
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemically modified electrodes with MOFs for the determination of inorganic and organic analytes via voltammetric techniques: a critical review
Anastasia D.
Pournara†
a,
Georgios D.
Tarlas†
b,
Giannis S.
Papaefstathiou
 *b and
Manolis J.
Manos
*a
*b and
Manolis J.
Manos
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry, University of Ioannina, 45110 Ioannina, Greece. E-mail: emanos@cc.uoi.gr
bLaboratory of Inorganic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, 157 71, Zografou, Greece. E-mail: gspapaef@chem.uoa.gr
First published on 2nd October 2019
Abstract
Voltammetric analytical techniques combine exceptional sensitivity, low cost, portability and capability for simultaneous determination of multiple analytes. The sensitivity of voltammetric analysis is largely determined by the efficiency of the working electrode. Electrodes modified with metal organic frameworks (MOFs) seem particularly promising for use in the analysis of a series of important inorganic and organic analytes. Nevertheless, research on chemically modified electrodes with MOFs is still in its infancy. In this critical review, we present the current status of research related to MOF-modified electrodes highlighting the respective MOF-modified electrodes which are based on MOFs that show exceptional chemical stability or/and sorption capability towards the targeted analytes. We also provide perspectives for future research aiming at motivating additional scientists to be involved in this exciting field of MOF-based electroanalytical sensors.
 Anastasia D. Pournara | Anastasia Pournara received her degree in Biology (2008) and her Ph.D. (2014) in Inorganic Chemistry from the University of Ioannina, Greece, under the supervision of Prof. D. Kovala-Demertzi. Since 2015, she has been a postdoctoral fellow in the group of Dr M. J. Manos at the University of Ioannina. Her research is focused on the synthesis of metal organic frameworks with interesting ion-exchange properties towards organic dyes and heavy metals. |
 Georgios D. Tarlas | Georgios D. Tarlas received his degree in Chemistry (2012), his Masters in Inorganic Chemistry and Technology (2015) and his Ph.D. (2019) from the Department of Chemistry of the National and Kapodistrian University of Athens under the supervision of Prof. G. S. Papaefstathiou. His research is focused on the synthesis, characterization and post-synthetic modification of functionalized metal–organic frameworks with interesting sorption and electronic properties. |
 Giannis S. Papaefstathiou | Giannis S. Papaefstathiou received his Chemistry degree in 1998 and his Ph.D. in Inorganic Chemistry in 2002 from the University of Patras, Greece. After two years’ postdoctoral work at the University of Iowa, he joined the Department of Chemistry of the National and Kapodistrian University of Athens in 2006 where he currently serves as an Associate Professor of Inorganic Chemistry. His research focuses on aspects of coordination chemistry and supramolecular chemistry. |
 Manolis J. Manos | Dr Manolis J. Manos is an Associate Professor of Inorganic Chemistry at the University of Ioannina, Greece. He completed his Ph.D. at the same university under the supervision of Prof. T. Kabanos. He was a post-doctoral researcher in the group of Prof. M. Kanatzidis at MSU and NU and in the group of Prof. A. Tasiopoulos at the University of Cyprus. His research interests are directed toward materials and composites with ion exchange and luminescence sensing properties. |
1. Introduction
Metal organic frameworks (MOFs) are polymeric metal complexes with potential voids that have received much attention over the last two decades. Besides their rich chemistry, MOFs are attractive for their potential applications in various fields.1 MOFs are particularly appealing as sensors since they can combine highly porous structures with a variety of functional groups leading to the fast diffusion of analytes into their pores and enhanced framework–analyte interactions. So far, the majority of research is directed towards the development of MOF-based luminescent sensors.2 Such sensory materials have shown excellent detection properties for a series of inorganic and organic pollutants, as a result of substantial modification of the emission properties of MOFs in the presence of trace amounts of specific analytes. A number of review articles discuss in detail the features of MOFs as luminescent sensors.3 Although luminescence-based sensing has several advantages (e.g. portability, low cost, and low detection limits), it also suffers from limitations such as limited selectivity and inability for simultaneous determination of multiple analytes.2,3More recently, MOFs have been utilized for the modification of electrodes to be used for the voltammetric determination of inorganic and organic species. As we will discuss in more detail below, chemically modified electrodes show enhanced accumulation of the analysed species, as a result of the high sorption capability of the modifier (e.g. MOF) towards the targeted analyte.4 Thus, voltammetric analysis using such modified electrodes offers a number of advantages such as exceptionally low detection limits, high selectivity and simultaneous determination of several analytes.4 Although MOFs that have been utilized so far in voltammetric determination show great promise, research in this field is still in its infancy. There are particular challenges to be addressed for the extended development of MOF-based electrodes.5 For example, up to now, there are several publications which describe the modification of electrodes with MOFs but in many cases there is limited information about the stability of the MOF in the working environment (e.g. they do not discuss if the MOFs are chemically stable in aqueous media).5 There is also limited information in many relevant publications about the selectivity and the sorption ability of MOFs towards specific analytes while the nature of the frameworks (pore size, charge of the frameworks, functional groups, etc.) does not always justify their choice as chemical modifiers for electrodes.
So far, this new field of MOF-based electroanalytical sensors has not been reviewed critically. Here we aim to present the current status of research on MOF-modified electrodes discussing selected examples and their applications related to voltammetric determination of important inorganic and organic analytes, highlighting those examples where unambiguous information on the chemical (and hydrolytic) stability of the MOFs6 or sorption studies have been reported. Prior to the presentation of examples of MOF-modified electrodes used in voltammetric analysis of inorganic and organic analytes, we discuss briefly the basic principles of voltammetric analytical techniques focusing on stripping voltammetry and provide a short introduction on chemically modified electrodes.4 We further suggest directions and perspectives for future research on the use of MOF-modified electrodes for analytical applications, focusing on the specific characteristics that MOFs should possess in order to be utilized as electrode modifiers for voltammetric determination. Our ambition is that the present review will inspire scientists with interest in MOFs to extend their research efforts towards this new field of MOF-based analytical applications.
2. Background information on stripping voltammetry and chemically modified electrodes
One of the most powerful voltammetric techniques is stripping voltammetry which combines particularly low detection limits, portability and capability for simultaneous determination of multiple analytes.4,7 Stripping voltammetry involves accumulation (pre-concentration) of the analyte on the working electrode followed by its release (stripping) to the solution. For example, stripping voltammetry analysis of metal ions (Mn+) includes deposition of Mn+ on the working electrode with simultaneous reduction of Mn+ to M0 (pre-concentration step) and a subsequent electrochemical stripping step of the accumulated analyte (oxidation of M0 to Mn+) producing a peak current proportional to the analyte's concentration in solution (Fig. 1). The three most commonly used variations of stripping voltammetry are anodic (in this method, used for most metal ions, stripping is achieved by scanning anodically towards a more positive potential), cathodic (in this method, used for several inorganic anions, stripping is achieved by scanning cathodically towards a more negative potential) and adsorptive stripping voltammetry (in this method, which is used for some metal ions and organic molecules, deposition involves adsorption of a metal complex or organic analyte on the working electrode's surface without electrolysis of the adsorbed species).8 In addition, depending on the type of the potential waveform used in the stripping step (mainly differential pulse and square wave), the voltammetric analytical technique is called Differential Pulse Stripping Voltammetry (DPSV) or Square Wave Stripping Voltammetry (SWSV).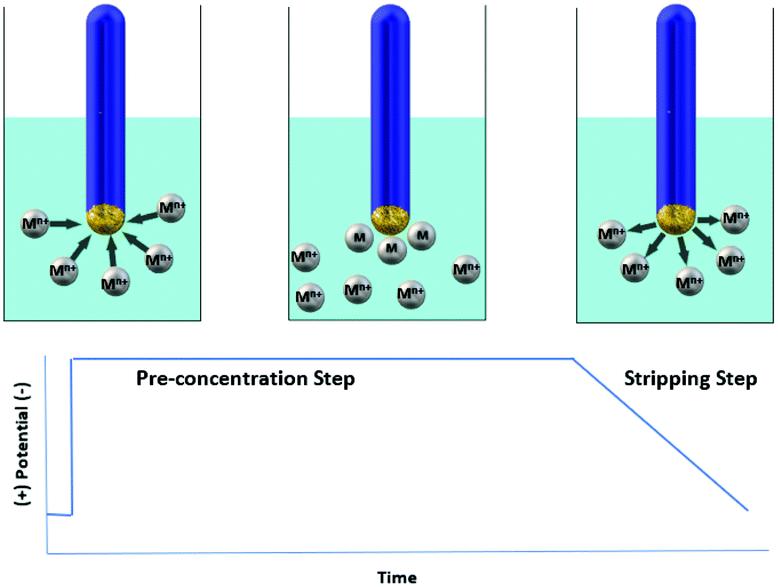 | ||
| Fig. 1 Schematic representation of the determination method for heavy metals via stripping voltammetry. | ||
A prerequisite for sensitive electrochemical measurements, which can achieve detection limits even at ppt levels, is the appropriate selection of the working electrode material. Traditionally, stripping voltammetry makes use of the Hanging Mercury Drop Electrode (HMDE), where mercury forms an amalgam with a metal ion and the current is measured after an oxidation reaction.4 However, health considerations and environmental limitations have led to intense research efforts towards the replacement of the highly toxic mercury-based electrode with other types of environmentally friendly electrodes. To this end, electrodes modified with several materials have been developed and tested for the voltammetric determination of various analytes. For example, as an alternative to mercury-based electrodes, carbon paste electrodes (CPEs) constitute a special class of heterogeneous carbon electrodes consisting of a mixture of conductive graphite powder and a suitable chemical modifier.9 The material that is chosen to serve as a chemical modifier in an electrode should at least have the following characteristics: (i) stability under the experimental conditions, (ii) stability after multiple pre-concentration–stripping cycles and (iii) high selectivity and sorption capacity for the targeted analyte. The latter is particularly important as it is directly related to the pre-concentration step (deposition of the analyte on the electrode) and therefore greatly influences the observed detection limits. Several already reported electrode modifiers include nanomaterials/ionophores,10 bismuth modified zeolite,11 antimony powder,12 multi-walled carbon nanotubes,13 double-stranded DNA and ferric oxide nanoparticles,14 Ca10(PO4)6(OH)2,15 tripolyphosphate-modified kaolinite clay,16 a mixture of titanium dioxide/zirconium dioxide,17 graphene,18 and other polymeric materials.19 Although there is a plethora of chemical modifiers reported in the literature, the search for new materials that would lead to enhanced analytical performance towards the targeted analytes is continuous.
MOFs seem to have high potential to be used for the modification of electrodes for voltammetric analyses. Indeed, they could be superior over other materials which are currently being tested for this purpose. The surface areas of MOFs can reach values as high as 7000–8000 m2 g−1, higher than those commonly found for other types of porous materials.20 Thus, various analytes can rapidly diffuse into the pores of certain highly porous MOFs thereby enhancing pre-concentration. In addition, MOFs can be derivatized with a variety of functional groups, something that is either very difficult or even impossible with conventional materials. By choosing appropriate functional groups, MOFs can display selective sorption of analytes in the presence of various competitive species, a fact that is also highly beneficial for the pre-concentration of analytes in the course of voltammetric analysis.
3. Determination of organic analytes with MOF-modified electrodes
3.1 General comments
In recent years, there has been a large number of literature reports describing the fabrication and utilization of MOF-based modified electrodes for the detection of organic molecules. The driving force behind these efforts is identified as the need for the detection and quantification of organic molecules that: (i) demonstrate biological action crucial to maintaining human health such as glucose,21 (ii) act as explosives such as 2,4,6-trinitrophenol22 and (iii) exist as traces in food, such as methyl parathion,23 which is one of the most widely used pesticides in agricultural practices in order to control a wide range of pests infesting various crops.It is apparent that MOFs which are either soluble or degradable in aqueous media (the most common working environment for an electrode) or lose their crystallinity and therefore their ordering and porosity, the main feature that classifies a coordination polymer as a MOF, are inappropriate to be used as electrode modifiers. Nevertheless, modified electrodes, based on HKUST-1 ([Cu3(BTC)2], H3BTC = trimesic acid) and MOF-5 ([Zn4O(BDC)3], H2BDC = terephthalic acid), which are among the best defined and most studied MOFs, have been used for the determination of organic molecules, despite the fact that the aforementioned materials are not stable in aqueous media.24,25 Recently, Li et al.22 and F.A. Sofi et al.26 described the development of a glassy carbon electrode modified with HKUST-1 as a sensitive electrochemical sensor for the determination of dopamine, an important neurotransmitter in the mammalian central nervous system, using Differential Pulse Voltammetry (DPV). They managed to detect the targeted analyte in a wide range of concentrations ranging between 5.0 × 10−7–1.0 × 10−4 M and 12.5 × 10−6–175 × 10−6 M, respectively, with an estimated detection limit of 1.5 × 10−7 M and 0.11 × 10−9 M, respectively. In addition, HKUST-1 and its analogues have been extensively utilized for the modification of electrodes, claiming selective and sensitive detection, with low detection limits, excellent stability and reproducibility for molecules such as 2,4,6-trinitrophenol,27 catechol,28–30 hydroquinone,28,29 2,4-dichlorophenol,30b glyphosate,31 resorcinol,29 bisphenol A,32,33 paracetamol,25,34,35 caffeine,34 glucose,36L-cysteine37 and methyl parathion.38 On the other hand, Li et al.39 demonstrated a new electrochemical sensor which was constructed by the in situ assembly of MOF-5 onto a PGN nanocomposite, for the determination of echinacoside, one of the natural ingredients extracted from the herb cistanche, with a limit of detection of 1.0 × 10−8 M.
3.2. Examples of chemically stable MOFs used as electrode modifiers for the determination of organic analytes
Despite the above, there are also several examples of chemically stable MOFs that have been utilized as modifiers for electrodes. As we will see in the following discussion, such MOFs have improved significantly the analytical performance of the electrodes, presumably due to their high surface area and their potential capability for rapid sorption of organic analytes into their pores. These MOFs are summarized in Table 1 and analyzed hereafter. Nevertheless, the published studies on MOF-modified electrodes for determination of organic analytes did not include sorption studies for the examined organic molecules.| MOF | Analyte | Linear range (M) | Detection limit (M) | Detection method | Ref. |
|---|---|---|---|---|---|
| UiO-66 | Paraoxon | (1.0–100) × 10−9 | 0.2 × 10−9 | SWV | 41 |
| Chlorpyrifos | (5–300) × 10−9 | 1.0 × 10−9 | |||
| Hydroquinone | (0.5–100) × 10−6 | 0.056 × 10−6 | DPV | 42 | |
| Catechol | (0.4–100) × 10−6 | 0.072 × 10−6 | |||
| Resorcinol | (30–400) × 10−6 | 3.51 × 10−6 | |||
| MOF-525 | Dopamine | (2–270) × 10−6 | 0.04 × 10−6 | DPV | 43 |
| MOF-525 | Luteolin | (0.005–5) × 10−6 | 0.35 × 10−9 | DPV | 47 |
| MIL-101-Cr | Catechol | (10–1400) × 10−6 | 4.1 × 10−6 | DPV | 48 |
| Hydroquinone | (4–1000) × 10−6 | 0.66 × 10−6 | |||
| Metronidazole | (0.5–900) × 10−6 | 0.24 × 10−6 | DPV | 49 | |
| Quercetin | (0.1–700) × 10−6 | 0.06 × 10−6 | DPV | 50 | |
| ZIF-8 | Acetaminophen | 3.5 × 10−6–0.56 × 10−6 | 1.02 × 10−6 | DPV | 51 |
| Cu-MOF-74 | 2.4.6 trichlorophenol | (0.01–9) × 10−6 M | 0.005 × 10−6 M | DPV | 55 |
| Ce(BTC)(H2O)6 | Bisphenol A | (0.005–50) × 10−6 M | 2.0 × 10−9 M | DPV | 58 |
| [In48(HImDC)96]48− | Acetaminophen | 1.0 × 10−8–2.0 × 10−5 M | 6.4 × 10−9 M | DPV | 59 |
A family of MOFs with remarkable and ultrahigh chemical stability under aqueous conditions constitute the Zr6 based MOFs.40 Recently, N. Karimian, H. Bagheri et al.41 described the fabrication of a glassy carbon electrode (GCE) modified with a composite consisting of TiO2 functionalized graphene oxide (TGO) and UiO-66 [Zr6O4(OH)4(BDC)6], for the simultaneous detection of paraoxon (POX) and chlorpyrifos (CPF). The latter are used as pesticides and insecticides, respectively, and are considered hazardous to human health. The UiO-66 MOF consists of hexanuclear clusters that serve as 12-coordinated (12-c) nodes extended in space through the phenyl rings of the ligands, creating a 3D framework with channels ∼6 Å in diameter. The composite (TGO@UiO-66) was synthesized by a similar method to that for the synthesis of UiO-66. The procedure includes the dispersion of TGO in DMF by sonication, followed by the addition of UiO-66's components in DMF solution. The resulting suspension was further sonicated and transferred into an autoclave at 120 °C for 12 h. The preparation of the TGO@UiO-66/GCE was achieved by dropping 6.0 μL of a suspension of TGO@UiO-66 in ethanol onto a bare GCE surface followed by air-drying. The phase purity of the prepared materials was investigated by X-ray diffraction analysis. The observed diffraction peaks of UiO-66 were in good agreement with those of the pristine material, while the Powder X-ray Diffraction (PXRD) pattern of TGO@UiO-66 indicated that the presence of TGO did not prevent the formation of UiO-66. N2 adsorption–desorption measurements confirmed the porosity of both UiO-66 and the TGO@UiO-66 composite. The modified TGO@UiO-66/GCE electrode was tested for the determination of POX and CPF separately and simultaneously (Fig. 2), using Square Wave Voltammetry (SWV). In the case of POX, the SWV curves were recorded at various concentrations ranging between 1.0 × 10−9 and 100 × 10−9 M and in the presence of 50 × 10−9 M CPF. The current peaks on SWV curves of POX indicated that the existence of 50 × 10−9 M CPF had no effect on the determination of POX. CPF determination within the 5–300 × 10−9 M range was achieved following the same route. The detection limits for the POX and CPF were estimated to be 0.2 × 10−9 M and 0.1 × 10−9 M, respectively. The optimized performance of the electrochemical assay was attributed to the high surface area and excellent conductivity of the TGO@UiO-66 composite which facilitated the electron transfer between the analyte and the electrode surface.
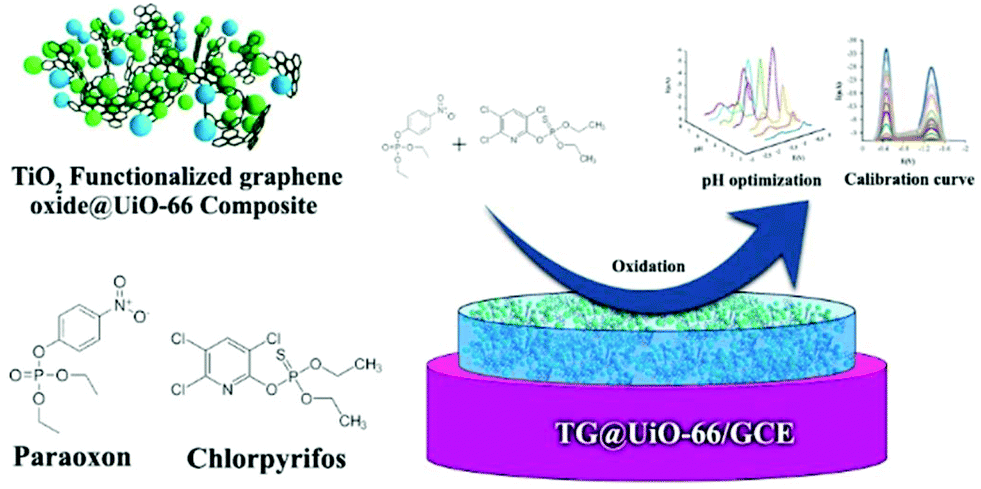 | ||
| Fig. 2 Schematic representation of the procedure for the determination of POX and CPF by the TGO@UiO-66/GCE modified electrode. Reproduced from ref. 41 with permission from the Royal Society of Chemistry. | ||
M. Deng, L. Guo et al.42, using the same MOF (UiO-66), synthesized under hydrothermal conditions a composite, combining UiO-66 with mesoporous carbon (MC). The UiO-66/MC composite was characterized by a plethora of techniques such as SEM, TEM, PXRD and N2 adsorption–desorption isotherms, which proved the stability and the porous nature of the composite. The preparation of the modified electrode was achieved by the dispersion of 5 μL of a DMF suspension of the UiO-66/MC composite on the mirror like surface of a GCE. The UiO-66 based electrode was utilized for the determination of several di-hydroxybenzene molecules such as hydroquinone (HQ), catechol (CT) and resorcinol (RS) which have been categorized as emerging contaminants in aqueous media. Quantitative determination was performed using DPV. Under optimized conditions, the electrochemical sensor showed a wide linear response for each targeted analyte, that is, 0.5–100 × 10−6 M, 0.4–100 × 10−6 M and 30–400 × 10−6 M for HQ, CT and RS, respectively, with the limit of detection reaching the values of 0.056 × 10−6, 0.072 × 10−6 and 3.51 × 10−6 M, respectively. The outstanding electrochemical performance of the modified electrode for the simultaneous determination of the di-hydroxybenzene isomers was attributed to the UiO-66/MC composite, which showed excellent electrochemical stability, larger pore size and good conductivity that enabled faster electron transfer and contributed to mass transfer.
The amino-functionalized derivative of UiO-66 (UiO-66-NH2 [Zr6O4(OH)4(NH2-BDC)6], NH2-BDCH2 = 2-aminoterephthalic acid) has emerged as a sorbent for heavy metal-ions such as Cd2+ and Pb2+ (the maximum sorption capacity was found 268 and 293 mg g−1 for Cd2+ and Pb2+, respectively).43a Due to the excellent adsorption ability, which is attributed to the –NH2 groups, materials such as UiO-66-NH2 are suitable for the construction of biosensors because they act as ideal signal carriers.43 There will be no further discussion on this class of electrochemical bio-sensors that incorporate MOFs44 because it is out of the scope of this review.
T.-Y. Huang, C.-W. Wu et al.45 demonstrated a dopamine sensor, composed of poly(3,4-ethylenedioxythiophene) nanotubes (PEDOT NTs) coated with MOF-525 [Zr6O4(OH)4(TCPP-H2)3] (H4TCPP-H2 = tetrakis(4-carboxyphenyl)porphyrin). MOF-525 is a porphyrin-containing MOF that is exceptionally chemically stable, maintaining its structure upon its treatment with water and various organic solvents and can be metalated with iron(III) and copper(II) to yield its metalated analogues without losing its high surface area and chemical stability.46 MOF-525 was synthesized from the reaction of zirconyl chloride with H4TCPP-H2 in a DMF solution at 65 °C for 3 days. The Zr6 clusters serve as 12-c nodes and the ligand as a 4-c node resulting in an ftw network. MOF-525 acted as an electrocatalytic surface, while PEDOT NTs acted as charge collectors that rapidly transported the electrons from the MOF-525 surface. The response of the electrode was measured by DPV with the linear concentration range of dopamine detection estimated at 2–270 × 10−6 M and the detection limit reaching the value of 0.04 × 10−6 M. The enhanced electrochemical performance was attributed to the synergistic effects originating from the simultaneous presence of MOF-525 nanocrystals, which function as electrode materials with numerous electrochemically active sites and the PEDOT NTs, which serve as charge collectors to efficiently transport electrons to the electrode.
MOF-525 was also combined, by M. Cao, L. Guo et al.,47 with a macroporous carbon (MPC), and the resulting MOF-525/MPC composite was successfully prepared by a solvothermal reaction and used for the determination of luteolin, which is a common flavonoid with neuroprotective and antioxidant effects, by DPV. According to the DPV curves, the luteolin sensor (MOF-525/MPC-2/GCE) showed two parts of linearity in the ranges of 0.005–0.1 × 10−6 and 0.1–5 × 10−6 M with a limit of detection of 0.35 × 10−9 M. The electrochemical performance was attributed to the large pore size, high BET surface area and good conductivity of the MOF-525/MPC composite, which can conduce mass transfer and quicken electron transfer.
H. Wang, F. Lu et al.,48 in order to overcome certain drawbacks associated with the majority of MOFs, such as the low electrical conductivity and poor chemical stability, combined MIL-101-Cr [Cr3O(F/OH)(H2O)2(BDC)3] with reduced graphene oxide (rGO), which is one of the most promising electroconductive materials. The resulting composite material was used as a chemical modifier in a carbon paste electrode (CPE) for the quantitative determination of CT and HQ. MIL-101-Cr is a mesoporous MOF (based on super-tetrahedral units, each one consisting of three chromium trimers that are bridged by the BDC2− linkers) and shows incredible stability (including stability against hydrolysis). MIL-101-Cr contains two types of cages with diameters of 34 and 29 Å. The corresponding cage windows are 16 and 12 Å in size and they are diffusion paths for adsorbates. The authors hypothesized that the incorporation of rGO in MIL-101-Cr crystals could enhance their conductivity and electron transfer and thus allow better performance in electrochemical sensing systems. The synthesis of the MIL-101-Cr/rGO composite included the dispersion of graphene oxide and terephthalic acid followed by the addition of a solution of tetramethylammonium hydroxide. Then, Cr(NO3)3·9H2O was added under vigorous stirring and transferred into an autoclave that was heated for 24 h at 150 °C. The obtained composites were denoted as MIL-101-Cr-rGO-1, MIL-101-Cr-rGO-2 and MIL-101-Cr-rGO-3, according to the amount of graphene oxide that was used (i.e. 1 wt%, 2 wt% and 5 wt%, respectively). The fabrication of the CPE included the grounding of the composite with graphite powder with a weight ratio of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by mixing with paraffin. Finally, a copper wire was inserted within the body of the electrode as inner electrical contact. The modified electrode after the characterization of the composites by IR, XRD, SEM and EIS was tested by recording cyclic voltammograms of HQ and CT (simultaneously) in phosphate saline solution, giving a broad oxidation peak, with the MIL-101-Cr-rGO modified electrode exhibiting the ability to identify the two isomers by increasing the oxidation and reduction peak currents by raising the rGO content in the electrode. The DPV results indicated that the oxidation peak shifted negatively with the increase of the pH value, due to the participation of H+ in the electrode reaction, with the quantitative detection of HQ and CT taking place in phosphate-buffered solution (PBS) with pH = 7.5. The determination of HQ and CT was performed in a single analyte solution, with the regression equation showing linearity in two ranges (HQ: 4–100 × 10−6 and 100–1000 × 10−6 M, CT: 10–60 × 10−6 and 60–1400 × 10−6 M). The detection limits were estimated at 0.66 × 10−6 M and 4.1 × 10−6 M for HQ and CT, respectively.
:1, followed by mixing with paraffin. Finally, a copper wire was inserted within the body of the electrode as inner electrical contact. The modified electrode after the characterization of the composites by IR, XRD, SEM and EIS was tested by recording cyclic voltammograms of HQ and CT (simultaneously) in phosphate saline solution, giving a broad oxidation peak, with the MIL-101-Cr-rGO modified electrode exhibiting the ability to identify the two isomers by increasing the oxidation and reduction peak currents by raising the rGO content in the electrode. The DPV results indicated that the oxidation peak shifted negatively with the increase of the pH value, due to the participation of H+ in the electrode reaction, with the quantitative detection of HQ and CT taking place in phosphate-buffered solution (PBS) with pH = 7.5. The determination of HQ and CT was performed in a single analyte solution, with the regression equation showing linearity in two ranges (HQ: 4–100 × 10−6 and 100–1000 × 10−6 M, CT: 10–60 × 10−6 and 60–1400 × 10−6 M). The detection limits were estimated at 0.66 × 10−6 M and 4.1 × 10−6 M for HQ and CT, respectively.
Another MIL-101-Cr/rGO composite electrochemical sensor, was developed by J. Gu, L. Guo et al.,49 for the detection of metronidazole, an oral drug that pertains to a class of nitroimidazole derivatives and is crucial for the treatment of anaerobic bacterial and protozoal diseases. They followed a similar pathway (to the previous MIL-101-Cr-rGO composite) for the MIL-101-Cr/rGO composite synthesis and were confronted with the same challenges, with the metronidazole detection achieved at an optimal operation pH value of 7.5 in the concentration ranges of 0.5–200 × 10−6 M and 200–900 × 10−6 M. The detection limit was estimated at 0.24 × 10−6 M. Again, the electrocatalytic ability of the MIL-101-Cr/rGO composite was attributed to the synergistic effect of the super electrical conductivity of rGO and the distinctive structural features of MIL-101-Cr.
The combination of MIL-101-Cr with MoS2 forms an MIL-101-Cr/MoS2 composite, which involves the Cr(III) MOF and exemplifies a synergetic advantage. W. Zhang, Y. Zhang et al.50 introduced the molecular imprinting technique (MIT) to design and synthesize molecularly imprinted polymers (MIP), which can effectively promote the selectivity of the MIL-101-Cr/MoS2/GCE towards quercetin (Qu), a member of the family of flavonoids with antioxidant activity. The MIL-101-Cr/MoS2/GCE was constructed by dropping 10 μL of a suspension consisting of 10 mg of MIL-101-Cr and MoS2 (in a 1:6 molar ratio) onto the surface of a pre-treated GCE electrode. To further stabilize the electrode, 10 μL of a 1 wt% Nafion solution was used. The aforementioned sensor was polymerized at 0.7 V for 100 s, using chronocoulometry when inserted into the polymerization solution (pyrrole in 0.1 M H2SO4/0.01 M Qu). The electrochemical behavior of the MIP/MIL-101-Cr/MoS2/GCE was investigated by DPV under different concentrations of Qu with the current peak increasing with the increase of the concentration of quercetin. The current–concentration of Qu ratio exhibited a linear relationship in two ranges, which were 0.1–10.5 × 10−6 M and 10.5–700 × 10−6 M. The detection limit of the sensor was estimated at 0.06 × 10−6 M in PBS and pH = 3.5. The results indicated that the MIL-101-Cr/MoS2 composite as an electrode modifier exhibited the advantages of the large surface area of MIL-101-Cr and the electrocatalytic performance of MoS2. Furthermore, the combination of MIL-101-Cr and MoS2 created a chemically catalytic environment on the electrode surface, which allowed specific interactions between Qu and the organic parts of MIL-101-Cr.
L. Wang, Y. Zhang et al.51 took advantage of the incredible chemical stability of the zeolitic imidazolate framework ZIF-8 ([Zn(MeIM)2], H-MeIM = 2-methylimidazolate)52 to decorate leaf-like ZIF-8 (ZIF-L) with Au nanoparticles, in order to modify a GCE for the effective determination of Acetaminophen, the most common over-the-counter antipyretic and analgesic drug. ZIF-8 is a MOF with a sod topology, exhibiting a nanopore structure formed by four-ring and six-ring ZnN4 moieties. The typical preparation procedure includes the addition of solid Zn(NO3)2·4H2O and H-MeIM in a glass vial containing DMF and keeping the reaction mixture at 140 °C for 24 h. The preparation of the Au nanoparticles and the Au/ZIF-L composite was achieved following previously published methods with a minor modification in the case of ZIF-L.53,54 The synthesis of the GCE's additives was followed by the extensive characterization of both ZIF-L and Au/ZIF-L. The size of the Au nanoparticles was estimated at 13.62 nm, while the porosity of ZIF-L and Au/ZIF-L was confirmed by BET measurements, which revealed the microporous nature of the materials. The electrode was tested by electrochemical impedance spectroscopy (EIS) to confirm that the modification had taken place and then its behavior as an electrocatalyst and sensor for Acetaminophen, using cyclic voltammetry (CV) and DPV, respectively, was studied. The results of Acetaminophen determination by DPV revealed two linear segments. The linear concentration segments were found in the ranges of 0.056–0.56 × 10−6 M and 3.50 × 10−6 to 0.056 × 10−3 M, with a limit of detection of 1.02 × 10−6 M. The long-term stability of the sensor, a key parameter for the development and application of sensors in electrochemistry, was confirmed by placing the Au/ZIF-L/GCE at 4 °C for two weeks. The stored sensor was tested again by DPV in the presence of 0.05 × 10−3 M of Acetaminophen, showing the same results. The electrode performance was attributed to the synergistic effect of the unique structures of ZIF-L with a leaf-like morphology and the strong electrocatalytic activity of Au NPs for small molecules. The combination of large specific surface area and porosity made ZIF-L a platform for loading the Au NPs. Thus, the Au/ZIF-L nanohybrids offered a favorable microenvironment for transferring species in solution, and were beneficial for accelerating electron transfer between the electrode and the species in solution.
In order to construct a sensitive electrochemical sensor for the detection of 2,4,6-trichlorophenol (2,4,6-TCP), which is commonly used in industry and agricultural chemicals, T. Zhang, J. Wang et al.55 developed an electrochemical sensor by immobilizing hemin into Cu-MOF-74 ([Cu2(DHTP)], H4DHTP = 2,5-dihydroxyterephthalic acid). Cu-MOF-74 was synthesized by adding H4DHTP and Cu(NO3)2·3H2O in a vial containing 20 mL DMF and 1 mL 2-propanol which was heated at 80 °C for 18 h. From the topological point of view MOF-74 is a 3D MOF with rod Secondary Building Units (SBUs) that is based on edge sharing polyhedra bridged to the other two dimensions by the phenyl rings of the ligand. This classifies MOF-74 as an I1O2 framework.56 The preparation of hemin/Cu-MOF-74 was achieved by the addition of 5 mg of Cu-MOF-74 into 20 ml of hemin solution (2.2 g hemin in 10 ml water and 15 ml MeOH) which was then stirred for 6 h. The hemin/Cu-MOF-74/GCE was prepared by coating the surface of a polished GCE (pre-treated with ethanol and double deionized water) with 5 μL of a DMF homogeneous solution of hemin/Cu-MOF-74. The modification of the electrode was completed by further coating of the surface of the hemin/Cu-MOF-74/GCE with 5 mL of 0.5% Nafion solution. The fully modified electrode, before use, was stored at 4 °C. The electrochemical behavior of the 2,4,6-trichlorophenol-hemin/Cu-MOF-74/GCE was investigated by recording cyclic voltammograms in 0.1 M PBS solution (pH = 7.0), which indicated the irreversible oxidation of the 2,4,6-trichlorophenol and the improved performance of the fully modified electrode against the MOF (only)-functionalized and bare electrodes. The quantitative detection of the 2,4,6-TCP was performed by DPV. The peak current increased linearly with the increase of the concentration of 2,4,6-trichlorophenol in the range of 0.01–9 × 10−6 M. In this range, two linear ranges were revealed (0.01–1.0 × 10−6 M and 1.0–9.0 × 10−6 M) while the limit of detection was found at 0.005 × 10−6 M. When hemin was immobilized in Cu-MOF-74, the current intensity at the hemin/Cu-MOF-74/GCE was higher than those at other composites, probably owing to the synergistic effect produced by hemin and Cu-MOF-74. The reason for the improved performance may be the contribution from the following properties: (i) Cu-MOF-74 consists of metal ions and organic ligands, which can adsorb more 2,4,6-TCP onto the electrode surface through π–π stacking interactions. (ii) Cu-MOF-74 possesses high surface areas, favoring high hemin loading. (iii) The possible interaction between hemin and metal ions in Cu-MOF-74 increased the catalytic activity of hemin.
The combination of the trivalent lanthanide ion Ce3+ with trimesic acid (H3BTC) afforded the 1D coordination polymer, namely [Ce(BTC)(H2O)6] (Ce-MOF), which extends to the second dimension through π–π interactions between the phenyl rings of the ligand.57 J. Zhang et al.58 prepared a modified electrode by combining the Ce-MOF with the cationic surfactant cetyltrimethylammonium bromide (CTAB) via electrostatic interactions, in their effort to fabricate an ultrasensitive electrochemical bisphenol A (BPA) sensor. BPA is an organic monomer which is employed in the production of food packaging and is found in the environment, food and bodies of living organisms. The preparation of the CTAB/Ce-MOF composite was achieved when an aqueous solution of Ce-MOF (1.0 mL, 2.0 mg mL−1) was added into 1.0 mL of 3.0 mg mL−1 CTAB. Before the modification, a bare GCE (3 mm diameter) was polished using 0.3 and 0.05 μm alumina slurry and rinsed thoroughly with distilled water. The GCE was successively sonicated in a nitric acid/acetone mixture (1:1 v/v) and distilled water, and afterwards the GCE was rinsed with distilled water and dried at room temperature. 4.0 μL of the CTAB/Ce-MOF suspension was dropped onto the pretreated GCE. After drying at room temperature, the CTAB/Ce-MOF modified GCE (CTAB/Ce-MOF/GCE) was successfully prepared. The PXRD measurements indicated that the nature of the as-synthesized Ce-MOF and the MOF that exist in the CTAB/Ce-MOF composite is unchanged which is evidence that the Ce-MOF is stable in aqueous media and that the electrode could be characterized as being successfully MOF-modified. The analytical performance of the modified electrode was studied by DPV. According to the DPV, the peak current gradually increased with the increase of BPA's concentration. The linear concentration range of BPA at CTAB/Ce-MOF/GCE was found to be between 0.005 × 10−6 and 50 × 10−6 M with the detection limit at 2.0 × 10−9 M. In this work, the CTAB/Ce-MOF was prepared by the modification of self-assembled CTAB monolayers on the surface of the Ce-MOF via electrostatic interactions with the assistance of ultrasonication. The CTAB/Ce-MOF/GCE showed improved electrochemical response because of the enhanced preconcentration of BPA of the long alkane chain in CTAB via hydrophobic interactions.
Z. Chang, Y. Li et al.59 prepared by a one-step procedure a ferrocene-immobilized MOF-modified graphite electrode for the detection of Acetaminophen. They combined the ability of ferrocene to oxidize Acetaminophen with the ability of the zeolitic type [In48(HImDC)96]48− (H3ImDC: 4,5-imidazoledicarboxylic acid) rho-ZMOF to host cationic species in its cavities due to its anionic nature. The synthesis and structure of the rho-ZMOF were first reported by Eddaoudi et al. and it was found to be chemically stable in aqueous media.60,61 It is based on 8-coordinated In3+ ions chelated by four HImDC2− ligands. The ferrocene functionalized MOF was prepared in the presence of ferrocene in a reaction mixture similar to the one resulting in the original rho-ZMOF. The preparation of the electrode was achieved by the immersion of a graphite electrode in the aforementioned mixture in a glass bottle, which was then sealed and placed in an oven at 100 °C for 36 h. The detection ability of the modified electrode was tested by DPV in an Acetaminophen solution with the concentration in the range of 1.0 × 10−8 to 2.0 × 10−5 M. The peak current increased gradually with the increase of the concentration of Acetaminophen and demonstrated good linearity in the abovementioned range with the detection limit estimated at 6.4 × 10−9 M. In this work, an electrode was modified with a MOF in which ferrocene was immobilized, thus overcoming the problems caused by the increased solubility of ferrocene in its oxidized form.
4. Determination of inorganic analytes with MOF-modified electrodes
4.1 General comments
Intense efforts have been made towards the development of analytical methods for determination of trace amounts of inorganic analytes, particularly heavy metals such as Hg2+, Pb2+, Cd2+, Cu2+etc. Heavy metals are highly toxic, having even lethal effects as they are easily accumulated in humans and other living organisms. Therefore, their accurate, fast and reliable analytical determination in aqueous media is of vital importance. Stripping voltammetry is highly efficient for the quantitative analysis of heavy metals at extremely low concentrations, even at ppt levels.4 Nevertheless, the use of HDME in this analytical method causes concerns due to the high toxicity of mercury. Therefore, there has been interest in developing MOF-modified electrodes that could be as efficient as HDME for the determination of heavy metals without having the toxicity issues of mercury-based electrodes. In the following section, we discuss selected examples of MOFs that have been successfully utilized as modifiers in electrodes for voltammetric analysis of heavy metals. We focus on MOFs (summarized in Table 2) that were demonstrated to be efficient sorbents for heavy metals or combine hydrolytic stability and appropriate structural characteristics that could potentially facilitate the sorption of heavy metal ions by these materials.| Sensor | Analyte | Linear range (M) | Detection limit (M) | Detection method | Ref. |
|---|---|---|---|---|---|
| MIL-101-Cr/GCE | Pb2+ | 1.0 × 10−9–1.0 × 10−6 | 5.0 × 10−10 | DPASV | 62 |
| Ni-MOF/GCE | Pb2+ | 5.0 × 10−7–6.0 × 10−6 | 5.08 × 10−7 | SWASV | 65 |
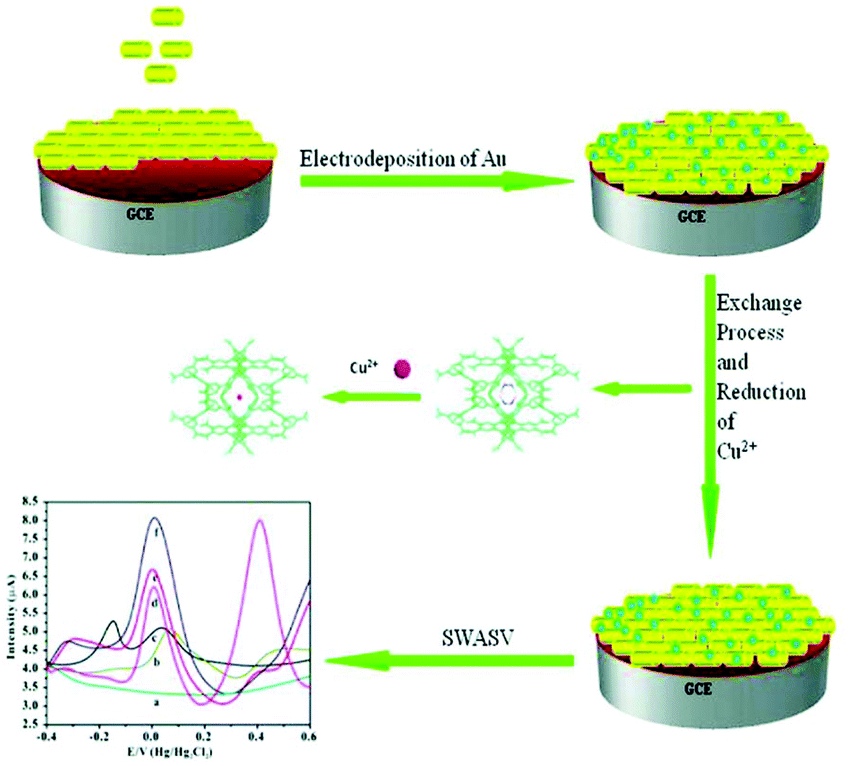
| Au/Me2NH2@MOF-1/GCE | Cu2+ | 5.0 × 10−12–9.0 × 10−7 | 1.0 × 10−12 | SWASV | 70 |
| TMU-16-NH2/Gr/CPE | Cd2+ | 6.2 × 10−12–1.0 × 10−9 | 1.7 × 10−12 | DPASV | 66 |
| UiO-66-NH2@PANI/GCE | Cd2+ | 4.4 × 10−12–5.3 × 10−9 | 2.6 × 10−12 | DPASV | 69 |
| SPAN@UIO-66-NH2/SPCE | Cd2+ | 4.4 × 10−12–8.9 × 10−10 | 1.5 × 10−12 | SWASV | 71 |
| NH2-MIL-125-Ti/CPE | Mn2+ | 1.0 × 10−8–1.0 × 10−5 | 4.0 × 10−9 | Photoirradiation-CSV | 72 |
| GA-UiO-66-NH2/GCE | Cd2+ | 6.0 × 10−8–3.0 × 10−6 | 2.0 × 10−8 | DPASV | 74 |
| Pb2+ | 1.0 × 10−8–4.0 × 10−6 | 1.5 × 10−9 | |||
| Cu2+ | 1.0 × 10−7–3.5 × 10−6 | 7.0 × 10−9 | |||
| Hg2+ | 5.0 × 10−9–3.0 × 10−6 | 2.0 × 10−9 | |||
| Ca-MOF/GPE | Pb2+ | 4.8 × 10−11–2.3 × 10−10 | 3.0 × 10−12 | SWASV | 78 |
| Cd2+ | 8.9 × 10−11–5.3 × 10−10 | 1.1 × 10−11 | |||
| Cu2+ | 1.5 × 10−10–9.4 × 10−10 | 2.2 × 10−11 | |||
| Zn2+ | 1.5 × 10−10–9.1 × 10−10 | 1.6 × 10−11 |
4.2 Single analyte determination
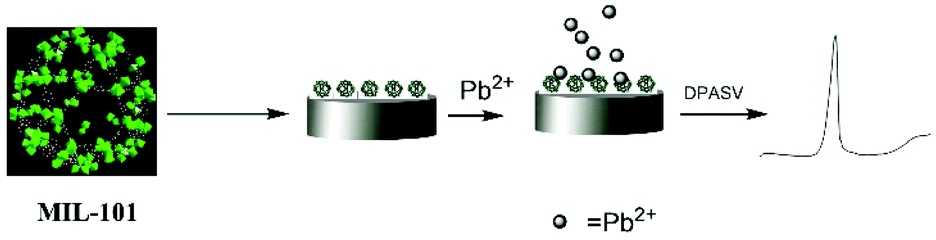 | ||
| Fig. 3 Schematic representation of the fabrication of the MIL-101-Cr modified electrode and the accumulation of Pb2+ ions on its surface for the voltammetric determination of lead ions. Reproduced from ref. 62 with permission from the Royal Society of Chemistry. | ||
A Ni2+ MOF-based modifier [Ni(NH2-BDC)(H2O)]n was synthesized through a facile one-pot hydrothermal reaction of Ni(NO3)2·6H2O with NH2-BDCH2 in a mixture of DMF/water and was incorporated on a GCE for the determination of Pb2+.65 PXRD data for this material confirm its crystalline nature; however, the exact structure of this compound has not been identified. Nevertheless, the PXRD patterns of this Ni-MOF, obtained after its treatment with aqueous solutions of various pH values, are similar to those of the as-prepared material, thus revealing its significant hydrolytic stability. The electrochemical response of this sensor, denoted as Ni-MOF/GCE, was investigated towards Pb2+via square wave anodic stripping voltammetry (SWASV). The oxidation current peak was increased linearly in a concentration range from 5.0 × 10−7 to 6.0 × 10−6 M. The limit of detection was estimated to be 5.08 × 10−7 M, which is 10 times higher than the established limit by U.S. EPA for Pb2+ ions in drinking water. The Ni-MOF modifier seems to play the key role in the electrochemical analysis of Pb2+, as indicated by the comparative study with the unmodified GCE, where the oxidation current peak and, consequently, the determination ability for lead ions decreased. It is possible that Pb2+ interacts with the NH2-groups of the Ni-MOF, thus resulting in enhanced pre-concentration for the Ni-MOF/GCE vs. the unmodified GCE. However, no Pb2+ sorption data were provided. The electrochemical characterization of the modified sensor was also performed, in which important operational parameters such as pH, deposition potential, and deposition time were optimized to detect trace amounts of Pb2+ ions. In addition, the ability of the electrode to detect selectively lead ions was studied in the presence of a 10-fold excess of Cr3+, Cu2+, and Cd2+. No significant modification of the peak current of Pb2+ was observed in the presence of the above interfering cations, indicating efficient selectivity of the sensor for Pb2+.
UiO-66-NH2 was coated with the polyaniline polymer (PANI) and was used as a modifier on a GCE for the detection of Cd2+via DPASV.67 UiO-NH2 is quite stable in aqueous solutions and also shows significant sorption capacity for heavy metals.68 On the other hand, PANI can effectively increase the conductivity of composite materials by inducing the electron transfer between the solution and the electrode surface. The contribution of these materials on the GCE resulted in a reliable and highly sensitive electrochemical sensor with a detection limit for cadmium ions as low as 2.6 × 10−12 M and a linear increase in the current peak, when the Cd2+ concentration was between 4.4 × 10−12 and 5.3 × 10−12 M. The repeatability and the reproducibility of the usage of the UiO-66-NH2@PANI/GCE for Cd2+ analysis were also studied. Moreover, the study on the interfering effects of several common coexisting ions revealed that only Cu2+ with a concentration higher than 7.87 × 10−10 M could affect the electrochemical response of the sensor, due to the competition between Cu2+ and Cd2+ at the active sorption sites.
Very recently, a screen-printed carbon electrode (SCPE) based on UiO-NH2 and self-doped polyaniline nanofibers (SPAN) copolymerized with aniline (AN) and m-aminobenzenesulfonic acid (SAN) was prepared for the detection of trace levels of Cd2+.69 In contrast to PANI, which is conductive only in its protonated form or under low pH values, SPAN display high electrical conductivities, large specific surface areas, and relevant hydrophilicities, which are particularly useful features for the construction of electrochemical sensors. The synthetic procedure of SPAN@UIO-66-NH2 consists of two steps: (1) preparation of UIO-66-NH2 and (2) coating of UIO-66-NH2 with SPAN at controlled low-temperature by self-assembly technology (Fig. 4). The efficiency of the modified electrochemical sensor SPAN@UIO-66-NH2/SPCE to determine Cd2+ was determined by the SWASV method. Under optimized conditions, the response current of Cd2+ increased linearly with an increase in the concentration in the range from 4.4 × 10−12 to 8.9 × 10−10 M, with the detection limit found to be as low as 1.5 × 10−12 M. The stability and the repeatability of the proposed sensor, as well as its applicability in real water and urine samples, were also demonstrated.
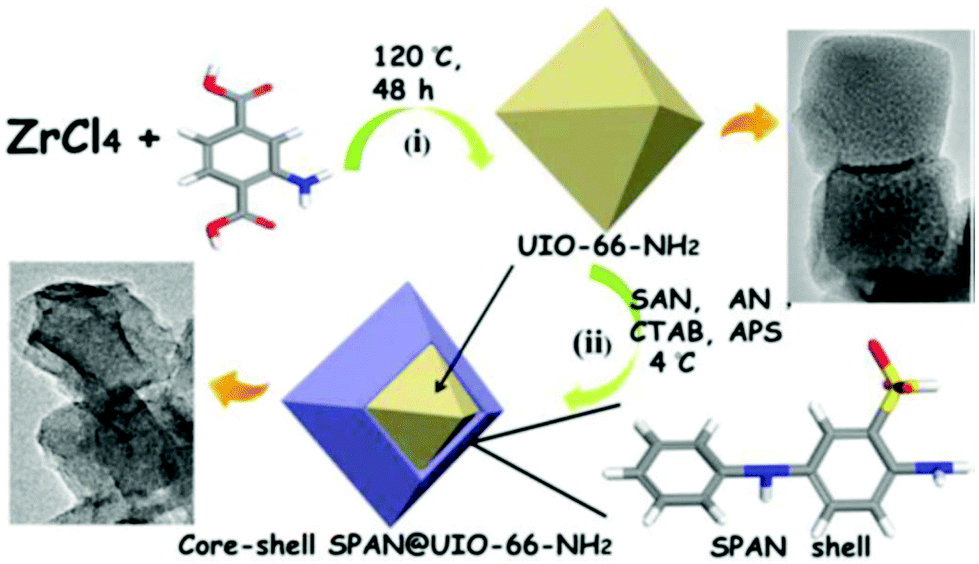 | ||
| Fig. 4 Schematic representation of the synthetic route of UiO-NH2, core–shell SPAN@UIO-66-NH2 and core–shell SPAN@UIO-66-NH2. Reprinted from ref. 69 with permission from The Electrochemical Society. | ||
 | ||
| Fig. 5 Schematic representation of the preparation process of the Au/Me2NH2@MOF-1/GCE and the procedures for determination of copper(II) via the cation exchange and nano-particle synergy effect enhanced electrochemical signal. Reproduced from ref. 70 with permission from the Royal Society of Chemistry. | ||
A linear increase in the peak current was observed for Cu2+ concentrations in the range of 5.0 × 10−12 to 9.0 × 10−7 M. The detection limit was found to be 1.0 × 10−12 M, which is quite lower than the established limit (1.57 × 10−5 M) for copper(II) by the U.S. EPA and the World Health Organization (WHO).71 A comparison study among the bare GCE, Au/GCE, Me2NH2@MOF-1/GCE and Au/Me2NH2@MOF-1/GCE revealed that the latter demonstrated greater electrochemical response to Cu2+. Moreover, the sensitivity and selectivity of the modified electrode were confirmed in the presence of several interfering ions and in river water samples, indicating a promising sensor for real environmental water sample analyses. According to the authors, anionic Me2NH2@MOF-1 can exchange [H2N(CH3)2]+ ions with Cu2+ thus enhancing the pre-concentration of Cu2+ traces and greatly increasing the analytical signal, while Au acts as a binder. Nevertheless, the proposed sensing mechanism is not supported by experimental data, as no Cu2+ sorption data are reported for Me2NH2@MOF-1 and the hydrolytic stability of the MOF has not been investigated.
Therefore, the determination of manganese is of vital importance. The modified NH2-MIL-125-Ti/CPE revealed favorable detection ability for Mn2+ in water, with a low detection limit of 4.0 × 10−9 M (well below the defined limit of 9.1 × 10−7 M by U.S. EPA) and a linear regression equation for manganese in the concentration range from 1.0 × 10−8 to 1.0 × 10−5 M. The determination of Mn2+ was achieved by CSV, after light irradiation of the samples for about 20 min. The proposed mechanism for the detection of manganese is based on (a) the oxidation of Mn2+ to MnO2 by the electron holes formed upon light irradiation of NH2-MIL-125-Ti and (b) reduction of the deposited MnO2 to Mn2+ during the cathodic stripping step (Fig. 6). Many important parameters to acquire the best electrochemical response were investigated in detail, including the mass percentage of the MOF, the pH of the supporting electrolytes and the photoirradiation time for the response of the sensor. Finally, the modified CPE was applied for determination of Mn2+ in various tea samples showing similar results to those obtained via the flame atomic absorption spectroscopic method (FAAS).
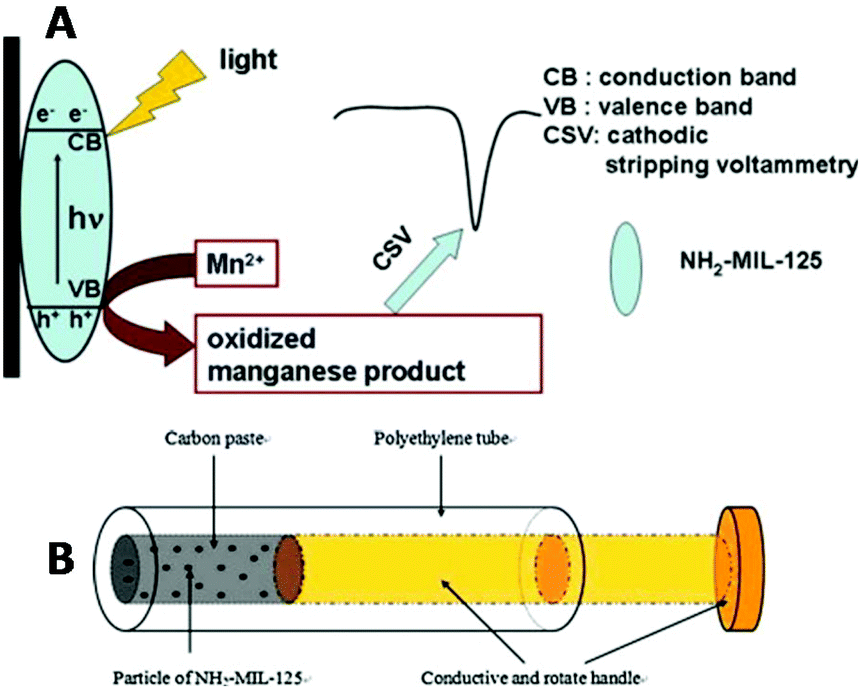 | ||
| Fig. 6 (A) The photooxidation procedure of Mn2+ at the NH2-MIL-125-Ti/CPE and its determination by CSV. (B) Schematic illustration of the structure of the NH2-MIL-125(Ti) modified carbon paste electrode. Reprinted from ref. 72d with permission from Elsevier. | ||
4.3 Multiple analyte determination
A UiO-66-NH2/GA (GA = graphene aerogel) composite was isolated via the in situ growth of the MOF on the GA matrix and the composite was applied on a GCE, for individual and for simultaneous determination of Cd2+, Pb2+, Cu2+ and Hg2+ in water samples (Fig. 7).74 GA enhances the conductivity of the composite material and speeds up the electron diffusion in the matrix. The determination of the examined heavy metals was achieved by DPASV.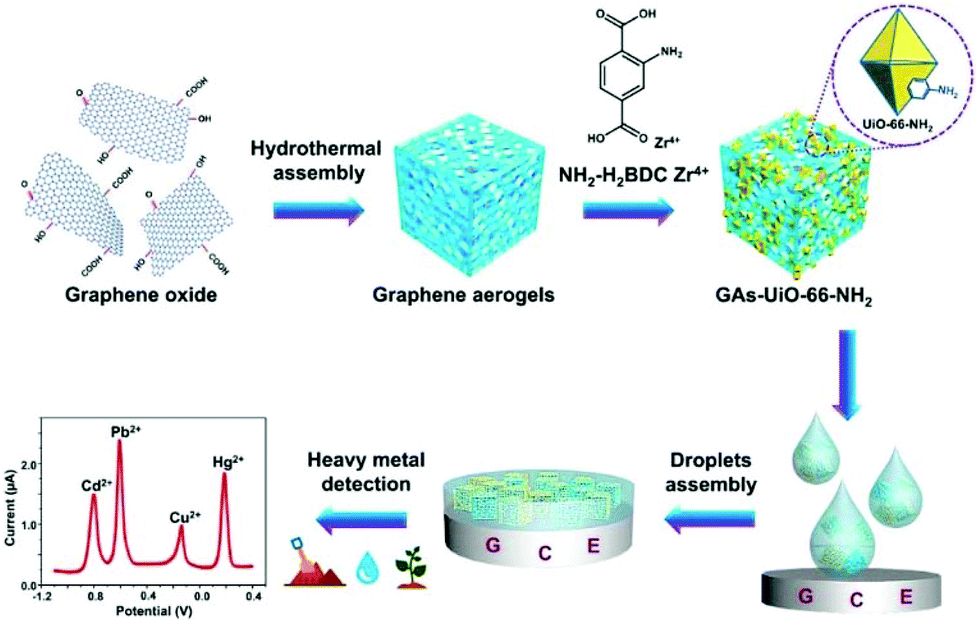 | ||
| Fig. 7 Synthesis of the GAs-UiO-66-NH2 composite via the in situ growth of the UiO-66-NH2 crystal on the GAs Matrix and the determination of heavy metal ions with the modified electrode. Reprinted from ref. 74 with permission from American Chemical Society. | ||
The linear ranges for the determination were 6.0 × 10−8 to 3.0 × 10−6 M for Cd2+, 1.0 × 10−8 to 4.0 × 10−6 M for Pb2+, 1.0 × 10−7 to 3.5 × 10−6 M for Cu2+, and 5.0 × 10−9 to 3.0 × 10−6 M for Hg2+. The limits of detection for each metal ion were found to be 2.0 × 10−8 M for Cd2+, 1.5 × 10−9 M for Pb2+, 7.0 × 10−9 M for Cu2+ and 2.0 × 10−9 M for Hg2+. The detection limits are below the acceptable limits of these metal ions in water. Furthermore, the electrochemical response of the GA-UiO-66-NH2/GCE was explored in the presence of all four examined ions. The results revealed lower sensitivity for simultaneous determination, since a narrower linear range, lower slope and in the case of Cu2+ a relatively higher limit of detection were observed, probably due to the interactions between different metals such as competitive deposition and formation of intermetallic compounds.75,76 Moreover, a weak peak that appeared between Pb2+ and Cu2+ may be attributed to the formation of Pb–Cu intermetallic compounds during the deposition step.76,77 Taking into account the excellent results which emerged from the studies in the presence of competitive ions and from studies with real samples, including river water, soil, and vegetables (spinach), the proposed sensor seems promising for accurate and reliable monitoring of toxic heavy metal ions under various conditions.
Very recently, our groups reported a simple ready-to-use electrochemical sensor based on graphite paste modified with the Ca-MOF {[Ca(H4L)(DMA)2]·2DMA, with H6L = N,N′-bis(2,4-dicarboxyphenyl)-oxalamide}, which can be used for the detection of trace levels of Pb2+, Cd2+, Cu2+ and Zn2+ in aqueous solutions via SWASV.78a Single crystals of the Ca-MOF were obtained after the reaction of Ca(NO3)3·4H2O with H6L in DMA under solvothermal conditions at 100 °C.78b The asymmetric unit of Ca-MOF comprises one half of H4L2−, one half of a Ca2+ ion and two DMA molecules. The H4L2− ligand bridges four Ca2+ ions through two carboxylate and two carboxylic O atoms to form a 2D framework (Fig. 8).
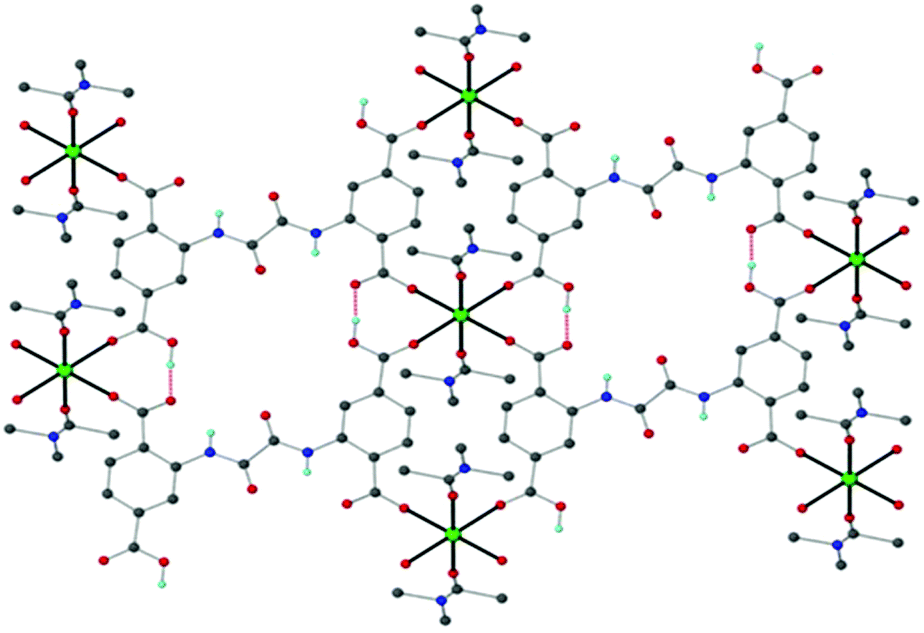 | ||
| Fig. 8 Schematic illustration of the structure of the Ca-MOF. Several H atoms have been removed for clarity. Color code: Ca, green; C, grey; O, red; N, blue; and H, cyan. Reproduced from ref. 78b with permission from the Royal Society of Chemistry. | ||
Our motivation to use the Ca-MOF as a chemical modifier in electrodes for the determination of heavy metals was the excellent capability of this material for efficient and selective sorption of Pb2+, Cd2+, Cu2+ and Zn2+ in aqueous media (due to the rapid exchange of Ca2+ ions by other metal ions) and its significant hydrolytic stability. Specifically, the Ca-MOF displays some of the highest sorption capacities reported for Pb2+ (522 mg g−1) and Cd2+ (220 mg g−1). In addition, the sorption capability of the Ca-MOF for these heavy metal ions is not influenced by the presence of common competitive ions (e.g. Ca2+, Na+). Furthermore, the Ca-MOF shows appreciable sorption capability for Zn2+ and Cu2+. The stability of the Ca-MOF in water was demonstrated via1H NMR data revealing no leaching of the H4L2− ligands, even in the presence of metal ions. The limits of detection using the Ca-MOF/CPE sensor were estimated to be 3.0 × 10−12 M for Pb2+, 2.2 × 10−11 M for Cu2+, 1.6 × 10−11 M for Zn2+ and 1.1 × 10−11 M for Cd2+, which were comparable to, or lower than, those of other electrochemical sensors (Fig. 9).79
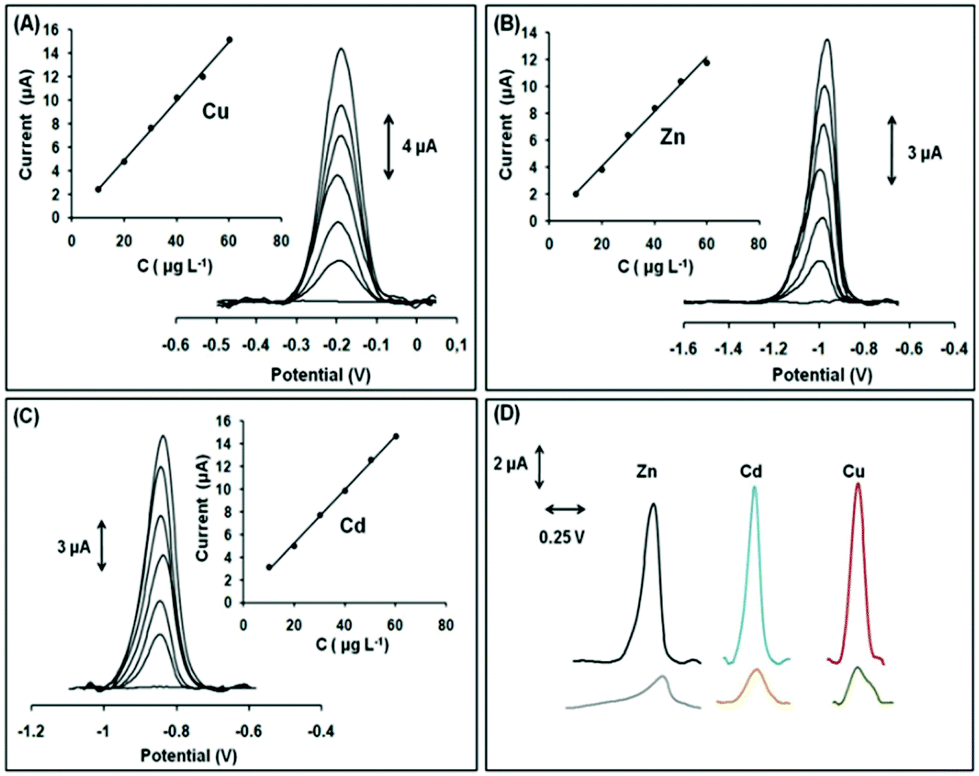 | ||
| Fig. 9 SWASV voltammograms and respective calibration plots in the range 10–60 μg L−1 (step: 10 μg L−1) of (A) Cu2+ in 0.1 mol L−1 acetate buffer, (B) Zn2+ in 1 × 10−3 mol L−1 KCl, and (C) Cd2+ in 0.1 mol L−1 acetate buffer (pH 4.5). (D) SWASV voltammograms of 30 μg L−1 of Zn2+, Cd2+, Cu2+ (black, blue, and red lines, respectively) obtained with the modified GPE with Ca-MOF and SWASV voltammograms of 30 μg L−1 of Zn2+, Cd2+, and Cu2+ (gray, orange, and green lines, respectively) obtained with the bare GPE. Reproduced from ref. 78a with permission from the Royal Society of Chemistry. | ||
Compared to conventional electrochemical sensors, this modified electrode can effectively detect Cu2+ (normally not possible since the oxidation peak of Cu appears outside the useful potential window of these electrodes), is able to detect simultaneously multiple heavy metal ions and is ready-to-use, i.e. it does not demand multistep reconstruction between the electrochemical measurements. Interestingly, the Ca-MOF modified electrode represents the first MOF-based sensor for the voltammetric determination of trace amounts of Zn2+.
5. Comparison of MOFs with other types of electrode modifiers
As mentioned in the introduction, there are a number of materials that have been tested as electrode modifiers, such as zeolites, clays, graphene, Au NPs, mesoporous silica and so on, for the fabrication of electrodes to be used for the determination of inorganic analytes (mainly heavy metal ions) via voltammetric techniques.80 These modifiers enhance the pre-concentration efficiency of the electrodes (mainly carbon paste or glassy carbon electrodes) resulting in particularly low detection limits (in the range 10−8–10−12 M) for the inorganic species.The analysis of the organic species is mainly done by modifying the glassy carbon electrodes (GCEs) which have been used as substrates for modification due to their inherent advantages such as a wide potential window, low background currents and chemical stability. The modification of GCEs takes place in order to construct more selective and more sensitive electrodes. For this purpose, there have been several reports on GCEs modified with carbon nanotubes,81 nanoparticles82 and polymers83 for the detection of organic molecules of biological interest such as dopamine, uric acid and ascorbic acid. For example, a typical single-walled carbon nanotube (SWCNT)/GCE81a exhibited a good linear working range of 0.01–0.20 × 10−6 M with the detection limit estimated to be 15 × 10−6 M for dopamine determination. A common nanoparticle modified electrode82d for the detection of the same analyte exhibited a linear range response of 0.5–30 × 10−6 M and a detection limit at 0.011 × 10−6 M. Organic polymer modified electrodes83d also responded well exhibiting a similar linear range for the detection of dopamine and almost the same limits of detection as those mentioned above were achieved.
MOFs have been recently employed as modifiers in electrodes to be used for trace analysis of heavy metal ions and organic species. MOF-modified electrodes have achieved detection limits comparable to those obtained with the other types of modified electrodes (see above). Nevertheless, the research on MOF-electrode modifiers is at an early stage since there is plenty of room for the optimization of their properties, as will be discussed in the next section. The fact that the properties of MOFs can be easily tuned by applying targeted modifications in the structures of known MOFs or designing new MOFs with appropriate structural features is a major advantage of MOFs compared to other types of modifiers.1 Furthermore, the high crystallinity of MOFs allows precise structural determination of MOFs loaded with analytes, which can facilitate the identification of analyte–MOF interactions and, thus, the design of MOFs with appropriate characteristics for efficient sorption capability and selectivity for the targeted analyte.84 The latter is particularly important for the utilization of a material as an electrode modifier, as it is directly related to the pre-concentration efficiency of the working electrode. The main disadvantage of MOFs for their extended use in electroanalytical applications is related to their poor electrical conductivities; however, this drawback of MOFs can be overcome by either mixing them with conductive materials (e.g. conductive graphite powder) or transforming them to composites with conductive polymers (e.g. PANI).69,71,78
6. Conclusions and prospects
The above discussion revealed that MOFs are highly promising as modifiers for electrodes that can be used for determination of trace amounts of inorganic and organic analytes via voltammetric techniques. The enhanced performance of MOF-modified electrodes is largely attributed to the intrinsic properties of MOFs such as their high surface area, larger pore size, ability to adsorb the analytes from solution, better distribution-exposure and loading, immobilization-stabilization of electrocatalytic centers (e.g. metal NPs) that may be utilized etc. All the above can be realized only in cases in which the MOFs maintain their structural stability during the electrode fabrication and performance. Nevertheless, in several cases, MOFs that were chosen to modify electrodes did not satisfy the basic features of appropriate electrode modifiers, i.e. stability under the conditions of analyses and sorption capability towards the targeted analyte. We thus propose that prior to the choice of a MOF to be used as an electrode modifier the following three steps should be followed (Scheme 1):1. Rational design and synthesis of a MOF possessing structural features (e.g. pore size and functional groups) that will lead to high affinity for a specific analyte.
2. Investigation of the structural stability of the MOF under the conditions of analyses (i.e. spectroscopic, analytical and PXRD studies of the MOF after its treatment with solutions having similar compositions to those of samples that will be analysed), in order to avoid using MOFs that either decompose or dissolve under the working conditions, thus leaching unwanted chemical species in the working media.
3. Evaluation of the sorption capacity of the MOF towards the analyte of interest, under the premise that the MOF has been proved to be stable under the conditions of analyses (step 2).
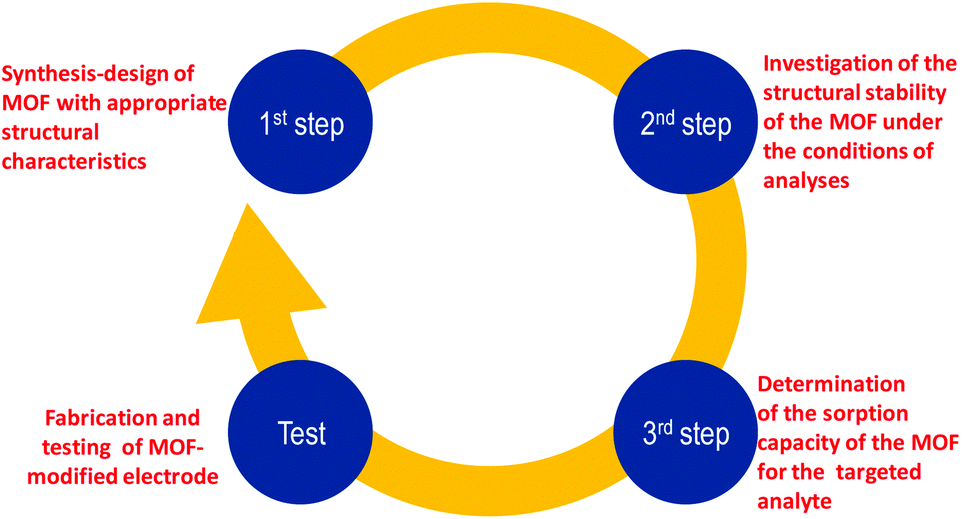 | ||
| Scheme 1 The proposed steps that should be followed prior to the fabrication and testing of a successful MOF-modified electrode. | ||
We should note that the stability and sorption studies (steps 2 and 3) are of vital importance since these two elements are closely related to the determination of the mechanism of the whole electrochemical procedure which can result in the design of new and improved MOFs as electrode modifiers.
Therefore, there is a lot of future research to be performed aiming at obtaining MOF-modified electrodes with optimum performance. Attention should be paid to known MOFs with demonstrated structural stability under the conditions of analysis and efficient sorption capacity for various organic or inorganic species. Such materials are particularly suitable to be tested as electrode modifiers. Furthermore, new MOFs can also be synthesized by employing polytopic organic ligands with functionalities having high affinity for specific analytes. In addition, new alkaline or alkaline earth MOFs as electrode modifiers may be involved in facile cation exchange with toxic heavy metal ions. All the above MOFs, showing efficient chemical stability and sorption capacity for the species of interest, may be then tested as electrode modifiers for voltammetric determination of trace analytes. We should also note that the development of optimum MOF-modified electrodes should involve a collaboration between inorganic synthetic chemists and analytical chemists with expertise in electroanalysis, since the optimization process requires both synthetic and analytical work. Overall, we believe that the research on MOF-modified electrodes will evolve rapidly over the coming years, as many researchers from various scientific disciplines will find this field of research particularly appealing for the development of new tools for trace analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Prof. M. Prodromidis (Department of Chemistry, University of Ioannina), Prof. A. Economou and Assistant Prof. C. Kokkinos (Department of Chemistry, National and Kapodistrian University of Athens) for fruitful discussions about voltammetric analysis.Notes and references
- (a) A. D. Katsenis, E. K. Brechin and G. S. Papaefstathiou, Encycl. Inorg. Bioinorg. Chem., 2014, 1–14 CAS; (b) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC; (c) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed; (d) S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed; (e) H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed; (f) C. Wang, X. Liu, N. Keser Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC; (g) P. Silva, S. M. F. Vilela, J. P. C. Tome and F. A. Almeida Paz, Chem. Soc. Rev., 2015, 44, 6774–6803 RSC; (h) P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill and M. J. Styles, Chem. Soc. Rev., 2014, 43, 5513–5560 RSC; (i) O. Shekhah, J. Liu, R. A. Fischer and C. Woell, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC; (j) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. A. Diamantis, A. Margariti, A. D. Pournara, G. S. Papaefstathiou, M. J. Manos and T. Lazarides, Inorg. Chem. Front., 2018, 5, 1493–1511 RSC.
- (a) W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC; (b) D. Zhao, Y. Cui, Y. Yang and G. Qian, CrystEngComm, 2016, 18, 3746–3759 RSC; (c) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC; (d) Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H. C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS; (e) P. Kumar, A. Deep and K. H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS.
- (a) D. Harvey, Modern analytical chemistry, McGraw-Hill, 2000, pp. 508–531 Search PubMed; (b) A. Economou and C. Kokkinos, Advances in Stripping Analysis of Metals, RSC Detection Science Series No. 6, Electrochemical Strategies in Detection Science, The Royal Society of Chemistry, 2016, pp. 1–18 Search PubMed.
- (a) Y. Wang, Y. Wu, J. Xie, H. Ge and X. Hu, Analyst, 2013, 138, 5113–5120 RSC; (b) Y. Wang, H. Ge, Y. Wu, G. Ye, H. Chen and X. Hu, Talanta, 2014, 129, 100–105 CrossRef CAS PubMed; (c) Y. Wang, Y. Wu, J. Xie and X. Hu, Sens. Actuators, B, 2013, 177, 1161–1166 CrossRef CAS.
- (a) N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed; (b) J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y. G. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 RSC.
- H. Xu, L. Zeng, S. Xing, Y. Xian, G. Shi and L. Jin, Electroanalysis, 2008, 20, 2655–2662 CrossRef CAS.
- (a) H. B. Herman, R. L. McNeely, P. Surana, C. M. Elliott and R. W. Murray, Anal. Chem., 1974, 46, 1258–1265 CrossRef; (b) Z. Yang, X. Huang, R. Zhang, J. Li, Q. Xu and X. Hu, Electrochim. Acta, 2012, 70, 325–330 CrossRef CAS.
- I. Švancara, K. Kalcher, A. Walcarius and K. Vytras, in Electroanalysis with carbon paste electrodes, CRC Press, 2012 Search PubMed.
- D. W. Pan, Y. E. Wang, Z. P. Chen, T. T. Lou and W. Qin, Anal. Chem., 2009, 81, 5088–5094 CrossRef CAS PubMed.
- L. Y. Cao, J. B. Jia and Z. H. Wang, Electrochim. Acta, 2008, 53, 2177–2182 CrossRef CAS.
- E. Svobodova-Tesarova, L. Baldrianova, M. Stoces, I. Svancara, K. Vytras, S. B. Hocevar and B. Ogorevc, Electrochim. Acta, 2011, 56, 6673–6677 CrossRef CAS.
- J. X. Guo, Y. Q. Chai, R. Yuan, Z. J. Song and Z. F. Zou, Sens. Actuators, B, 2011, 155, 639–645 CrossRef CAS.
- P. P. Dai and Z. S. Yang, Microchim. Acta, 2012, 176, 109–115 CrossRef CAS.
- M. A. El Mhammedi, M. Achak and A. Chtaini, J. Hazard. Mater., 2009, 161, 55–61 CrossRef CAS PubMed.
- Y. Gómez, L. Fernández, C. Borrás, J. Mostany and B. Scharifker, Talanta, 2011, 85, 1357–1363 CrossRef.
- P. K. Q. Nguyen and S. K. Lunsford, Talanta, 2012, 101, 110–121 CrossRef CAS.
- W. Wonsawat, S. Chuanuwatanakul, W. Dungchai, E. Punrat, S. Motomizu and O. Chailapakul, Talanta, 2012, 100, 282–289 CrossRef CAS PubMed.
- (a) M. F. Mousavi, A. Rahmani, S. M. Golabi, M. Shamsipur and H. Sharghi, Talanta, 2001, 55, 305–312 CrossRef CAS; (b) Z. M. Hu, C. J. Seliskar and W. R. Heineman, Anal. Chim. Acta, 1998, 369, 93–101 CrossRef CAS.
- (a) O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS; (b) D. A. Gómez-Gualdrón, P. Z. Moghadam, J. T. Hupp, O. K. Farha and R. Q. Snurr, J. Am. Chem. Soc., 2016, 138, 215–224 CrossRef PubMed; (c) I. M. Hönicke, I. Senkovska, V. Bon, I. A. Baburin, N. Bönisch, S. Raschke, J. D. Evans and S. Kaskel, Angew. Chem., Int. Ed., 2018, 57, 13780–13783 CrossRef PubMed.
- Y. Sun, Y. Li, N. Wang, Q. Q. Xu, L. Xu and M. Lin, Electroanalysis, 2018, 30, 474–478 CrossRef CAS.
- J. Li, J. Xia, F. Zhang, Z. Wang and Q. Liu, J. Chin. Chem. Soc., 2018, 65, 743–749 CrossRef CAS.
- Z. Wang, B. Ma, C. Shen and L.-Z. Cheong, Talanta, 2019, 197, 356–362 CrossRef CAS PubMed.
- Y. Ming, N. Kumar and D. J. Siegel, ACS Omega, 2017, 2, 4921–4928 CrossRef CAS.
- L. N. McHugh, M. J. McPherson, L. J. McCormick, S. A. Morris, P. S. Wheatley, S. J. Teat, D. McKay, D. M. Dawson, C. E. F. Sansome, S. E. Ashbrook, C. A. Stone, M. W. Smith and R. E. Morris, Nat. Chem., 2018, 10, 1096–1102 CrossRef CAS.
- F. A. Sofi, M. A. Bhat and K. Majid, New J. Chem., 2019, 43, 3119–3127 RSC.
- Y. Wang, W. Cao, L. Wang, Q. Zhuang and Y. Ni, Microchim. Acta, 2018, 185, 315 CrossRef PubMed.
- (a) Q. Chen, X. Li, X. Min, D. Cheng, J. Zhou, Y. Li, Z. Xie, P. Liu, W. Cai and C. Zhang, J. Electroanal. Chem., 2017, 789, 114–122 CrossRef CAS; (b) J. Li, J. Xia, F. Zhang, Z. Wang and Q. Liu, Talanta, 2018, 181, 80–86 CrossRef CAS PubMed.
- Y. Yang, Q. Wang, W. Qiu, H. Guo and F. Gao, J. Phys. Chem. C, 2016, 120, 9794–9803 CrossRef CAS.
- (a) D. Brondani, E. Zapp, R. da Silva Heying, B. de Souza and I. Cruz Vieira, Electroanalysis, 2017, 29, 2810–2817 CrossRef CAS; (b) S. Dong, G. Suo, N. Li, Z. Chen, L. Peng, Y. Fu, Q. Yang and T. Huang, Sens. Actuators, B, 2016, 222, 972–979 CrossRef CAS.
- Y. Cao, L. Wang, C. Shen, C. Wang, X. Hu and G. Wang, Sens. Actuators, B, 2019, 283, 487–494 CrossRef CAS.
- C. Li, Y. Zhou, X. Zhu, B. Ye and M. Hu, Int. J. Electrochem. Sci., 2018, 4855–4867 CrossRef CAS.
- C. T. P. da Silva, F. R. Veregue, L. W. Aguiar, J. G. Meneguin, M. P. Moisés, S. L. Fávaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, New J. Chem., 2016, 40, 8872–8877 RSC.
- T. T. Minh, N. H. Phong, H. Van Duc and D. Q. Khieu, J. Mater. Sci., 2017, 53, 2453–2471 CrossRef.
- Y. Shi, Y. Zhang, Y. Wang, H. Huang and J. Ma, Int. J. Electrochem. Sci., 2018, 13, 7643–7654 CrossRef CAS.
- P. Arul and S. A. John, J. Electroanal. Chem., 2017, 799, 61–69 CrossRef CAS.
- H. Hosseini, H. Ahmar, A. Dehghani, A. Bagheri, A. Tadjarodi and A. R. Fakhari, Biosens. Bioelectron., 2013, 42, 426–429 CrossRef CAS PubMed.
- L. Hu, N. Wu, J. Zheng, J. Xu, M. Zhang and P. He, Anal. Sci., 2014, 30, 663–668 CrossRef CAS PubMed.
- S. Li, Y. Duan, S. Lei, J. Qiao, G. Li and B. Ye, Sens. Actuators, B, 2018, 274, 218–227 CrossRef CAS.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- N. Karimian, H. Fakhri, S. Amidi, A. Hajian, F. Arduini and H. Bagheri, New J. Chem., 2019, 43, 2600–2609 RSC.
- M. Deng, S. Lin, X. Bo and L. Guo, Talanta, 2017, 174, 527–538 CrossRef CAS PubMed.
- (a) M. Chen, N. Gan, Y. Zhou, T. Li, Q. Xu, Y. Cao and Y. Chen, Sens. Actuators, B, 2017, 242, 1201–1209 CrossRef CAS; (b) Y. Yang, J. Cheng, B. Wang, Y. Guo, X. Dong and J. Zhao, Microchim. Acta, 2019, 186, 101–111 CrossRef PubMed.
- (a) X. Jiang, Z. Wang, H. Wang, Y. Zhuo, R. Yuan and Y. Chai, Chem. Commun., 2017, 53, 9705–9708 RSC; (b) Z.-H. Zhang, F.-H. Duan, J.-Y. Tian, J.-Y. He, L.-Y. Yang, H. Zhao, S. Zhang, C.-S. Liu, L.-H. He, M. Chen, D.-M. Chen and M. Du, ACS Sens., 2017, 2, 982–989 CrossRef CAS; (c) F. Su, S. Zhang, H. Ji, H. Zhao, J.-Y. Tian, C.-S. Liu, Z. Zhang, S. Fang, X. Zhu and M. Du, ACS Sens., 2017, 2, 998–1005 CrossRef CAS PubMed; (d) C.-S. Liu, C.-X. Sun, J.-Y. Tian, Z.-W. Wang, H.-F. Ji, Y.-P. Song, S. Zhang, Z.-H. Zhang, L.-H. He and M. Du, Biosens. Bioelectron., 2017, 91, 804–810 CrossRef CAS; (e) C. Gu, C. Guo, Z. Li, M. Wang, N. Zhou, L. He, Z. Zhang and M. Du, Biosens. Bioelectron., 2019, 134, 8–15 CrossRef CAS PubMed; (f) M. Wang, M. Hu, Z. Li, L. He, Y. Song, Q. Jia, Z. Zhang and M. Du, Biosens. Bioelectron., 2019, 142, 111536 CrossRef CAS PubMed.
- T. Y. Huang, C. W. Kung, Y. T. Liao, S. Y. Kao, M. Cheng, T. H. Chang, J. Henzie, H. R. Alamri, Z. A. Alothman, Y. Yamauchi, K. C. Ho and K. C.-W. Wu, Adv. Sci., 2017, 4, 1700261 CrossRef PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- M. Cao, X. Yin, X. Bo and L. Guo, J. Electroanal. Chem., 2018, 824, 153–160 CrossRef CAS.
- H. Wang, Q. Hu, Y. Meng, Z. Jin, Z. Fang, Q. Fu, W. Gao, L. Xu, Y. Song and F. Lu, J. Hazard. Mater., 2018, 353, 151–157 CrossRef CAS.
- J. Gu, X. Yin, X. Bo and L. Guo, ChemElectroChem, 2018, 5, 2893–2901 CrossRef CAS.
- W. Zhang, L. Zong, G. Geng, Y. Li and Y. Zhang, Sens. Actuators, B, 2018, 257, 1099–1109 CrossRef CAS.
- L. Wang, T. Meng, Y. Fan, C. Chen, Z. Guo, H. Wang and Y. Zhang, J. Colloid Interface Sci., 2018, 524, 1–7 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- L. Chen, Y. Peng, H. Wang, Z. Gu and C. Duan, Chem. Commun., 2014, 50, 8651–8654 RSC.
- J. Guan, Y. Hu, Y. Wang, H. Li, Z. Xu, T. Zhang, P. Wu, S. Zhang, G. Xiao, W. Ji, L. Li, M. Zhang, Y. Fan, L. Li, B. Zheng, W. Zhang, W. Huang and F. Huo, Adv. Mater., 2017, 29, 1606290 CrossRef.
- T. Zhang, L. Wang, C. Gao, C. Zhao, Y. Wang and J. Wang, Nanotechnol., 2018, 29, 074003 CrossRef.
- G. D. Tarlas, A. D. Katsenis and G. S. Papaefstathiou, Eur. J. Inorg. Chem., 2018, 4458–4464 CrossRef CAS.
- K. Liu, H. You, G. Jia, Y. Zheng, Y. Huang, Y. Song, M. Yang, L. Zhang and H. Zhang, Cryst.Growth Des., 2010, 10, 790–797 CrossRef CAS.
- J. Zhang, X. Xu and L. Chen, Sens. Actuators, B, 2018, 261, 425–433 CrossRef CAS.
- Z. Chang, N. Gao, Y. Li and X. He, Anal. Methods, 2012, 4, 4037–4041 RSC.
- M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639–12641 CrossRef CAS.
- Y. Liu, V. C. Kravtsov, R. Larsen and M. Eddaoudi, Chem. Commun., 2006, 14, 1488–1490 RSC.
- Y. Wang, K. Du, Y. Chen, Y. Li and X. He, Anal. Methods, 2016, 8, 3263–3269 RSC.
- D. Y. Hong, Y. K. Hwang, C. Serre, G. Ferey and J. S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- Y. K. Seo, J. W. Yoon, J. S. Lee, J. K. Hwang, C. H. Jun, J. S. Chang, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, S. Bourrelly, P. L. Llewellyn, P. Horcajada, C. Serre and G. Ferey, Adv. Mater., 2012, 24, 806–810 CrossRef CAS.
- H. Guo, Z. Zheng, Y. Zhang, H. Lin and Q. Xu, Sens. Actuators, B, 2017, 248, 430–436 CrossRef CAS.
- M. Roushani, A. Valipour and Z. Saedi, Sens. Actuators, B, 2016, 233, 419–425 CrossRef CAS.
- Y. Wang, L. Wang, W. Huang, T. Zhang, X. Hu, J. A. Perman and S. Ma, J. Mater. Chem. A, 2017, 5, 8385–8393 RSC.
- M. Chen, N. Gan, Y. Zhou, T. Li, Q. Xu, Y. Cao and Y. Chen, Sens. Actuators, B, 2017, 242, 1201–1209 CrossRef CAS.
- X. Zhao, W. Bai, Y. Yan, Y. Wang and J. Zhang, J. Electrochem. Soc., 2019, 166, B873–B880 CrossRef CAS.
- J. C. Jin, J. Wu, G. P. Yang, Y. L. Wu and Y. Y. Wang, Chem. Commun., 2016, 52, 8475–8478 RSC.
- S. D. Richardson, Anal. Chem., 2011, 84, 747–778 CrossRef.
- (a) S. N. Kim, J. Kim, H. Y. Kim, H. Y. Cho and W. S. Ahn, Catal. Today, 2013, 204, 85–93 CrossRef CAS; (b) F. Jeremias, V. Lozan, S. K. Henninger and C. Janiak, Dalton Trans., 2013, 42, 15967–15973 RSC; (c) S. Vaesen, V. Guillerm, Q. Y. Yang, A. D. Wiersum, B. Marszalek, B. Gil, A. Vimont, M. Daturi, T. Devic, P. L. Llewellyn, C. Serre, G. Maurin and G. De Weireld, Chem. Commun., 2013, 49, 10082–10084 RSC; (d) Q. Xu, Y. Wang, G. Jin, D. Jin, K. Li, A. Mao and X. Hu, Sens. Actuators, B, 2014, 201, 274–280 CrossRef CAS.
- R. Witholt, R. H. Gwiazda and D. R. Smith, Neurotoxicol. Teratol., 2000, 22, 851–861 CrossRef CAS PubMed.
- M. Lu, Y. Deng, Y. Luo, J. Lv, T. Li, J. Xu, S. W. Chen and J. Wang, Anal. Chem., 2018, 91, 888–895 CrossRef PubMed.
- (a) B. Cheng, L. Zhou, L. Lu, J. Liu, X. Dong, F. Xi and P. Chen, Sens. Actuators, B, 2018, 259, 364–371 CrossRef CAS; (b) A. A. Ensafi, P. Nasr-Esfahani and B. Rezaei, Sens. Actuators, B, 2018, 270, 192–199 CrossRef CAS.
- Z. Guo, D. D. Li, X. K. Luo, Y. H. Li, Q. N. Zhao, M. M. Li, Y. T. Zhao, T. S. Sun and C. J. Ma, J. Colloid Interface Sci., 2017, 490, 11–22 CrossRef CAS.
- Y. Wei, C. Gao, F. L. Meng, H. H. Li, L. Wang, J. H. Liu and X. J. Huang, J. Phys. Chem. C, 2012, 116, 1034–1041 CrossRef CAS.
- (a) A. Pournara, A. Margariti, G. D. Tarlas, A. Kourtellaris, V. Petkov, C. Kokkinos, A. Economou, G. S. Papaefstathiou and M. J. Manos, J. Mater. Chem. A, 2019, 7, 15432–15443 RSC; (b) A. Margariti, S. Rapti, A. D. Katsenis, T. Friščić, Y. Georgiou, M. J. Manos and G. S. Papaefstathiou, Inorg. Chem. Front., 2017, 4, 773–781 RSC.
- (a) L. Xiao, H. Xu, S. Zhou, T. Song, H. Wang, S. Li, W. Gan and Q. Yuan, Electrochim. Acta, 2014, 143, 143–151 CrossRef CAS; (b) N. Serrano, J. M. Díaz-Cruz, C. Ariño and M. Esteban, Anal. Bioanal. Chem., 2009, 396, 1365–1369 CrossRef PubMed; (c) P. S. Adarakatti, V. Gangaiah and A. Siddaramanna, Mater. Sci. Semicond. Process., 2018, 84, 157–166 CrossRef CAS.
- (a) C. Mousty, Appl. Clay Sci., 2004, 27, 159–177 CrossRef CAS; (b) D. R. Rolison, Chem. Rev., 1990, 90, 867–878 CrossRef CAS; (c) B. J. Sanghavi, P. K. Kalambate, S. P. Karna and A. K. Srivastava, Talanta, 2014, 120, 1–9 CrossRef CAS; (d) K. Kalcher, Electroanalysis, 1990, 2, 419–433 CrossRef CAS.
- (a) S. Alwarappan, G. Liu and C.-Z. Li, Nanomedicine, 2010, 6, 52–57 CrossRef CAS PubMed; (b) D. P. Quan, D. P. Tuyen, T. D. Lam, P. T. N. Tram, N. H. Binh and P. H. Viet, Colloids Surf., B, 2011, 88, 764–770 CrossRef CAS PubMed; (c) C.-L. Sun, C.-T. Chang, H.-H. Lee, J. Zhou, J. Wang, T.-K. Sham and W. F. Pong, ACS Nano, 2011, 5, 7788–7795 CrossRef CAS PubMed; (d) D. Jia, J. Dai, H. Yuan, L. Lei and D. Xiao, Talanta, 2011, 85, 2344–2351 CrossRef CAS PubMed; (e) X. Liu, Y. Peng, X. Qu, S. Ai, R. Han and X. Zhu, J. Electroanal. Chem., 2011, 654, 72–78 CrossRef CAS; (f) S. He, Y. Yu, Z. Chen, Q. Shi and L. Zhang, Anal. Lett., 2014, 48, 248–258 CrossRef.
- (a) C. F. Tang, S. A. Kumar and S. M. Chen, Anal. Biochem., 2008, 380, 174–183 CrossRef CAS; (b) J. Huang, Y. Liu, H. Hou and T. You, Biosens. Bioelectron., 2008, 24, 632–637 CrossRef CAS; (c) M. S. Hsu, Y. L. Chen, C. Y. Lee and H. T. Chiu, ACS Appl. Mater. Interfaces, 2012, 4, 5570–5575 CrossRef CAS; (d) J. Du, R. Yue, F. Ren, Z. Yao, F. Jiang, P. Yang and Y. Du, Gold Bull., 2013, 46, 137–144 CrossRef; (e) C. L. Sun, H. H. Lee, J. M. Yang and C. C. Wu, Biosens. Bioelectron., 2011, 26, 3450–3455 CrossRef CAS; (f) M. Hadi and A. Rouhollahi, Anal. Chim. Acta, 2012, 721, 55–60 CrossRef CAS; (g) S. Palanisamy, Mater. Res. Express, 2014, 1, 045020 CrossRef.
- (a) H. Filik, A. A. Avan, S. Aydar and R. B. Arpacı, Int. J. Electrochem. Sci., 2014, 9, 2775–2789 Search PubMed; (b) D. Han, T. Han, C. Shan, A. Ivaska and L. Niu, Electroanalysis, 2010, 22, 2001–2008 CrossRef CAS; (c) X. Liu, X. Ou, Q. Lu, J. Zhang, S. Chen and S. Wei, RSC Adv., 2014, 4, 42632–42637 RSC; (d) Y. Wang and C. Bi, J. Mol. Liq., 2013, 177, 26–31 CrossRef CAS; (e) C. Wang, R. Yuan, Y. Chai, Y. Zhang, F. Hu and M. Zhang, Biosens. Bioelectron., 2011, 30, 315–319 CrossRef CAS PubMed.
- (a) S. Rapti, D. Sarma, S. A. Diamantis, E. Skliri, G. S. Armatas, A. C. Tsipis, Y. S. Hassan, M. Alkordi, C. D. Malliakas, M. G. Kanatzidis, T. Lazarides, J. C. Plakatouras and M. J. Manos, J. Mater. Chem. A, 2017, 5, 14707–14719 RSC; (b) S. Rapti, S. A. Diamantis, A. Dafnomili, A. Pournara, E. Skliri, G. S. Armatas, A. C. Tsipis, I. Spanopoulos, C. D. Malliakas, M. G. Kanatzidis, J. C. Plakatouras, F. Noli, T. Lazarides and M. J. Manos, J. Mater. Chem. A, 2018, 6, 20813–20821 RSC; (c) P. Kumar, A. Pournara, K. H. Kim, V. Bansal, S. Rapti and M. J. Manos, Prog. Mater. Sci., 2017, 86, 25–74 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2019 |