
Significance of surface charge and shell material of superparamagnetic iron oxide nanoparticle (SPION) based core/shell nanoparticles on the composition of the protein corona†
Usawadee
Sakulkhu
a,
Morteza
Mahmoudi
bc,
Lionel
Maurizi
a,
Geraldine
Coullerez
a,
Margarethe
Hofmann-Amtenbrink
d,
Marcel
Vries
e,
Mahdi
Motazacker
f,
Farhad
Rezaee
e and
Heinrich
Hofmann
*a
aLaboratory of Powder Technology, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland. E-mail: heinrich.hofmann@epfl.ch
bNanotechnology Research Center and Department of Nanotechnology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
cDivision of Cardiovascular Medicine, Stanford School of Medicine, Stanford University, CA, USA
dMat Search Consulting Hofmann, Ch. Jean Pavillard 14, 1009 Pully, Switzerland
eUniversity Medical Center Groningen (UMCG)University of Groningen, Groningen, The Netherlands
fDepartment for Experimental and Molecular Medicine, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands
First published on 25th September 2014
Abstract
As nanoparticles (NPs) are increasingly used in many applications their safety and efficient applications in nanomedicine have become concerns. Protein coronas on nanomaterials’ surfaces can influence how the cell “recognizes” nanoparticles, as well as the in vitro and in vivo NPs’ behaviors. The SuperParamagnetic Iron Oxide Nanoparticle (SPION) is one of the most prominent agents because of its superparamagnetic properties, which is useful for separation applications. To mimic surface properties of different types of NPs, a core–shell SPION library was prepared by coating with different surfaces: polyvinyl alcohol polymer (PVA) (positive, neutral and negative), SiO2 (positive and negative), titanium dioxide and metal gold. The SPIONs with different surfaces were incubated at a fixed serum![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nanoparticle surface ratio, magnetically trapped and washed. The tightly bound proteins were quantified and identified. The surface charge has a great impact on protein adsorption, especially on PVA and silica where proteins preferred binding to the neutral and positively charged surfaces. The importance of surface material on protein adsorption was also revealed by preferential binding on TiO2 and gold coated SPION, even negatively charged. There is no correlation between the protein net charge and the nanoparticle surface charge on protein binding, nor direct correlation between the serum proteins’ concentration and the proteins detected in the coronas.
:nanoparticle surface ratio, magnetically trapped and washed. The tightly bound proteins were quantified and identified. The surface charge has a great impact on protein adsorption, especially on PVA and silica where proteins preferred binding to the neutral and positively charged surfaces. The importance of surface material on protein adsorption was also revealed by preferential binding on TiO2 and gold coated SPION, even negatively charged. There is no correlation between the protein net charge and the nanoparticle surface charge on protein binding, nor direct correlation between the serum proteins’ concentration and the proteins detected in the coronas.
Introduction
The SuperParamagnetic Iron Oxide Nanoparticle (SPION) has promising biomedical applications. The biomedical applications of coated SPION can be divided into 3 major groups: separation (e.g. cellular proteomics, cell sorting,1 purification), therapy (e.g. hyperthermia,2 drug delivery3) and diagnosis (e.g. Magnetic Resonance Imaging (MRI),4,5 cell tracking6–9). Coating of SPION with a variety of polymers has been approved for some clinical applications. The nanoparticles coated with dextran i.e. ferucarbotran have been used as contrast agents in MRI.10 Coating with polyelectrolyte PEI-PEG-chitosan copolymer was used for gene delivery.11 Moreover, coating of NPs with other hydrophilic polymers such as polyethylene glycol (PEG), polyvinyl alcohol (PVA) or poly (acrylic acid) has been used for imaging and drug delivery.12 In addition to polymeric coating, inorganic coatings of SPION with silica or gold have interested researchers and industry for many years towards further surface derivatization of NPs and because of their influences on colloidal stability and the biological behaviour of SPION in biomedical applications.13,14 SPION is also of interest in the discovery of biomarker proteins for example in blood plasma, serum or urine, which are easily accessible in the acquisition of proteins secreted or released from cells and various interconnecting tissues. These proteins may indicate a disease status if detected and by this serve as biomarkers. On the other hand, as the majority of the nanoparticle formulation, SPION must also be safe for any biomedical applications. The safety of the nanoparticles used in biomedical applications mainly relates to NP stability, biodistribution and toxicity.15 Without coating, naked SPIONs are not stable and become aggregated at physiological pH. In order to overcome this shortcoming, various materials are employed as coatings on the surface of SPION. Coating of SPION does not only improve the colloidal stability but also increases the opportunity to functionalize molecules (e.g. fluorescent dyes, polymers, radiotracers, drugs or targeting biomolecules such as antibodies) onto the SPION surface.Once the NPs are exposed to biological environments, biomolecule adsorption immediately occurs. If NPs were injected into the body, blood proteins are the biomolecules that have the highest chance to interact with the nanoparticles.16 Protein adsorption forms a protein corona on the nanoparticles. Protein adsorptions can be categorized by their physiological location into 2 groups: (i) extracellular and (ii) inter-/intracellular (effects between cell–cell junctions and within cells). Extracellular protein adsorption depends on the nanoparticle characteristics (e.g. size, shape, surface area, surface charge, roughness and porosity, functional groups, ligands, crystallinity and hydrophobicity–hydrophilicity) and on the suspending media.17 The largest factors of influence on the adsorption arise from acids and bases, salts and multivalent ions, and natural or synthetic organic matter (e.g. proteins, lipids, surfactants, polymers, and polyelectrolytes). All of these factors determine nanoparticle stability and behavior in biological fluids.18 The extracellular protein adsorption will consequently influence the protein adsorption at the inter-/intracellular level.
Protein adsorption is one of the major driving forces in the selection of a nanoparticle destination for theranostic applications, which will further affect cells, tissues and finally a body system. Common proteins like albumin, fibrinogen, IgG, complement C3, apolipoprotein A-I and apolipoprotein E19–22 bind to various semi-solid nanoparticles, for instance liposomes, and solid nanoparticles of polymeric or inorganic nature, such as iron oxide, silica, titanium oxide, etc.; some of them also covered by polymer coatings or carbon based nanoparticles such as nanotubes. All of the proteins mentioned are highly abundant in blood plasma, in which at least 99% of the protein mass is covered by 22 of the most abundant proteins.23–25 It is now a challenge of biomarker development to find the specific but very rare protein disease markers (tissue-derived proteins) within the 1% of the remaining proteins of the blood plasma, already diluted on their way to the blood stream.
The constitution of the protein corona depends on two main parameters: (i) composition of the biological milieu and (ii) the surface property of SPION. Further cellular/tissue responses arise due to the composition of the corona. The uptake of nanoparticles is determined by particle–protein and corona–membrane interactions, receptor–ligand binding interactions, membrane wrapping, biomolecule interaction and conformational change in the biomolecules.17 It is reported that the protein coating reduces the targeting capability of surface engineered NPs by blocking the active sites of the targeting ligands.26 It was revealed that the presence of the corona (protein layer) inhibits the formation of Aβ fibrils (which is the main cause of Alzheimer's diseases)27 for all of the tested nanomaterials (e.g. carbon nanotubes and silica nanoparticles). All of these factors finally determine either the activation of or damage to physiological responses towards, for example, oxidant injury or mitochondrial and lysosomal damage.28 If the cellular damage is severe, it might cause an adverse effect on the body at the system level. This is the reason why the synthesis and surface modification of NPs to eliminate or minimize the negative effects on cell behavior, as well as to reduce the clearance of NPs in vivo, has become one of the most interesting topics for medical applications. Recently Giri et al.29 have presented a detailed study of the composition of the protein coronas of gold nanoparticles at incubation times between 15 and 24 h. They showed that proteins adsorbed in the first 15 min have a significantly higher isoelectric point than the mean theoretical isoelectric point of all proteins present in the incubation media. Regarding the influence of the protein secondary structure on adsorption, Fleischer et al.30 showed that BSA adsorbed onto negatively charged (carbocylate modified) and positively charged (amine) polystyrene particles. Circular dichroism spectra show no changes for BSA on negatively charged particles whereas a significant change was observed for BSA adsorbed on positively charged NPs. As a consequence, the amount of adsorbed proteins and interaction with receptors at the cell surface is different for negatively and positively charged particles.
Research on the interactions between plasma proteins and nanoparticles has been increasingly reported. Many ex situ methods have been applied to investigate the protein coronas on NPs, all showing advantages and limitations, such as centrifugation, gel filtration, or membrane-based microfiltration.31,32 Centrifugation assays have been reported as an efficient way to retrieve enough protein for safe identification of protein adsorption patterns, if conducted with care and accompanied with other methods to avoid false positive results due to insufficient washing of high abundance proteins. Moreover, sedimentation of large proteins, protein aggregates, and co-precipitation may further complicate the picture. SPION-containing nanoparticles offer the possibility to be rapidly isolated from biofluids by magnetic separation, therefore preventing contamination or degradation during the purification process and with significant improvement in the recovery yield of proteins and particles.22 A magnetic separation technique with a high gradient magnetic separation column offers in particular the greatest advantage in the simplicity of operation, in the possibility of optimizing the separation efficiency upon flow rate under reduced shear forces and in controlled elution conditions.33,34
Nonspecific protein adsorption on the surface of nanoparticles and the formation of protein coronas are widely seen as negative side effects, screening the targeting ability of functional nanoparticles towards specific receptors on cells in biological fluids in vitro or in vivo.35 On the other hand the formation of a biomolecular corona is also relevant in the creation of bionanoconjugates that can stabilize the nanoparticle,36 promote interactions with biological systems37,38 or as a way to capture therapeutic biomarkers through an engineered nanoparticle–protein interface.39 The advantages and disadvantages of the protein–nanoparticle interaction, with the corresponding in vitro or in vivo biological impacts, are strongly dependent on the type and amount of corona protein content. In the current study, SPION coated with PVA polymer, varying the surface charge from highly positive (amine modified), to positive (mixture of amine modified PVA copolymer and unmodified plain PVA), neutral (unmodified plain PVA), and negative (carboxylic acid modified PVA), as well as SPION coated with inorganic coatings such as silica shell using tetraethyl orthosilicate (TEOS), further modified with 3-aminopropyltriethoxysilane (APTES) and with gold layer, as well as with TiO2, were synthesized (Fig. 1). The different surface properties of the NPs were expected to influence the formation of the protein corona in fetal bovine serum (FBS) commonly used in in vitro cell culture. As it is essential to identify the protein corona that may ultimately play a biological role, a high resolution LC-MS/MS analysis was applied to allow a precise quantification of all adsorbed proteins. The aim of this study was to determine whether the different surface coatings influence the preferential binding of certain serum proteins (low and high abundance proteins) and to determine the relative abundance of the adsorbed proteins.
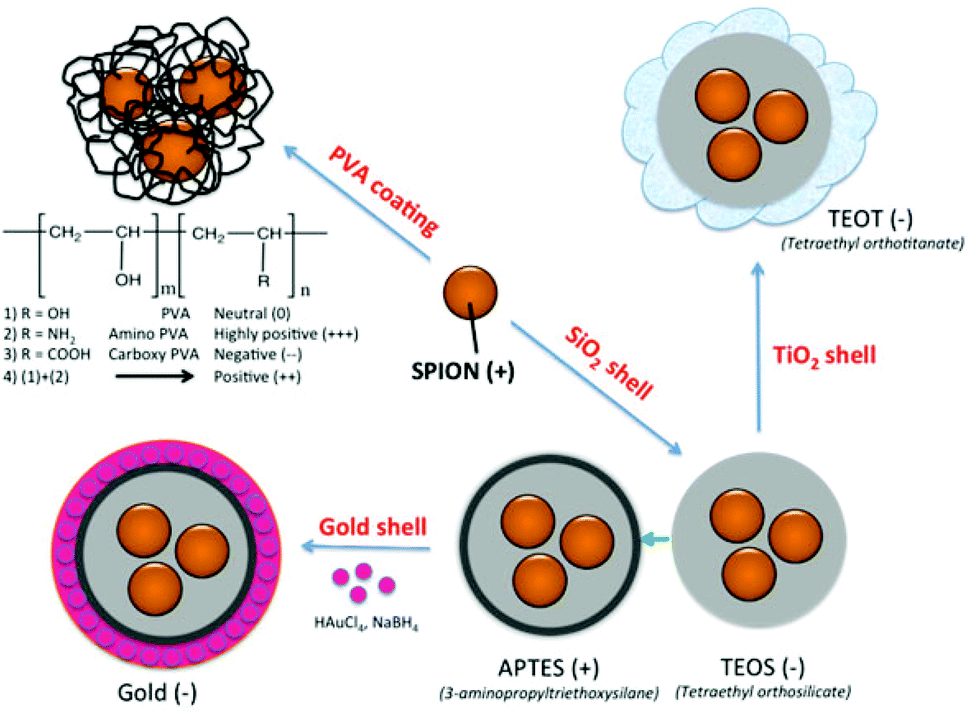 | ||
| Fig. 1 Outlining of the various coatings employed on the surface of SPION. | ||
Results and discussion
Coated SPION characterization (sizes and zeta potentials)
The 9 different core/shell iron oxide nanoparticles were produced to investigate FBS protein adsorption. The characteristics of the produced nanoparticles are shown in Fig. 2 and Table 1. The particle diameter size of naked SPION, inorganic and metallic core–shell NPs were measured by Transmission Electron Microscopy (TEM), while the hydrodynamic diameter of PVA coated NPs and the zeta potential of all NPs were measured in deionized water by Photon Correlation Spectroscopy (PCS). The nanoparticles used in this study were all less than 150 nm in diameter. The variation in the size of highly positively charged PVA(NH2)–SPION, positively charged PVA(NH2)/(OH)–SPION, neutral PVA(OH)–SPION and negatively charged PVA(COOH)–SPION (71, 47, 113, and 79 nm respectively), suggested that the coverage of the polymer on the surface of the NPs and the differing hydration degree of the PVA layer should be considered in protein adsorption. The polydispersity index was between 0.2 and 0.25. The mean diameter of SiO2, TiO2 and Au coated SPION, measured by TEM, varied from 50 to 144 nm (with the iron oxide core ∼8 nm). | ||
| Fig. 2 Transmission electron microscopy (TEM) images of naked SPION (a), SiO2-SPION (b), TiO2-SPION (c) and gold-SPION (d). | ||
| Sample | Hydrodynamic mean diameter (by number) (nm) | TEM (nm) | Zeta potential (mV) | Point of zero charge (pzc) |
|---|---|---|---|---|
| Naked SPION | 28 ± 3 | 7.7 ± 2.2 | 27.5 ± 3.0 | 7.0 |
| PVA(NH2)–SPION (++++) | 71.8 ± 0.143 | — | 34.6 ± 2.1 | 9.0 |
| PVA(NH2)/(OH)–SPION (+++) | 46.8 ± 0.088 | — | 13.6 ± 1.3 | 8 |
| PVA(OH)–SPION (0) | 112.7 ± 0.155 | — | 5.6 ± 0.9 | 7 |
| PVA(COOH)–SPION (−) | 78.5 ± 0.006 | — | −15.5 ± 1.0 | 5 |
| SiO2(TEOS)–SPION | — | 102.1 ± 25.2 | −36.6 ± 1.4 | 3 |
| SiO2(APTES)–SPION | — | 100.7 ± 28.2 | 36.8 ± 1.9 | 9 |
| TiO2–SPION | — | 102.1 ± 25.2 | −24.9 ± 0.5 | 4 |
| Gold–SPION | — | 143.9 ± 18.4 | −48.2 ± 1.7 | 6 |
Coating of an initial naked SPION (zeta potential: ∼27 mV) with different materials led to NPs with different surface charges. SPION coated with PVA containing amino and carboxylic acid groups showed positive and negative charges of ∼13 and ∼−15 mV, respectively. SPION coated with a plain PVA (–OH) showed a slightly positive charge of 6 mV and was used as representative of a neutral particle. The increasing size and the different surface charges of these 3 particles suggested successful coating of polymer onto a SPION surface when compared to a naked SPION core. The inorganic SiO2 shell nanoparticles showed negative and positive charges after coating with TEOS and APTES, respectively. The particles were spherical in shape with multiple core SPIONs (bead form). TiO2 and gold coated SPION showed strongly negatively charged surfaces with zeta potentials of −24.9 and −48 mV, respectively, confirming successful coating of these two materials onto SPION as reported in previous works.40,41 In addition, the presence of the inorganic shell was confirmed by TEM images (Fig. 2). The SPION core is represented by dark 8 nm nanoparticles (a) inside a lighter shell of silica SiO2 (b) and TiO2 (c), while the gold shell was confirmed by a larger nanoparticle size with a highly electron-dense shell (d).
Nanoparticle–protein adsorption
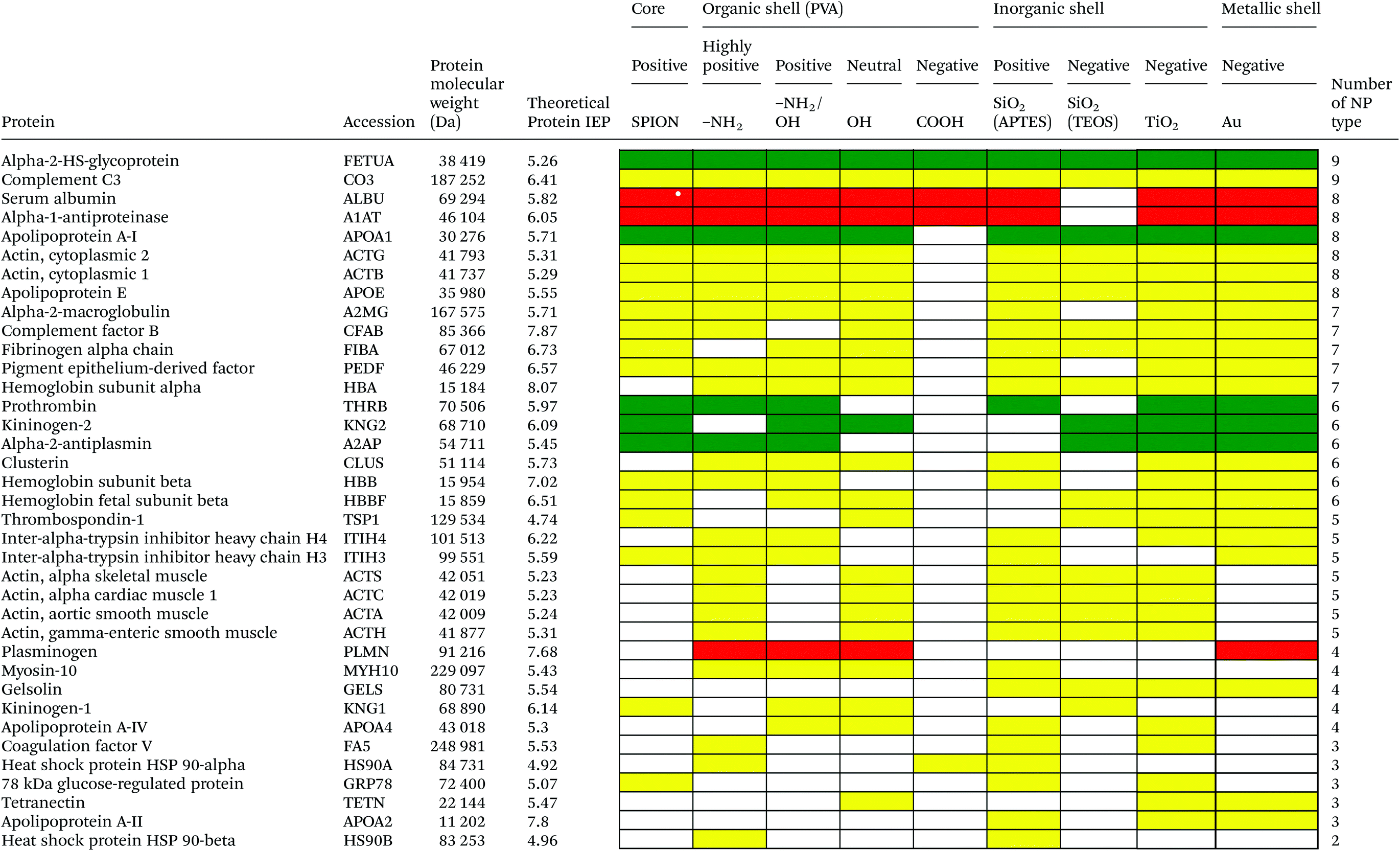
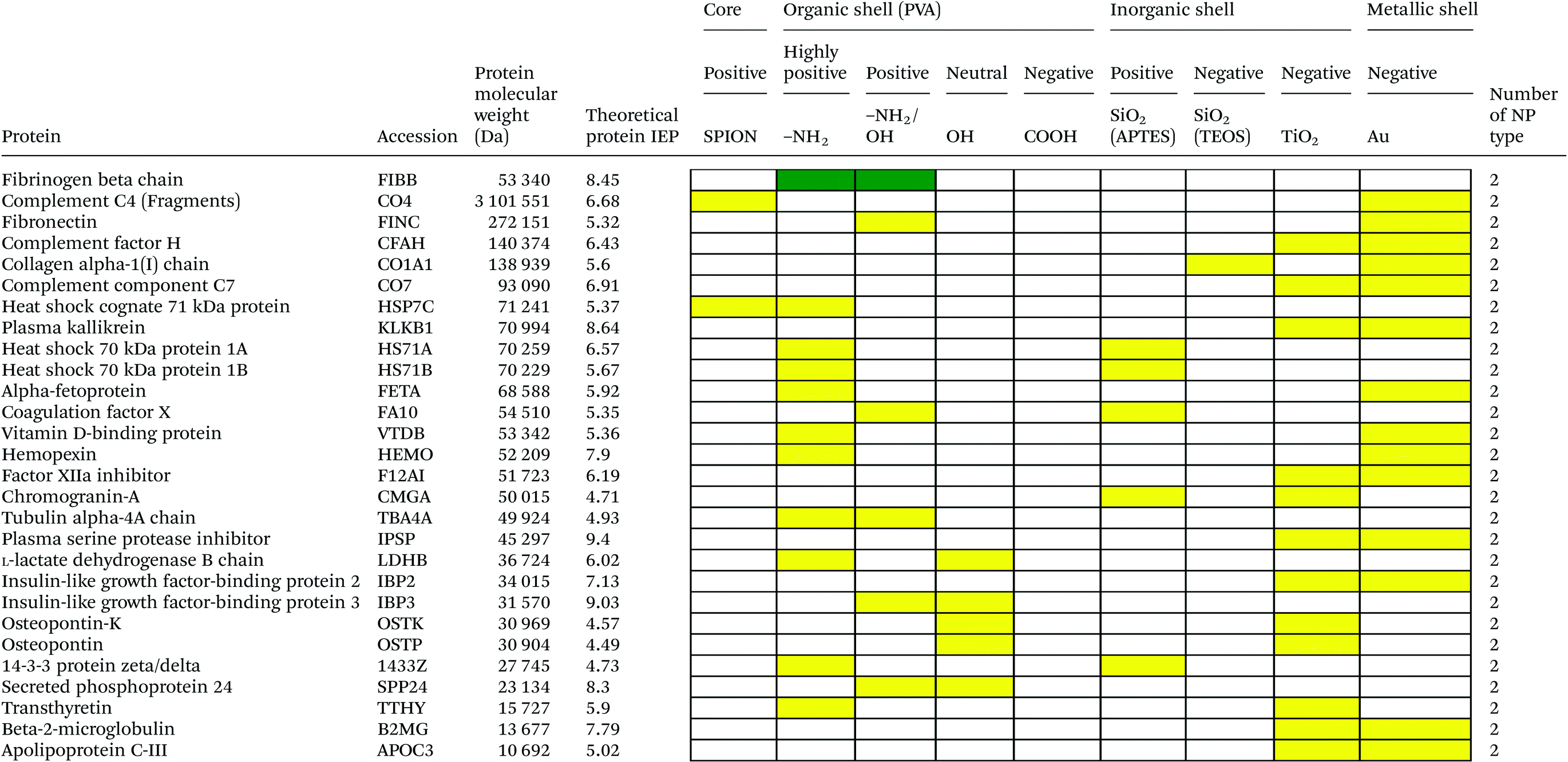
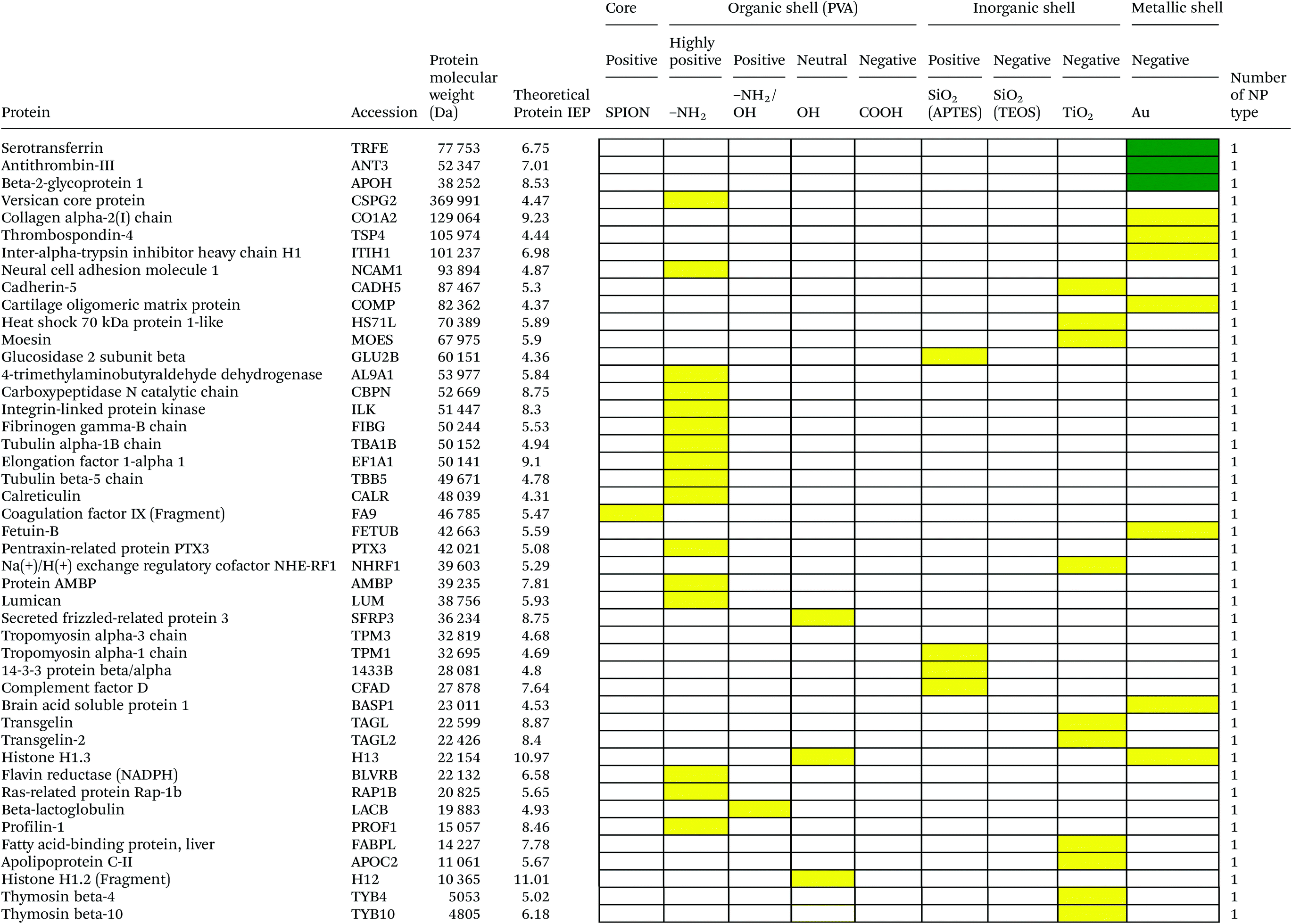
Serum is a complex fluid that contains for human about 3700 different proteins with concentrations up to 70 mg ml−1; in which the most abundant proteins such as albumin, immunoglobulin G (IgG) and α2-macroglobulin represent 97% of the total protein content whereas the remaining 3% is a mixture composed of low abundance proteins. Identification of all the proteins is therefore a challenging task where proteomic techniques, e.g. electrophoretic methods, chromatography or mass spectrometry, can provide qualitative and quantitative analysis of protein patterns and enable the detection of potentially relevant biomarkers at very low concentrations in protein coronas when combined with nanomaterials (<1 ng cm−2).42–44 Based on the available proteomic information of fetal bovine serum (FBS) reported by Zheng et al.45 the table of abundant proteins was adapted (Table 2). Fetal bovine serum proteins were therefore categorized, as a percentage relative concentration of proteins, into 3 groups: high (≥6%), medium (≥3%) and low (≤3%). Tables 3–5 show the list of proteins adsorbed on the polymer, inorganic and metallic coated SPION with increased specificity for nanoparticle surface, i.e. proteins common to several surface types (>2), common to 2 surface types and common to only 1 surface type, respectively. A color code was used to show the adsorbed protein given their relative abundance in FBS, e.g. red (high), green (medium) and yellow (low abundance).| Level of abundance | Protein | % Relative concentration of protein |
|---|---|---|
| High (>6%) | Serum albumin | 18.7 |
| Alpha-1-antiproteinase | 7.9 | |
| Plasminogen | 6.4 | |
| Cone cGMP-specific 3′,5′-cyclic phosphodiesterase alpha-subunit | 6.1 | |
| Lactoperoxidase | 6.1 | |
| Medium (3% < X ≤ 6%) | NADH-ubiquinone oxidoreductase 75 | 5.2 |
| Alpha-2-HS-glycoprotein | 5.2 | |
| Kininogen, LMW II | 4.7 | |
| Hemiferrin | 4.7 | |
| Integrin beta-1 | 4.7 | |
| Prothrombin | 4.4 | |
| Apolipoprotein A-I | 3.8 | |
| Antithrombin-III | 3.8 | |
| Beta-2-glycoprotein I | 3.5 | |
| Alpha-2-antiplasmin | 3.2 | |
| Low (≤3%) | Alpha-1-1-microglobulin and inter alpha-trypsin inhibitor light chain | 2.9 |
| Hemoglobin beta fetal chain | 2.9 | |
| Alpha 1 antichymotrypsin | 2.3 | |
| Apolipoprotein A-II | 1.7 | |
| Hemoglobin R chain | 1.7 |
|
|
|
Effect of nanoparticle surface charge on the protein corona
As depicted in Table 3, based on the theoretical protein isoelectric point from http://web.expasy.org/compute_pi/, no clear correlation between protein isoelectric point (IEP) and protein adsorption on nanoparticles with different surface charge could be observed. For instance, alpha-2-HS-glycoprotein (IEP 5.26) tends to have a negative charge at physiological pH 7.4, so according to electrostatic binding the protein should preferentially adsorb onto the positively charged nanoparticles. However, alpha-2-HS glycoprotein bound on all types of nanoparticles irrespective of their surface charge. The electrostatic effect alone, between protein and NPs of opposite surface charge, is therefore not the only driving force that can explain the protein adsorption. It is well known that proteins have an inhomogeneous distribution of charges at their surfaces. Therefore, even if the overall net charge of the protein is negative, positive charge domains may allow an electrostatic interaction with the particle surface. In addition, the formation of the protein corona will involve simultaneously a combination of protein–particle and protein–protein interactions. Adsorption models of coronas consisting of sequential attachment and multiple layers of proteins are also proposed, which consist of primary binding proteins on the NP surface followed by secondary binders mediated by protein–protein interactions,46e.g. oppositely charged anionic to cationic proteins, or by specific recognition of molecular cues on the primary protein layer. Protein conformational change and denaturation could additionally occur after adsorption onto a solid interface driven by a favorable protein–surface interaction and an entropy gain due to loss of ordered secondary structure. M. Rankl et al. (2006) have proven that immunoglobulin G (IgG) undergoes conformational changes during non-specific binding once it is exposed to different surface models.47 Particle size, i.e. surface curvature, can also have a significant effect on the protein conformational change48,49 and ultimately on the protein corona with variability in the surface chemistry and surface charge. Size dependent protein adsorption was for example shown on polystyrene (PS) nanoparticles of two different sizes (50 nm, 200 nm) with a more pronounced trend in amine- and carboxylic acid-containing particles compared to neutral surfaces.50 Moreover, protein adsorption on NP surfaces also depends on the length of time of the protein–particle incubation. The adsorption/desorption process could happen via either reversible or irreversible protein conformational change during incubation.51The protein corona appears to follow a general structure with few proteins adsorbed at high abundance and many more at low abundance although the composition in the corona does not necessarily correlate with the relative abundances of the proteins in the biological milieu. The proteins compete for the surface through a dynamic process (“Vroman Effect”) based on protein abundances, affinities, and incubation time with the nanoparticles. Upon exposure of the NPs to the serum the most abundant and smaller molecular weight proteins first cover the NPs, e.g. albumin, IgG, or fibrinogen in plasma and are replaced by proteins with slower adsorption rates but higher affinity, e.g. apolipoproteins although the phenomenon is still debated for nanomaterials.52 Protein adsorption is a dynamic system. The adsorption/desorption process could happen via either reversible or irreversible protein conformational change during incubation51 and the protein corona is therefore hypothesized to consist of a long-lived hard shell of proteins assumed to be irreversibly bound. The corona is not immediately established but changes over time until equilibrium is reached.53,54 Temporal studies showed that equilibrium takes place in few minutes for nanoparticles incubated with full protein serum.55
Based on competitive binding between serum proteins, high abundance proteins were generally expected to bind to the NPs with higher probability than low abundance proteins and to all nanoparticle surfaces. Surprisingly, from the 5 listed high abundance proteins, 2 proteins were not detected on any particle surfaces, i.e. cone cGMP-specific 3′,5′-cyclic phosphodiesterase alpha-subunit and lactoperoxidase. The highly abundant plasminogen binds to only 4 of the 9 investigated NPs. The surface specific binding of high abundance proteins reveals that the need of a specific surface character for protein adsorption is not ignorable. Only serum albumin and alpha-1-antiproteinase were found on 8 of the 9 different particles; both did not adsorb onto negatively charged silica coated SPION.
It is obvious that proteins preferred to bind onto positively charged NPs rather than on negatively charged NPs. Positively charged PVA coated and positively charged SiO2(APTES) coated NPs showed similar patterns of adsorbed proteins indicating an influence of surface charge on protein adsorption. However, the presence of alpha-2-antiplasmin and plasminogen on highly positively charged PVA coated SPION (–NH2), but not on SiO2(APTES)–SPION is clear evidence for the effect of the surface materials. Previous studies showed that the main proteins that associated with large particles (and hydrophobic surfaces) were albumin, IgG and antibodies, complement proteins, and apolipoproteins.22,56 Notably, these proteins belong to the group of high abundance plasma proteins (some being complement factors), with apolipoproteins as hydrophobic proteins. Surprisingly, none of the high abundance proteins, e.g. serum albumin, alpha-1-antiproteinase and plasminogen, bound onto negatively charged silica coated SPION (SiO2(TEOS)–SPION), while these high abundance proteins (except plasminogen) bound to all PVA coated nanoparticles. One of the reasons could be that albumin (IEP 4.7–4.8) is negatively charged at physiological pH. Although TiO2 and gold coated SPION had negative charges, more than 50 proteins bound onto these particles. This indicates that the surface materials play an important role in protein adsorption. There are some proteins bound specifically to certain nanoparticles (Table 5). This knowledge would be useful in further experimental design for a biomarker study.
Effect of different surface charge of the polymer coating on protein adsorption
PVA has been widely used in biomedicine for biomedical devices and pharmaceutical applications because of suitable properties, e.g. it is hydrophilic, biocompatible, nontoxic, non carcinogenic, non immunogenic, inert in body fluids and confers biopassive properties to surfaces. Although those surfaces show reduced protein adsorption, a large number of proteins can still adsorb from full serum and vary with the polymer physicochemical properties and coating.57,58 The total number of proteins that bind to PVA–SPION decreased upon changing the PVA coating on SPION in the order PVA(NH2) > PVA(OH) > PVA(NH2/OH) > PVA(COOH). Neutral to highly positively charged NPs bound a wide range of proteins and showed similar protein binding patterns whereas negatively charged PVA coated SPION bound a lower number of proteins (Table 3). Only 5 protein types bound onto the negatively charged PVA coated SPION while 24, 55, 31 and 34 proteins were found on naked SPION, highly positive, positive and neutral PVA coated SPION, respectively. Serum albumin, alpha-1-antiproteinase, alpha-2-HS-glycoprotein were high abundance proteins which bound to naked SPION and to all PVA coated SPION. In addition, complement C3, a low abundance protein also binds on all types of PVA surfaces. Serum albumin is a highly abundant protein which plays a role in opsonization and increasing nanoparticle half-life in blood.59 Alpha-1-antiproteinase is an inhibitor of serine proteases. Its primary target is elastase, but it also has a moderate affinity for plasmin and thrombin and inhibits trypsin, chymotrypsin and plasminogen activators.60 Alpha-2-HS glycoprotein is more abundant in fetal than in adult blood. It is involved in several functions, such as endocytosis, brain development and the formation of bone tissue.61 Hirsch et al.34 also found these three abundant proteins on positively, neutral and negatively charged PVA coated SPION. Deviation in the total amount of proteins found on all investigated PVA-SPION surfaces described in the work by Hirsch et al. could be due to different protein content fluids performed at a lower concentration of serum (10% v/v in PBS) versus full FBS serum in our study and sample preparation for MS analysis.The protein coronas on polymer coated particles did not reflect the relative abundance of proteins in the serum. Albumin, the most abundant protein in serum, is well-detected on all the surfaces but the composition of the protein coronas also revealed enrichment in apolipoprotein versus albumin in all of the PVA coated SPION. Albumin can indeed be displaced by other proteins in serum and results in a different protein corona, e.g. around positive and negative charged particles.34 Apolipoprotein A-I has furthermore been detected in the coronas of many other silica, lipid and polymeric nanoparticles, e.g. PS and polyNIPAM copolymers,20,31,50 suggesting that lipid coating is a general feature of nanoparticles in physiological conditions.62 Apolipoprotein might be affected in different ways by the surface characters of the particles and may undergo in particular conformational changes on surfaces of different charges63 that can explain the clear difference of protein composition between negatively charged PVA–SPION and other PVA–SPION. Multiple studies have however also reported that surface hydrophobicity played an important role in protein adsorption onto nanoparticles e.g. mediated by interaction with the lipid binding domain. Difference in the relative abundance of apolipoprotein might reflect the relative abundance of amino, alcohol, and carboxylic acid functional groups and the relative hydrophobicity/hydrophilicity of the different polymer layers. The central role of lipoproteins was also shown in the fouling of protein plasma on various polymeric biomaterials on flat surfaces.64
The absence of kininogen on positively charged nanoparticles was already observed for amine-modified silica nanoparticles in the work from Lundqvist et al.50 Interestingly, kininogen (high molecular weight) has been reported as a surface binding protein on iron oxide through histidine-rich sequences and is a possible marker of an incompletely masked iron oxide core as shown with loose dextran coating of ferumoxides.46
The negatively charged PVA–SPION is the only particle type that adsorbs almost no proteins, or surprisingly specifically those high abundance proteins showing an isoelectric point (IEP) < 7 and therefore net negatively charged under physiological conditions, e.g. the pH present in 100% serum. These findings are consistent with previous reports that found that the protein coronas of negatively charged silica NPs were preferentially composed of negatively charged proteins with IEP < 7.65 Interestingly, the low protein adsorption onto negatively charged PVA–SPION is supported by a greater colloidal stability and formation of smaller aggregates than for other PVA–SPION in FBS-supplemented cell culture medium.34 The reduced fouling of PVA(COOH) coated SPION suggests an enhanced stability of the PVA polymer layer, indeed mediated by carboxylate–iron coordination in addition to multiple electrostatic and hydrogen bonding interactions with the iron oxide surface. The exchange of the PVA dispersant by proteins might therefore be reduced and the SPION core less accessible for protein binding. The decrease in total protein adsorption for carboxylated PVA was also demonstrated for PVA membranes exposed to human plasma as a result of negative surface potentials and anionic substitution on PVA.57
Given that many abundant proteins bind to all different PVA–SPION independently of the surface charges and that the protein coronas of the negatively charged particles did not correlate with protein charge either, electrostatic interaction is likely not the only effect in modulating the protein adsorption on PVA–SPION. Difference in protein corona composition might also be induced by variation in the effective size of the particle (hydrodynamic diameter), such that it was not possible to effectively decouple in our study the net surface charge effect from size variation.50,66 To date, very few surfaces were found to be resistant to protein adsorption onto nanoparticles exposed to full serum, since the polymer surface density and the conformation might additionally influence the pattern of adsorbed proteins.67,68 The adsorption patterns will therefore result in a combination of physicochemical properties: in this case, the charge, size, the functional groups, molecular structures and the polymer conformation on the SPION surface.
Effect of different surface charge of inorganic and metallic coated SPION on protein adsorption
Here we investigated the adsorption of proteins on SPION core–solid shell nanoparticles, i.e. silica, titanium dioxide and gold, with special focus on the surface charge and effect of functionalities, e.g. amino, on the total amount of adsorbed proteins. Metal oxide nanoparticles were also selected as a model system as they are widely used for life science applications and are exposed to the general public in many commercial applications. Numerous high to medium abundance serum proteins were absorbed onto the oxide and metal nanoparticles.There were 40, 19, 53 and 50 proteins bound on SiO2(APTES)–SPION, SiO2(TEOS)–SPION, TiO2–SPION and gold–SPION, respectively. For SiO2 coated NPs, as observed in PVA–SPION, proteins preferentially bound onto positively charged silica coated nanoparticles rather than the negatively charged NPs, indicating the influence of the surface charge of SiO2 on protein adsorption. Alpha-2-HS-glycoprotein, apolipoprotein A-I, complement 3, fibrinogen alpha chain, cytoplasmic actin 1 & 2, apolipoprotein E, hemoglobin subunit alpha and complement factor B were proteins that were found on all investigated inorganic and metallic nanoparticles. The presence of kininogen-2 on SiO2(TEOS)–SPION, but not on SiO2(APTES)–SPION showed a surface charge specific binding of this protein on the silica surface. Kininogen-2 is an inhibitor of thiol proteases which plays an important role in blood coagulation, inhibiting the thrombin- and plasmin-induced aggregation of thrombocytes.69 Protein adsorption on silica NPs studied by Monopoli et al. showed some of the same proteins as in our work, such as alpha-2-SH-glycoprotein, apolipoprotein E, apolipoprotein AI, kininogen-1 and thrombospondin-1.70 In Monopoli's work, serum albumin was observed on the particles after washing 3 times with PBS. It could be interpreted that serum albumin is not a loosely bound protein. In our work, however, serum albumin was eluted out after washing with high ionic strength solutions. This indicated that serum albumin is neither a loosely bound protein nor a tightly bound protein; it is an intermediate.
Interestingly, TiO2 coated and gold coated NPs which also have highly negative charges showed binding of 53 and 50 proteins, respectively. The high numbers of proteins observed on both positive and highly negatively charged particles indicated a requirement of sufficient surface charge for protein adsorption. In addition, protein adsorption markedly depends on the surface materials. Qualitatively, high and low abundance adsorbed serum proteins were generally common to all negatively and positively charged surfaces. However a lower number of proteins were common to 2 types of surfaces (Table 4) and few low abundant proteins were found on only one material surface (Table 5) suggesting that the different particle surfaces, bare metal and metal oxide, bound proteins in a more specific manner and not simply in proportion of their abundance in the serum. Although most studies investigated the adsorption of single proteins on nanoparticle surfaces, it was shown that the protein amount adsorbed at physiological pH correlated well with the zeta-potentials and IEPs of surfaces.71 For Si, Ti and Fe oxide NPs, the points of zero charge are in the order of SiO2 (pH ∼ 2) < TiO2 (pH ∼ 5) < Fe2O3 (pH ∼ 7) also reflecting the order of acid strength of the hydroxylated surfaces, e.g. at physiological pH silica and titanium dioxide are negatively charged while maghemite is positively charged (Table 1). However, in a complex composition of FBS, adsorption ability of each inorganic particle does not correlate to the IEP and decreases in the order of TiO2 > Fe2O3 > SiO2. This was also confirmed by work of Horie et al.72 The difference of protein adsorption results from FBS and one protein system might be explained by the influence of protein–protein interaction and other biomolecule interference such as lipids on protein–nanoparticle complex formation.
Tedja et al., reported the influence of serum protein adsorption on TiO2 increasing nanoparticle stability and cellular uptake by A549 cells. TiO2 was found to be taken up inside the cell by an endocytosis mechanism.73 Although in our work, the crystalline type of TiO2 was not studied. The TiO2 surface area has been reported to play a more important role than crystallinity.72 Gold NPs directly interact with lipid membranes, enhancing internalization of the particles74 and revealing the possibility of gold NPs binding onto the lipid related compartment. Gold NPs are also able to induce protein conformational changes.75 A high number of proteins bound on gold coated NPs can also be explained by preferential binding of the –SH group of proteins on the gold NPs.
Based on the results presented herein and in the recent publication from Giri29 it seems that the surface charge of the particles and of the proteins could play an important role in the formation of the protein corona. Because the surface charge of proteins is inhomogeneously distributed over the surface of the protein, we have chosen the isoelectric point (IEP) as a characteristic parameter to investigate the interaction in detail. The difference between the IEP value for a protein and the pH of the media determines the charge (positive if IEP < pH, negative if IEP > pH) but is also in a first estimation proportional to the difference of charges. The same is valid for the IEP of the nanoparticles. In the case of opposed charges of proteins and NP we expect attraction as vice versa to NPs and proteins with the same sign of charge. Therefore we propose an electrostatic interaction index (ESII) which is defined as follows:
| ESII = (IEPprotein − pHmedia)(IEPNanoparticle − pHmedia) |
ESII is positive in the case of both differences being negative or positive, meaning the particles have the same sign of charge (positive or negative); ESII is negative if we have a positive and negative difference. In the first case we can expect repulsion between protein and NP in the second case attraction (this is analogous to colloidal chemistry where attraction has a negative and repulsion a positive potential). In the supplement†, the ESII for all investigated particles is given, taking 7.4 as the pH in the media and the molecular weight of the proteins as a further parameter. The calculation of ESII allows in a further step to determine to what degree the adsorption of proteins is driven by the electrostatic interaction. When the percentage of adsorbed proteins with a negative and positive ESII per particle type taking into account all detected proteins was taken as 100%, it is evident that with increasing IEP the amount of proteins adsorbed increases (Fig. S1†). At low IEP (COOH coated particles), PVA coated and naked SPION (all with IEP < 7.4), it is not specific what has been adsorbed, meaning the electrostatic interaction is not significantly important. If the IEP > 7.4 and/or the particles are functionalized with amino groups, the electrostatic interaction is predominant and the amount of adsorbed proteins is increased. Interestingly, a very similar behavior could be observed with particles with inorganic coating. Negatively charged particles (IEP < 7.4) have a low number of adsorbed proteins and the IEP is not relevant, whereas particles with IEP near to the pH of the media show increased adsorption of proteins but this adsorption is indifferent regarding the ESII. Finally, the particles with NH2 groups at the surface clearly attract proteins with a negative charge (IEP < 7.4) and negative ESII.
Experimental
Materials section is detailed in the ESI.†Methods
In order to obtain highly positively, positively, neutral and negatively charged PVA coated SPION, the different polymer solutions (i.e. 0.2% w/v M12, a mixture of Mowiol®3–85 and M12 at a mass ratio of 45, 10% w/v Mowiol®3–85 and 6% w/v KL-506 solutions: see ESI†) were mixed with 10 mgFe ml−1 SPION suspension at a v/v ratio of 1. The particle suspensions were stored for at least one week and kept at 4 °C until further use. The particle suspension was adjusted to pH 7.4 by 1 M NaOH at least one day before used.
The silica particles were functionalized with amines to produce positively charged silica coated SPION (see ESI†). The obtained particles (SiO2(APTES)–SPION) were washed twice and dispersed in DI water (final concentration approx. 0.1 mgFe ml−1). The nanoparticles were stored at 4 °C.
000g for 45 min. The pellet was washed 3 times with absolute ethanol before being resuspended in DI water and kept at 4 °C.
where NpSpCk is the normalized percentage of spectral count for protein k, SpC is the spectral count identified, and Mw is the molecular weight (in kDa) of the protein k. The MS analysis including the NpSpCk of tightly bound protein eluted from each type of nanoparticle was represented in the supplementary information (Tables S1 to S9†).
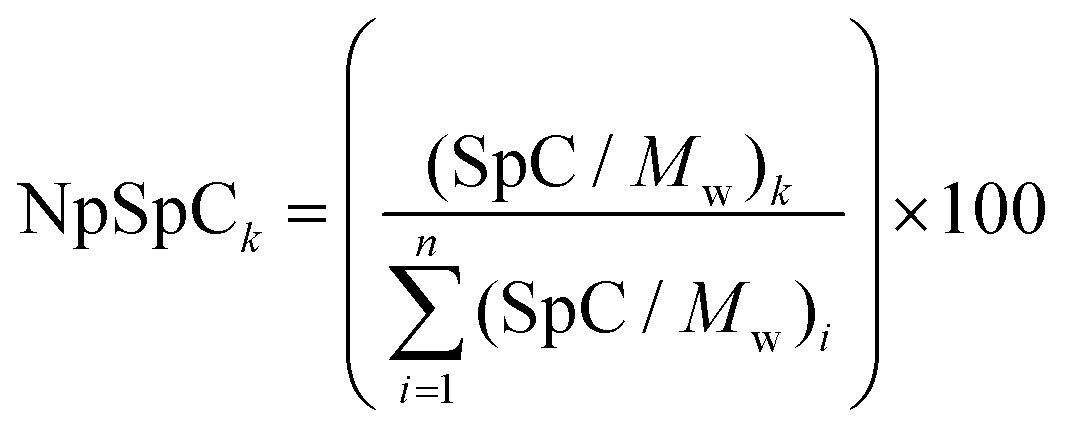
Conclusions
This study provides important information about SPIONs with different nanomaterial coating and their link with the protein composition of coronas derived from a biological fluid (i.e. serum). Also, our results shed more light on the application of SPION with a specific surface meant for the binding of a specific cluster of proteins (in vitro and in vivo) in nanomedicine and nanobiology. The results presented in this work indicated that the composition of the protein corona is very difficult to predict. Highly abundant serum proteins are not always the most abundant proteins in the nanoparticle–protein corona. Some highly abundant serum proteins are even missing in the tightly bound protein corona. Also the charge of the particles is not the only decisive factor. It is likely to be a combination of surface chemistry and charge, which determines protein adsorption. One of the reasons for the absence of correlation between the net charge of adsorbed proteins and the surface charge of the nanoparticles is the complexity of protein nature such as protein conformation, charge distribution and the hydrophobicity/hydrophilicity grade of the protein. We showed clearly that nanoparticle–protein binding strongly depends on the exact surface chemistry of the nanoparticles more than the abundancy level of protein in the biological system. The surface charges, especially on polymeric coated SPION, play a dominant role in protein adsorption. The importance of the material can clearly be seen by comparing polymeric to inorganic and metallic coatings. To quantify this observed relationship between the charge of the proteins and that of the nanoparticles at a given pH of the media, an electrostatic interaction index was introduced and applied with success to the investigated particles. The results presented in this work indicated that the corona composition could be generated for specific biomedical engineering, using NPs with specific surface properties. Moreover understanding of nanoparticle–protein adsorption could finally lead to the prediction of nanoparticle behavior in vitro.Acknowledgements
This work has been supported by the NanoDiaRA project; grant agreement number 228929, funded by the EC Seventh Framework Programme FP7-NMP-2008-L, and the Swiss National Science Foundation (SNSF) fund number 205321-120161/1.Notes and references
- J. H. Clement, M. Schwalbe, N. Buske, K. Wagner, M. Schnabelrauch, P. Görnert, K. O. Kliche, K. Pachmann, W. Weitschies and K. Höffken, J. Cancer Res. Clin. Oncol., 2006, 132, 287–292 CrossRef CAS PubMed
.
- A. Ito, K. Tanaka, K. Kondo, M. Shinkai, H. Honda, K. Matsumoto, T. Saida and T. Kobayashi, Cancer Sci., 2003, 94, 308–313 CrossRef CAS PubMed
- C. Alexiou, R. Jurgons, R. J. Schmid, C. Bergemann, J. Henke, W. Erhardt, E. Huenges and F. Parak, J. Drug Targeting, 2003, 11, 139–149 CrossRef CAS
- D. Artemov, J. Cell. Biochem., 2003, 90, 518–524 CrossRef CAS PubMed
- C. Corot, P. Robert, J.-M. Idée and M. Port, Adv. Drug Delivery Rev., 2006, 58, 1471–1504 CrossRef CAS PubMed
- P. Tartaj, M. P. Morales, S. Veintemillas-Verdaguer, T. González-Carreno and C. J. Serna, J. Phys. Appl. Phys., 2003, 36, R182 CrossRef CAS
-
R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, John Wiley & Sons, 2006 Search PubMed
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed
- C. Chouly, D. Pouliquen, I. Lucet, J. J. Jeune and P. Jallet, J. Microencapsulation, 1996, 13, 245–255 CrossRef CAS PubMed
- S. Saito, M. Tsugeno, D. Koto, Y. Mori, Y. Yoshioka, S. Nohara and K. Murase, Int. J. Nanomedicine, 2012, 7, 5415–5421 CAS
- F. M. Kievit, O. Veiseh, N. Bhattarai, C. Fang, J. W. Gunn, D. Lee, R. G. Ellenbogen, J. M. Olson and M. Zhang, Adv. Funct. Mater., 2009, 19, 2244–2251 CrossRef CAS PubMed
- M. Mahmoudi, A. Simchi, M. Imani and U. O. Häfeli, J. Phys. Chem. C, 2009, 113, 8124–8131 CAS
-
C. S. S. R. Kumar, Magnetic nanomaterials, ed. C. Kumar, Wiley-VCH, Weinheim, 2009 Search PubMed
-
K. T. Thanh Nguyen, Magnetic nanoparticles: from fabrication to clinical applications: theory to therapy, chemistry to clinic, bench to bedside, CRC Press, Taylor and Francis Group, LLC, Boca Raton, FL, Theory edn, 2012 Search PubMed
- R. Landsiedel, L. Ma-Hock, A. Kroll, D. Hahn, J. Schnekenburger, K. Wiench and W. Wohlleben, Adv. Mater., 2010, 22, 2601–2627 CrossRef CAS PubMed
- I. Lynch, T. Cedervall, M. Lundqvist, C. Cabaleiro-Lago, S. Linse and K. A. Dawson, Adv. Colloid Interface Sci., 2007, 134–135, 167–174 CrossRef CAS PubMed
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed
- E. Amstad, M. Textor and E. Reimhult, Nanoscale, 2011, 3, 2819–2843 RSC
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, Colloids Surf., B Biointerfaces, 2000, 18, 301–313 CrossRef CAS
- T. Cedervall, I. Lynch, M. Foy, T. Berggård, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed
- C. Salvador-Morales, E. Flahaut, E. Sim, J. Sloan, M. L. H. Green and R. B. Sim, Mol. Immunol., 2006, 43, 193–201 CrossRef CAS PubMed
- K. Thode, M. Lück, W. Semmler, R. H. Müller and M. Kresse, Pharm. Res., 1997, 14, 905–910 CrossRef CAS
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845–867 CAS
- S. Lista, F. Faltraco and H. Hampel, Prog. Neurobiol., 2013, 101–102, 18–34 CrossRef CAS PubMed
- Y. Shen, J. Kim, E. F. Strittmatter, J. M. Jacobs, D. G. Camp 2nd, R. Fang, N. Tolié, R. J. Moore and R. D. Smith, Proteomics, 2005, 5, 4034–4045 CrossRef CAS PubMed
- V. Mirshafiee, M. Mahmoudi, K. Lou, J. Cheng and M. L. Kraft, Chem. Commun., 2013, 49, 2557–2559 RSC
- S. Krol, R. Macrez, F. Docagne, G. Defer, S. Laurent, M. Rahman, M. J. Hajipour, P. G. Kehoe and M. Mahmoudi, Chem. Rev., 2013, 113, 1877–1903 CrossRef CAS PubMed
- A. Elsaesser and C. V. Howard, Adv. Drug Delivery Rev., 2012, 64, 129–137 CrossRef CAS PubMed
- K. Giri, K. Shameer, M. T. Zimmermann, S. Saha, P. K. Chakraborty, A. Sharma, R. R. Arvizo, B. J. Madden, D. J. Mccormick, J.-P. A. Kocher, R. Bhattacharya and P. Mukherjee, Bioconjugate Chem., 2014, 25(6), 1078–1090 CrossRef CAS PubMed
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2014, 47(8), 2651–2659 CAS
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed
- J. Klein, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2029–2030 CrossRef CAS PubMed
- B. Steitz, J. Salaklang, A. Finka, C. O'Neil, H. Hofmann and A. Petri-Fink, Bioconjugate Chem., 2007, 18, 1684–1690 CrossRef CAS PubMed
- V. Hirsch, C. Kinnear, M. Moniatte, B. Rothen-Rutishauser, M. J. D. Clift and A. Fink, Nanoscale, 2013, 5, 3723–3732 RSC
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed
- H. T. R. Wiogo, M. Lim, V. Bulmus, J. Yun and R. Amal, Langmuir, 2011, 27, 843–850 CrossRef CAS PubMed
- C.-C. You, A. Chompoosor and V. M. Rotello, Nano Today, 2007, 2, 34–43 CrossRef
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed
- R. R. Arvizo, K. Giri, D. Moyano, O. R. Miranda, B. Madden, D. J. McCormick, R. Bhattacharya, V. M. Rotello, J.-P. Kocher and P. Mukherjee, PLoS One, 2012, 7 Search PubMed
- K. Suttiponparnit, J. Jiang, M. Sahu, S. Suvachittanont, T. Charinpanitkul and P. Biswas, Nanoscale Res. Lett., 2010, 6, 27 Search PubMed
- C. Schulze, C. Schulze, A. Kroll, C. Schulze, A. Kroll, C.-M. Lehr, U. F. Schäfer, K. Becker, J. Schnekenburger, C. Schulze Isfort, R. Landsiedel and W. Wohlleben, Nanotoxicology, 2008, 2, 51–61 CrossRef CAS
- F. Rezaee, B. Casetta, J. H. M. Levels, D. Speijer and J. C. M. Meijers, Proteomics, 2006, 6, 721–730 CrossRef CAS PubMed
- K. C. S. Queiroz, R. A. Tio, C. J. Zeebregts, M. F. Bijlsma, F. Zijlstra, B. Badlou, M. de Vries, C. V. Ferreira, C. A. Spek, M. P. Peppelenbosch and F. Rezaee, J. Proteome Res., 2010, 9, 6052–6059 CrossRef CAS PubMed
- S. Ray, P. J. Reddy, S. Choudhary, D. Raghu and S. Srivastava, J. Proteomics, 2011, 74, 2660–2681 CrossRef CAS PubMed
- X. Zheng, H. Baker, W. S. Hancock, F. Fawaz, M. McCaman and E. Pungor Jr., Biotechnol. Prog., 2006, 22, 1294–1300 CrossRef CAS PubMed
- D. Simberg, J.-H. Park, P. P. Karmali, W.-M. Zhang, S. Merkulov, K. McCrae, S. N. Bhatia, M. Sailor and E. Ruoslahti, Biomaterials, 2009, 30, 3926–3933 CrossRef CAS PubMed
- M. Rankl, T. Ruckstuhl, M. Rabe, G. R. J. Artus, A. Walser and S. Seeger, ChemPhysChem, 2006, 7, 837–846 CrossRef CAS PubMed
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed
- W. Shang, J. H. Nuffer, J. S. Dordick and R. W. Siegel, Nano Lett., 2007, 7, 1991–1995 CrossRef CAS PubMed
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed
-
P. Vilaseca, K. A. Dawson and G. Franzese, Understanding surface-adsorption of proteins: the Vroman effect, 2012 Search PubMed
- D. Dell'Orco, M. Lundqvist, C. Oslakovic, T. Cedervall and S. Linse, PLoS One, 2010, 5, e10949 Search PubMed
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggård, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef CAS PubMed
- H. Zhang, K. E. Burnum, M. L. Luna, B. O. Petritis, J.-S. Kim, W.-J. Qian, R. J. Moore, A. Heredia-Langner, B.-J. M. Webb-Robertson, B. D. Thrall, D. G. Camp, R. D. Smith, J. G. Pounds and T. Liu, Proteomics, 2011, 11, 4569–4577 CrossRef CAS PubMed
- A. Gessner, R. Waicz, A. Lieske, B. Paulke, K. Mäder and R. H. Müller, Int. J. Pharm., 2000, 196, 245–249 CrossRef CAS
- K. E. Ryu, H. Rhim, C. W. Park, H. J. Chun, S. H. Hong, J. J. Kim and Y. M. Lee, Macromol. Res., 2003, 11, 451–457 CrossRef CAS
- D. A. Barrett, M. S. Hartshome, M. A. Hussain, P. N. Shaw and M. C. Davies, Anal. Chem., 2001, 73, 5232–5239 CrossRef CAS
- K. Ogawara, K. Furumoto, S. Nagayama, K. Minato, K. Higaki, T. Kai and T. Kimura, J. Controlled Release, 2004, 100, 451–455 CrossRef CAS PubMed
- G. Döring, Pediatr. Pulmonol., 1999, 28, 363–375 CrossRef
- W. Jahnen-Dechent, C. Schäfer, M. Ketteler and M. D. McKee, J. Mol. Med., 2008, 86, 379–389 CrossRef CAS PubMed
- E. Hellstrand, I. Lynch, A. Andersson, T. Drakenberg, B. Dahlbäck, K. A. Dawson, S. Linse and T. Cedervall, FEBS J., 2009, 276, 3372–3381 CrossRef CAS PubMed
- R. Cukalevski, M. Lundqvist, C. Oslakovic, B. Dahlbäck, S. Linse and T. Cedervall, Langmuir, 2011, 27, 14360–14369 CrossRef CAS PubMed
- G. Gunkel and W. T. S. Huck, J. Am. Chem. Soc., 2013, 135, 7047–7052 CrossRef CAS PubMed
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed
- D. Hühn, K. Kantner, C. Geidel, S. Brandholt, I. De Cock, S. J. H. Soenen, P. Rivera_Gil, J.-M. Montenegro, K. Braeckmans, K. Müllen, G. U. Nienhaus, M. Klapper and W. J. Parak, ACS Nano, 2013, 7, 3253–3263 CrossRef PubMed
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed
- C. Sacchetti, K. Motamedchaboki, A. Magrini, G. Palmieri, M. Mattei, S. Bernardini, N. Rosato, N. Bottini and M. Bottini, ACS Nano, 2013, 7, 1974–1989 CrossRef CAS PubMed
- N. Kitamura, Y. Takagaki, S. Furuto, T. Tanaka, H. Nawa and S. Nakanishi, Nature, 1983, 305, 545–549 CrossRef CAS
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed
- K. Rezwan, A. R. Studart, J. Vörös and L. J. Gauckler, J. Phys. Chem. B, 2005, 109, 14469–14474 CrossRef CAS PubMed
- M. Horie, K. Nishio, K. Fujita, S. Endoh, A. Miyauchi, Y. Saito, H. Iwahashi, K. Yamamoto, H. Murayama, H. Nakano, N. Nanashima, E. Niki and Y. Yoshida, Chem. Res. Toxicol., 2009, 22, 543–553 CrossRef CAS PubMed
- R. Tedja, M. Lim, R. Amal and C. Marquis, ACS Nano, 2012, 6, 4083–4093 CrossRef CAS PubMed
- J. Lin, H. Zhang, Z. Chen and Y. Zheng, ACS Nano, 2010, 4, 5421–5429 CrossRef CAS PubMed
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed
- M. Chastellain, A. Petri and H. Hofmann, J. Colloid Interface Sci., 2004, 278, 353–360 CrossRef CAS PubMed
- A. Petri-Fink, M. Chastellain, L. Juillerat-Jeanneret, A. Ferrari and H. Hofmann, Biomaterials, 2005, 26, 2685–2694 CrossRef CAS PubMed
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef
- R. C. Mehrotra, J. Non-Cryst. Solids, 1988, 100, 1–15 CrossRef CAS
- E. Scolan and C. Sanchez, Chem. Mater., 1998, 10, 3217–3223 CrossRef CAS
- M. R. Rasch, K. V. Sokolov and B. A. Korgel, Langmuir, 2009, 25, 11777–11785 CrossRef CAS PubMed
- U. Sakulkhu, L. Maurizi, M. Mahmoudi, M. Motazacker, M. Vries, A. Gramoun, M.-G. O. Beuzelin, J.-P. Vallée, F. Rezaee and H. Hofmann, Nanoscale, 2014, 6, 11439–11450 RSC
- U. Sakulkhu, M. Mahmoudi, L. Maurizi, J. Salaklang and H. Hofmann, Sci. Rep., 2014, 4, 5020 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4bm00264d |
| This journal is © The Royal Society of Chemistry 2015 |