
A nanoporous MgO based nonenzymatic electrochemical sensor for rapid screening of hydrogen peroxide in milk†
Xiu-xiu Dongab,
Mei-ying Lia,
Nan-nan Fengb,
Yuan-ming Suna,
Chi Yang*b and
Zhen-lin Xu*a
aThe Guangdong Provincial Key Laboratory of Food Quality and Safety, College of Food Science, South China Agriculture University, Guangzhou 510642, China. E-mail: jallent@163.com; Tel: +86 20 8528 3448, +86 513 85051728
bDepartment of Pharmacy, Nantong University, Nantong 226001, China. E-mail: toyangchi@ntu.edu.cn; Tel: +86 20 513 85051728
First published on 30th September 2015
Abstract
In this work, a nonenzymatic electrochemical sensor based on nanoporous magnesium oxide (MgO) was developed for the rapid screening of hydrogen peroxide (H2O2) in milk. The nanoporous MgO, which was synthesized by a novel one-pot reaction process at low temperature, showed a large specific surface area and was favorable for constructing biosensors. Based on the nanoporous MgO, a nano-sensor for H2O2 was developed. The sensor exhibited extremely high electrocatalytic activity toward the oxidation of H2O2 with a detection limit of 3.3 μM and a wide linear range from 0.05 to 0.2 and 0.2 to 10 mM. The averaged recovery was determined to be from 94.3% to 119% for spiked milk samples. The proposed method is ideally suited for the fast screening of H2O2 misuse in milk at low cost.
1. Introduction
Food additives are commonly used in food processing, transportation, and storage to maintain the nutritional value, taste and flavor of the food and reduce the risk of foodborne disease; they are indispensable for the production and processing of many foods.1,2 As a good antiseptic and stabilizer, hydrogen peroxide (H2O2) is widely used in the fields of foods, pharmaceuticals, and dental products. However, the excessive addition of H2O2 in food is not completely safe for people’s health. In the USA, H2O2 is approved for treating milk and its weight cannot exceed 0.05% of the milk weight.3 For the government standard of China, the qualitative detection limit of H2O2 is 0.5 mg kg−1 in food. According to the World Food and Agriculture Organization and the United Nations Agriculture Organization, the addition of H2O2 in milk may be permitted at 0.05–0.25%.4 Packing boxes for milk are usually sterilized with H2O2 in China and some other countries, and residual H2O2 molecules can transfer to the milk and maintain their strong oxidizing properties. Even though it has not been reported that people have died from H2O2 directly, H2O2-induced oxidative stress in endothelial cells5 and H2O2-induced DNA damage in human leucocytes and cell death in PC12 cells6 have been reported in recent years. In China, the misuse of H2O2 in milk has been commonly reported. Thus, the development of a technique for the fast, reliable and on-line monitoring of residual H2O2 in foods is very urgent.Numerous analytical methods for H2O2 determination have been developed, such as FTIR,7 fluorescence,3 chemiluminescence,8 and electrochemical methods.9–12 Among these methods, the electrochemical method has received considerable attention due to its high sensitivity, simple instrumentation, and excellent compatibility with miniaturization technologies. Commonly used electrochemical methods for H2O2 determination include enzyme-based and nonenzymatic electrochemical sensors. Enzyme-based electrochemical biosensors have been widely used because of their remarkable selectivity, low detection limit and wide linear range.13 However, their main shortcoming is that enzymes would not maintain their activity under the detection conditions because of their intrinsic nature.14 In contrast, nano-material based nonenzymatic sensors could endure harsh physicochemical environments.9 Nanometer materials, such as precious metal nanoparticles, carbon materials, transition metal nanoparticles, transition metal oxides and alkaline earth metal oxides,15–20 are the most important options for building nonenzymatic sensors to solve the problems of low long-term operational stability and poor reproducibility. Magnesium oxide (MgO) has been applied in ascorbic acid, dopamine, glucose, and uric acid sensors21,22 due to its environmental friendliness, electroconductivity, electrocatalysis, and the other general characteristics of nanometer materials.23–26 However, the traditional synthesis method for nanometer-sized MgO is under a super high temperature (∼950 °C), which makes it difficult to prepare in a bioanalysis laboratory. Expensive equipment and professional operators were also required for the preparation of MgO.23 Furthermore, nanometer-sized MgO with low aspect ratios such as nanorods or nanodisks have been reported.21,22
In this work, nanoporous MgO with a high aspect ratio was synthesized by a facile method under a lower temperature, and then dropped onto the surface of an electrode to establish a nonenzymatic electrochemical sensor for H2O2. The results showed that the nanoporous MgO had extraordinary advantages for H2O2 determination compared to previously reported works. The nanoporous MgO based electrochemical sensor exhibits good sensitivities for H2O2 of 30.729 μA mM−1 at 0.05–0.2 mM and 6.602 μA mM−1 at 0.2–10 mM, and the limit of detection is 3.3 μM. Meanwhile, there was no obvious catalytic ability for ascorbic acid, folic acid and glucose at a potential of 0.9 V. In addition, the prepared sensor was applied to the detection of H2O2 in real milk samples, and it exhibited good sensitivity and specificity. Therefore, this work would provide an excellent platform for H2O2 screening in foods.
2. Experimental section
2.1 Reagents and materials
Magnesium acetate tetrahydrate (Mg (CH3COO)2·4H2O), sodium citrate dihydrate (C6H5Na3O7·2H2O) and ascorbic acid (C6H8O6) were purchased from Aladdin, hexamethylenetetramine (C6H16N4, HMT) and folic acid (C19H19N7O6) were purchased from Sigma (USA). A 0.1 M pH 7.0 phosphate buffer (PBS) was prepared by mixing stock solutions of KH2PO4 and Na2HPO4. Other reagents were of analytical grade and used as received without further purification. All solutions were prepared using double distilled water.2.2 Apparatus
All electrochemical measurements were recorded using a CHI630D electrochemical workstation (Shanghai Chenhua Co., China) controlled by a microcomputer. Electrochemical studies were carried out using a conventional three-electrode system, in which the working, auxiliary and reference electrodes were a glassy carbon electrode (GCE) (Φ = 3 mm) or modified GCE, a Pt-rod and Ag/AgCl (3.0 mol L−1 in KCl), respectively. All electrochemical experiments were recorded when the surface of the electrode was stabilised by 10 cycles over a potential range from −0.2 to +1.2 V (vs. Ag/AgCl). Cyclic voltammetry (CV) experiments were carried out in PBS solution (pH 7.0) at a scan rate of 50 mV s−1. Magnesia was prepared using an autoclave (Shanghai Hanjun Co., China) and characterized using field emission scanning electron microscopy with energy-dispersive X-ray analysis (SEM, EDX, and JSM-6700F).2.3 Synthesis of nanoporous MgO
Nanoporous MgO was synthesized through a low-temperature hydrothermal process. 50 mM Mg (CH3COO)2·4H2O, 50 mM C6H16N4 and 10 mM C6H5Na3O7·2H2O were dissolved in an aqueous solution. The mixed aqueous solution was sonicated at room temperature to dissolve the compounds completely. Then the solution was transferred to a Teflon-lined stainless steel autoclave and heated at 90 °C for 3 h. A white precipitate was collected by centrifugation for ∼10 min at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 081 × g to remove the supernatant, washed with distilled water and ethanol, and finally dried at 120 °C for 10 h.
081 × g to remove the supernatant, washed with distilled water and ethanol, and finally dried at 120 °C for 10 h.
2.4 Preparation of the H2O2 sensor
The bare GCE was polished successively with 0.3 and 0.05 μm alumina slurry, and thoroughly washed ultrasonically in ethanol and distilled water. Then the electrode was rinsed with distilled water, and dried in the air. 1.0 mg of nanoporous MgO synthesized by the method described above was dispersed in a mixed solution of 200 μL of isopropanol and 200 μL of Nafion (0.5 wt%) and ultrasonicated (∼30 min) to form a homogeneous suspension. The working electrode was prepared by dropping 4 μL of suspension on a polished GCE, and then covering the electrode with 4 μL of Nafion solution (0.5 wt%). A similar method for sensor fabrication in electrochemical catalysis has been applied and reported before.27 The addition of a Nafion layer over the MgO nanocomposite modified electrode resulted in a significant increase in the sensor signal due to the enhanced accumulation of protons generated by the catalytic reaction at the electrode surface. Another advantage of using the Nafion polymer for sensor fabrication is that it prevented the loss of the MgO nanocomposite. The obtained nanoporous MgO/GCE was then allowed to dry slowly. Based on a unique nanomaterial modified electrode, the nonenzymatic sensor expressed different sensitivity and specificity to the unmodified GCE; a schematic illustration of the stepwise procedure for the biosensor fabrication is shown in Fig. 1. For H2O2 detection, the electrochemical curves were measured using the CHI630D electrochemical workstation and the MgO/GCE was used as the working electrode.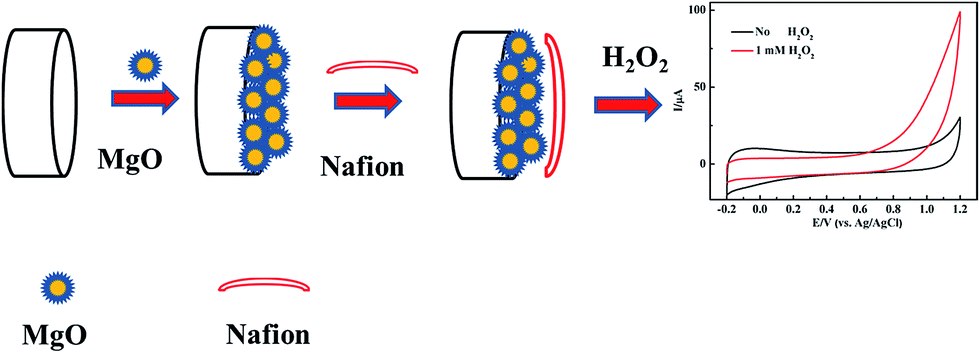 | ||
| Fig. 1 Schematic diagram of the electrochemical H2O2 sensor. | ||
3. Results and discussion
3.1 Synthesis and characterization of nanoporous MgO
Numerous methods for the synthesis of MgO have been reported. DC arc plasma jet chemical vapor deposition (DCPJCVD) has been used the most. The main deposition parameters are as follows: an arc power of 18 kW, an argon flow rate of 1.5 L min−1, a hydrogen flow rate of 10 L min−1, the substrate temperature maintained at 950 °C, and the pressure in the reaction chamber maintained at 4.0 kPa.21 This process comprises four stages: preparation, starting, running, and shutdown, and every stage needs continuous monitoring to ensure the normal operation of the instrument. The other method is a thermal evaporation process,22 and the resulting MgO based sensor could detect glucose with oxidation by glucose oxidase but not magnesium oxide. Therefore, these methods were known for the synthesis of nano MgO in earlier research, but the disadvantages have become more and more obvious due to the demand for plenty of product and some specialised properties.The band gap energy for MgO can be made smaller by decreasing the reaction temperature,28 with higher conductivity, which is beneficial to the structure of a nonenzymatic H2O2 sensor.21 In this work, therefore, we tried to synthesize nanoporous MgO at low temperature by a hydrothermal method. The morphology of the MgO was characterized through SEM (Fig. 2A). It was observed that the MgO nanostructure has a uniform distribution with a nanoporous shape and the average diameter of the MgO nanopores is about 450 nm. Apparently, the nanostructure could provide a high specific surface area. EDX analysis was used to study the components of the nanoporous MgO on an N type silicon wafer substrate (Fig. 2B). It reveals only Mg and O as the constituent elements (the Si, N, and C come from the N type silicon wafer substrate). Therefore, nanoporous MgO was synthesized successfully.
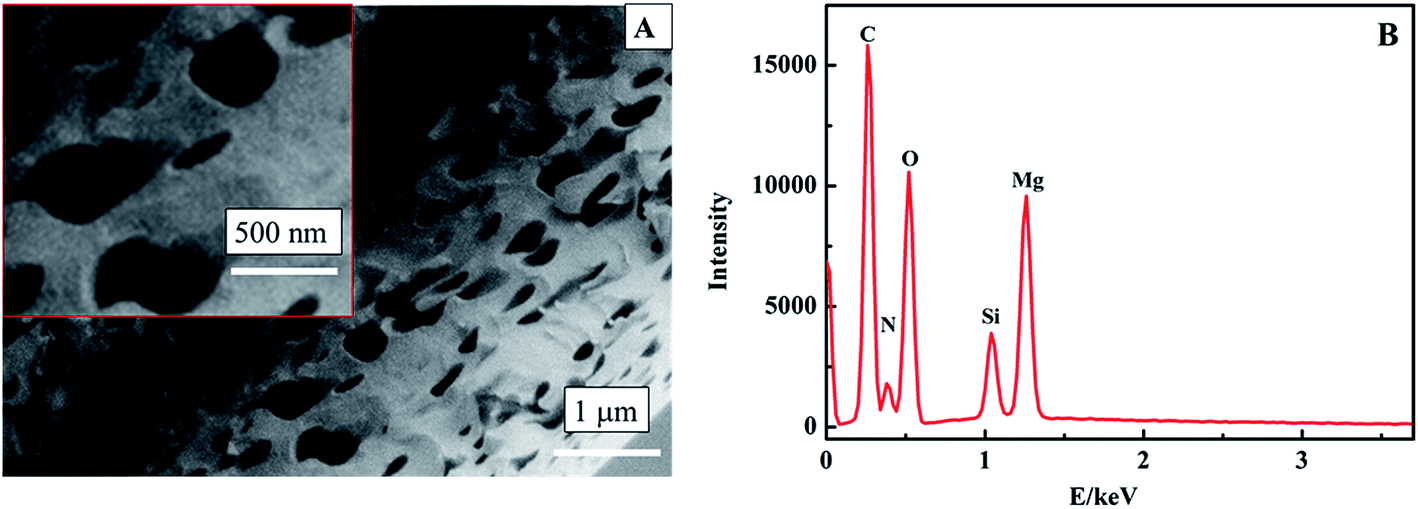 | ||
| Fig. 2 SEM image (A) and EDX analysis (B) of the nanoporous MgO. | ||
3.2 Construction of biosensor and electrochemical response of nanoporous MgO/GCE to H2O2
In a general way, the nano-material based sensors show selective electrocatalysis at different potentials to some molecules. So we modified the GCE with the pretreated nanoporous MgO and Nafion solution to judge the appropriate potential for H2O2 by cyclic voltammetry (CV) measurements. Fig. 3 shows the typical CV responses of the nanoporous MgO/GCE between the potentials of −0.2 and +1.2 V in the presence of 1.0 mM H2O2. As shown in Fig. 3b and b′, a dramatic change in the CV curve was observed with the MgO/GCE (red line). In contrast, almost no change was observed with the GCE, especially below 0.9 V (Fig. 3a and a′). H2O2 has a wide electrocatalytic oxidation range from 0.75 to 1.2 V (from the CV electrocatalysis, as seen in Fig. 3). Meanwhile, the selectivity of the sensor for the detection of H2O2 showed that the oxidation of interference substances did not occur at a potential of 0.9 V (from the selectivity of the sensor, as shown in Fig. S2†). On the other hand, many interference substances could not be catalyzed at lower potential. Therefore, for fulfilling both the selectivity and sensitivity of the present biosensor, 0.9 V was selected as the working potential. The above results demonstrated that the nanoporous MgO modified GCE attained terrific electrochemical performance for the detection of H2O2. To illustrate the performance of the nanoporous MgO based sensor, other measurements were also taken. For example, the relationship between the response current of the oxidation of H2O2 and the scan rate is shown in Fig. S1A and B,† and the effect of pH on the electrochemical response to H2O2 at the MgO/GCE is shown in Fig. S1C and D.† A linear regression equation Ip (μA) = −0.232 + 3.132 × V1/2 (mV1/2 s−1/2), with a correlation coefficient of 0.998, was obtained. This result indicated that the electrochemical reaction of the modified electrode belonged to diffusion controlled electrochemical processes. Meanwhile, the non-linearity of the pH vs. current relationship showed that a weakly alkaline environment could accelerate the oxidization of H2O2 because of the existence of hydroxide ions in the solution, but an exorbitant concentration (pH > 7.8) did not have an obvious influence. As shown in Fig. S1D,† there is a linear relationship between the current and pH (6.4–7.4). Otherwise, the linear relationship is inconsistent. The major reason for this phenomenon may be that the H2O2 molecule is sensitive to the existence of hydroxide ions in low concentration. Considering the sensitive determination of H2O2 and the pH of milk, a scan rate of 50 mV s−1 and a solution of pH = 7.0 were chosen as the optimal experimental conditions for the subsequent electrochemical detection. In each case the potential was 0.9 V.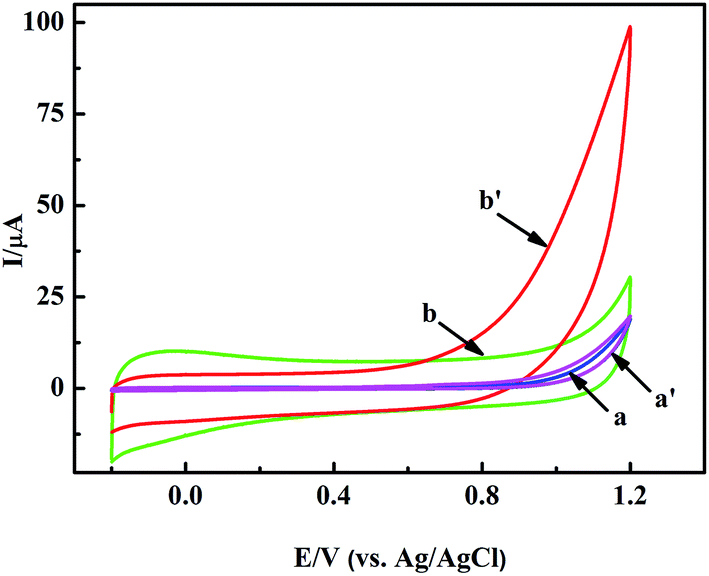 | ||
| Fig. 3 CVs of the GCE (a and a′), and nanoporous MgO/GCE (b and b′) in PBS solution in the absence (a and b) and presence (a′ and b′) of 1 mM H2O2. | ||
3.3 Calibration curve and interferences
Fig. 4A shows the CV curves of the nanoporous MgO modified electrode in 0.1 M PBS solution (pH 7.0) in the presence of H2O2. The concentrations of H2O2 were 0.05, 0.075, 0.1, 0.15, 0.2, 0.5, 1.0, 2.0, 5.0, and 10.0 mM. We obtained two linear regression equations at 0.9 V: y = 30.729x + 9.435 (R2 = 0.984, 0.05–0.2 mM) and y = 6.602x + 15.729 (R2 = 0.997, 0.2–10 mM), and a detection limit of 3.3 μM was calculated using the equation DL = 3 × SD (standard deviation)/slope (Fig. 4B). At low H2O2 levels the local concentration at the electrode surface is rapidly depleted as the substrate is converted into product29 by the catalytic action of the nanoporous MgO, resulting in a high sensitivity of the electrode response. At higher H2O2 concentrations, the nano-material is supplied with substrate for a longer period of time and the reaction proceeds over a larger time window. This, together with the possibility of fouling of the electrode surface by the reaction products, results in a lower slope. Also, it attains a saturation level at higher concentration. Thus, the sensor showed different linear correlations at different concentration ranges. A related summary of the merit of recently reported methods for the determination of H2O2 is shown in Table 1. It reveals that the proposed sensor has a wider linear range, lower detection limit and higher sensitivity.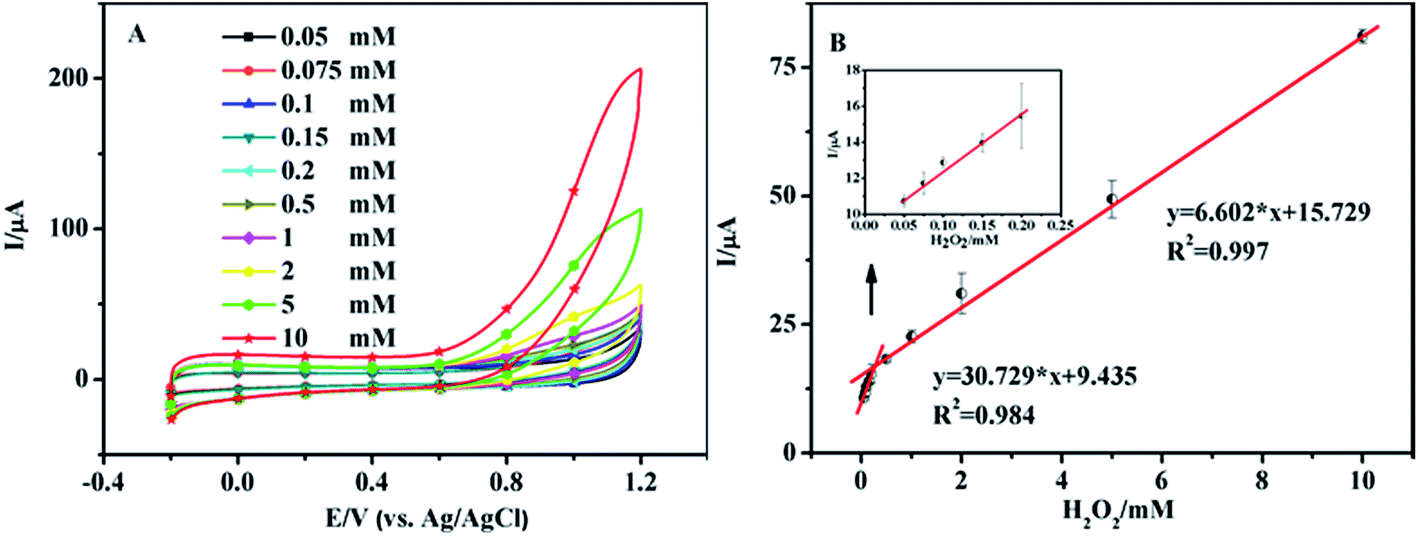 | ||
| Fig. 4 (A) CV curves of the nanoporous MgO modified electrode in PBS with different concentrations of H2O2; the scan rate is 50 mV s−1. (B) Sensor response curve between the magnitude of the anodic current and the H2O2 concentration in a range from 0.05 to 0.2 mM and 0.2 to 10 mM. The inset is the linear relationship between the magnitude of the anodic current and the H2O2 concentration in a range from 0.05 to 0.2 mM. The potential is 0.9 V. | ||
| Method | Material | Linear ranges (mM) | LODs (μM) | Ref. |
|---|---|---|---|---|
| Colorimetry | Nitrogen-doped grapheme quantum dots | 0.02–1.17 | 5.3 | 30 |
| Photoelectrochemistry | Fluorescent gold nanoclusters | 0–4 | 35 | 31 |
| Electrochemistry | Amino acid-based chiral coordination polymer | 0.01–7.69 | 10 | 32 |
| Electrochemistry | Manganese dioxide | 1–5 | — | 33 |
| Electrochemistry | Cu2O/GNs/GCE | 0.3–7.8 | 20.8 | 18 |
| Electrochemistry | Nanoporous Au films | 0.02–9.74 | 10 | 34 |
| Electrochemistry | Urchin-like Co3O4 | 0.0001–0.05 | 0.145 | 35 |
| Electrochemistry | Nanoporous magnesium oxide | 0.05–10 | 3.3 | This work |
As shown in Fig. 4A, the response current increased sharply along with the increase in the applied potential. On the other hand, the modified GCE and bare GCE can be catalyzed without H2O2 at high potential above 0.9 V in this work (Fig. 3). Considering some of the interfering substances in milk, we chose three representative interferents: ascorbic acid, folic acid and glucose, which reflected the milk composition and content of the different substances within it. To illustrate the influence of the interferences individually, the amperometric measurements were investigated at 0.9 V and the result showed that no appreciable signal was observed for the successive injections of interferent into the electrolyte solution (Fig. S2†).
3.4 Reproducibility and stability of the sensor
The repeatability of the developed biosensor was evaluated by using the same electrode for 3 successive measurements. The coefficients of variance (CVs) were 0.25%, 0.85%, 0.61%, 0.75%, 0.35% and 1.44% for 0.1, 0.15, 1, 1.5, 3 and 6 mM H2O2, respectively, indicating the reproducibility of the method. The stability of the developed biosensor was investigated by measuring the current shift after different storage times. The experimental results indicated that the current response could retain 95% of its initial current response to H2O2 after storage for 5 days at 20 °C (Fig. S3†).3.5 Samples analysis
To investigate the feasibility of the application of the MgO/GCE for real samples, we chose milk as the base solution. Various concentrations of H2O2 were initially spiked into the three different sterilized milk samples in the absence of endogenous H2O2. These results are listed in Table S1.† As shown in Table S1,† the recovery was determined to be from 94.3% to 119% for the spiked milk samples, and the average recovery of the biosensor was 105.2% (n = 3). Moreover, the milk sample without spiked H2O2 did not show any detectable signal (data not shown).4. Conclusions
In summary, we have demonstrated that nanoporous MgO can be synthesized by a facile method at low temperature with a high aspect ratio. The nanoporous MgO exhibits excellent electro-oxidation activity toward H2O2 with a linear range of 0.05 to 2.0 and 2.0 to 10 mM, and a lower detection limit of 3.3 μM. Furthermore, the H2O2 biosensor based on nanoporous MgO also exhibited excellent long-term stability and reproducibility. Therefore, the excellent response to H2O2 of the biosensor developed here makes it possible to be applied in food safety determinations.Acknowledgements
This study was funded by National Basic Research Program of China (973 Program, 2012CB720803), National Natural Science Foundation (61404075, BK20130394), Guangdong Natural Science Funds for Distinguished Young Scholar (2014A030306026) and the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University.References
- T. Ariga, BioFactors, 2004, 21, 197–201 CrossRef CAS PubMed.
- J. D. Fernstrom, S. D. Munger, A. Sclafani, I. E. de Araujo, A. Roberts and S. Molinary, J. Nutr., 2012, 142, 1134S–1141S CrossRef CAS PubMed.
- M. E. Abbas, W. Luo, L. H. Zhu, J. Zou and H. Q. Tang, Food Chem., 2010, 120, 327–331 CrossRef CAS PubMed.
- D. L. Young, C. A. Mihaliak, S. D. West, K. A. Handelman, R. A. Collins, A. M. Phillips and C. K. Robb, J. Agric. Food Chem., 2000, 48, 5146–5153 CrossRef CAS PubMed.
- C. H. Coyle, L. J. Martinez, M. C. Coleman, D. R. Spitz, N. L. Weintraub and K. N. Kader, Free Radical Biol. Med., 2006, 40, 2206–2213 CrossRef CAS PubMed.
- H. J. Lee, M. Y. Yoon, J. Y. Kim, Y. Kim, H. R. Park and E. Park, Food Sci. Biotechnol., 2008, 17, 343–348 Search PubMed.
- U. Sansal and G. Somer, Food Chem., 1999, 65, 259–261 CrossRef CAS.
- S. Yamashoji, N. Yoshikawa, M. Kirihara and T. Tsuneyoshi, Food Chem., 2013, 138, 2146–2151 CrossRef CAS PubMed.
- S. H. Chen, R. Yuan, Y. Q. Chai and F. X. Hu, Microchim. Acta, 2013, 180, 15–32 CrossRef CAS.
- B. B. Luo, X. M. Li, J. C. Yang, X. L. Li, L. P. Xue, X. L. Li, J. K. Gu, M. Z. Wang and L. Jiang, Anal. Methods, 2014, 6, 1114–1120 RSC.
- Y. Y. Zhang, X. Y. Bai, X. M. Wang, K. Shiu, Y. L. Zhu and H. Jiang, Anal. Methods, 2014, 86, 9459–9465 CAS.
- S. L. Yang, G. Li, G. F. Wang, J. H. Zhao, M. F. Hua and L. B. Qu, Sens. Actuators, B, 2015, 208, 593–599 CrossRef CAS PubMed.
- Y. L. Wang, Z. C. Wang, Y. P. Rui and M. G. Li, Biosens. Bioelectron., 2015, 64, 57–62 CrossRef CAS PubMed.
- M. Ammam, Biosens. Bioelectron., 2014, 58, 121–131 CrossRef CAS PubMed.
- T. T. Han, Y. Zhang, J. Q. Xu, J. P. Dong and C. C. Liu, Sens. Actuators, B, 2015, 207, 404–412 CrossRef CAS PubMed.
- H. F. Liu, X. L. Chen, L. H. Huang, J. Wang and H. B. Pan, Electroanalysis, 2014, 26, 556–564 CrossRef CAS PubMed.
- X. M. Chen, B. Y. Su, Z. X. Cai, X. Chen and M. Oyama, Sens. Actuators, B, 2014, 201, 286–292 CrossRef CAS PubMed.
- M. M. Liu, R. Liu and W. Chen, Biosens. Bioelectron., 2013, 45, 206–212 CrossRef CAS PubMed.
- B. B. Jiang, X. W. Wei, F. H. Wu, K. L. Wu, L. Chen, G. Z. Yuan, C. Dong and Y. Ye, Microchim. Acta, 2014, 181, 1463–1470 CrossRef CAS.
- M. Wayua, R. Spidle, T. Devkota, A. Deb, R. Delong, K. Ghosh, A. Wanekay and C. Chusuei, Electrochim. Acta, 2013, 97, 99–104 CrossRef PubMed.
- M. J. Li, W. L. Guo, H.J. Li, D. Wei and B. H. Yang, Sens. Actuators, B, 2014, 204, 629–636 CrossRef CAS PubMed.
- A. Umar, M. Rahman and Y. B. Hahn, Electrochem. Commun., 2009, 11, 1353–1357 CrossRef CAS PubMed.
- T. Wang, Y. F. Xu, Q. Y. Su, R. Yang, L. F. Wang, B. Liu, S. Shen, G. H. Jiang, W. X. Chen and S. Wang, Mater. Lett., 2014, 116, 332–336 CrossRef CAS PubMed.
- G. Martra, T. Cacciatori, L. Marchese, J. Hargreaves, I. Mellor, R. Joyner and S. Coluccia, Catal. Today, 2001, 71, 121–130 CrossRef.
- T. Selvamani, A. Sinhamahapatra, D. Bhattacharjya and I. Mukhopadhyay, Mater. Chem. Phys., 2011, 129, 853–861 CrossRef CAS PubMed.
- M. J. Li, X. F. Wang, H. J. Li, D. Wei, G. J. Qiu, F. D. Liu and B. H. Yang, Mater. Lett., 2013, 106, 45–48 CrossRef CAS PubMed.
- J. P. Wang, H. Gao, F. L. Sun, Q. Hao and C. X. Xu, Biosens. Bioelectron., 2013, 550–555 CrossRef PubMed.
- M. S. Mastuli, R. Rusdi, A. M. Mahat, N. Saat and N. Kamarulzaman, Adv. Mater. Res., 2012, 545, 137–142 CrossRef CAS.
- A. Gholizadeh, S. Shahrokhian, A. Iraji zad, S. Mohajerzadeh, M. Vosoughi, S. Darbari, J. Koohsorkhi and M. Mehran, Anal. Chem., 2012, 84, 5932–5938 CrossRef CAS PubMed.
- L. P. Lin, X. H. Song, Y. Y. Chen, M. C. Rong, T. T. Zhao, Y. R. Wang, Y. Q. Jiang and X. Chen, Anal. Chim. Acta, 2015, 869, 89–95 CrossRef CAS PubMed.
- J. X. Zhang, L. P. Tu, S. Zhao, G. H. Liu, Y. Y. Wang, Y. Wang and Z. Yue, Biosens. Bioelectron., 2015, 67, 296–302 CrossRef CAS PubMed.
- B. Zhou, L. M. Liang and J. Yao, J. Solid State Chem., 2015, 223, 152–155 CrossRef CAS PubMed.
- C. Revathi, R. G. Mohan and T. R. Ramasamy, Mater. Sci. Semicond. Process., 2015, 31, 709–714 CrossRef PubMed.
- X. Ke, Z. H. Li, L. Gan, J. Zhao, G. F. Cui, W. Kellogg, D. Materae, D. Higgins and G. Wu, Electrochim. Acta, 2015, 170, 337–342 CrossRef CAS PubMed.
- S. Barkaoui, M. Haddaoui, H. Dhaouadi, N. Raouafi and F. Touati, J. Solid State Chem., 2015, 228, 226–231 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18560b |
| This journal is © The Royal Society of Chemistry 2015 |