was treated with serum-free DMEM medium containing fluorescent R and S NPs (0.05 mg mL−1) and incubated to 0–180 minutes at 37 °C in CO2 incubator. Successively, the cells were washed three times with cold PBS 1×, pH 7.4. Subsequently, all the cells were lysed by 0.5% Triton X-100 in a 0.2 N NaOH solution. The amount of R and S NPs (excitation wavelength of 550 nm and an emission wavelength of 645 nm) in the cells was fluorometrically evaluated for the lysate using a fluorescence spectrometer. The cellular uptake efficiency (%) was showed as the fluorescence associated with the cells vs. the ones present in the positive control solution. Representative measurements of three distinct sets of data have been reported (Student's t-test, P < 0.05). Uptake and intracellular quantification: for fluorescence confocal analysis, cells were fixed for 5 minutes in 3.7% formaldehyde and mounted using fluoroshield with DAPI. Intracellular localization of R and S NPs after 1 hour of incubation were performed with LysoTracker Green, MitoTracker Green and ER-Tracker Green (Life technology) immunostaining, according to the manufacturer's instructions, to label lysosomes, mitochondria and ER, respectively. Confocal micrographs were taken with a Leica confocal scanning system mounted on a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany), using a 40× and 63× oil immersion objective and spatial resolution of 200 nm in x–y and 100 nm in z. Quantitative co-localization analysis of ten fields, within three different experiments, were casually selected for each sample. Coefficient of correlation (CC or Pearson's r),29 intensity correlation quotient (ICQ)30 and overlap coefficient (OC)31 as frequency indices of co-localization between NPs and LysoTracker, MitoTracker and ER-Tracker were used. Coefficients and quotients were calculated from each confocal image, as well as pixel shuffling and calculation of the 999/1000 quantile of intensity, were conducted with custom-made plug-in programs (available at http://www.mbs.med.kyoto-u.ac.jp/imagej/index.html) combined with ImageJ software (National Institute for Health, Bethesda, MD, USA). Numerical data was processed with Excel (Microsoft, Redmond, WA, USA) for further calculation. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05). In vitro cytotoxicity of chiral NPs: in vitro cytotoxicity of R and S NPs in solution and deposited on glass coverslip (0.05 mg mL−1) against mouse fibroblasts (NIH-3T3) was evaluated via MTT assay after 24–48–72 hours of incubation, according to the manufacturer's instructions (Sigma-Aldrich, USA). The absorbance was spectrophotometrically measured at a wavelength of 570 nm and the background absorbance measured at 690 nm was subtracted. The percentage viability was expressed as the relative growth rate (RGR) by following equation:
000 rpm for 10 min in order to eliminate hard corona and then resuspended in PBS 1×. Washing process was performed three times prior to SDS-PAGE analysis. Eluted hard corona proteins from all samples were mixed with SDS-sample buffer and boiled at 100 °C for 10 min. The samples were subjected to 4–12% SDS-PAGE for 90 min at 120 V. Coomassie blue staining was used for detection of protein bands. Proteins concentration on NPs was determined by using Bradford reagent (BCA, Sigma Aldrich) at 590 nm. Representative results of three independent experiments are reported. NPs cellular uptake and intracellular localization: mouse fibroblast cells (NIH-3T3) were cultured in DMEM supplemented with 10% fetal bovine serum at 37 °C in 5% CO2 in a humidified incubator. Quantification of cellular uptake: for cellular uptake efficiency (%), NIH-3T3 fibroblasts (105 cells) was treated with serum-free DMEM medium containing fluorescent R and S NPs (0.05 mg mL−1) and incubated to 0–180 minutes at 37 °C in CO2 incubator. Successively, the cells were washed three times with cold PBS 1×, pH 7.4. Subsequently, all the cells were lysed by 0.5% Triton X-100 in a 0.2 N NaOH solution. The amount of R and S NPs (excitation wavelength of 550 nm and an emission wavelength of 645 nm) in the cells was fluorometrically evaluated for the lysate using a fluorescence spectrometer. The cellular uptake efficiency (%) was showed as the fluorescence associated with the cells vs. the ones present in the positive control solution. Representative measurements of three distinct sets of data have been reported (Student's t-test, P < 0.05). Uptake and intracellular quantification: for fluorescence confocal analysis, cells were fixed for 5 minutes in 3.7% formaldehyde and mounted using fluoroshield with DAPI. Intracellular localization of R and S NPs after 1 hour of incubation were performed with LysoTracker Green, MitoTracker Green and ER-Tracker Green (Life technology) immunostaining, according to the manufacturer's instructions, to label lysosomes, mitochondria and ER, respectively. Confocal micrographs were taken with a Leica confocal scanning system mounted on a Leica TCS SP5 (Leica Microsystem GmbH, Mannheim, Germany), using a 40× and 63× oil immersion objective and spatial resolution of 200 nm in x–y and 100 nm in z. Quantitative co-localization analysis of ten fields, within three different experiments, were casually selected for each sample. Coefficient of correlation (CC or Pearson's r),29 intensity correlation quotient (ICQ)30 and overlap coefficient (OC)31 as frequency indices of co-localization between NPs and LysoTracker, MitoTracker and ER-Tracker were used. Coefficients and quotients were calculated from each confocal image, as well as pixel shuffling and calculation of the 999/1000 quantile of intensity, were conducted with custom-made plug-in programs (available at http://www.mbs.med.kyoto-u.ac.jp/imagej/index.html) combined with ImageJ software (National Institute for Health, Bethesda, MD, USA). Numerical data was processed with Excel (Microsoft, Redmond, WA, USA) for further calculation. Representative measurements of three distinct sets of data have been reported (Student t-test, P < 0.05). In vitro cytotoxicity of chiral NPs: in vitro cytotoxicity of R and S NPs in solution and deposited on glass coverslip (0.05 mg mL−1) against mouse fibroblasts (NIH-3T3) was evaluated via MTT assay after 24–48–72 hours of incubation, according to the manufacturer's instructions (Sigma-Aldrich, USA). The absorbance was spectrophotometrically measured at a wavelength of 570 nm and the background absorbance measured at 690 nm was subtracted. The percentage viability was expressed as the relative growth rate (RGR) by following equation: 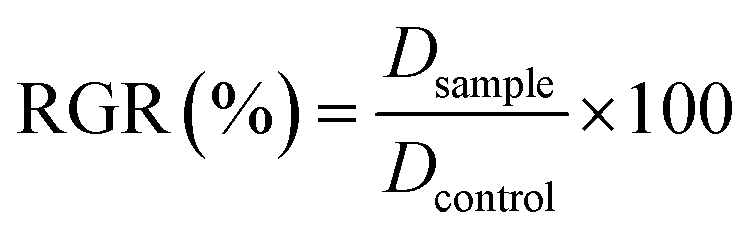 where Dsample and Dcontrol were the absorbance of the sample and the negative control. Each experiment was repeated three times in triplicate (Student's t-test, P < 0.05). Antibacterial activity of chiral NPs: Gram-negative DH5-Alpha E. coli and Gram-positive S. aureus bacteria were growth in Luria–Bertani (LB) broth. For antibacterial assay, growth bacterial inoculum (104 cells per mL) was incubated with different concentrations (50–1000 μg mL−1) of R and S NPs. Growth of E. coli and S. aureus was indexed by measuring the optical density (OD) at a wavelength of 600 nm using a UV-visible spectrophotometer (Varian Cary® 300 Scan; Varian Instruments, CA, USA) every 1 hour. In addition, samples treated with different formulation of NPs and control (CTR) were spread on solid LB plates and after 24 hours to 37 °C the number of colony forming units (CFU) were counted using a Miles–Misra method. Antibacterial tests on cotton substrates treated with R and S NPs: cotton gauzes were impregnated with R and S NPs (1 mg mL−1). The antibacterial capability of the R and S NPs were evaluated through agar diffusion tests performed according to standard ‘SNV 195920-1994’.32 The procedure consists of placing the samples in contact with bacteria on an agar plate and evaluating the width of the free-bacteria zone around and under the sample after 24 hours incubation at 37 °C. The antibacterial activity of the material can be labelled as “good” if an inhibition zone to bacterial proliferation can be observed under and close to the sample and if it results larger than 1 mm. On the other hand, the antibacterial activity of the sample can be labelled as “insufficient” if the material is totally colonized by bacteria. A “sufficient” antibacterial capability is associated to samples showing free bacteria zone under their surface only. Representative measurements of three different sets of data are reported (Student's t-test, P-values < 0.05).
where Dsample and Dcontrol were the absorbance of the sample and the negative control. Each experiment was repeated three times in triplicate (Student's t-test, P < 0.05). Antibacterial activity of chiral NPs: Gram-negative DH5-Alpha E. coli and Gram-positive S. aureus bacteria were growth in Luria–Bertani (LB) broth. For antibacterial assay, growth bacterial inoculum (104 cells per mL) was incubated with different concentrations (50–1000 μg mL−1) of R and S NPs. Growth of E. coli and S. aureus was indexed by measuring the optical density (OD) at a wavelength of 600 nm using a UV-visible spectrophotometer (Varian Cary® 300 Scan; Varian Instruments, CA, USA) every 1 hour. In addition, samples treated with different formulation of NPs and control (CTR) were spread on solid LB plates and after 24 hours to 37 °C the number of colony forming units (CFU) were counted using a Miles–Misra method. Antibacterial tests on cotton substrates treated with R and S NPs: cotton gauzes were impregnated with R and S NPs (1 mg mL−1). The antibacterial capability of the R and S NPs were evaluated through agar diffusion tests performed according to standard ‘SNV 195920-1994’.32 The procedure consists of placing the samples in contact with bacteria on an agar plate and evaluating the width of the free-bacteria zone around and under the sample after 24 hours incubation at 37 °C. The antibacterial activity of the material can be labelled as “good” if an inhibition zone to bacterial proliferation can be observed under and close to the sample and if it results larger than 1 mm. On the other hand, the antibacterial activity of the sample can be labelled as “insufficient” if the material is totally colonized by bacteria. A “sufficient” antibacterial capability is associated to samples showing free bacteria zone under their surface only. Representative measurements of three different sets of data are reported (Student's t-test, P-values < 0.05).
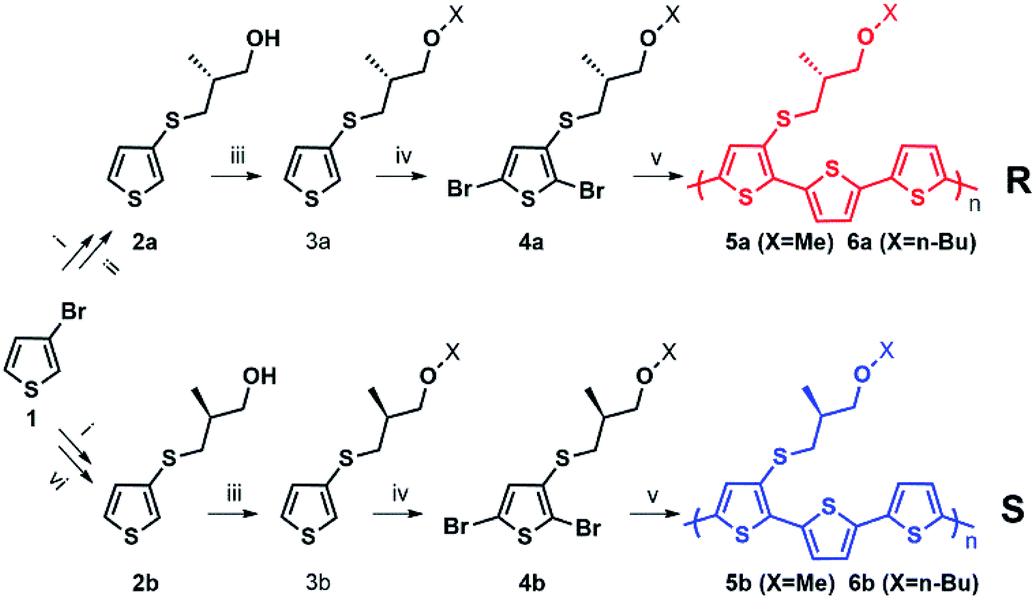 | ||
| Scheme 1 (i) BuLi (1 eq.), S8 (1 eq.), Et2O; (ii) (CH3)3COK (2 eq.), (R)-(−)-3-bromo-methyl-1-propanol (1.05 eq.) or (R)-(−)-3-bromo-n-butyl-1-propanol (1.05 eq.), EtOH; (iii) NaH (1.05 eq.), CH3I (1.5 eq.), DMF (4 mL), 0 °C, nitrogen; (iv) N-bromosuccinimide (2.2 eq.), CHCl3; (v) 5,5′-bis(tributylstannyl)-2,2′-bithiophene (1 eq.), Pd (PPh3)4 (0.08 eq.), toluene, reflux overnight; (vi) (CH3)3COK (2 eq.), (S)-(+)-3-bromo-methyl-1-propanol (1.05 eq.), EtOH or (S)-(+)-3-bromo-n-butyl-1-propanol (1.05 eq.). | ||
3. Results and discussion
3.1 Synthesis and characterization of polymers and nanoparticles
Enantiopure chiral nanoparticles were prepared starting from polymers 5a, 6a and 5b, 6b which contain the thiophene rings 3a or 3b bearing an R and S chiral centre, respectively, in the side chain. The synthetic pattern for the preparation of the polymers is shown in Scheme 1.The detailed synthesis and characterization of all compounds in Scheme 1 are reported in ESI (Pages S2–S5†). Molecular weights and thermal properties of the polymers are shown in Table S1 and Fig. S3–S5† and are in line with those already obtained for analogous compounds.33 The number of repeating units varies from 9 to 8 (i.e., 27 and 24 thiophene rings) for 5a and 5b, and from 4 to 5 (i.e. 12 and 15 thiophene rings) for 6a and 6b, respectively. All polymers are characterized by good thermal stability in air. The DSC thermograms (Fig. S3–S5†) indicate only second order thermal transitions attributable to glass transitions (Tg) with the absence of any crystallization peaks, indicative of an essentially amorphous structure in the solid state. As expected, polymers 6a and 6b, having longer side chains are more soluble in organic solvents compared to 5a and 5b. Nanoparticles were obtained from the polymers by the reprecipitation method (see Scheme S1†), as already reported for the preparation of poly(3-hexylthiophene) nanoparticles.20–22 Scanning Electron Microscopy (SEM) images of the nanoparticles are shown in Fig. S6.† Several sets of nanoparticles starting from different solution concentrations were prepared and characterized by Dynamic Light Scattering (DLS) and laser doppler electrophoresis. As an example, representative of the results obtained, the Z-potentials and the diameters of different sets of NPs obtained from solutions of 6a, 6b with different starting polymer concentrations are reported in Table 1.
| Item | Concentration (mg mL−1) | Size (nm) | Z-potential (mV) |
|---|---|---|---|
| 6a (R) | 2.68 | 90 ± 15 | −22.1 |
| 6b (S) | 0.74 | 110 ± 15 | −19.5 |
| 6a (R) | 1 | 80 ± 20 | −25.6 |
| 6b (S) | 1 | 290 ± 65 | −22.0 |
In this example all the particles show a Z-potential ranging from −20 to −26 mV, indicative of a good stability of the dispersion. Table 1 shows that there is no simple relationship between the starting solution concentration and the final diameter of the NPs. Nonetheless, NPs with comparable sizes can be obtained for both the R and S polymers by choosing appropriate polymer concentrations in order to compare the optical properties of nanoparticles having similar surface to volume ratios. In Fig. 1 the absorption and emission spectra of polymers 6a, 6b and of the corresponding nanoparticles of similar size (90 and 110 nm) are compared. Fig. 1A shows that the absorption spectrum of the S polymer is blue shifted with respect to that of the R polymer, probably due to a lower aggregation in solution, as suggested by the presence of a weak shoulder at 600 nm. The fluorescence of the two polymers reveals that their emission is composed by three main peaks: ∼600, ∼650 and ∼700 nm.
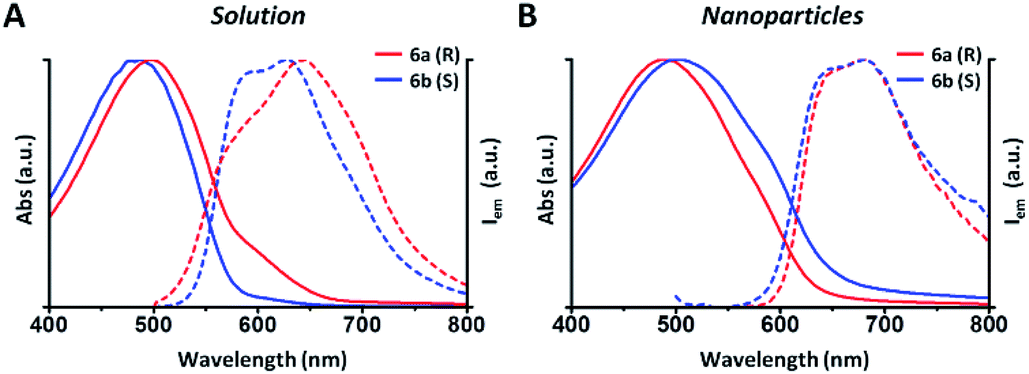 | ||
| Fig. 1 (A) Absorption (solid lines) and PL (dashed lines) of polymers 6a (R) and 6b (S) in THF solution. In blue, the spectra of S polymer (0.04 g L−1) and in red, the spectra of R polymer (0, 13 g L−1). (B) Absorption (solid lines) and PL (dashed lines) of nanoparticles of similar size (90 and 110 nm). In blue, the spectra of S nanoparticles and in red the spectra of R nanoparticles. | ||
The 700 nm peak is almost absent in the S polymer while it is more visible in the R one. On the contrary, the 600 nm peak is much stronger in the S polymer than in the R one. The difference is again indicative of lower aggregation in the S polymer probably due to the lower concentration.34 Fig. 1B shows that the NPs display similar spectra but now the R ones show a blue shifted absorption compared to that of the S ones. Moreover, if we compare the NPs spectra with the solution spectra, we observe the growth of an absorption peak at ∼580 nm, probably due to aggregation in the nanoparticles cores. Table 2 shows the quantum yield (QY) of R and S polymers 6a and 6b in THF and of the corresponding nanoparticles in water suspension.
| Sample | Quantum yield |
|---|---|
| 6a in THF solution | ∼10% |
| 6a NPs in water suspension | <1% |
| 6b in THF solution | ∼10% |
| 6b NPs in water suspension | <1% |
We notice that S and R polymer solutions have similar QY that, as expected, sensibly drop when NPs are formed, in agreement with what has already been observed for fluorescent organic nanoparticles (FONs).35 Nevertheless, despite the low quantum yield, the fluorescence intensity of the nanoparticles remains sufficiently high inside living cells (see figures below) owing to the large optical absorption of the conjugated polymers.36 Fig. 2C and D shows circular dichroism spectra of polymers 5a (R) and 5b (S) in CHCl3 solution and of the corresponding nanoparticles in water suspension. Z-potentials and the diameters of different sets of NPs are reported in Table S2.† Circular dichroism measurements show that R and S polymers in solution display a mirrored Cotton effect in the region of the π–π* transition (Fig. 2A). As shown in Fig. 2B, a similar mirror-image CD profile is also observed for the NPs in water dispersion, indicating that the chirality present in the side chains has been transferred to the conjugated chains. Analogous results were obtained for polymers 6a, 6b (Fig. S6†). Passing from the polymers in solution to the suspension of the corresponding NPs in water, two observations can be made. First, the nanoparticles of both polymers exhibit a markedly enhanced Cotton effect with respect to the polymers in solution for comparable absorption intensity and, second, the absorption and CD spectra of the NPs vary with the size of the nanoparticles. Examples of size-dependent circular dichroism of polymer nanoparticles have already been reported.37 Since the properties of polymers 5 and 6 are very similar, to study the effect of NP chirality in biological systems we performed experiments only with polymers 5a, 5b because with these polymers NPs batches of the same size were obtained.
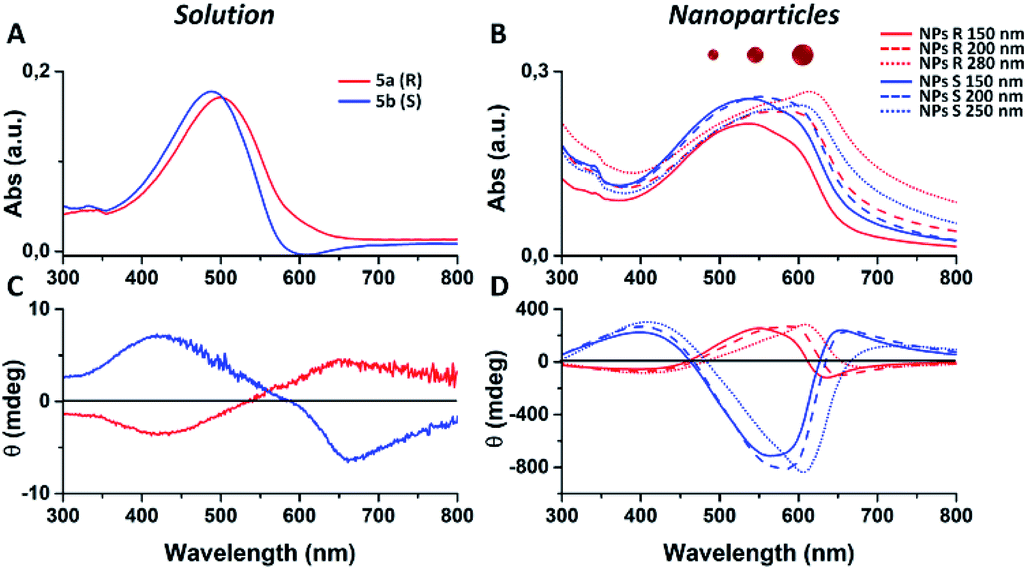 | ||
| Fig. 2 (A and C) Absorption and circular dichroism spectra of polymers 5a (R, red line) and 5b (S, blue line) in CHCl3. (B and D) Absorption and circular dichroism spectra of NPs obtained from polymers 5a (R, red lines) and from polymer 5b (S, blue lines). | ||
3.2 Cytotoxicity analysis of chiral NPs
Cytotoxicity analysis was performed using mouse fibroblast cells (NIH-3T3) and S and R nanoparticles of 150 nm, obtained from polymers 5a, 5b with or without surfactant, in solution and deposited on glass coverslips to a NPs final concentration of 0.05 mg mL−1 at different time points (24–72 hours) using a metabolic MTT assay (Fig. 3). The test item is considered non-cytotoxic if the percentage of viable cells is equal to or greater than 70% of the untreated control (CTR).38 As shown in Fig. 3A, S and R NPs, with or without surfactant (0.05 mg mL−1 TRITON-X 102), showed a similar high degree of cytocompatibility resulting in cell viability of at least 70% over all three time periods. Similar results were observed with the S and R NPs deposited on glass coverslips (Fig. 3B), but with a major viability when fibroblasts were seeded on film of R NPs compared to film of S NPs. This discrepancy can be attributed to different exposition of chiral groups, and this behaviour was confirmed by similar analysis using a film of R and S polymer deposited on glass coverslips (Fig. S8†).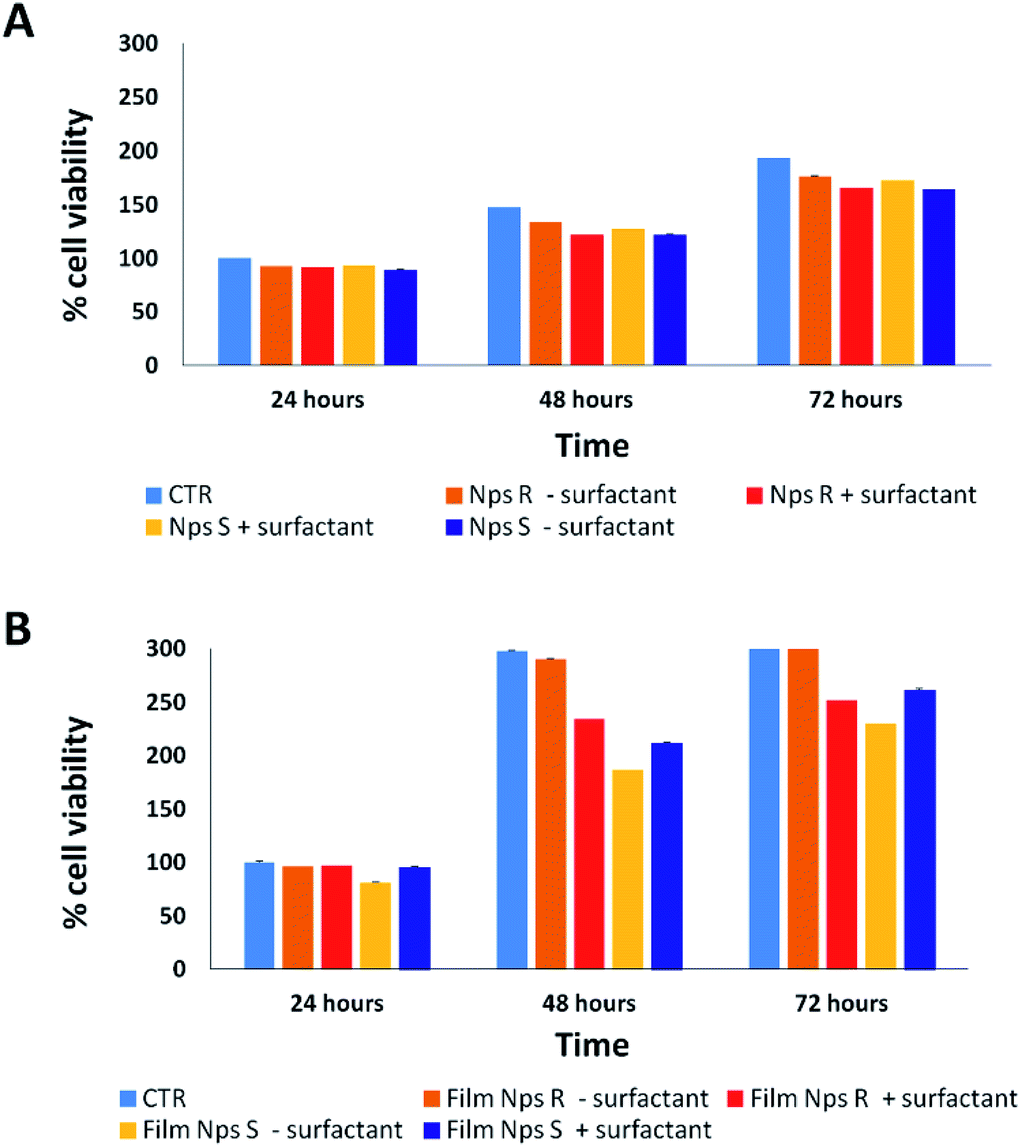 | ||
| Fig. 3 Cytotoxicity analysis on fibroblast cells by MTT assay treated for 24, 48, 72 hours in the absence (CTR) or in the presence of S and R NPs obtained from polymers 5a, 5b with or without surfactant (0.05 mg mL−1) in solution (A) and deposited glass coverslips (B) to final concentration of 0.05 mg mL−1. The test item is considered non-cytotoxic if the percentage of viable cell is equal to or greater than 70% of the untreated control. Representative measurements of three distinct sets of data have been reported (P-values of <0.05 for t-Student test). | ||
3.3 Protein corona analysis of chiral NPs
It is well known that live cells-NPs interactions depend not only on the chemical structure and the physical properties of NPs but also (when not predominantly) on the formation of NPs-protein ‘corona’ complexes due to proteins-NPs surface interactions in the biological environment.39,40 In the cell medium nanoparticle surfaces absorb biomolecules, proteins in particular, and form a new adsorbed biomolecule–nanoparticle interface which influences the cellular response. The ‘corona’ is a supramolecular dynamic layer where proteins vary in nature and concentration with time and it is this nanoparticle-protein ‘corona’ complex that drives the interaction with the cells.Chiral S and R NPs were incubated with complete cell culture medium with 10% FBS for 1 hour in agitation at 37 °C. After wash, an SDS-PAGE was performed to analyse the NPs-hard corona. The amount of hard protein corona (see Fig. 4A and B) on the surface of R NPs appeared to be higher (lane 2) compared to S NPs (lane 1). This was confirmed by the data from Bradford protein assay (Fig. 4B).
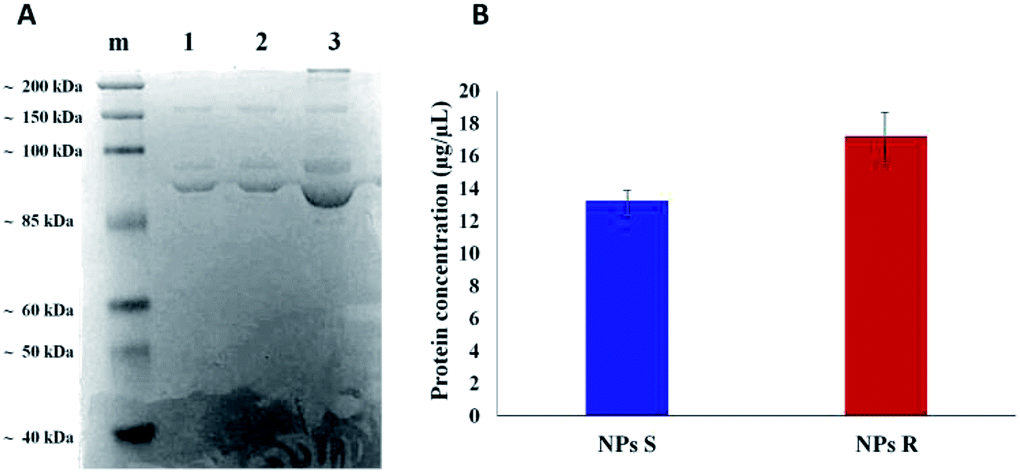 | ||
| Fig. 4 SDS-PAGE of hard protein corona on S NPs (A, lane 1), R NPs (A, lane 2) from polymers 5a, 5b, after incubation with complete culture medium (A, lane 3) for 1 hour at 37 °C; marker (lane m). Quantification of the adsorbed proteins on S and R NPs by the Bradford protein assay (B). A representative result of three independent experiments is shown (P-values < 0.05). | ||
The dissimilar protein absorption is assigned to the different chirality of NPs. Our hypothesis is that, since the proteins in the milieu are prevalently of one chirality sign, their absorption by the surface of chiral nanoparticles and the force of the interaction will depend on NPs' handedness, i.e. there will be enantioselective adsorption of proteins on the surface of chiral NPs. Thus, enantiomeric NPs will have a different ‘corona’ shell depending on their chirality sign. One of the difficulties in investigating chiral selectivity in biological systems is that the interactions generated by opposite chirality are generally small and their origin poorly understood. In the present case we do not know whether the prevalent interactions are of homo- or heterochiral type. However, what we observe is that the formation of protein corona does not flatten the chirality difference between R and S NPs but, on the contrary, enhances this difference since R NPs absorb a greater amount of proteins from the milieu. Dissimilar protein corona arrangement will influence the cellular uptake and the localization of chiral NPs in cells, as we described below.
3.4 NPs behaviour in live NIH-3T3 cells
NPs generally enter cells through one of the several pathways of endocytosis, although different mechanisms as passive diffusion (i.e. the mechanism by which molecules generally enter the cells) cannot be excluded.41,42 The internalization mechanism is guided by the NPs size and shape, surface charge and hydrophobicity/hydrophilicity ratio. NPs can be favorably internalized via caveolin/clathrin-mediated endocytosis mechanisms.43,44 In particular, NPs of size 20–40 nm are internalized by caveolin-mediated endocytosis, while NPs with variable shape and size are internalized by clathrin-mediated endocytosis mechanisms.45,46 Since the NPs we have used for uptake experiments had an average diameter of about 300 nm, we assume a clathrin-mediated endocytosis as an accepted internalization mechanism, as we previously reported for NPs of similar size.47,48 Cell uptake and intracellular fate of chiral fluorescent S and R NPs in fibroblast cell line was studied by laser scanning confocal microscopy (LSCM). The results are shown in Fig. 5.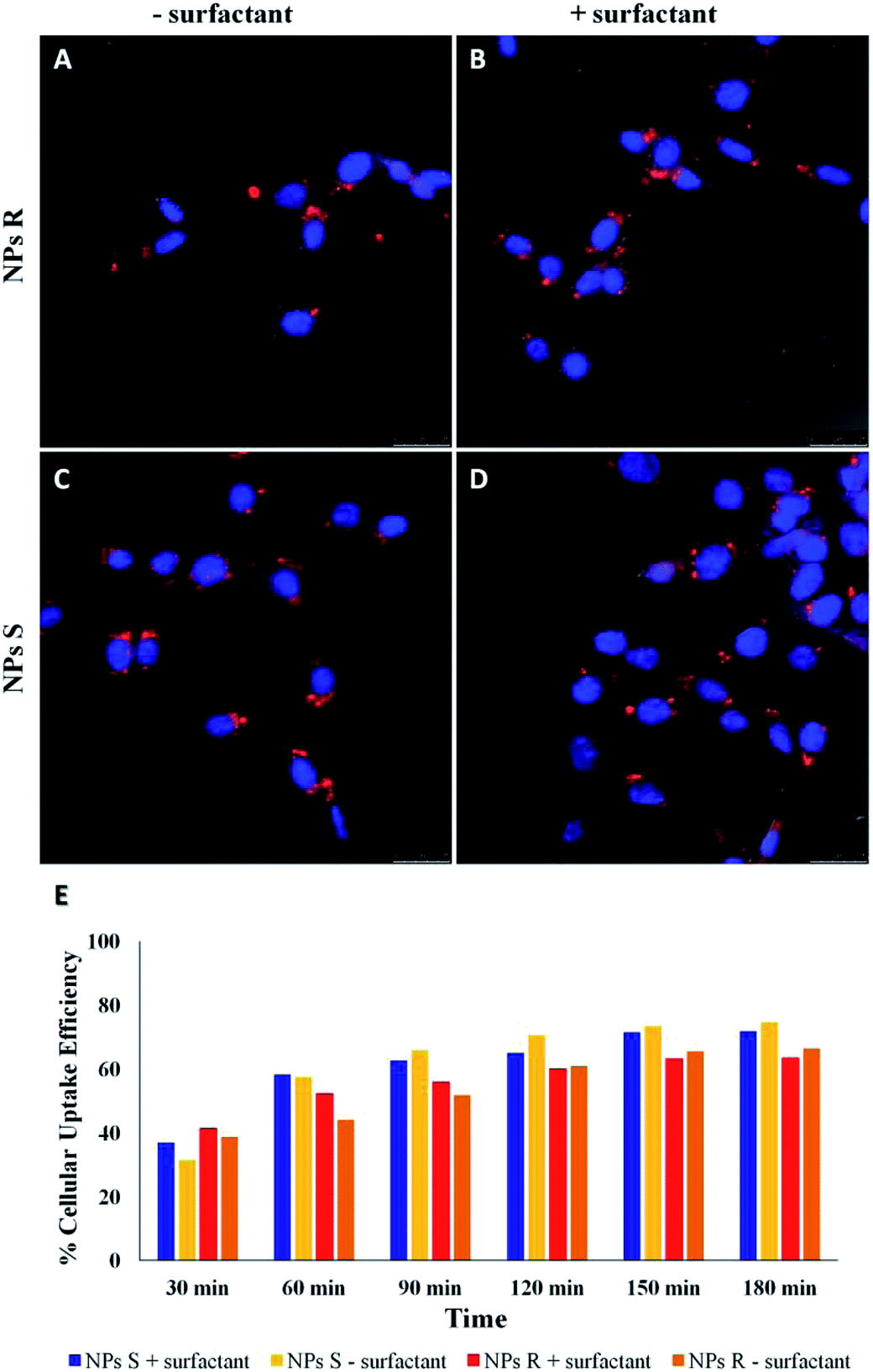 | ||
| Fig. 5 LSCM images of cellular uptake of S and R NPs from polymers 5a, 5b with and without surfactant after 24 h of incubation with fibroblasts (A–D in red). Cell nuclei were labelled with DAPI (blue). Scale bars: 25 μm. (E) Time-dependent cellular uptake efficiency of S and R NPs with and without surfactant. Representative measurements of three independent experiments have been reported (P-values < 0.05 for Student's t-test between different time points). | ||
After 24 hours of incubation, S and R NPs with and without surfactant appeared as red fluorescent spots with a predominant distribution in the cytoplasm (Fig. 5A–D). The efficiency of cellular uptake was also quantitatively evaluated by fluorimeter study after a period frame of 3 h. As shown in Fig. 5E the efficiency of cellular uptake was time dependent in all NPs formulations tested. Moreover, Fig. 5E shows that regardless of the formulation (with or without the surfactant) and the side chain length, the uptake efficiency is systematically (∼3–10%) larger for the S than for the R enantiomeric NPs. Thus, we conclude that the cells discriminate between R and S nanoparticles and the use of specific enantiomeric nanoparticles impacts cell uptake efficiency. As mentioned above, the mechanisms of biorecognition events are far from having been fully elucidated. However, very recently, a mechanism for chiral biorecognition – based on both experimental and theoretical evidence – has been proposed.49,50 Putting in a very simple way, non-bonding interactions between biomolecules cause a molecular electron reorganisation similar to that occurring upon application of an electrical field. In chiral biomolecules, such electron reorganisation causes in turn polarization of the spins related to the displaced charge. Spin polarization dictates symmetry constraints inducing enantioselectivity. Calculations indicate that homochiral interactions have a different energy from heterochiral ones and the interaction energy for two molecules of the same handedness are comparable with the available thermal energy. Consequently, following a cascade of biorecognition events inside the cells, enantioselectivity could be observed, as in the case of our chiral NPs. To get further experimental evidence on the ability of living mouse fibroblasts to discriminate between NPs of opposite chirality we have also carried out co-localization experiments using R and S NPs with surfactant. The results are shown in Fig. 6.
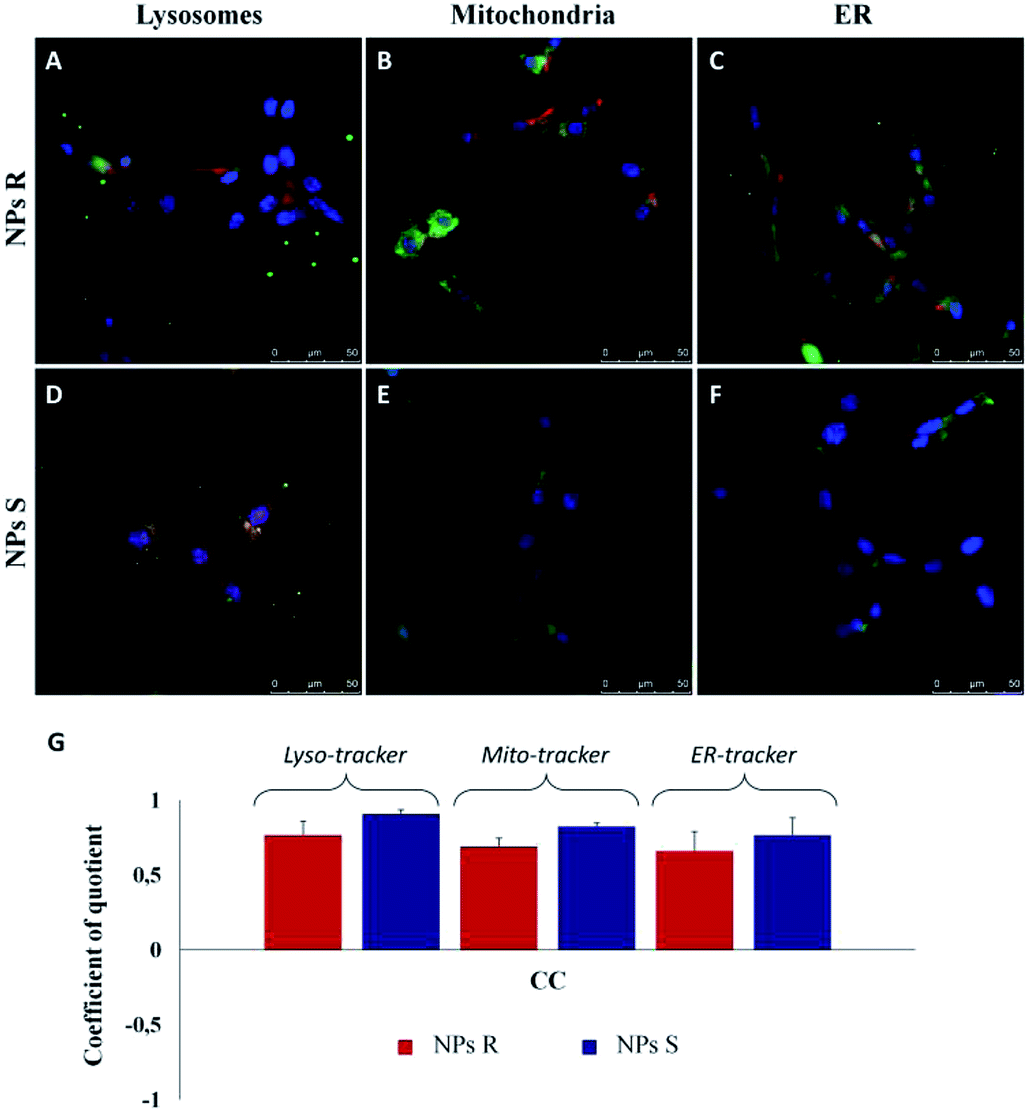 | ||
| Fig. 6 Merged LSCM images of intracellular localization of R (A–C, red) and S (D–F, red) NPs from polymers 5a, 5b with surfactant in fibroblast cells after 3 h of incubation. Cell nuclei were counterstained with DAPI (blue). Lysosomal compartment was marked with Lyso-tracker (A and D, lysosome marker, green); mitochondria were labelled with Mito-Tracker (B and E, green) and ER with ER-Tracker (C and F, green). Scale bars: 50 μm. (G) Quantitative co-localization analysis between S and R NPs with surfactant and Lyso-Tracker, Mito-Tracker and ER-Tracker for fibroblasts. For the identical LSCM images, the CC (coefficient of correlation) was calculated and plotted. Ten different fields were randomly selected for each sample and three distinct experiments were performed (P-values of <0.05 for t-Student test). | ||
In order to quantify the intracellular localization of our NPs, we extrapolated the coefficient of correlation (CC), the overlap coefficient (OC) and the intensity correlation quotient (ICQ) from LSCM images. We have found that S NPs were predominantly transported into lysosomal compartment (CC about 0.90) and mitochondria (CC around 0.82). By contrast, CC between S NPs and ER-Tracker was around 0.76 confirming a slightly lower co-localization. On the contrary, R NPs showed a low co-localization with lysosomal, mitochondria and endoplasmic reticulum (ER) compartments (CC about of 0.76, 0.68 and 0.65, respectively). Clearly, the cellular response to the R and S enantiomeric form of the nanoparticles is quite different.
3.5 Antimicrobial activity of chiral NPs
Finally, to verify the possible different behavior of chiral NPs on prokaryotic cells, such as Gram-negative E. coli DH5-Alpha and Gram-positive S. aureus bacteria strain, we have performed different antibacterial tests.51 A key difference between Gram negative and Gram positive bacteria is the thickness of the cell wall composed by peptidoglycan, in particular Gram positive present a thick layer (20–80 nm) and Gram negative a single layer (1.5–10 nm). In addition, Gram negative present an outer membrane with several pores and appendices. These differences in cellular envelope confer different responses to external stimuli.52 In Fig. 7 it is possible to note a different antibacterial activity of chiral NPs. In particular, R NPs with respect to S NPs show good antibacterial properties against Gram-negative E. coli DH5-Alpha (green box of Fig. 7), in contrast to S NPs showing good antibacterial activity against Gram-positive S. aureus (yellow box of Fig. 7). A bacterial growth reduction was evident when the E. coli DH5-Alpha strain (green box of Fig. 7) were treated with R NPs, both in function of optical density (OD) measured every 1 hour for a time window of 5 hours (Fig. 7A, green box) and when performed a CFU counting on solid LB broth as a function of NPs concentrations after an overnight (Fig. 7B, green box). On the contrary, we have observed a reduction of S. aureus growth (yellow box of Fig. 7) when treated with S NPs, both as a function of the optical density (OD) (Fig. 7A, yellow box) and when performed a CFU counting as a function of NPs concentrations after an overnight (Fig. 7B, yellow box). In addition, the antibacterial properties of S and R NPs were evaluated by agar diffusion tests performed according to standard ‘SNV 195920-1994’.31 Cotton gauzes were impregnated with S and R NPs and the antibacterial capability of the samples was evaluated. The procedure consists in placing the samples in contact with bacteria on an agar plate and evaluating the width of the free-bacteria zone around and under the sample after 24 hours incubation at 37 °C. The antibacterial activity of the material can be labelled as ‘good’ if an inhibition zone to bacterial proliferation can be observed under and close to the sample and if it results larger than 1 mm. On the other hand, the antibacterial activity of the sample can be labelled as “insufficient” if the material is totally colonized by bacteria. A ‘good’ antibacterial capability is associated to samples showing free bacteria zone under their surface only (see green and yellow of Fig. 7C–E). Gram negative E. coli DH5-Alpha samples treated with R and S NPs exhibited ‘good’ antibacterial capabilities with an inhibition zone of about 3 mm for R NPs and 1.5 mm for S NPs. In contrast, Gram-positive S. aureus samples exhibited ‘good’ antibacterial abilities with an inhibition zone of about 1 mm for R NPs and 2 mm for S NPs.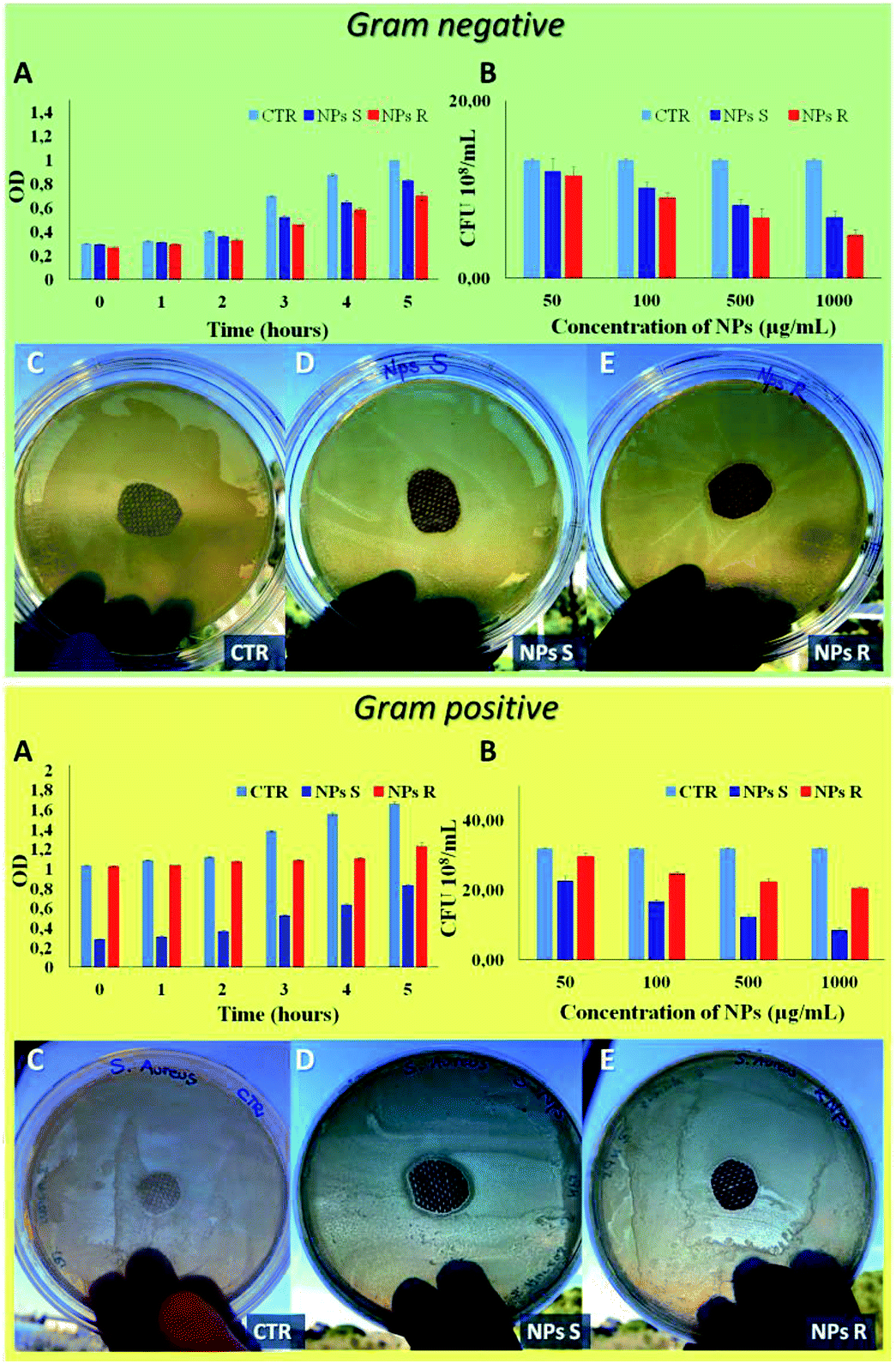 | ||
| Fig. 7 Antibacterial analysis of Gram negative E. coli DH5-Alpha (green box) and Gram positive S. aureus (yellow box). (A) Bacterial growth in function of optical density (OD) measured every 1 hour for a time window of 5 hours, treated with S and R NPs obtained from polymers 5a, 5b with surfactant (1 mg mL−1). (B) Antibacterial analysis of S and R NPs with surfactant by CFU counting on solid LB broth as a function of NPs concentrations after an overnight. Agar diffusion test on E. coli and S. aureus according standard ‘SNV 195920-1994’ (C) untreated sample used as control (CTR), (D) sample treated with S NPs with surfactant (1 mg mL−1), (E) sample treated with R NPs with surfactant (1 mg mL−1). Representative measurements of three different sets of data have been reported (Student's t-test, P-values < 0.05). | ||
4. Conclusions
In this paper we show that enantiopure thiophene based R and S nanoparticles are not toxic to living mouse fibroblast cells (NIH-3T3). By means of SDS-PAGE experiments we demonstrate that the formation of hard protein corona on the surface of chiral NPs does not flatten but, on the contrary, enhances the chirality difference between the NPs. Through cellular uptake measurements and co-localization experiments we show that the cells are able to discriminate between NPs of opposite chirality. Finally, thanks to experiments carried out on Gram negative and Gram positive bacteria we demonstrate that the enantiomeric NPs display different antibacterial activity. This is the first time that fully polymeric nanoparticles of opposite chirality are proved to interact selectively with live cells, as already observed for metallic nanoparticles coated with chiral organic molecules. It is worth noting that the NPs described here display different activity towards live cells only by virtue of the presence of a chiral R or S carbon in the side chains of the polymers. In other words, they are not functionalized with chiral groups favouring specific intracellular interactions, as in the reported cases of metallic nanoparticles. The importance of this result is related to the simplicity of functional modification inherent to polymeric NPs whose structure can be finely tuned via organic synthesis. Careful modification of the structure and the chirality of the precursor polymers could help to optimize the properties of the corresponding NPs and their enantioselectivity towards biological events inside living cells, for example in drug delivering. Similarly, since the efficiency of the uptake of chiral NPs by living cells is related to the sign of their chirality, one could target NPs where one enantiomer enters the cells and the other not at all. Note that owing to the complexity of biochemical processes, the uptake mechanism of nanoparticles by living cells and their distribution inside the cells is far from being fully understood and NPs chirality studies could help to shed light and optimize the process. Currently, nanoparticles are object of wide interest in biology and nanomedicine. We believe that the chirality of NPs could act as a filter for many properties of importance in these fields and that further and deeper studies on the enantioselective interaction of chiral NPs with living organism could make possible new applications difficult to foresee at this stage of knowledge.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by FISR project – C.N.R. ‘Tecnopolo di Nanotecnologia e Fotonica per la Medicina di Precisione’ – CUP B83B17000010001 Tecnomed. We are grateful to Dr Federica Paladini for supplying Gram positive S. aureus. F. D. M. acknowledges financial support from the Project Molecular Nanotechnologies for Human Health and Environment (PON R&C 611 2007–2013, code PON02_00563_3316357), and the UE Project INFUSION (Engineering optoelectronic interfaces: a global action intersecting fundamental concepts and technology implementation of self-organized organic materials, proposal number: 734834).References
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041 CrossRef CAS PubMed.
- J. G. Ibanez, M. E. Rincón, S. Gutierrez-Granados, M. Chahma, O. A. Jaramillo-Quintero and B. A. Frontana-Uribe, Chem. Rev., 2018, 118, 4731 CrossRef CAS PubMed.
- H. Jiang, L. Zhang and M. Liu, ChemNanoMat, 2018, 4, 720 CrossRef CAS.
- Z. Tang, Chiral Nanomaterials: Preparation, Properties and Applications, Wiley-VCH Verlag GmbH & Co. KgaA, 1st edn, 2018 Search PubMed.
- X. Shang, I. Song, H. Ohtsu, Y. H. Lee, T. Zhao, T. Kojima, J. H. Jung, M. Kawano and J. H. Oh, Adv. Mater., 2017, 29, 1605828 CrossRef PubMed.
- L. Dong, Y. Zhang, X. Duan, X. Zhu, H. Sun and J. Xu, Anal. Chem., 2017, 89, 9695 CrossRef CAS PubMed.
- K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904 CrossRef CAS PubMed.
- J. J. Giner-Casares, M. Henriksen-Lacey, M. Coronado-Puchau and L. M. Liz-Marza, Mater. Today, 2016, 19, 19 CrossRef CAS.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218 RSC.
- N. Seda Kehra and J. Joseb, Appl. Surf. Sci., 2017, 425, 432 CrossRef.
- I. V. Martynenko, V. A. Kuznetsova, I. K. Litvinov, A. O. Orlova, V. G. Maslov, A. V. Fedorov, A. Dubavik, F. Purcell-Milton, Y. K. Gun'ko and A. V. Baranov, Nanotechnology, 2016, 27, 075102 CrossRef CAS PubMed.
- J. Deng, S. Wu, M. Yao and C. Gao, Sci. Rep., 2016, 6, 31595 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Wang, R. Zhou and B. Xu, ChemNanoMat, 2017, 3, 17 CrossRef CAS PubMed.
- Q. Xin, Q. Liu, L. Geng, Q. Fang and J. R. Gong, Adv. Healthcare Mater., 2017, 6, 1601011 CrossRef PubMed.
- H. Kim, Y. J. Jin, B. S. I. Kim, T. Aoki and G. Kwak, Macromolecules, 2015, 48, 4754 CrossRef CAS.
- M. Li, C. Zhang, L. Fang, L. Shi, Z. Tang, H. Y. Lu and C. F. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 8225 CrossRef CAS PubMed.
- S. Arias, M. Nunez-Martinez, E. Quinoa, R. Riguera and F. Freire, Small, 2017, 13, 1602398 CrossRef PubMed.
- F. Di Maria, A. Zanelli, A. Liscio, A. Kovtun, E. Salatelli, R. Mazzaro, V. Morandi, G. Bergamini, A. Shaffer and S. Rozen, ACS Nano, 2017, 11, 1991 CrossRef CAS PubMed.
- L. P. Fernando, P. K. Kandel, J. Yu, J. McNeill, P. C. Ackroyd and K. A. Christensen, Biomacromolecules, 2010, 11, 2675 CrossRef CAS PubMed.
- E. Zucchetti, M. Zangoli, I. Bargigia, C. Bossio, F. Di Maria, G. Barbarella, C. D'Andrea, G. Lanzani and M. R. Antognazza, J. Mater. Chem. B, 2017, 5, 565 RSC.
- M. Zangoli, F. Di Maria, E. Zucchetti, C. Bossio, M. R. Antognazza, G. Lanzani, R. Mazzaro, F. Corticelli, M. Baroncinia and G. Barbarella, Nanoscale, 2017, 9, 9202 RSC.
- F. Di Maria, F. Lodola, E. Zucchetti, F. Benfenati and G. Lanzani, Chem. Soc. Rev., 2018, 47, 4757 RSC.
- M. M. Bournan and E. W. Meijer, Adv. Mater., 1995, 7, 385 CrossRef.
- M. Kiel, K. Duncker, C. Hagendorf and W. Widdra, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195439 CrossRef.
- K. P. R. Nilsson, J. Rydberg, L. Baltzer and O. Inganäs, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11197 CrossRef CAS PubMed.
- S. Arnaboldi, T. Benincori, R. Cirilli, S. Grecchi, L. Santagostini, F. Sannicolò and P. R. Mussini, Anal. Bioanal. Chem., 2016, 408, 7243 CrossRef CAS PubMed.
- F. Di Maria, M. Zangoli, M. Gazzano, E. Fabiano, D. Gentili, A. Zanelli, A. Fermi, G. Bergamini, D. Bonifazi, A. Perinot, M. Caironi, R. Mazzaro, V. Morandi, G. Gigli, A. Liscio and G. Barbarella, Adv. Funct. Mater., 2018, 28, 1801946 CrossRef.
- E. M. Manders, J. Stap, G. J. Brakenhoff, R. van Driel and J. A. Aten, J. Cell Sci., 1992, 103, 857 CAS.
- L. Qi, A. Lau, T. J. Morris, L. Guo, C. B. Fordyce and E. F. Stanley, J. Neurosci., 2004, 24, 4070 CrossRef PubMed.
- E. M. M. Manders, F. J. Verbeek and J. A. Aten, J. Microsc., 1993, 169, 375 CrossRef.
- Technical Committee CEN/TC 248: ISO20645:2004, Textile Fabrics - Determination of the Antibacterial Activity - Agar Diffusion Plate Test, 1994, p. 195920 Search PubMed.
- K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011 CrossRef CAS.
- B. Ferreira, P. Ferreira da Silva, J. S. Seixas de Melo, J. Pina and A. Macanita, J. Phys. Chem. B, 2012, 116, 2347 CrossRef CAS PubMed.
- C. Mastrodonato, P. Pagano, J. Daniel, M. Vaultier and M. Blanchard-Desce, Molecules, 2016, 21, 1227 CrossRef PubMed.
- M. Vezie, S. Few, I. Meager, G. Pieridou, B. Dörling, R. S. Ashraf, A. R. Goñi, H. Bronstein, I. McCulloch, S. C. Hayes, M. Campoy-Quiles and J. Nelson, Nat. Mater., 2016, 15, 746 CrossRef CAS PubMed.
- C. Dai, D. Yang, W. Zhang, B. Bao, Y. Cheng and L. Wang, Polym. Chem., 2015, 6, 3962 RSC.
- International Organization for Standardization, 2009 ISO 10993-5. Biological evaluation of medical devices-part 5: tests for in vitro cytotoxicity, ISO, Geneve, 2009, p. 34 Search PubMed.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40 CrossRef CAS.
- E. Miceli, M. Kara and M. Calderón, J. Mater. Chem. B, 2017, 5, 4393 RSC.
- S. Behzadi, V. Serpooshan, W. Taoa, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany and O. C. Farokhzad, Chem. Soc. Rev., 2017, 46, 4218 RSC.
- R. Misra and S. K. Sahoo, Eur. J. Pharm. Sci., 2010, 39, 152 CrossRef CAS PubMed.
- E. B. Garon, L. Marcu, Q. Luong, O. Tcherniantchouk, G. M. Crooks and H. P. Koeffler, Leuk. Res., 2007, 31, 643 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12 CrossRef CAS PubMed.
- M. Ehrlich, W. Boll, A. Van Oijen, R. Hariharan, K. Chandran, M. L. Nibert and T. Kirchhausen, Cell, 2004, 118, 591 CrossRef CAS PubMed.
- L. Pelkmans and A. Helenius, Traffic, 2002, 3, 311 CrossRef CAS PubMed.
- B. Cortese, S. D'Amone and I. Palamà, Pharmaceutics, 2018, 10, 52 CrossRef PubMed.
- I. E. Palamà, A. M. Coluccia and G. Gigli, Nanomedicine, 2014, 9, 2087 CrossRef PubMed.
- K. Michaeli, N. Kantor-Uriel, R. Naaman and D. H. Waldeck, Chem. Soc. Rev., 2016, 45, 6478 RSC.
- A. K. E. Capua, M. K. Kesharwani, J. M. L. Martin, E. Sitbon, D. H. Waldeckd and R. Naaman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2474 CrossRef PubMed.
- S. Löffler, H. Antypas, F. X. Choong, K. P. R. Nilsson and A. Richter-Dahlfors, Front. Chem., 2019, 7, 265 CrossRef PubMed.
- W. Vollmer, D. Blanot and M. A. De Pedro, FEMS Microbiol. Rev., 2008, 32, 149 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04782d |
| This journal is © The Royal Society of Chemistry 2019 |