
Metal–organic framework-derived heterojunctions as nanocatalysts for photocatalytic hydrogen production
Lele
Lu
,
Boyuan
Wu
,
Wei
Shi
 * and
Peng
Cheng
* and
Peng
Cheng
Key Laboratory of Advanced Energy Materials Chemistry (MOE), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: shiwei@nankai.edu.cn
First published on 30th September 2019
Abstract
Based on various well-defined components and structures, high specific surface areas and adjustable pore sizes, metal–organic frameworks (MOFs) have not only been widely used for gas sorption and separation as well as catalytic and sensing materials but have also been applied as the precursors for nanomaterials with promising applications such as in energy storage and conversion. In this review, we reported a summary of the recent progress of MOF-derived heterojunctions as nanocatalysts for photocatalytic hydrogen production from water splitting, which is organized in two main categories: (i) using the pore chemistry of MOFs to accommodate small species for MOF-derived heterojunctions; (ii) using the surface chemistry of MOFs to combine with other functional species for MOF-derived heterojunctions. The fundamentals of the rational design and synthesis of MOF-derived heterojunctions for photocatalytic hydrogen production were discussed in detail.
1 Introduction
With the intensification of the global energy dilemma, the massive burning and overuse of fossil fuels, it has been imminent to find new, green, renewable and efficient energy sources, among which hydrogen energy is considered to be an ideal alternative source in the 21st century owing to its characteristics of extensive sources, high calorific value of combustion, pollution-free nature and so on. To produce hydrogen, photocatalytic water splitting is a very eco-friendly technology with abundant water used as the raw material and the endless solar energy as the driving force.1,2In principle, the overall water splitting contains two half-reactions: the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). As an uphill reaction with a high Gibbs free energy change of 237.13 kJ mol−1, it requires photons with energy higher than 1.23 eV.3 There are two types of water splitting catalysts, i.e., homogeneous catalysts and heterogeneous catalysts, in which the semiconductor catalysts as heterogeneous catalysts have received significant attention because of their adjustable catalytic properties, high thermal stability and strong anti-toxicity.4 The photocatalytic water splitting process by semiconductor-based photocatalysts mainly consists of three steps: (i) when the photoenergy absorbed by the photocatalyst exceeds its band-gap, the electrons in the valence band (VB) can be excited to the conduction band (CB) and simultaneously leave behind positive holes. (ii) The separated charge carriers migrate to the surface of the photocatalyst. (iii) The protons of water molecules accept photo-generated electrons and are reduced to hydrogen; meanwhile, the holes and oxygen atoms of the water molecules combine, causing the production of oxygen (Fig. 1).5 For these two half-reactions, the bottom potential of CB is required to be more negative than the redox potential of the H+/H2 energy level (0 V vs. NHE, pH = 0), while the top potential of VB must be higher than the oxidation potential of the O2/H2O energy level (1.23 V vs. NHE, pH = 0). As a result, the minimum band-gap required for photocatalytic water splitting is 1.23 eV.6 Furthermore, it should be noted that the potential positions of CB and VB of a photocatalyst determine the ability of reduction and oxidation; thus, an appropriate band structure in well-designed semiconductors is the key for the better performance of the photocatalytic system.
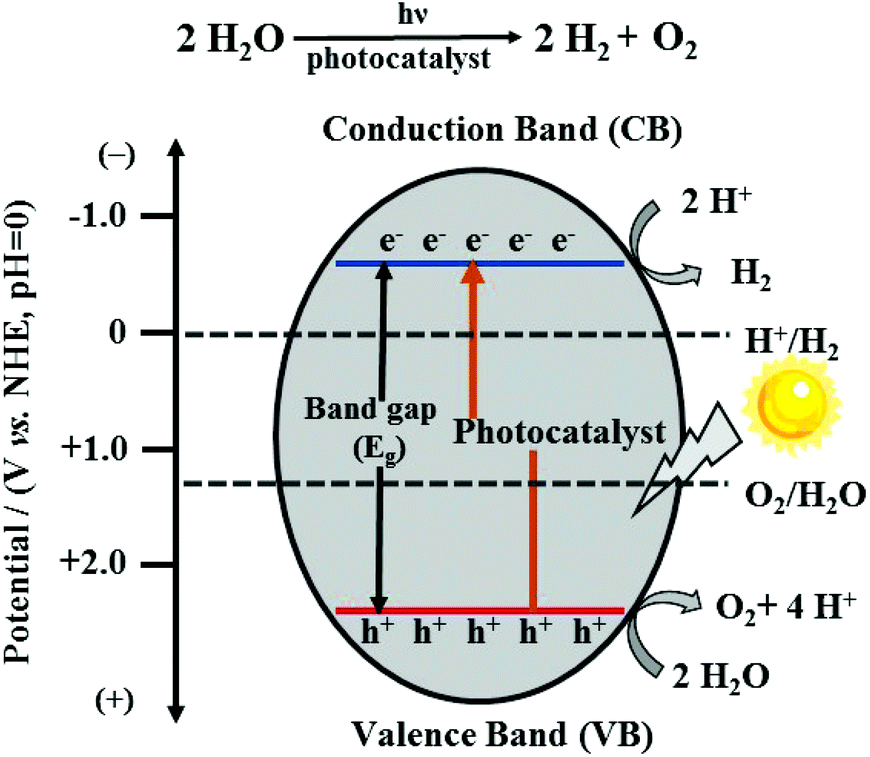 | ||
| Fig. 1 The basic principle of semiconductor-based photocatalyst for water splitting. | ||
However, because of the inefficiency of light utilization, the lack of a suitable energy-band structure and the rapid recombination of photo-generated electron–hole pairs, it is still vital to obtain highly effective nanocatalysts for water splitting under sunlight, which demands a deeper understanding of the fundamentals for the rational design and synthesis of high-performance water splitting nanocatalysts.
In a practical system, semiconductor photocatalysts,7 such as TiO2,8 ZnO,9 CdS,10 and g-C3N4,11,12 have greatly attracted researchers’ attention for their catalytic properties. Nevertheless, the efficiency of semiconductor photocatalysts remains low because of weak light harvesting properties, which are mostly in the ultraviolet region, and also the fast electron–hole recombination rate.13 To solve these problems, the construction of a heterojunction by the combination of two semiconductors is a promising way. Heterojunctions can be divided into conventional heterojunctions (Type-I,14 Type-II,15 and Type-III16), p–n heterojunctions17 and Z-scheme heterojunctions,18 as shown in Fig. 2.19,20 However, the synthesis of these heterojunctions usually needs complicated processes as well as fine-tuning of the compositions and morphologies of these nanomaterials to optimize the catalytic properties, which is highly challenging.21 Under these circumstances, the development of new synthetic methods for high-performance heterojunction-based photocatalysts for water splitting is desired.
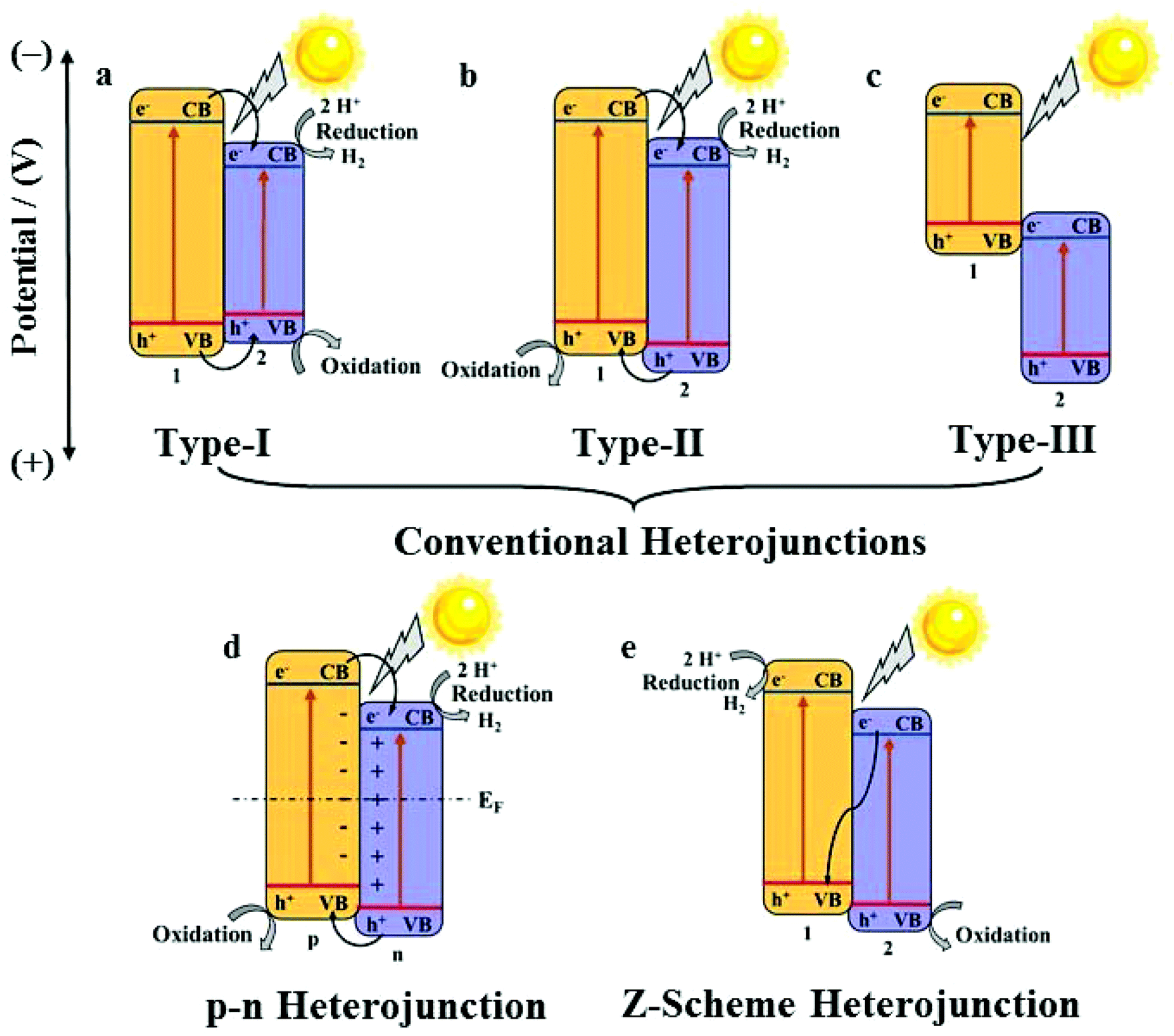 | ||
| Fig. 2 Diagrams of Type-I (a); Type-II (b); Type-III (c), p–n heterojunction (d), Z-scheme (e) heterojunction. 1, 2, p, and n refer to semiconductor 1, semiconductor 2, p-semiconductor, and n-semiconductor, respectively. | ||
Metal–organic frameworks (MOFs), as a set of crystalline porous materials fabricated by metal nodes and organic linkers, have shown great potentials in numerous fields such as gas sorption and separation, catalysis, and sensors due to their varied and customizable structures, size/shape adjustable nanopores or nanochannels and high surface areas.22,23 As water splitting catalysts, MOFs are used as catalysts directly24 or as precursors for nanocatalysts.25 For example, in 2009, Mori et al. reported the first example of MOF, namely, [Ru2(1,4-BDC)2]n (1,4-BDC = 1,4-benzene-dicarboxylic acid) as a photocatalyst for HER under the irradiation of visible light.26 By the calcination of the nanoscale MIL-101 octahedral particles coated with a titanium dioxide shell, a core–shell nanostructure of Fe2O3@TiO2 was obtained for HER by Lin et al.27 A timeline depicting the milestones of MOF-derived heterojunctions as the nanocatalysts for photocatalytic hydrogen production is illustrated in Fig. 3.27–36 Because of the unique structures and morphologies of the nanocatalysts derived from MOFs, the catalytic performances of these nanomaterials have been significantly improved.37 In the literature, reviews of the nanomaterials derived from MOFs have been summarized.38–40 In 2018, Xu et al. summarized MOF-based nanocatalysts for hydrogen production from water and for batteries.41,42 Tang et al. summarized MOF-based heterogeneous catalysts for CO2 conversion.43 Wu et al. summarized MOFs and their derived materials as electro- or photo-catalysts for CO2 reduction.44 In this review, we summarized the developing strategy of using the pore chemistry and surface chemistry of MOFs to make MOF-based composites and then via the pyrolysis of the composites to obtain heterojunctions as nanocatalysts for photocatalytic hydrogen production from water splitting. The advantages of this strategy and the applications of these photocatalysts were discussed.
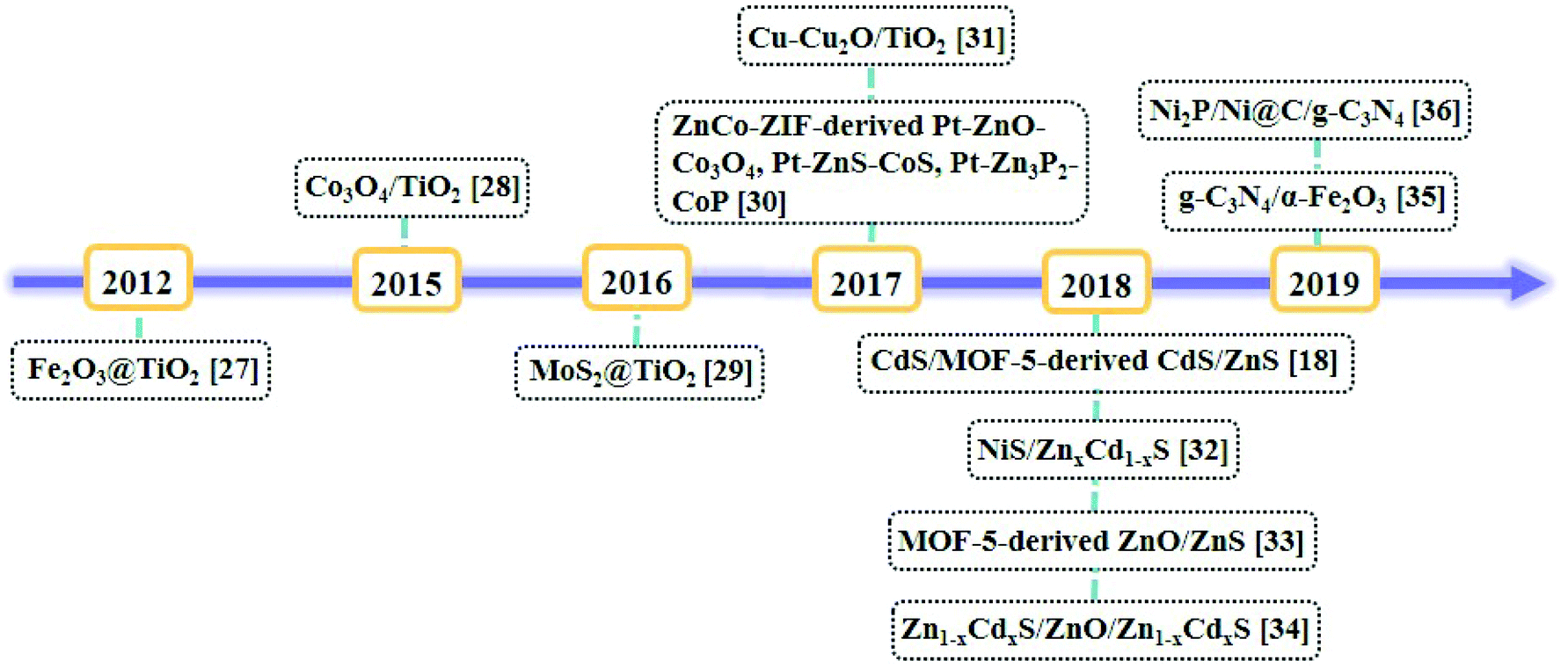 | ||
| Fig. 3 Timeline of the milestones of MOF-derived heterojunctions as nanocatalysts for photocatalytic hydrogen production. | ||
2. Mechanism of heterojunctions for photocatalytic hydrogen production
A heterojunction generally refers to the interfacial region formed by two different semiconductors, satisfying lattice matching and thermal matching; it is considered to be one of the most applicable photocatalysts for water splitting.19 Because of the differences in band structures and material properties, when a heterojunction is formed, an interfacial potential difference will also be created, leading to the generation of a built-in electric field. Under the influence of this electric field, the photo-generated electrons and holes will migrate rapidly, promoting the separation of electron–hole pairs and effectively suppressing recombination.The energy-band structure of a Type-I heterojunction is defined by the VB and CB of semiconductor 2 located in the forbidden band range of semiconductor 1. The CB potential of semiconductor 1 is more negative than that of semiconductor 2; thus, the photoelectrons on semiconductor 1 can easily pass on to the CB of semiconductor 2. Meanwhile, the holes will also migrate from the VB of semiconductor 1 to the VB of semiconductor 2 (Fig. 2a).
In a Type-II heterojunction, both the CB and VB potentials of semi-conductor 1 are more negative than that of semiconductor 2. This band structure facilitates the transport of electrons from the CB of semiconductor 1 to the CB of semiconductor 2, and the holes from the VB of semiconductor 2 migrate to the VB of semi-conductor 1 simultaneously (Fig. 2b).
The CB and VB of a type-III heterojunction have similarities to those of a type-II heterojunction except the extremely staggered gap, leading to the mismatch of the band-gaps. Hence, the electron–hole separation and migration between the two semiconductors cannot take place, making it unfavourable for the separation of the electron–hole pairs for photocatalytic applications (Fig. 2c).
For a p–n heterojunction, due to the different concentrations of the electrons and holes in the two semiconductors, the reversible diffusion of the electrons and holes occurs at the interface of the p–n heterojunction; this results in the formation of a spatial charge region that creates a built-in electric field from the n-region to the p-region, guiding the electrons to quickly move from the p-type semiconductor to the n-type semiconductor. The holes also rapidly migrate backwards, effectively implementing the spatial separation of the electron–hole pairs (Fig. 2d).
The Z-scheme heterojunction was first proposed in 1979.45 Its unique energy structure and charge transport mode allow electrons and holes to be spatially segregated, and the recombination rate of the electrons and holes is low in the photocatalytic process. As shown for the band structure in Fig. 2e, the electrons on the CB of semiconductor 2 can rapidly migrate to the VB of semiconductor 1 and recombine with holes, thereby ensuring that the holes on the VB of semiconductor 2 and the electrons on the CB of semiconductor 1 do not recombine easily, hence increasing the catalytic efficiency significantly.
The construction of a heterojunction by the combination of two semiconductors can notably broaden the light-harvesting range, enhance the spatial separation and moreover suppress the recombination of electron–hole pairs; thus, attention should be paid to each type of a heterojunction. However, the specific band structure and obviously different charge migration modes have made the Z-scheme photocatalyst the most promising system.46 In addition, the p–n heterojunction requires p- and n-type semiconductors, which has distinct limitations in semiconductor selection.
3. MOFs for MOF-derived nanocatalysts
In the past few decades, MOFs have been developed very rapidly in most of the fields in chemistry and materials science due to their powerful pore chemistry. Nevertheless, MOFs are less documented in the study of photocatalysis on account of the relatively poor stability and low electronic conductivity.47 However, recent studies have shown that some MOFs can be excellent precursors for the synthesis of heterojunction-based nanocatalysts, exhibiting a high photocatalytic HER performance due to the following features: (i) the compositions and the ordered framework structures of MOFs can not only be the sources of metal compounds of the heterojunctions but also the in situ generated allotropes of carbon to enhance the electrical conductivity; (ii) the porosity of MOFs permits small species to be quantificationally accommodated in the pores or channels of MOF-based composites with well-defined compositions as precursors; (iii) the surfaces of the nanoparticles of MOFs can be fine-tuned by structural modifications to be linked with specific species to form MOF-based composites with well-defined compositions as precursors (Fig. 4).22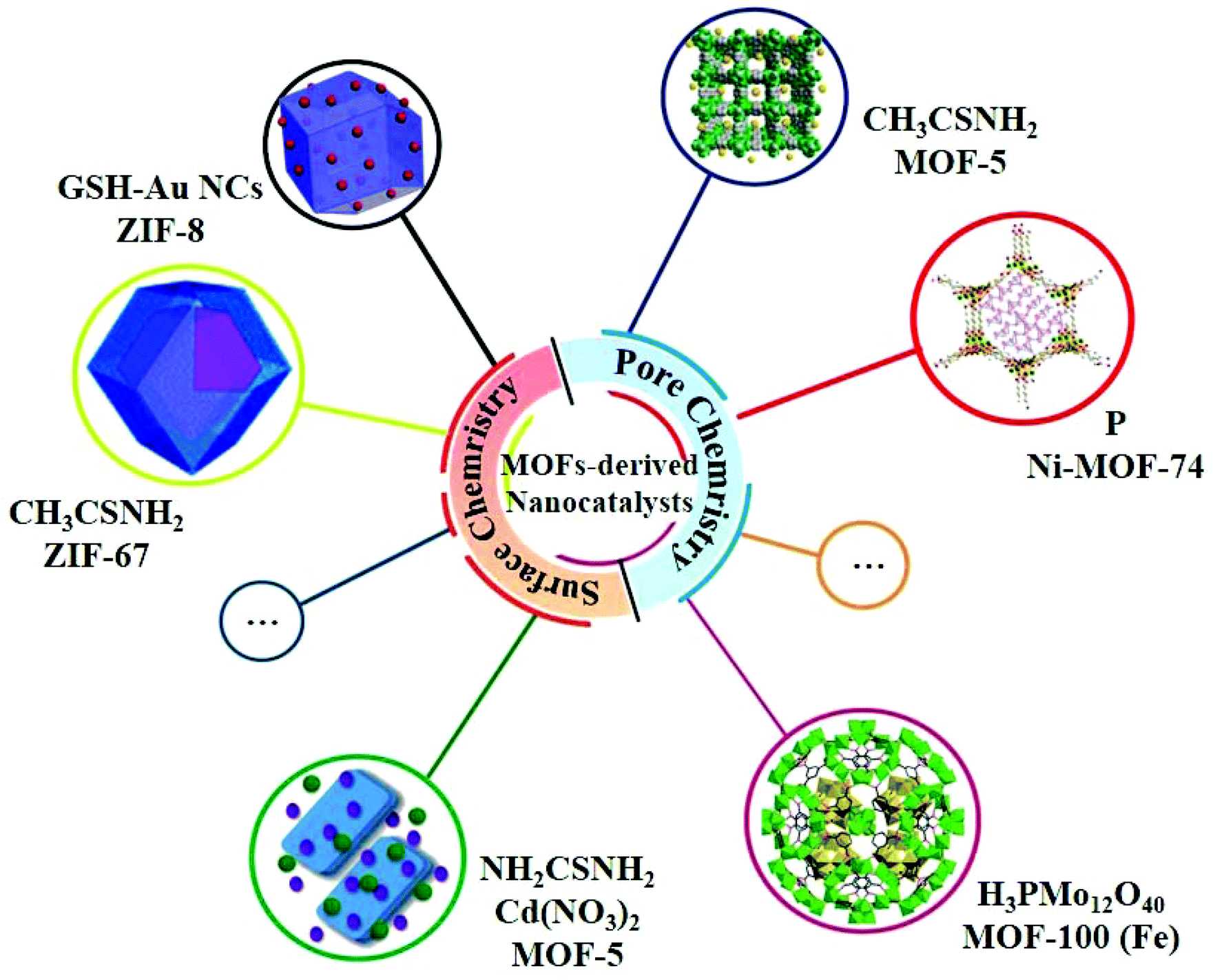 | ||
| Fig. 4 Selected examples of MOFs for MOF-derived nanocatalysts using the pore chemistry and surface chemistry of MOFs. | ||
To use these features of MOFs for the heterojunction-based nanocatalysts, the synthesis methods of the selected MOFs should be facile and performed on a large scale. Organic bridging ligands are usually the costliest part of the total cost of MOFs; well-used and relatively cheap ligands such as 1,4-benzenedicarboxylate (H2BDC), 1,3,5-benzenetricarboxylic acid and 2-methylimidazole (2-mIM) were selected for discussing in detail in the following section.32,35,48
4. MOF-derived nanocatalysts for photocatalytic hydrogen production
Pyrolysis of the well-designed composites is a useful way for the synthesis of nanocatalysts.49 For the heterojunction-based nanocatalysts, due to the relatively complex compositions, it is highly challenging to combine different compositions uniformly and control the morphology of the products by calcination. Based on the well-defined framework components, long-range ordered structures, and varied pore and surface chemistry of MOFs, the chemical compositions of the MOF-based precursors can be well adjusted at the molecular level, which is beneficial for the chemical fine-tuning of the compositions and morphology of the nanocatalysts.4.1 Using pore chemistry of MOFs for MOF-derived nanocatalysts
The pores or channels of MOFs can accommodate specific small species and hence have been well studied for gas storage and separation, catalysis, and so on.50 For chemistry, the nano-sized pores or channels of MOFs can also be used to combine with specific species, such as thioacetamide,32,33 phosphorus51 and H3PMo12O40,52 to produce well-defined composites followed by pyrolysis to generate nanomaterials. Very recently, this strategy was successfully applied to synthesize nanocatalysts for photocatalytic HER (Table 1).| Photocatalyst | Template | Light | Sacrificial agent | Cocatalyst/sensitizer | Catalyst dose (mg) | Activity (μmol g−1 h−1) | Stability/AQE | Ref. |
|---|---|---|---|---|---|---|---|---|
| CdS@Cd(II)-MOF@TiO2 (7) | Cd-MOF | Visible light (λ > 400 nm) | 0.025 M Na2SO3/0.1 M Na2S | Pt | 20 | 1416 | 2.05% at 420 nm | 54 |
| CdS@Cd(II)-MOF@TiO2 (8) | 1604 | 2.31% at 420 nm | ||||||
| Pt-ZnO-Co3O4 | ZnCo-ZIF | UV-Vis light | 10% methanol | Pt | 100 | 7800 | 5 times recycled | 30 |
| Pt-ZnS-CoS | 8210 | 30 h | ||||||
| Pt-Zn3P2-CoP | 9150 | |||||||
| Co-Doped Zn0.5Cd0.5S | ZnCo-ZIF | Visible light (λ > 420 nm) | 0.35 M Na2S/0.25 M Na2SO3 | — | 5 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 360 360 |
6 times recycled | 55 |
| 30 h | ||||||||
| 39.2% at 420 nm | ||||||||
| 30 wt% CdS/ZnxCo3−xO4 | ZnCo-ZIF | Visible light (λ > 420 nm) | 5 mL lactic acid | — | 10 | 3978.6 | 4 times recycled | 56 |
| 16 h | ||||||||
| NiS/Zn0.5Cd0.5S | Ni-ZnxCd1−x-MOF | Visible light (λ > 420 nm) | 35 mM Na2S/25 mM Na2SO3 | — | 50 | 16780 |
5 times recycled | 32 |
| 20 h | ||||||||
| Zn0.5Cd0.5S/ZnO/Zn0.5Cd0.5S cages | ZIF-8 | 300 W Xe lamp | 0.75 M Na2S/1.05 M Na2SO3 | — | 20 | 28600 |
6 times recycled | 34 |
| 30 h | ||||||||
| ZnO/ZnS-30 | MOF-5 | Visible light (λ > 420 nm) | 10 mM Na2S/Na2SO3 | — | 50 | 435 | 8 times recycled | 33 |
| 40 h | ||||||||
| NiS/CdS/h-TiO2 | NH2-MIL-125 | Visible light (λ > 420 nm) | 0.35 M Na2S/0.25 M Na2SO3 | — | 50 | 2149.15 | 4 times recycled | 53 |
| 16 h |
Using newly developed bimetallic ZnCo-ZIF as template, Pt-ZnO-Co3O4, Pt-ZnS-CoS, and Pt-Zn3P2-CoP photocatalysts were prepared through the oxidation, sulfurization, and phosphorization of ZnCo-ZIF with Pt doping.30 More importantly, suitable band matching and strong electron coupling were achieved in these heterojunctions, greatly facilitating effective electron–hole separation and transmission. Moreover, the Pt nanoparticles dispersed on porous heterojunctions as electron traps can provide abundant redox active sites for HER. Correspondingly, the rates of HER for three photocatalysts were found to be 7800, 8210, and 9150 μmol g−1 h−1, which were higher than those of the respective monometallic ZIF-8 or ZIF-67-based derivatives.
In 2018, a non-noble metal-based cocatalyst/solid solution heterojunction of NiS/ZnxCd1−xS with superior photocatalytic HER activity was reported by us (Fig. 5a).32 The chemical composition and band-gap of ZnxCd1−xS were well-tuned, and the light capturing capacity and photocatalytic activity were also optimized by regulating the doped Zn2+ in the precursor of Cd-MOF (Fig. 5b–e). As a cocatalyst, NiS played a crucial role in further improving the catalytic activity of the heterojunction. NiS/Zn0.5Cd0.5S exhibited an optimum HER rate (16780 μmol g−1 h−1) and retained excellent stability and recyclability under visible-light irradiation (Fig. 5f–i) because of its high light absorption, suitable band gap as well as the position of CB and structural stability.
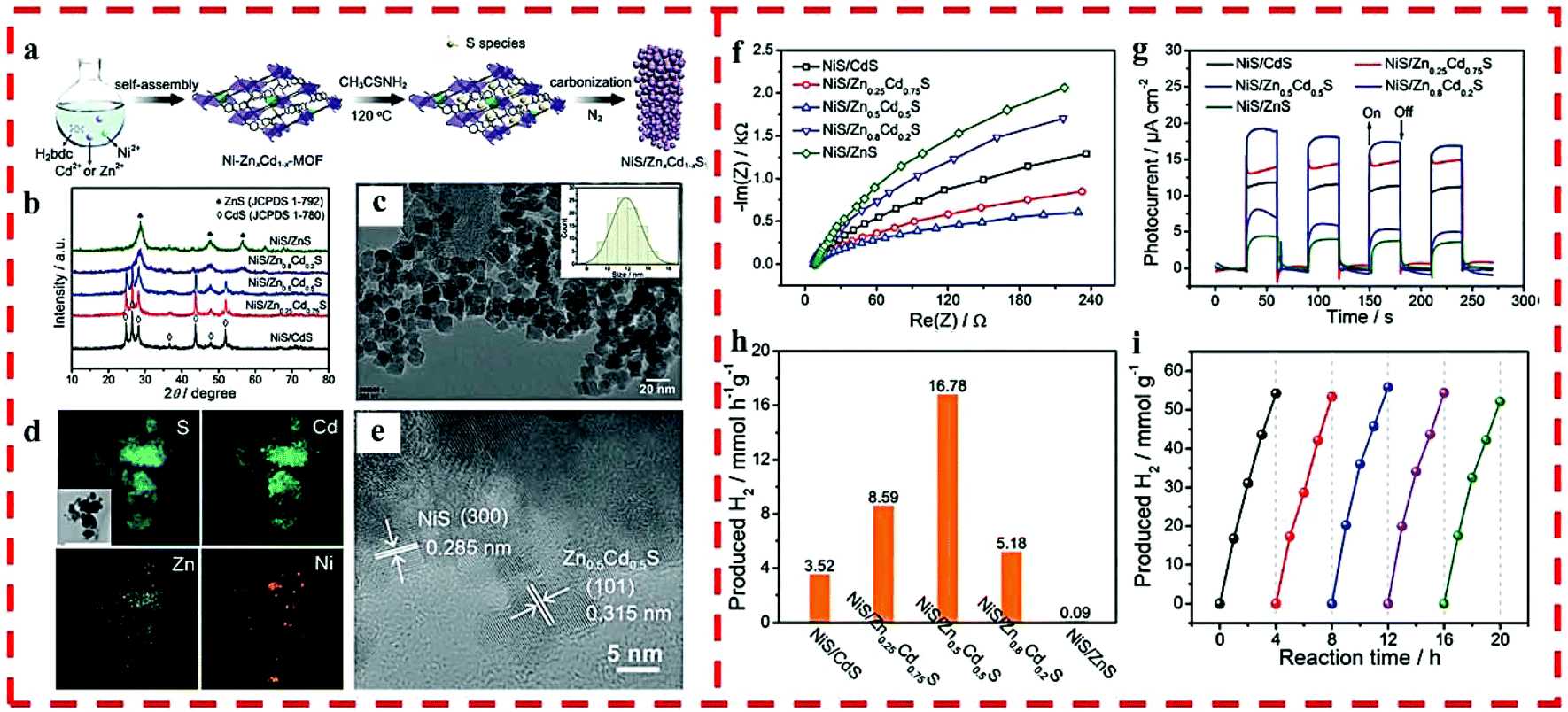 | ||
| Fig. 5 (a) The synthetic route of NiS/ZnxCd1−xS. (b) PXRD of NiS/ZnxCd1−xS. (c) TEM and particle size distribution (inset), (d) elemental mapping and (e) HRTEM of NiS/Zn0.5Cd0.5S. (f) EIS and (g) time-dependent photocurrent of NiS/ZnxCd1−xS. (h) Photocatalytic HER rates of NiS/ZnxCd1−xS under visible light. (i) HER cycling test for NiS/Zn0.5Cd0.5S under visible light. Reprinted with permission from ref. 32. | ||
A set of ZnO/ZnS nanostructured heterojunctions was fabricated for photocatalytic HER under visible light by coupling solvothermal synthesis with MOF-5 as the host and thioacetamide (TAA) as the sulfur source via two-step calcination in different atmospheres.33 In the heterojunctions, ZnO and ZnS were evenly integrated due to the homogeneous sulfidation of the MOF-5 precursor at the molecular level and then the oxidization of ZnS@C (Fig. 6a–g). This facile synthesis method increased the interfacial catalytic active sites and the light absorption capability as well as charge separation efficiency by optimizing the calcination time of ZnS@C in air, thus obtaining optimized ZnOS-30, which possessed excellent photocatalytic HER activity (435 μmol g−1 h−1) and recyclability without any co-catalyst (Fig. 6h–k).
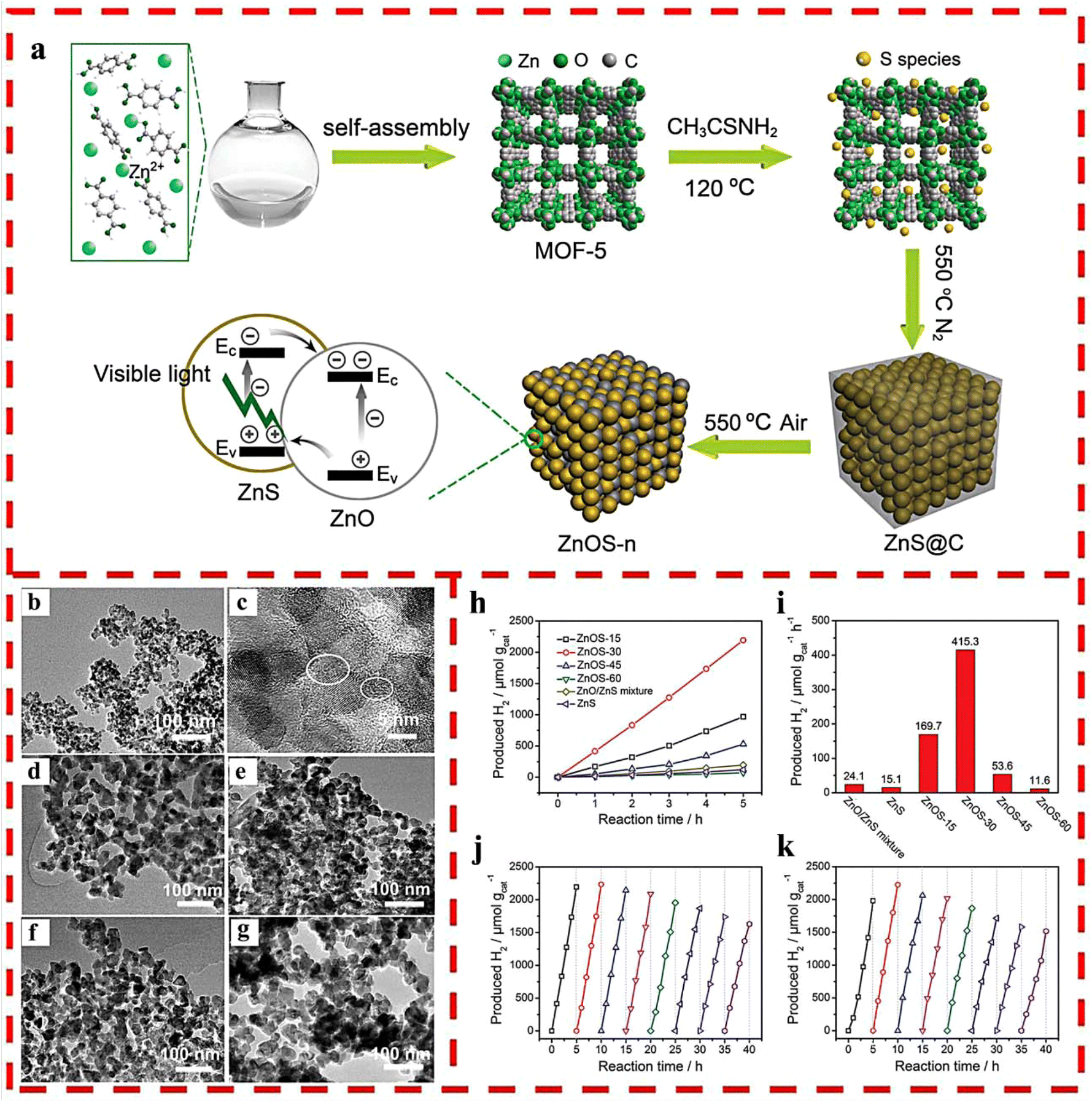 | ||
| Fig. 6 (a) The synthetic route of ZnO/ZnS heterostructures. (b) TEM and (c) HRTEM images of ZnS@C. TEM images of (d) ZnOS-15; (e) ZnOS-30; (f) ZnOS-45 and (g) ZnOS-60. (h) Time-dependent photocatalytic HER under visible-light irradiation of ZnOS-n. (i) Comparison of photocatalytic HER of ZnS/ZnO mixture, ZnS and ZnOS-n under visible-light irradiation. (j) Cycling test of HER for ZnOS-30 under visible-light irradiation. (k) Cycling test of HER for recycled and reprocessed ZnOS-30 after 40 h cycling test. Reprinted with permission from ref. 33. | ||
Sandwich-shelled ZnCdS/ZnO/ZnCdS cages were synthesized based on ZIF-8.34 ZnS and ZnO were produced concurrently during sulfidation by TAA, in which ZnS acted as an obstruction to centralize the ZnO particles filling up the spaces between the adjacent ZnS layers. For photocatalysis, ZnCdS resulting from the cation-exchanged ZnS and ZnO can be more advantageous for the formation of staggered band structures by fine-tuning the composition. Benefiting from the enhanced light utilization and high surface area, the Zn0.5Cd0.5S/ZnO/Zn0.5Cd0.5S cage showed an HER rate of 28600 μmol g−1 h−1 and long-time steadiness, resulting in supreme activities among the ZnCdS and ZnO families.
In 2019, Zhou et al. reported non-noble metal-containing hollow CdS/TiO2 (CdS/h-TiO2) decorated with an NiS cocatalyst nanohybrid photocatalyst obtained by the use of NH2-MIL-125 as the MOF template.53 It presented no prominent impact on the morphology of the composites after loading few NiS species. The final material not only maintained the original framework of pristine MOF, but also formed hollow morphology. Owing to the porous structure, the as-prepared composite exhibited enhanced photocatalytic HER efficiency under visible-light irradiation in contrast to TiO2, CdS/TiO2, or even CdS/TiO2 with Pt as the cocatalyast. By regulating the weight ratio of CdS and NiS, the obtained NiS/CdS/h-TiO2 photocatalyst with optimized amounts of 30% CdS and 0.3% NiS showed the highest photocatalytic HER activity of 2149.15 μmol g−1 h−1 due to the synergistic effect between the heterojunctions for quick charge separation and accelerated surface redox reaction by the NiS cocatalyst.
Using the pores or channels of MOFs to accommodate specific small species for photocatalytic water splitting is developing rapidly in the last few years because of the size and structure of the pores or channels and compositional diversities of MOFs. Further efforts should be put for developing high-efficiency and well-defined heterojunction-based photocatalysts that cannot be easily constructed from a direct reaction of the two semiconductor components from the well-matching of the pores or channels of the MOFs and guest species.
4.2. Using surface chemistry of MOFs for MOF-derived nanocatalysts
Besides, for the pore chemistry of MOFs, the surface of the nanoparticles of MOFs can also have affinity for specific species to produce MOF-based nanocomposites. The pyrolysis of the well-defined nanocomposites can generate heterojunction-based nanocatalysts for photocatalytic HER (Table 2).| Photocatalyst | Template | Light | Sacrificial agent | Cocatalyst/sensitizer | Catalyst dose (mg) | Activity (μmol g−1 h−1) | Stability/AQE | Ref. |
|---|---|---|---|---|---|---|---|---|
| Fe2O3@TiO2/Pt | MIL-101(Fe) | Visible light (λ > 420 nm) | 20/1 v/v H2O/TEOA | Pt | 0.5 | 625 | 3 times recycled | 27 |
| 48 h | ||||||||
| Hollow α-Fe2O3-TiO2-PtOx | MIL-88B | Visible light (λ > 420 nm) | Lactic acid | — | 20 | 1100 | 5 times recycled | 58 |
| 25 h | ||||||||
| NC-PA 2 wt%Co3O4/TiO2 | Co-MOF | 400 W xenon arc lamp | 15 vol% CH3OH | — | 20 | 6400 | 2.4% at 420 nm | 28 |
| CdS/2 wt% MCTMPs | ZIF-67 | Simulated solar light | 3 mL lactic acid | — | 1 | 136770 |
5 times recycled | 57 |
| 60 h | ||||||||
| 40.6% at 425 nm | ||||||||
| 14.6 wt% MoS2@TiO2 | NH2-MIL-125(Ti) | Visible light (λ ≥ 420 nm) | TEOA | Fluorescein | 5 | 10046 |
3 times recycled | 29 |
| 30 h | ||||||||
| 1 wt% Cu/TiO2-400 | MOF-199 | UV-light | 5 vol% glycerol | — | 2.5 | 15130 |
4 times recycled | 31 |
| 16 h | ||||||||
| 20 wt% CdS/Co-C@Co9S8 | ZIF-67 | Simulated solar light | 3 mL lactic acid | — | 1 | 26690 |
4 times recycled | 59 |
| 20 h | ||||||||
| 7.82% at 425 nm | ||||||||
| CdS/MOF-5 (25) | MOF-5 | Visible light (λ ≥ 420 nm) | 0.35 M Na2S/0.25 M Na2SO3 | — | 50 | 11620 |
4 times recycled | 18 |
| 16 h | ||||||||
| 11.09% at 470 nm | ||||||||
| g-C3N4/MOF/MoS2 (20%) | ZIF-67 | 5 W LED white light | 15% TEOA | 10 mg Eosin Y | 20 | 4012.5 | 3 times recycled | 60 |
| 12 h | ||||||||
| 5.04% at 425 nm | ||||||||
| Ni-MOF-74/CdS/Co3O4 (10 wt% Co) | Ni-MOF-74 | M300 xenon lamp | 10% lactic acid | — | 20 | 5810 | 4 times recycled | 61 |
| 20 h | ||||||||
| 14.7% at 475 nm | ||||||||
| Core–shell CdS/MoS2-0.6 | Cd-MOF (Cd-ZIF-8) | 300 W Xe lamp | 35 mM Na2S/18 mM Na2SO3 | — | 30 | 5587 | 2 times recycled | 62 |
| Mixed MoS2-CdS | 91 | — | ||||||
| CdS/MoS2-DP hybrid | 2102 | |||||||
| g-C3N4/α-Fe2O3-2 | MIL-100(Fe) | Visible light (λ > 420 nm) | 10 vol% TEOA | 3 wt% Pt | 10 | 2066.2 | 5 times recycled | 35 |
| 20 h | ||||||||
| 7.43% at 420 nm | ||||||||
| Ni2P/Ni@C/g-C3N4-550 | Ni-MOF | Visible light (λ ≥ 420 nm) | 10% TEOA | 1.0 mM Eosin Y | 10 | 18040 |
5 times recycled | 36 |
| 25 h | ||||||||
| 58.1% at 420 nm | ||||||||
| — | 210 | — | ||||||
| TiO2-Ti3C2-CoSx | TiO2-ZIF-67 | UV-Vis light | 10 mL methanol | — | 20 | 950 | 5 times recycled | 63 |
| 15 h |
For photocatalytic HER, an ideal system of Z-scheme heterojunctions can separate the photo-induced charges for exceedingly stronger catalytic ability. Wang et al. adopted a technique to synthesize Z-scheme CdS/ZnS heterojunctions with Zn vacancies derived from CdS/MOF-5, with a diverse loading weight of CdS by a one-step hydrothermal method, in which MOF-5 could be directly converted into ZnS with a large number of zinc vacancy defects in the aqueous solution with Na2S and Na2SO3 as the sacrificial agents at an ambient temperature.18 Its two-photon absorption behavior could be realized by the zinc vacancies of ZnS under visible-light irradiation. The interfacial charge transfer of CdS/ZnS transformed from a traditional type-I heterojunction to Z-scheme because of the ohmic contact caused by zinc vacancy defects. The maximum photocatalytic HER rate of 11620 μmol g−1 h−1 was obtained with AQE of 11.09% at 470 nm of the CdS/MOF-5(25) heterojunction under visible-light irradiation.
A facile, tunable and inexpensive MOF-templated strategy for synthesizing Fe2O3@TiO2 was reported by the use of MIL-101(Fe) as a template (Fig. 7a), which showed a higher H2 production yield than single Fe2O3 and TiO2 materials.27 For synthesis, the amorphous shell of titania was coated on the outside of the MIL-101 nanoparticles by the acid-catalyzed hydrolysis and condensation of a titanium(IV) bis-(ammonium lactato)-dihydroxide water solution, in which the concentration of the acid and the reaction time determined the thickness of the TiO2 coating. HRTEM showed that each octahedral shell consisted of roughly spherical single crystalline domains of 10 nm TiO2 and Fe2O3 blended together. Lattice fringes were visible with the lattice d-spacings of 0.354 nm of the {101} plane of anatase and 0.251 nm of the {110} plane of hematite (Fig. 7b–e). The optimized HER rate was 625 μmol g−1 h−1 with Pt as the cocatalyst (Fig. 7f).
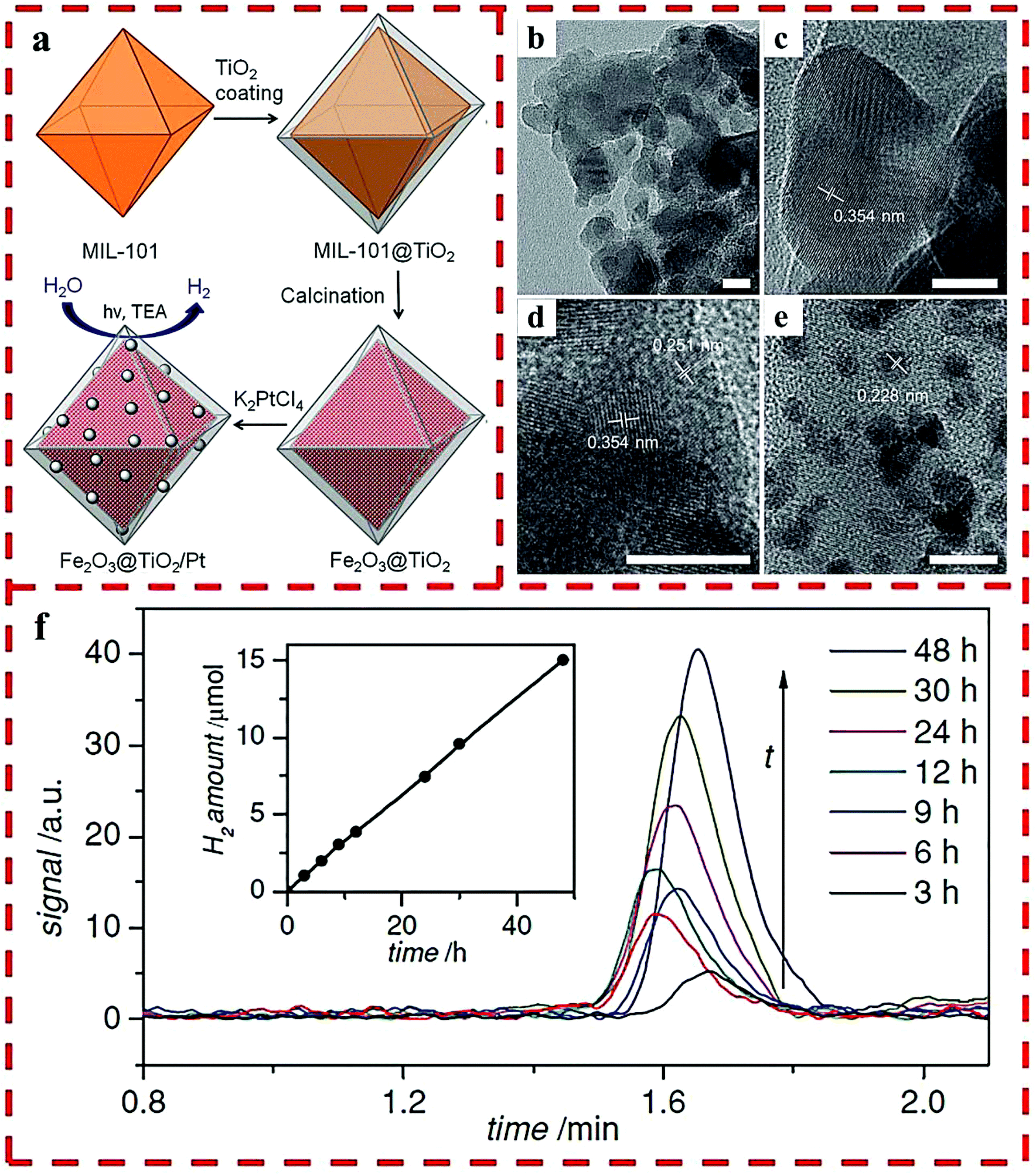 | ||
| Fig. 7 (a) The synthetic route of Fe2O3@TiO2 composite with Pt particles. HRTEM images of Fe2O3@TiO2: as-synthesized (b–d) and after Pt particles deposited (e). (f) H2 produced by Fe2O3@TiO2 with a 420 nm filter. Reprinted with permission from ref. 27. | ||
A Co3O4/TiO2 p–n heterojunction was prepared using Co-MOFs as sacrificial template by Bala et al.28 They demonstrated that their strategy of using MOFs can construct high-performance p–n Co3O4/TiO2 heterojunctions for photocatalytic HER, in which the nanocomposite containing 2 wt% Co loading showed supreme activity (6400 μmol g−1 h−1) under UV-vis light irradiation. The formation of the pony-size heterojunction and the cocatalytic character of Co3O4 should be ascribed to fast interfacial charge transfer and effective electron–hole separation in the photocatalytic reaction.
Another Z-scheme heterojunction of g-C3N4/α-Fe2O3 was prepared by using MIL-100 (Fe) as a self-sacrificing template. By changing the loading amount of MIL-100 (Fe), nanocomposites with diverse α-Fe2O3 and g-C3N4 ratios were obtained (Fig. 8a).35 The proportion of α-Fe2O3 was also regulated by changing the calcination temperature of g-C3N4/MIL-100-2 in air atmosphere. As shown in Fig. 8b, granular α-Fe2O3 nanoparticles tightly adhere to the flakes of g-C3N4. In addition, the obvious lattice spacing of 0.367 nm can be attributed to the {012} plane of α-Fe2O3 (Fig. 8c and d). The elemental mapping images of g-C3N4/α-Fe2O3-2 show that all the elements are homogeneously dispersed throughout the composite, and the α-Fe2O3 nanomaterials evenly adhere to g-C3N4 (Fig. 8e and f). Owing to the well separated photo-excited charge carriers, the HER performance of the optimized g-C3N4/α-Fe2O3-2 photocatalyst was 2066.2 μmol g−1 h−1 (Fig. 8g–j).
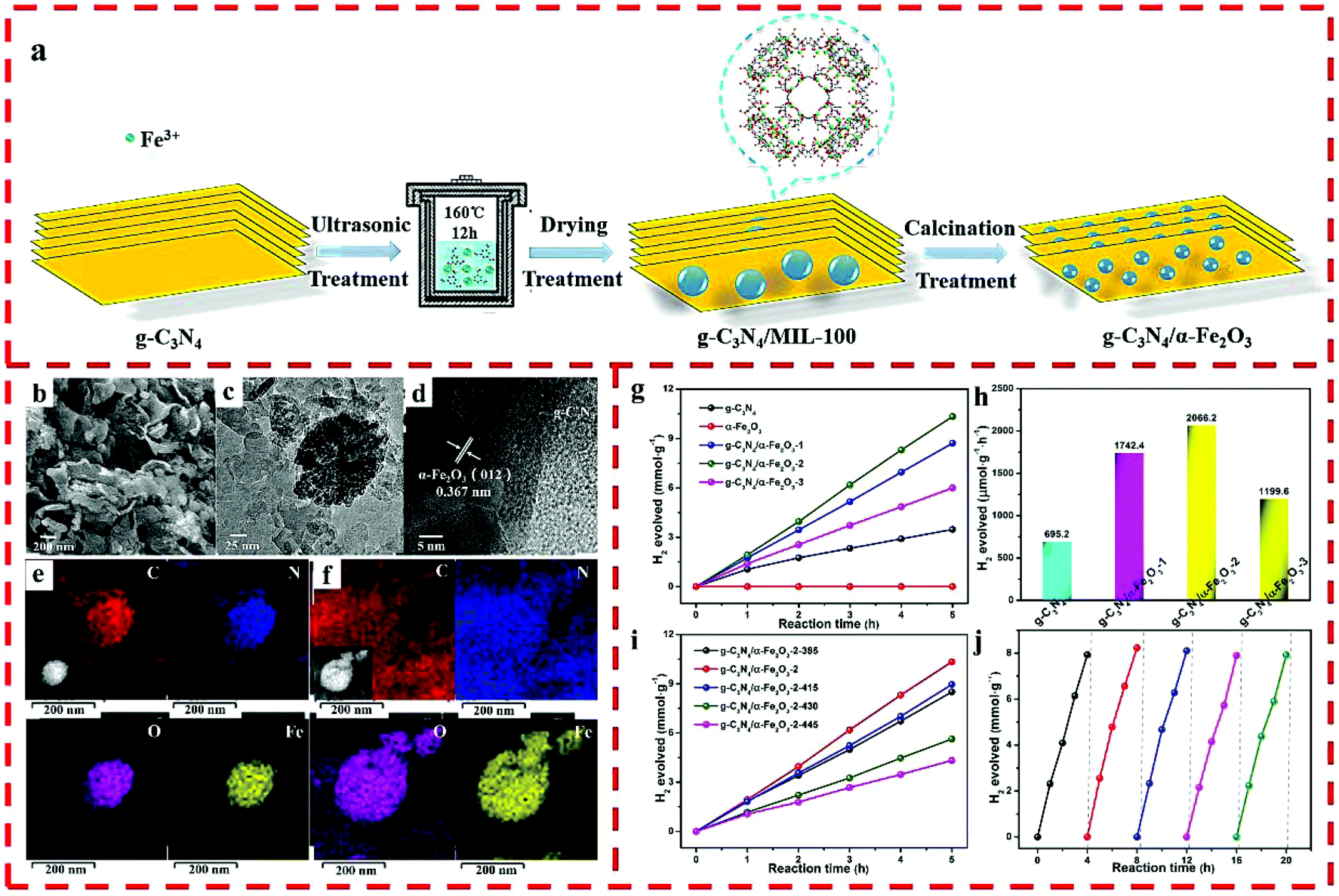 | ||
| Fig. 8 (a) The synthesis route of the g-C3N4/α-Fe2O3 hybrid. (b) SEM, (c) TEM, (d) HRTEM, and (e) elemental mapping images before reaction and (f) after 5 cycling tests of g-C3N4/α-Fe2O3-2. (g) Photocatalytic HER of g-C3N4 and g-C3N4/α-Fe2O3 hybrids. (h) Photocatalytic HER of pure g-C3N4 and g-C3N4/α-Fe2O3 hybrids. (i) Photocatalytic performance of g-C3N4/α-Fe2O3-2-T hybrids. (j) HER cycling test for g-C3N4/α-Fe2O3-2. Reprinted with permission from ref. 35. | ||
Ni2P/Ni nanoparticles (NPs) were wrapped in carbon/g-C3N4 hybrids, originated from the in situ pyrolysis and phosphatization of the Ni-MOF/g-C3N4 precursor, which served as the photocatalyst for HER under visible-light irradiation.36 By adjusting the pyrolysis temperature under an N2 atmosphere of the Ni-MOF/g-C3N4 powder, a set of Ni@C/g-C3N4 composites was obtained. The highest HER rate of Ni2P/Ni@C/g-C3N4-550 was 18040 μmol g−1 h−1 with 1.0 mM of EY-sensitization, while 210 μmol g−1 h−1 was obtained without EY-sensitization, and AQE at 420 nm was 58.1%. This improved photocatalytic activity can be assigned to the effective and quick transfer of the photo-generated charges from excited EY and g-C3N4 to Ni2P/Ni, where carbon as an electron transfer bridge, each component in close contact, interlaced band alignment among g-C3N4, Ni and Ni2P, and the acceleration of the proton reduction reaction by Ni2P/Ni NPs all play synergistic roles.
Kim's group fabricated polyporous multicomponent transition metal phosphides (MCTMPs) from layered double hydroxide double-shelled nanocages through MOF templates.57 CdS semiconductor nanorods combined with these unique MCTMP nanostructures could exhibit exceptionally high stability and prominent HER rate up to 136770 μmol g−1 h−1, with quantum efficiency of 40.6% for the sample with 2 wt% MCTMP loading, which can be ascribed to the large surface area, superior light absorption, and outstanding separation ability of the electron–hole pairs.
In light of the structural multiformity and tailorability of MOFs, the controllable surface modification of MOFs may inaugurate a new pathway to the synthesis of advanced MOF-based nanocatalysts for enhancing photocatalytic H2 production. This strategy will give more opportunities for the construction of efficient photocatalysts to inspire the research on new photocatalysts and to obtain a more efficient visible-light response for sunlight-driven hydrogen evolution by water splitting.
5. Summary and outlook
The recent progress of MOF-derived heterojunction-based nanocatalysts using their pore chemistry or surface chemistry for photocatalytic water splitting was summarized. These MOF-derived nanocatalysts have the following advantages: (i) well-defined composition from the parent MOFs or MOF-based composites; (ii) high porosity with ordered crystallized pores and (iii) effective integration of multifarious functional materials.However, there are still problems that need particular attention, such as how to realize the controllable and facile synthesis of MOF-derived nanocomposites with desired pore structures and active sites and how to achieve the efficient use of visible even near-infrared light by the combination of two or more components to improve the utilization of light energy.
In addition, in photocatalytic water splitting, HER is a half-reaction and takes advantage of sacrificial reagents to accomplish sacrificing holes as the oxidation half-reaction; thus, the following two valuable applications are suggested to be studied by taking the synthetic advantages of the above-mentioned MOF-derived nanocatalysts: (i) Splitting water by sunlight without sacrificial reagents. For example, Wang's team reported that the RGO nanosheets can serve as a solid-state electron mediator to link Fe2O3 nanoparticles with PCN nanosheets by chemical bonding and π–π stacking, forming an Fe2O3/RGO/PCN ternary heterojunction and thus enhancing the photocatalytic efficiency.64 (ii) Replacing the sacrificial agent consumption with organic compound oxidation to realize the coupling of H2 production with useful organic reactions. As the first example of MOF-based photocatalysts to simultaneously perform HER and organic conversion, Jiang's group used 2.3 wt% Pt/PCN-777 as the photocatalyst for HER and selected benzylamine instead of TEOA as the sacrificial agent to produce N-benzylbenzaldimine.65
Overall, the MOF-templated strategy with the intrinsic features of MOFs has become a powerful method for the synthesis of nanocomposites and their related nanocatalysts, which can convert solar energy into hydrogen energy effectively and become the most potential alternatives to fossil fuels.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21622105, 21861130354 and 21421001), the Natural Science Foundation of Tianjin (18JCJQJC47200) and the Ministry of Education of China (B12015). W. S. acknowledges the receipt of a Newton Advanced Fellowship from Royal Society.References
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- W. Wang, X. M. Xu, W. Zhou and Z. P. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef PubMed.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. L. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- M. Z. Ge, Q. S. Li, C. Y. Cao, J. Y. Huang, S. H. Li, S. N. Zhang, Z. Chen, K. Q. Zhang, S. S. Al-Deyab and Y. K. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef PubMed.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- Y. Xu, W. W. Zhao, R. Xu, Y. M. Shi and B. Zhang, Chem. Commun., 2013, 49, 9803–9805 RSC.
- D. J. Martin, K. P. Qiu, S. A. Shevlin, A. D. Handoko, X. W. Chen, Z. X. Guo and J. W. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- A. Khurshida, M. Md, B. Nurlan, K. Sarkyt and N. Nurxat, J. Mater. Chem. A, 2018, 6, 21696–21718 RSC.
- Y. P. Xie, Z. B. Yu, G. Liu, X. L. Ma and H. M. Cheng, Energy Environ. Sci., 2014, 7, 1895–1901 RSC.
- D. P. Kumar, J. H. Choi, S. Hong, D. A. Reddy, S. Lee and T. K. Kim, ACS Sustainable Chem. Eng., 2016, 4, 7158–7166 CrossRef CAS.
- J. K. Hyun, S. Zhang and L. J. Lauhon, Annu. Rev. Mater. Res., 2013, 43, 451–479 CrossRef CAS.
- Y. Z. Chen, C. Y. Zhang, X. J. Zhang, X. M. Ou and X. H. Zhang, Chem. Commun., 2013, 49, 9200–9202 RSC.
- X. Q. Hao, Z. W. Cui, J. Zhou, Y. C. Wang, Y. Hu, Y. Wang and Z. G. Zou, Nano Energy, 2018, 52, 105–116 CrossRef CAS.
- J. X. Low, J. G. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- W. W. Shi and N. Chopra, Nanomater. Energy, 2013, 2, 158–178 CrossRef CAS.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- Z. B. Liang, R. Zhao, T. J. Qiu, R. Q. Zou and Q. Xu, Energy Chem., 2019, 1, 100001 CrossRef.
- Y. J. Xiao, Y. Qi, X. L. Wang, X. Y. Wang, F. X. Zhang and C. Li, Adv. Mater., 2018, 30, 1803401 CrossRef PubMed.
- J. K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 RSC.
- Y. Kataoka, K. Sato, Y. Miyazaki, K. Masuda, H. Tanaka, S. Natio and W. Mori, Energy Environ. Sci., 2009, 2, 397–400 RSC.
- K. E. deKrafft, C. Wang and W. B. Lin, Adv. Mater., 2012, 24, 2014–2018 CrossRef CAS PubMed.
- S. Bala, I. Mondal, A. Goswami, U. Pal and R. Mondal, J. Mater. Chem. A, 2015, 3, 20288–20296 RSC.
- B. Ma, P. Y. Guan, Q. Y. Li, M. Zhang and S. Q. Zang, ACS Appl. Mater. Interfaces, 2016, 8, 26794–26800 CrossRef CAS PubMed.
- M. Lan, R. M. Guo, Y. B. Dou, J. Zhou, A. W. Zhou and J. R. Li, Nano Energy, 2017, 33, 238–246 CrossRef CAS.
- I. Majeed, M. A. Nadeem, A. Badshah, F. K. Kanodarwala, H. Ali, M. A. Khan, J. A. Stride and M. A. Nadeem, Catal. Sci. Technol., 2017, 7, 677–686 RSC.
- X. X. Zhao, J. R. Feng, J. Liu, W. Shi, G. M. Yang, G. C. Wang and P. Cheng, Angew. Chem., Int. Ed., 2018, 57, 9790–9794 CrossRef CAS PubMed.
- X. X. Zhao, J. R. Feng, J. W. Liu, J. Lu, W. Shi, G. M. Yang, G. C. Wang, P. Y. Feng and P. Cheng, Adv. Sci., 2018, 5, 1700590 CrossRef PubMed.
- J. M. Chen, Z. R. Shen, S. M. Lv, K. Shen, R. F. Wu, X. F. Jiang, T. Fan, J. Y. Chen and Y. W. Li, J. Mater. Chem. A, 2018, 6, 19631–19642 RSC.
- J. Liu, X. X. Zhao, P. Jing, W. Shi and P. Cheng, Chem. – Eur. J., 2019, 25, 2330–2336 CAS.
- J. X. Xu, Y. H. Qi and L. Wang, Appl. Catal., B, 2019, 246, 72–81 CrossRef CAS.
- S. Dang, Q. L. Zhu and Q. Xu, Nat. Rev. Mater., 2018, 3, 17075 CrossRef CAS.
- J. N. Hao, X. Y. Xu, H. H. Fei, L. C. Li and B. Yan, Adv. Mater., 2018, 30, 1705634 CrossRef PubMed.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937 RSC.
- Y. Z. Chen, R. Zhang, L. Jiao and H. L. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS.
- B. J. Zhu, R. Q. Zou and Q. Xu, Adv. Energy Mater., 2018, 8, 1801193 CrossRef.
- R. Zhao, Z. B. Liang, R. Q. Zou and Q. Xu, Joule, 2018, 2, 1–25 CrossRef.
- Z. D. Lei, Y. C. Xue, W. Q. Chen, W. H. Qiu, Y. Zhang, S. Horike and L. Tang, Adv. Energy Mater., 2018, 8, 1801587 CrossRef.
- H. G. Zhang, J. Z. Li, Q. Tan, L. L. Lu, Z. B. Wang and G. Wu, Chem. – Eur. J., 2018, 24, 18137–18157 CrossRef CAS PubMed.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- Q. L. Xu, L. Y. Zhang, J. G. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS.
- W. W. Zhan, L. M. Sun and X. G. Han, Nano-Micro Lett., 2019, 11, 1 CrossRef CAS PubMed.
- Q. Mao, J. M. Chen, H. R. Chen, Z. J. Chen, J. Y. Chen and Y. W. Li, J. Mater. Chem. A, 2019, 7, 8472–8484 RSC.
- J. D. Xiao and H. L. Jiang, Small, 2017, 13, 1700632 CrossRef PubMed.
- M. Alhamami, H. Doan and C. H. Cheng, Materials, 2014, 7, 3198–3250 CrossRef PubMed.
- G. H. Li, H. Yang, F. C. Li, J. Du, W. Shi and P. Cheng, J. Mater. Chem. A, 2016, 4, 9593–9599 RSC.
- J. S. Li, Y. J. Tang, C. H. Liu, S. L. Li, R. H. Li, L. Z. Dong, Z. H. Dai, J. C. Bao and Y. Q. Lan, J. Mater. Chem. A, 2016, 4, 1202–1207 RSC.
- N. X. Lia, H. L. Huang, R. Bibi, Q. H. Shen, R. Ngulube, J. C. Zhou and M. C. Liu, Appl. Surf. Sci., 2019, 476, 378–386 CrossRef.
- C. W. Zhao, Y. A. Li, X. R. Wang, G. J. Chen, Q. K. Liu, J. P. Ma and Y. B. Dong, Chem. Commun., 2015, 51, 15906–15909 RSC.
- X. Tang, J. H. Zhao, Y. H. Li, Z. J. Zhou, K. Li, F. T. Liu and Y. Q. Lan, Dalton Trans., 2017, 46, 10553–10557 RSC.
- W. X. Chen, J. S. Fang, Y. W. Zhang, G. L. Chen, S. Zhao, C. Zhang, R. Xu, J. H. Bao, Y. M. Zhou and X. Xiang, Nanoscale, 2018, 10, 4463–4474 RSC.
- D. A. Reddy, H. K. Kim, Y. Kim, S. Lee, J. H. Choi, M. J. Islam, D. P. Kumar and T. K. Kim, J. Mater. Chem. A, 2016, 4, 13890–13898 RSC.
- M. H. Pham, C. T. Dinh, G. T. Vuong, N. D. Ta and T. O. Do, Phys. Chem. Chem. Phys., 2014, 16, 5937–5941 RSC.
- D. A. Reddy, H. Park, M. Gopannagari, E. H. Kim, S. Lee, D. P. Kumar and T. K. Kim, ChemSusChem, 2018, 11, 245–253 CrossRef CAS PubMed.
- Z. J. Wang, Z. L. Jin, G. R. Wang and B. Z. Ma, Int. J. Hydrogen Energy, 2018, 43, 13039–13050 CrossRef CAS.
- Y. K. Zhang, G. R. Wang, W. Ma, B. Z. Ma and Z. L. Jin, Dalton Trans., 2018, 47, 11176–11189 RSC.
- L. Lin, S. Y. Huang, Y. X. Zhu, B. Du, Z. H. Zhang, C. Chen, X. W. Wang and N. Zhang, Dalton Trans., 2019, 48, 2715–2721 RSC.
- J. H. Zhao, L. W. Liu, K. Li, T. Li and F. T. Liu, CrystEngComm, 2019, 21, 2416–2421 RSC.
- Z. M. Pan, G. G. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef.
- H. Liu, C. Y. Xu, D. D. Li and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 5379–5383 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2019 |