Prussian blue: a new framework of electrode materials for sodium batteries†
Yuhao
Lu‡
,
Long
Wang‡
,
Jinguang
Cheng
and
John B.
Goodenough
*
Texas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: jgoodenough@mail.utexas.edu
First published on 8th May 2012
Abstract
Prussian blue and its analogues consisting of different transition-metal ions (Fe, Mn, Ni, Cu, Co and Zn) have been synthesized at room temperature. Insertion of Na into KFe2(CN)6 in a carbonate electrolyte exhibited a reversible capacity near 100 mA h g−1 with no capacity fade in 30 cycles. The data indicate that a Na-ion battery with a Prussian blue framework as a cathode will be feasible.
The Li-ion battery has played a dominant role in portable electronic devices and power tools; it is being developed actively for plug-in hybrid vehicles. Its success places an increasing demand on sources of lithium, which has raised a desire to build a Na-ion battery as an alternative.1,2 The sodium–sulfur battery of Kummer and Weber3 operates at 300 to 350 °C, which creates maintenance problems and elevated operational costs. Therefore, there is motivation to find a room-temperature rechargeable Na-ion battery. Present Li-ion rechargeable batteries rely on an oxide host of the cathode from/into which Li can be extracted/inserted reversibly.4–11 The Li+ ion is small enough to have an acceptable mobility and a solid-solution range in close-packed oxide–ion arrays; but the larger Na+ ion requires a more open framework in which to move reversibly with an acceptable mobility. The hexagonal framework of Na1+3xZr2(P1−xSixO4)3, commonly referred to as NASICON, exhibits fast Na+-ion conductivity;12 its framework has the hexagonal Fe2(SO4)3 structure. Unfortunately, the Na+ mobility is greatly reduced where the M2(XO4)3 units of the framework are rotated with respect to one another to give a monoclinic structure, and substitution of a transition-metal of interest for Zr(IV) gives the monoclinic structure if Na+ is the guest alkali ion.13 Therefore, there is interest in identifying another open host framework containing a suitable transition-metal ion.
KFe2(CN)6, Prussian blue, has a cubic framework (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with Fe(II) and Fe(III) on alternate corners of a cube of corner-shared octahedra bridged by linear (C
m) with Fe(II) and Fe(III) on alternate corners of a cube of corner-shared octahedra bridged by linear (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)− anions (Fig. 1); the low-spin Fe(III) bond only with C atoms, the high-spin Fe(II) only with N atoms,14–16 and the CN bond opens the faces of the elementary cubes for Na+ to move between half-filled body-center positions.
N)− anions (Fig. 1); the low-spin Fe(III) bond only with C atoms, the high-spin Fe(II) only with N atoms,14–16 and the CN bond opens the faces of the elementary cubes for Na+ to move between half-filled body-center positions.
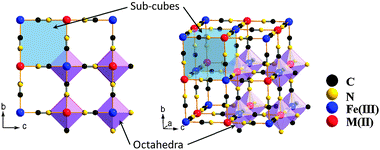 | ||
| Fig. 1 Framework of Prussian blue analogues. | ||
Other transition-metal M(II) ions can replace the Fe(II) of Prussian blue in the family KMFe(CN)6. Widman et al.17 and later Wessells et al.18–20 have investigated Na and K insertion into KMFe(CN)6 with M = Cu and Ni using a Pt counter electrode and an Ag–AgCl reference electrode with an aqueous electrolyte. The cell had low potentials owing to the limitation of the aqueous electrolyte and the reference electrode. Nevertheless, their data invite exploration of reversible insertion of Na+ into a Prussian blue analogue with an aprotic electrolyte. Therefore, we have synthesized KMFe(CN)6 compounds (MFHC) with M = Mn, Fe, Co, Ni and Zn; and we show that in an organic liquid-carbonate electrolyte and a Na anode, KFe(II)Fe(III)(CN)6 can exhibit a reversible capacity of ca. 100 mA h g−1.
In this communication, MFHC are synthesized at room temperature. Briefly, the M(II) salt solution of 0.1 M was slowly added into the K3Fe(CN)6 solution of 0.05 M with a strong magnetic stirring. The total mole of the M(II) salt is twice that of K3Fe(CN)6. After 30 minutes of magnetic stirring, the mixed solution was moved to ultrasonic stirring. The process was kept for 2 hours. The products of KM(II)FHC were filtered and washed with DI-water three times. The final products were dried in a vacuum box at 70 °C overnight. These compounds were stored in a vacuum desiccator.
The structures of the Prussian blue compounds were characterized by Powder X-ray diffraction (XRD) obtained with a Philips X-ray diffractometer equipped with Cu Kα radiation (λ = 1.5418 Å). The angular resolution in 2θ scans was 0.02° over a 2θ range of 10–70°. The powder X-ray diffraction (XRD) patterns of Fig. 2a for the KM(II)Fe(III) (CN)6 compounds show that KZnFe(CN)6 forms a hexagonal lattice (space group Rc) as has been reported.21 Whereas all the other M(II) cations simply change the cubic lattice parameter; a0 = 10.170 Å, 10.151 Å, 10.095 Å, 10.119 Å, 10.112 Å, respectively, for M = Mn, Fe, Co, Ni, and Cu. It is noticed that with M = Co, the lattice parameter is quite close to the high temperature phase of cobalt–iron cyanide, indicating that the compound mostly consists of FeIII(t52ge0g, S = 1/2)–CN–CoII(t52ge2g, S = 3/2).22 In order to further determine the electron configuration, magnetic susceptibility of all products was studied. Fig. 2b shows the inverse molar magnetic susceptibility (χm−1) as a function of temperature; μB calculated from this plot indicates that Fe(III) remains in the low-spin with S = 1/2 while M = Mn, Co, Ni, Cu, Zn have, respectively, spins S ≈ 5/2, 3/2, 1, 1/2, 0.
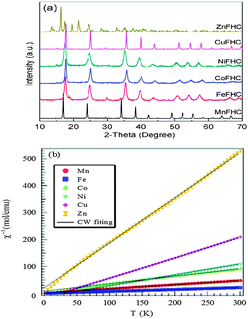 | ||
| Fig. 2 (a) XRD patterns of Prussian blue compounds. (b) χm−1–T plot of Prussian blue compounds. | ||
Whether a localized-electron configuration on an octahedral-site cation is in a high-spin or a low-spin state depends on the relative magnitudes of the cubic-field splitting Δc of the π-bonding t2 and σ-bonding e orbitals of the d-state manifold versus the exchange energy Δex responsible for Hund's highest-spin rule in the free atom; a low-spin configuration has a Δc > Δex, a high-spin configuration has Δc < Δex. From second-order perturbation theory appropriate for the covalent interaction to the bonding of localized 3d electrons, Δε = |b|2/ΔE, where b = 〈Ψd|H′|Ψp〉 ≈ ε(Ψd, Ψp) varies as the overlap integral (Ψd, Ψp) of the cation-d and anion-p orbitals and ΔE is the energy difference of the lowest empty cation-d and highest filled anion-p orbitals in the absence of covalent bonding. The (CN)− ion has its pπ orbitals primarily active in the internal triple bond whereas there is a strong pσ overlap with the cation-d orbitals. Therefore, since ΔE is the same for both the π-bonding t and σ-bonding e orbitals, the difference in the magnitude of the pσ and pπ overlap integrals makes Δc larger for a (CN)− ion than that for an O2− ion. In addition, ΔE for the M–C bond is smaller than that for the M–N bond, so Δc for the Fe(III) bonding to C is much larger than Δc for the M(II) bonding to N. As a result, the Fe(III) is in its low-spin state despite a large S = 5/2 in the high-spin state whereas the Fe(II), which bond to N, remains in its high-spin state despite a smaller S = 2. Nevertheless, we should anticipate a Δc only a little smaller than Δex for the Fe(II) of Prussian blue.
Fig. 3 shows the electrochemical behavior of KMFe(CN)6–Na cells at the third cycle with a current of C/20 in the range 2.0 to 4.0 V vs. Na+–Na. In these cells, Na is being inserted reversibly into the KMFe(CN)6 cathode and the low-spin Fe(III) is being converted to low-spin Fe(II). However, during charge, some K+ ions are removed as well as Na+ ions, so more than one Na+ ion per formula unit is inserted during the second and third discharge. Therefore, on the third discharge–charge cycle, the KFe2(CN)6 cathode shows two peaks at 2.97 and 3.69 V on its charge curves and two peaks at 2.92 and 3.58 V on its discharge curves. The peaks at 2.97/2.92 V correspond to oxidation–reduction of the high-spin Fe(III)–Fe(II) couple bonding to N and those at 3.69/3.58 V to the low-spin Fe(III)–Fe(II) couple bonding to C.17 The M = Co, Ni, and Cu show that the voltages of the low-spin Fe(III)–Fe(II) couple are all similar, but lower than that of KFe2(CN)6; there is little, if any, removal of K+ ions on charge where the lattice parameter a0 is smaller. Although more K+ is removed from KMnFe(CN)6 which has the largest lattice parameter, nevertheless relatively little participation of the Mn(III)–Mn(II) couple is found at 3.82/3.56 V; instead it exhibits a somewhat larger discharge capacity on the low-spin Fe(III)–Fe(II) couple. These features of the redox potentials and capacities of the low-spin redox energies are summarized in Fig. 4a. The KFe2(CN)6–Na battery in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC (1:1 v/v) with 1 M NaClO4 as an electrolyte was cycled at C/20 30 times at room temperature. Fig. 4b shows that the PB framework exhibits excellent capacity retention; more than 99% discharge capacity is retained after 30 cycles. Meanwhile, the cell shows a low coulombic efficiency at the initial cycling. A most probable cause for such a low coulombic efficiency is crystalline water in the PB compound. The large interstitial spaces in PB readily absorb water molecules.23 It is doubtful that the water molecules can be totally removed from the lattice. Decomposition of the residual water during the charge process would cause a low efficiency. With subsequent cycles, the amount of residual water would decrease and the efficiency increase as shown in Fig. 4b.
:DEC (1:1 v/v) with 1 M NaClO4 as an electrolyte was cycled at C/20 30 times at room temperature. Fig. 4b shows that the PB framework exhibits excellent capacity retention; more than 99% discharge capacity is retained after 30 cycles. Meanwhile, the cell shows a low coulombic efficiency at the initial cycling. A most probable cause for such a low coulombic efficiency is crystalline water in the PB compound. The large interstitial spaces in PB readily absorb water molecules.23 It is doubtful that the water molecules can be totally removed from the lattice. Decomposition of the residual water during the charge process would cause a low efficiency. With subsequent cycles, the amount of residual water would decrease and the efficiency increase as shown in Fig. 4b.
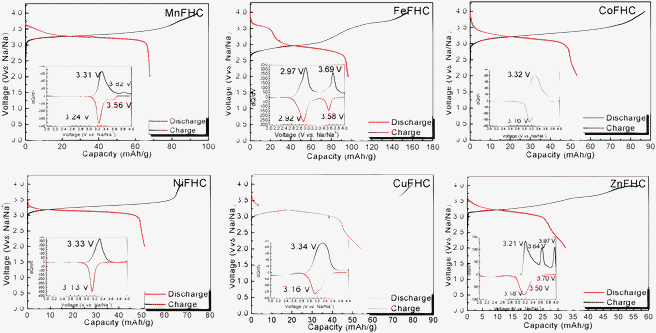 | ||
| Fig. 3 The charge–discharge curves of Prussian blue analogues KMFe(CN)6–Na cells at the third cycle with a current of C/20. Insets show their corresponding chronoamperograms. | ||
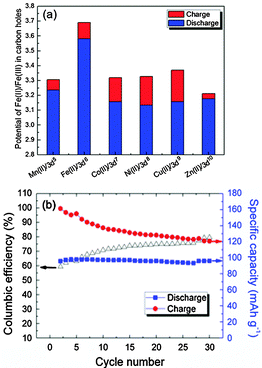 | ||
| Fig. 4 (a) The redox potentials of the low-spin Fe(II)–Fe(III) in the Prussian blue compounds. (b) Cycling performance of KFeFHC–Na battery cycled at C/20 between 2.0–4.0 V. | ||
We are left to ask why the voltage of the low-spin Fe(III)–Fe(II) couple is significantly larger in the KFe2(CN)6 than in the other KMFe(CN)6 compounds. A larger voltage means a more stable redox energy and, therefore, a weaker Fe–C bond since the redox couple is an antibonding state with respect to the anion-p orbitals. Clearly a quantitative calculation of the effect of the strength of the M(II)–N bonding on the strength of the Fe–C bonding as well as a measure of the variation of the Fe–C bond length with changing of the M(II) cation is needed to clarify this question.
In summary, the hexacyanides with the Prussian blue structure, KMFe(CN)6, are easily synthesized and have a low cost. We have shown that the linear (CN)− molecule gives an M(II)–NC–Fe(III) bond length that allows Na+ ions to be inserted reversibly into the empty large-ion sites. Moreover, the strong π-bond component in the linear CN bond enhances the crystal-field splitting Δc of the π-bonding t and σ-bonding e orbitals of the 3d manifolds on the transition-metal cations, particularly for the Fe–C bond, which favors stabilization of low-spin state Fe(III): 3d5 configuration in an octahedral site of C atoms whereas the Fe(II): 3d6 configuration coordinated to N atoms is in a high-spin state. A flat discharge capacity of over 70 mA h g−1 with good reversibility can be realized for a Na-ion battery using these cathodes, which warrants further investigation of exchanging Na+ for K+ in these structures.
Notes and references
- J.-M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS
.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Tojo, Energy Environ. Sci., 2012, 5, 5884 CAS
- J. Kummer and N. Weber, SAE [Tech. Pap.], 1967, 670179 Search PubMed
- R. Berthelot, D. Carlier and C. Delmas, Nat. Mater., 2011, 10, 74 CrossRef CAS
- X. Ma, H. Chen and G. Ceder, J. Electrochem. Soc., 2011, 158, A1307 CrossRef CAS
- D. Kim, S.-H. Kang, M. Slater, S. Rood, J. T. Vaughey, N. Karan, M. Balasubramanian and C. S. Johnson, Adv. Energy Mater., 2011, 1, 333 CrossRef CAS
- K. T. Lee, T. N. Ramesh, F. Nan, G. Botton and L. F. Nazar, Chem. Mater., 2011, 23, 3593 CrossRef CAS
- P. Moreau, D. Guyomard, J. Gaubicher and F. Boucher, Chem. Mater., 2010, 22, 4126 CrossRef CAS
- O. Tillement, J. Angenault, J. C. Couturier and M. Quarton, Solid State Ionics, 1992, 53–56, 391 CrossRef CAS
- S. I. Park, I. Gocheva, S. Okada and J.-i. Yamaki, J. Electrochem. Soc., 2011, 158, A1067 CrossRef CAS
- Z. Jian, L. Zhao, H. Pan, Y.-S. Hu, H. Li, W. Chen and L. Chen, Electrochem. Commun., 2012, 14, 86 CrossRef CAS
- J. B. Goodenough, H. Y. P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203 CrossRef CAS
- A. K. Padhi, K. S. Nanjundaswamy, C. Masquelier and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 2581 CrossRef CAS
- A. Kumar and S. M. Yusuf, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 054414 CrossRef
- R. Martínez-Garcia, E. Reguera, J. Balmaseda and H. Yee-Madeira, Powder Diffr., 2004, 19, 284 CrossRef
- N. Shimamoto, S. Ohkoshi, O. Sato and K. Hashimoto, Inorg. Chem., 2002, 41, 678 CrossRef CAS
- A. Widmann, H. Kahlert, I. Petrovic-Prelevic, H. Wulff, J. V. Yakhmi, N. Bagkar and F. Scholz, Inorg. Chem., 2002, 41, 5706 CrossRef CAS
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421 CrossRef CAS
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2012, 159, A98 CrossRef CAS
- A. Widmann, H. Kahlert, H. Wulff and F. Scholz, J. Solid State Electrochem., 2005, 9, 380 CrossRef CAS
- T. Yokovama, M. Kiguchi and T. Ohta, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 9340 CrossRef
- M. Ware, J. Chem. Educ., 2008, 85, 612 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cc31777j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |