
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon conduction and phase behaviour in dual cation polyelectrolyte blends for sodium-ion batteries†
Sneha
Malunavar
a,
Luca
Porcarelli
*ab,
Patrick C.
Howlett
a,
David
Mecerreyes
bc and
Maria
Forsyth
*abc
aInstitute for Frontier Materials (IFM) Deakin University, Burwood, Victoria 3125, Australia. E-mail: maria.forsyth@deakin.edu.au
bPolymat, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda, Tolosa 7, 20018 Donostia-San Sebastian, Spain
cIkerbasque, Basque Foundation for Science, 48013, Bilbao, Spain
First published on 31st January 2024
Abstract
Emerging battery technologies such as solid-state sodium batteries can benefit from new polymer electrolytes with improved sodium ion transport to optimise electrochemical performance. In this work, we propose, for the first time, the use of polyelectrolyte blends utilising a dual cation approach with a common polyanion backbone, poly(1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethanesulfonyl)imide) (polyMTFSI). Thus, three new anionic polyelectrolytes were synthesised based on polyMTFSI having three different counter cations such as sodium (Na) (polyMTFSI-Na), trimethyl(isobutyl)phosphonium (poly-MTFSIP111i4) and diethyl (isobutyl)(methyl)phosphonium (polyMTFSI-P122i4). The miscibility between the polyelectrolytes in blends was determined by observing a single glass transition, Tg, for different compositions. Upon the addition of bulky organic cations, an increase in the dynamics and ionic conductivity was observed. Finally, we investigated the effect of NaFSI as an additional component in a ternary electrolyte system, whereby the salt acted as a plasticizer, decreasing Tg, and further enhancing the ionic conductivity.
Introduction
Research into new energy storage systems is gaining increasing importance compared with any time in the past. In particular, efforts to improve the lithium (Li)/sodium (Na) ion battery technology are seen as paramount due to their high specific energy and energy density (per unit volume)1,2 compared to other technologies. With the electrolyte being the key component of any electrochemical device, research has been focusing on the development of new electrolytes with improved performance in addition to being cost effective and safe in batteries. One effective way to improve the safety of batteries is by developing solid polymer electrolytes (SPEs) having both good ion transport and robust mechanical properties.3–5 Solid-state electrolytes are promising compared to liquid electrolytes in terms of non-flammability, no leakage issues, and good mechanical properties, and they serve as both the electrolyte and the separator. The different classes of solid-state electrolytes are solid polymer electrolytes, gel polymer electrolytes, inorganic electrolytes, and composites. Despite their advantages, they are still in the early stages of research and development compared to liquid electrolytes, and the current solid-state Na electrolytes have low ionic conductivity at room temperature, and there have been some attempts to increase their ionic conductivity by incorporating various additives. There is a lot of research around the development of solid-state electrolytes for lithium-ion batteries but not as much progress has been made for SIB applications. Therefore, developing a solid-state electrolyte for SIB applications is the main goal of this work.8–10In the 1970s, Wright et al.6 reported SPEs comprised of PEO and alkali salts such as NaI. Later, Armand et al.7 reported SPEs for Li-ion conducting systems with weakly coordinating anions (bis(trifluoromethanesulfonyl)imide, TFSI−) forming a flexible molecular structure, hypothesising that the selection of such anions for making Li conducting SPEs would be beneficial for practical lithium metal batteries (LMBs).
Traditional SPEs usually consist of Li/Na salts dissolved in a polymer host; these are commonly recognised as dual-ion conductors, in which case both alkali metal cations and the accompanying anions will be mobile. In most cases anions are more mobile than the metal cations, which accounts for the low transference number in dual ion conducting SPEs, usually being much lower than 0.5, as has been demonstrated in extensive studies on these SPEs.8,10–15 This kind of dual ion conducting SPE tends to have drawbacks during the cycling of the cell, as both the ions are moving and the anions tend to accumulate at the positive electrode causing a concentration gradient which leads to cell polarization and results in poor cell performance, voltage loses, high internal impedance along with unexpected reactions during cycling and finally leading to cell death.16 One way to overcome these issues would be to develop SPEs which possess a metal ion transference number equal to unity, so-called single ion conductors. Fuller et al.17 demonstrated that polymer electrolytes with a transference number equal to 1 showed overall good performance even with a conductivity decrease by one order. Therefore, there is interest in developing single ion conducting SPEs to overcome the above-mentioned challenges caused by the dual ion conducing SPEs, at the very least to reduce or eliminate anion conductivity in SPEs.
Bannister et al.,18 in the 1980s, came up with two ways to design single Li-ion conducting SPEs. One way is to tether the anion to the polymer backbone; in this way the anion will be covalently bound to the polymer backbone, eliminating its contribution to conductivity (and hence its transference number is zero) and allowing the counter cation to be the sole charge carrier in the electrolyte. Another way is to use Li salts where the mobility of the anion is inhibited. Since then, there have been various reports on such SPEs with Armand et al.19 having extensively reviewed the various synthetic strategies and designs of single ion conducting SPEs with a high transference number and high ionic conductivities for lithium-ion batteries. Zheng et al.20 reported sodium ion single ion conducting SPEs consisting of poly(sodium 2-[μ-metharyloyloligo(oxyethylene)] ethylsulfonate); in this system, the oligo(oxyethylene) chain assists in the dissociation of the sodium cation from sulfonate and Na+ ion transport is facilitated by flexible macromolecular chains.
In all cases, the ion motion in these SPEs is governed by the local segmental motion of the polymer as reflected in the glass transition (Tg). However, it is difficult to achieve both high ionic conductivity and good mechanical properties if the design requires Tg to be low. Another approach is to design SPEs where ions are mobile below the Tg and hence decoupling ionic conductivity from polymer segmental motion. Using this strategy, Mohd Noor et al.21 reported SPEs composed of poly-([triethylmethylammonium][2-acrylamido-2-methyl-1-propanesulfonicacid]-co-sodium[vinyl sulfonate]), poly([N1222][AMPS]-co-Na[VS]); here some fractions of sodium ions were replaced by a bulky quaternary ammonium cation. The intention here was to decrease the strong interactions between the Na cations and the anions on the polymer backbone by replacing some of the interacting cations with bulky organic cations. They found this approach to be partially successful with considerable ionic conductivity measured below the glass transition temperature of the polymers; this indicated that the ion transport mechanism was indeed decoupled from the structural relaxations of the polymer. The hypothesis was that the addition of the bulky cation increased the free volume in the polymer matrix and created a ‘looser’ ionic structure facilitating sodium ion hopping. They further extended their study to another system, poly(2-acryl-amido-2-methyl-1-propane-sulfonate) homopolymer (PAMPS), where they also reported significant conductivity below the Tg, with the highest conductivity being achieved by 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 mol% of Na+/triethylmethylammonium (N1222) cations.22 They observed similar trends of decoupling in all these co-cation systems and found that conductivity is improved when the organic cation is bulkier. Unfortunately, the ionic conductivities reported were much lower than required for practical device applications due to the strong bonding between the sulfonate anions on the backbone and the metal cations. Interestingly, when a small amount of tetraglyme (∼10 wt%) was incorporated as a plasticizer, this resulted in a jump in ionic conductivity by several orders while still retaining a high Tg. There are additional reports in the literature based on this hypothesis; however, all rely on the sulfonate anion on the polyelectrolyte,23,24 and designing systems with a more delocalised anion to decrease the interionic binding energy is hypothesised as a possible way forward with this concept to develop Na+ conducting electrolytes for Na battery applications.
:90 mol% of Na+/triethylmethylammonium (N1222) cations.22 They observed similar trends of decoupling in all these co-cation systems and found that conductivity is improved when the organic cation is bulkier. Unfortunately, the ionic conductivities reported were much lower than required for practical device applications due to the strong bonding between the sulfonate anions on the backbone and the metal cations. Interestingly, when a small amount of tetraglyme (∼10 wt%) was incorporated as a plasticizer, this resulted in a jump in ionic conductivity by several orders while still retaining a high Tg. There are additional reports in the literature based on this hypothesis; however, all rely on the sulfonate anion on the polyelectrolyte,23,24 and designing systems with a more delocalised anion to decrease the interionic binding energy is hypothesised as a possible way forward with this concept to develop Na+ conducting electrolytes for Na battery applications.
Therefore, in the present work, we investigated mixed co-cation SPEs where the 1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethanesulfonyl)imide anion is tethered to the polymer backbone. The structure of this moiety is related to the bis(trifluoromethanesulfonyl) or TFSI anion that is known to have a more delocalised charge and leads to improved conductivity in dual ion conductors. Furthermore, instead of copolymerizing different monomers, we investigated the use of blends between different polyelectrolytes having different counter cations. For this purpose, three methacrylic sulfonamide polyelectrolytes with different counter cations, namely sodium and two bulky phosphonium cations, were synthesized as shown in Scheme 1. The blends between the different polyelectrolytes were investigated with a focus on their miscibility, glass transition temperature, ionic conductivity, mechanical behaviour, and their inter-relationships. We also investigated the effect of adding NaFSI to the most conductive polyelectrolyte blend composition to further enhance the ionic conductivity and present preliminary electrochemical properties for this material.
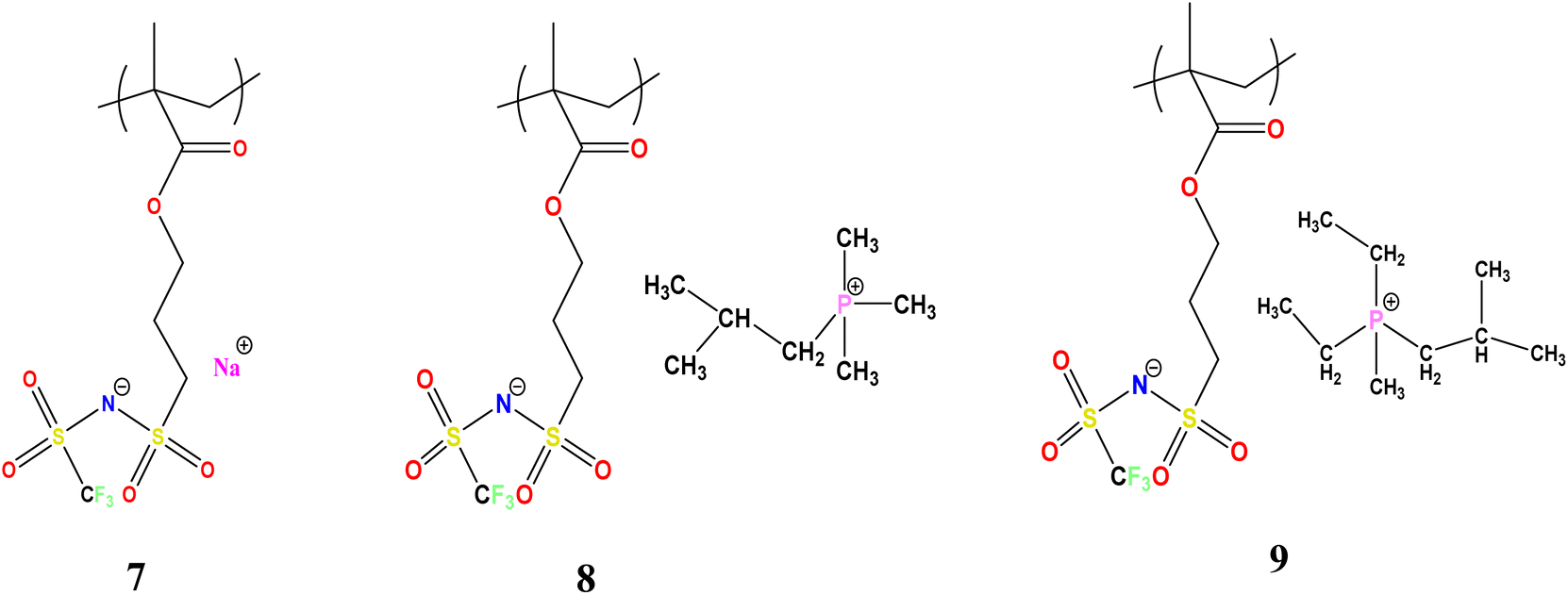 | ||
| Scheme 1 Structure of polymers NaMTFSI 7, P111i4MTFSI 8 and P122i4MTFSI 9. | ||
Results and discussion
The synthetic route for the preparation of the monomers is shown in Scheme 1. The NaMTFSI monomer was synthesised by following 3 steps as reported previously for the LiMTFSI monomer by Porcarelli et al. and Shaplov et al.25,26 Then, the corresponding three homopolymers were synthesized by free-radical polymerization. The chemical structure is shown in Scheme 1, with a common polyanion backbone, poly(1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethanesulfonyl)imide) (polyMTFSI), having three different counter cations such as sodium (Na) (polyMTFSI-Na), trimethyl(isobutyl)phosphonium (poly-MTFSIP111i4) and diethyl (isobutyl)(methyl)phosphonium (polyMTFSI-P122i4).In this work, a series of blends between two different polyelectrolytes having the same polymer backbone but different counterions were systematically prepared and characterised. Specifically, poly-NaMTFSI 7 was blended by simple co-casting from a common solvent (acetonitrile) with two different phosphonium based polyelectrolytes 8 and 9. The ratios of the polymer blends that were considered are provided in Table 1.
| Poly-NaMTFSI (mol%) | Poly-P111i4MTFSI (mol%) OR poly-P122i4MTFSI (mol%) |
|---|---|
| 100 | 0 |
| 75 | 25 |
| 50 | 50 |
| 25 | 75 |
| 0 | 100 |
Characterization of polyelectrolyte blends
The miscibility of the polyelectrolyte blends was investigated by measuring the glass transition temperature using differential scanning calorimetry (DSC). Fig. 1(a) shows the DSC thermograms of the homopolymer poly-NaMTFSI and its polymer blend with 50:50 (mol%) poly-P111i4MTFSI and poly-P122i4MTFSI. The Tg mid-point was determined and plotted in Fig. 1b. It can be observed that neat poly-NaMTFSI has a Tg of around 121 °C, much higher than that of the polymer with counter ion Li+.26 The polymer blends show only one Tg by DSC, which confirms the miscibility between the individual polymer components in these polymer blends. Compared to the poly-NaMTFSI homopolymers, the blends show a lower Tg in the case of poly-P122i4MTFSI (66 °C) compared to poly-P111i4MTFSI (72 °C). This is the expected behaviour as P122i4 is a bulkier group than P111i4 and previous studies with ammonium cations in related polyelectrolyte systems have shown a similar trend of decreasing Tg.22–24 Furthermore, a previous study by Tudryn et al.28 shows that the Tg decreases in polyester-sulfonate polyelectrolytes with increasing counter cation size; the two factors suggested by Tudyryn et al. are the following: (i) bulkier cations act as plasticizers, which help in lowering the Tg of the mixtures and increasing the free volume for ion dynamics, and (ii) bulkier cations are less associated with anions in the backbone, which in turn form weak physical crosslinks that lead to a decrease in the Tg. The Tg broadens with the addition of the bulky cation, but P122i4 shows more prominent broadening, inferring the possible heterogeneity in this polymer blend, i.e. while the system can be considered miscible as per the evidence of a single Tg, the molecular level there will still be heterogeneous. This is also discussed later in the context of the DMA measurement.
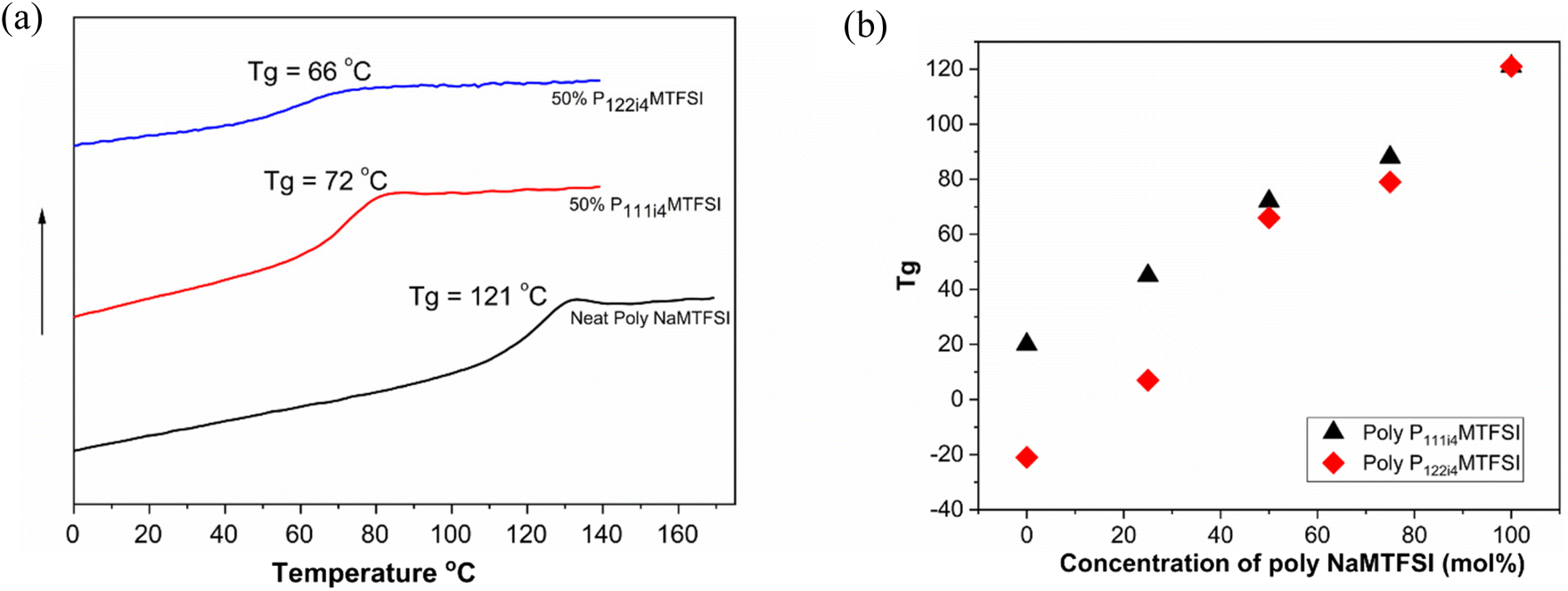 | ||
| Fig. 1 (a) DSC thermograms of neat poly-NaMTFSI and 50 mol% poly-P111i4MTFSI and poly-P122i4MTFSI polymer blends. (b) Tg of different polymer blends with poly-NaMTFSI with both poly-P111i4MTFSI and P122i4MTFSI at different compositions. Scan rate: 10 K min−1. | ||
Fig. 1(b) shows the composition dependence of the blend Tg for both P111i4 and P122i4. With increasing concentrations of these two cations in the blend with poly-NaMTFSI, a decrease in the Tg is clearly observed. The Tg for P122i4 over the entire concentration is lower compared to that of P111i4. The onset, mid-point and end-point temperatures of neat poly-NaMTFSI, 50 mol% P111i4MTFSI and 50 mol% P122i4MTFSI polymer blends are also shown in Table 2. These data more clearly indicate likely heterogeneity in the blends with the P122i4 counterion where the Tg spans 35 °C from the onset to the endpoint compared to only 17 and 18 °C for the poly-NaMTFSI homopolymer and 50 mol% poly-P111i4MTFSI, respectively. Furthermore, the composition dependence appears to deviate more from the ideal behaviour for this bulkier cation.
| Polymers | T g onset/°C | T u midpoint/°C | T g endpoint/°C |
|---|---|---|---|
| Neat poly-NaMTFSI | 110 | 121 | 128 |
| 50% P111i4MTFSI | 62 | 72 | 80 |
| 50% P122i4MTFSI | 41 | 66 | 76 |
Fig. 2 presents the conductivity data as a function of polyelectrolyte blend composition for different concentrations of poly-NaMTFSI with both poly-P111i4MTFSI and poly-P122i4MTFSI as well as a detailed study of the 50 mol% polymer blends. It can be observed that, as the concentration of the bulky phosphonium cation increased, the ionic conductivity also increased in both systems, P111i4MTFSI and P122i4MTFSI, as shown in Fig. 2(a) and (b), respectively. This suggests that the addition of the bulky cation leads to increased free volume and a looser packing of the polymer chains, which facilitates ion motion. Despite the lower Tg for P122i4 relative to the P111i4 containing blend, the ionic conductivity was higher in the latter polymer blend for all compositions, which was unexpected given that the larger cation would be expected to further enhance the free volume between the chains. Fig. 2(c) compares the conductivity for the two 50 mol% polymer blends, with the respective Tg for each system indicated by an arrow, clearly showing that a lower Tg does not necessarily lead to higher ionic conductivity in these cases. Also evident from Fig. 2(c) is the considerable ion conduction at and below Tg for both the polymer blend chemistries at 50 mol%; in the case of the P111i4 cation at Tg, the conductivity approaches 10−6 S cm−1, which indicates significant decoupling of cation motion from structural dynamics, i.e., even though the overall dynamics in the material was decreasing as the glass transition temperature was approached, some fractions of the cations were still mobile, thus contributing to the measured ionic conductivity. To better display this, the ionic conductivity is plotted against scaled temperature whereby the influence of Tg is removed (i.e. Tg/T), as shown in Fig. 2(d). In the polymer blends investigated here, we see that the conductivity is up to 3 orders of magnitude higher (at Tg) than what would be expected if ionic mobility was coupled to polymer dynamics. These findings are comparable to previous reports,22–24 which show that ionic conductivity increases with the increasing size of the bulky cation in the system and is highly decoupled from the Tg. What we cannot ascertain here is the relative contributions of the Na+ and the phosphonium cations to the conductivity. It is likely that the organic cations are also mobile although one would imagine that the larger size would make them less likely to contribute to conductivity at or near Tg; the mechanism of conduction in a glass would require a hopping or structural rearrangement for ion motion. We can observe that decoupling is an order of magnitude greater for the smaller P111i4 cation containing blends, which is consistent with a greater contribution of the smaller organic cation to the measured ionic conductivity. In traditional SPEs both cations and anions move and make it difficult to analyse the mobility of the cation of interest (Li/Na) in the electrolyte system. The Li/Na cations are less mobile than the counter anions, and this can cause concentration polarisation during cell cycling. Furthermore in the traditional SPE, the ionic conductivity is strongly coupled to the polymer segmental dynamics, whereby the ion motion relies on segmental relaxation, which decreases with increasing Tg.19,30 The work here is consistent with previous reports that showed that, using mixtures of co-cations, we can decouple ionic conductivity from polymer segmental motions compared to when a single counter ion is present (either only a Na/Li ion or a bulky cation) as studied in previous reports.28–30 Interestingly, in blends with increasing organic cation composition, the conductivity is significantly lower even though the Tg is also significantly lower, suggesting increased coupling to structural relaxation and indirectly suggesting that the Na+ cation is contributing substantially to the ionic conductivity in the 25 mol% and 50 mol% P122i4/P111i4 containing materials.
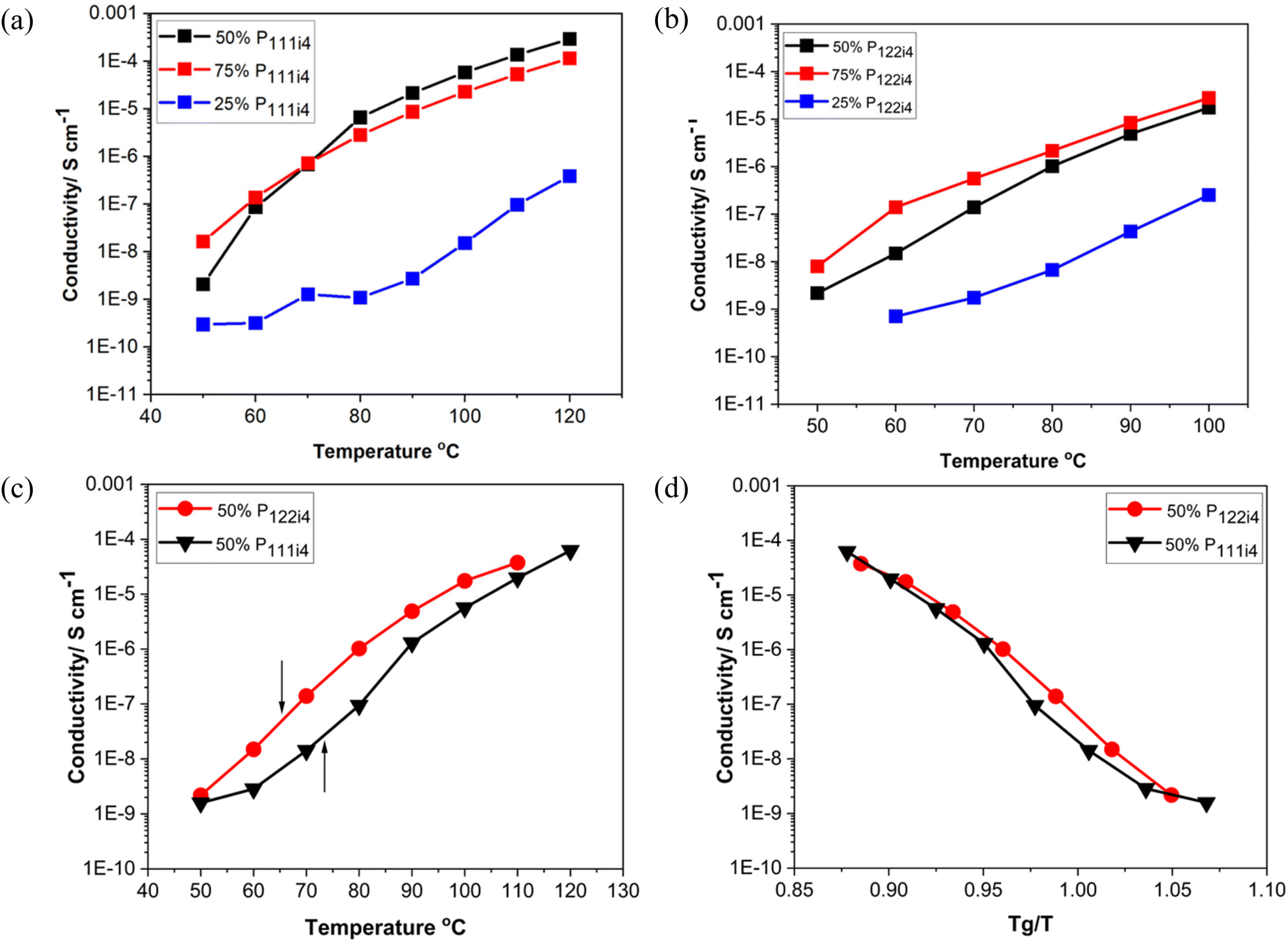 | ||
| Fig. 2 Ionic conductivity at different concentrations of poly-NaMTFSI with (a) poly-P111i4MTFSI and (b) poly-P122i4MTFSI, (c) ionic conductivity of 50 mol% polymer blends of the mixed co-cation of poly-P111i4 and poly-P122i4 with poly-NaMTFSI. (d) Ionic conductivity as a function of the bulky cation with inverse temperature normalized by Tg for each polymer blend. | ||
Dynamic mechanical analysis (DMA) is a technique that is used to measure mechanical properties and structural relaxations in materials. Herein DMA was carried out to understand the thermomechanical properties of 50 mol% polymer blends with the P111i4 and P122i4 cations. Several notable characteristics of the different materials can be observed in Fig. 3. Firstly, both polymers display a high modulus (of the order of 108 Pa) even up to 80 °C where the ionic conductivity is significant (approaching 10−5 S cm−1) and the polymer blend containing P111i4 has a slightly higher value compared to the P122i4 based polymer blend. This shows that significant ionic conductivity is possible in these materials without compromising the mechanical properties,31 and indeed this high modulus is maintained until 100 °C, within the required temperature range that might be considered for high temperature Na-ion battery operation. The loss modulus and tanδ also show interesting features, both being quite broad and consistent with a broad Tg, and the P122i4 based polymer blend shows two features which are indicative of two different relaxation processes. This also suggests that there is some degree of phase separation at the molecular level in this material, which was also hypothesised in previous di-cation polymers reported by Noor et al. and could be more prominent for the larger phosphonium with a greater degree of alkyl substitution.
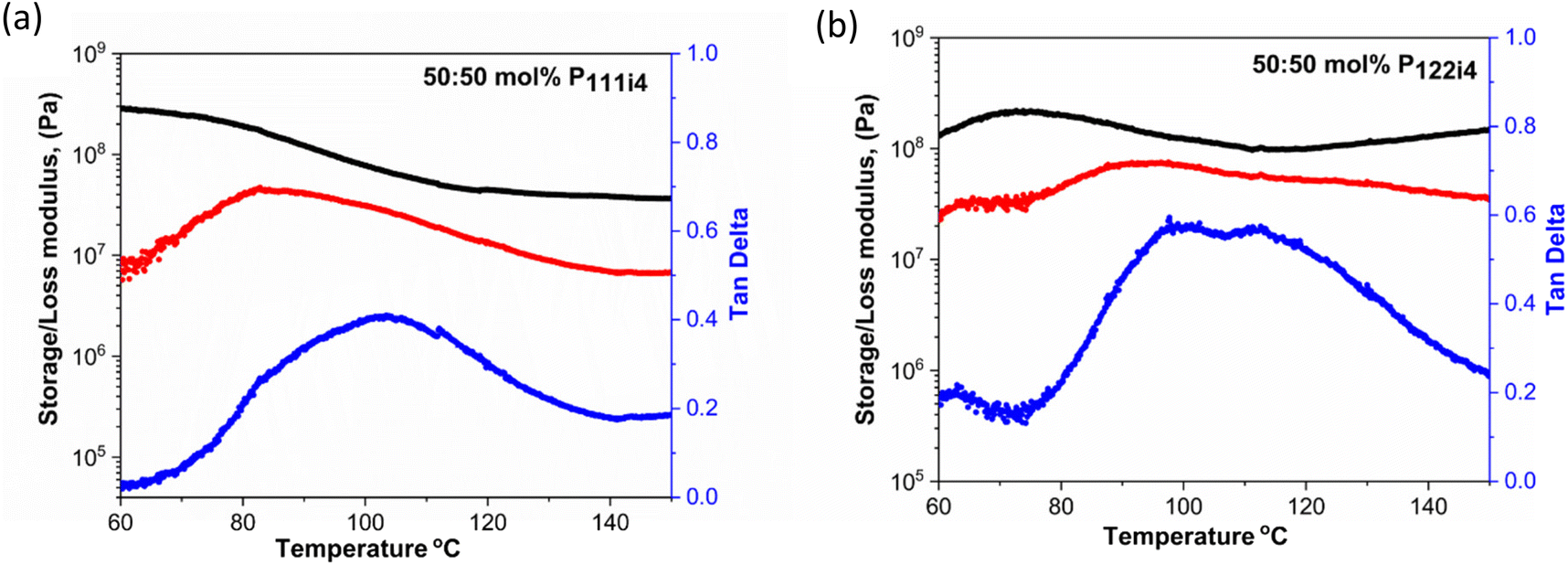 | ||
| Fig. 3 Storage modulus (E′, black), loss modulus (E′′, red) and tanδ (blue) as a function of temperature for the 50 mol% polymer blends of the mixed co-cation of poly-NaMTFSI with (a) P111i4MTFSI and (b) P122i4MTFSI. | ||
The relaxation temperature (tanδ) observed in Fig. 3(a) and (b) is due to the weak interactions in the ionic polymer chains which cause a loss of mechanical strength;32 this is associated with the plasticizing effects caused by the addition of bulky cations in the polymer blends.
The data so far suggest that blending anionic polyelectrolytes based on the PMTFSI backbone but with different counterions can be a promising technique to yield significant conductivity below the glass transition temperature of the electrolyte materials and with good mechanical properties, i.e., decoupling ion motion from structural relaxation processes. However, these conductivity values are still not high enough for device applications, so an alternative approach is necessary to boost ion transport.
Previously Mohd Noor et al.33 reported on the effect of the addition of 10% of an organic ether plasticizer to poly([N1222][AMPS]-co-Na[VS]) and poly([N1222][AMPS]-Na)22 polyelectrolytes. In both cases the ionic conductivity increased significantly, although the effect on Tg was not consistent between the samples, decreasing by 40 °C from the neat copolymer material whereas there was an insignificant change in the ionic conductivity. In another work,34 the addition of NaFSI salt has been shown to plasticize cationic polyelectrolyte systems based on poly(diallydimethylammonium)bis(trifluoromethanesulfonyl)imide (poly-DADMA) leading to high conductivity values. Such an approach has not been reported previously for the case of anionic polyelectrolytes, which we explore below.
The effect of NaFSI salt in the 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :50 mol% polymer blend of poly-P111i4MTFSI and poly-NaMTFSI
:50 mol% polymer blend of poly-P111i4MTFSI and poly-NaMTFSI
The polymer blend of poly-P111i4MTFSI and poly-NaMTFSI was selected to further study the effect of adding NaFSI to the system as this had the highest overall conductivity and decoupled properties as shown above. The main goal was to investigate whether the polyelectrolyte blend could act as a good solvent and further boost ionic conductivity.
The influence of NaFSI salt addition on the Tg of the 50:50 mol% polymer blend was studied. All the materials appeared to be brittle solids at room temperature. While the Tg of these systems did decrease even with 10% NaFSI addition (10 wt% NaFSI reduces Tg by 20 °C (56 °C) compared to the neat 50:50 polymer blend (72 °C)), the Tg did not vary much with a higher salt concentration, again in contrast to the cationic polyelectrolyte. The determination of Tg is shown in Fig. S2.† Although a broadening of Tg for 20 and 30 mol% salt concentrations is observed, less homogeneity is suggested in these materials, as shown in Fig. 4. This contrasting behaviour between polycationic polyelectrolytes and the present systems suggests a lesser salt solubility in the polyanionic systems and likely different coordination microstructure(s) and phase heterogeneity as areas for possible future investigation in these materials.
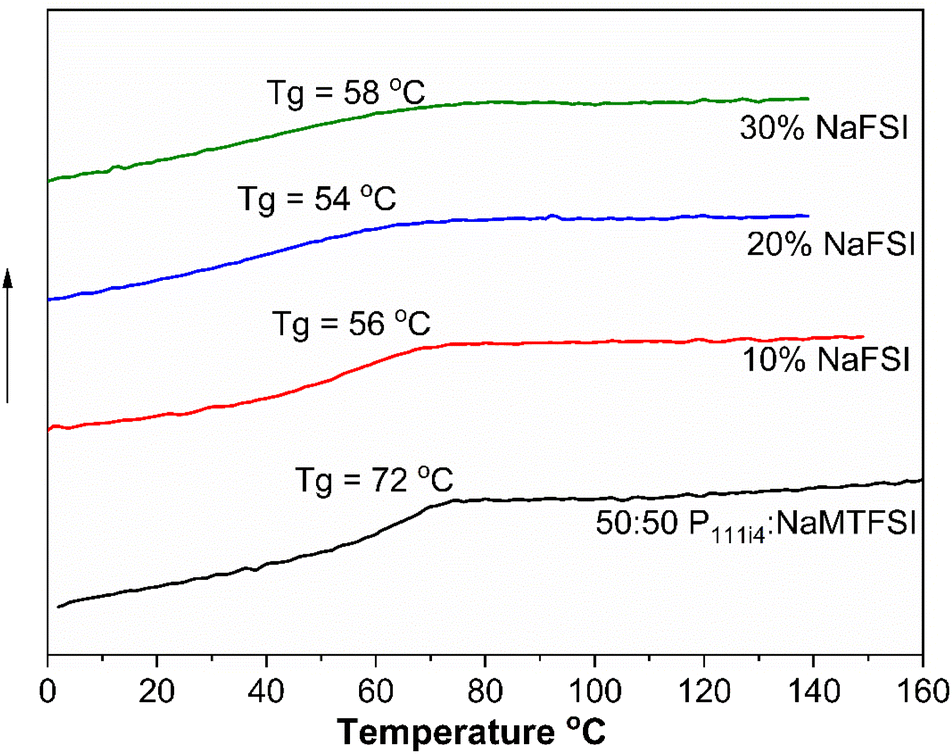 | ||
| Fig. 4 DSC plot of 50:50 mol% polymer blends and the addition of NaFSI salt in wt% at 10 K min−1. | ||
Fig. 5(a) shows the ionic conductivity upon the addition of 10, 20 and 30 wt% NaFSI salt to the 50:50 polymer blend based on poly-P111i4MTFSI and poly-NaMTFSI. It is clear that the ion conductivity increased over the entire temperature range for all the mixtures with the largest increase in conductivity seen for 30 wt% salt (10−6 S cm−1). It is interesting to note that even though the Tg did not vary much from 10 to 30 wt%, the conductivity varied dramatically for each system. When the ionic conductivity was scaled to Tg(Fig. 5(b)), the polymer blend with salts showed recoupling with the polymer backbone, suggesting a change in the conduction mechanism as NaFSI was added. This observation suggests that, whilst the addition of NaFSI salt increased the conductivity, it did not show any improvement in terms of decoupling from the polymer backbone. Thus, further addition of salt was not attempted.
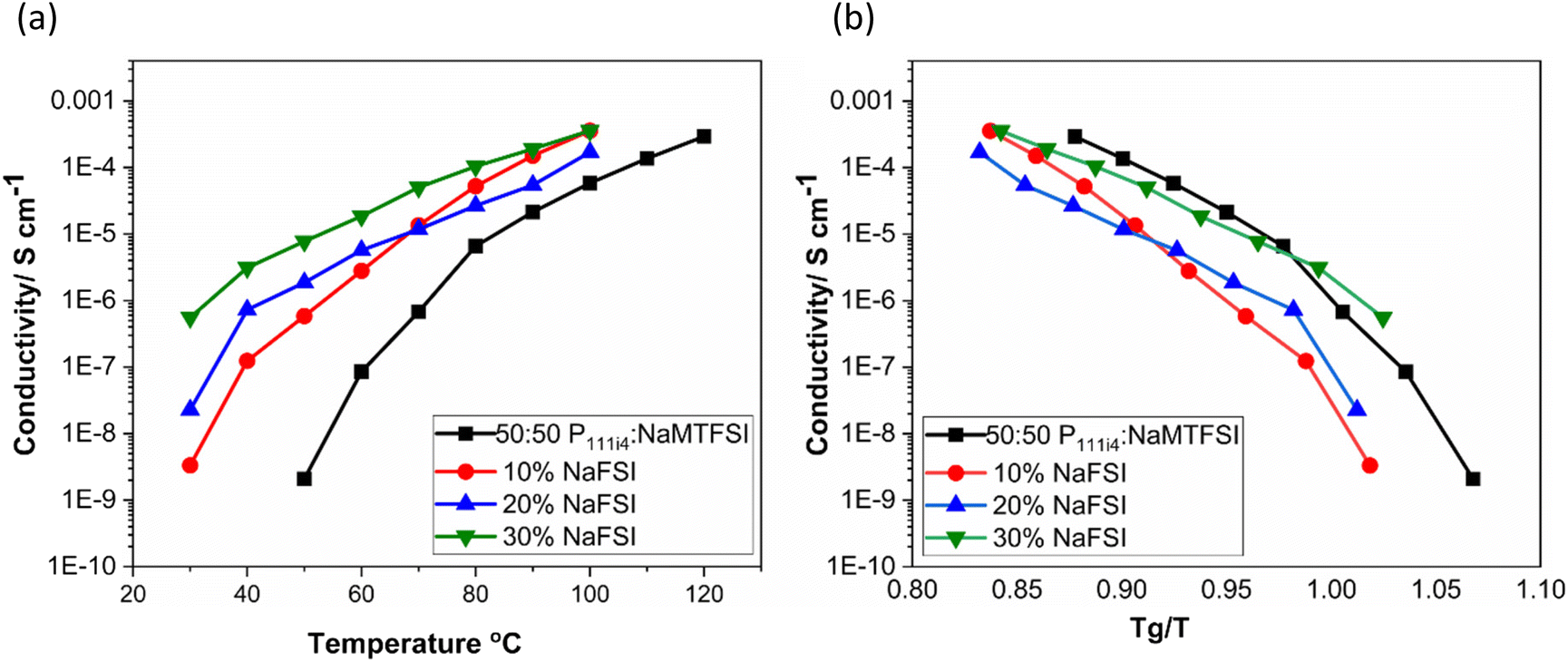 | ||
| Fig. 5 (a) Ionic conductivity of 50:50 mol% polymer blends and the addition of NaFSI salt in wt% at an elevated temperature and (b) ionic conductivity as a function of NaFSI salt concentration with inverse temperature normalized by Tg for each polymer blend. | ||
With these peculiar observations, it is difficult to understand which ions are conducting in the system and the relative ion mobility of FSI−, Na+ and the phosphonium cation. Thus, the Na electrochemical behaviour was investigated using the Na/Na symmetric cells discussed below.
The Na/Na symmetric cells were assembled to study the electrochemical properties of the polymer blend in the presence of 30 wt% added NaFSI salt. The sodium transport number, TNa+, was studied here following the Bruce Vincent method35 and is shown in Fig. S1.† The result of the electrochemical test showed excellent stability with almost no change in the impedance spectrum before and after polarisation and a value of TNa+ of 0.3 at 70 °C was obtained, indicating that only 30% of the current is attributed to the Na+ ion. This is a little higher than most PEO-based solvent free electrolytes,36 although still lower compared to other dual cation or single ion conducting polymers. Thus, the addition of NaFSI to this polyanionic electrolyte system leads to a dominant FSI anion conductivity and potentially ‘restricts’ sodium ion transport, which is again in contrast to previous PDADMAFSI/NaFSI34 based solid polymer electrolytes and emphasises the importance of molecular composition and architecture in designing next generation solid state polymer electrolyte systems.
Conclusion
We reported herein the use of mixed cation polyelectrolyte blends showing strong decoupling behaviour of conductivity from Tg and mechanical properties. In this work, three methacrylic sulfonamide polyanions with three different counter cations were synthesized, one with a sodium counter cation and two with phosphonium cations of different sizes. The polyelectrolyte blends were miscible in all compositions showing only one glass transition temperature, Tg, and such blending was seen to be a useful method to obtain materials with different counter-cation ratios in a simple way. While the ionic conductivity values for the blends of poly-NaMTFSI with 50 mol% poly-P111i4MTFSI and 50 mol% poly-P122i4MTFSI were the highest among all the samples prepared, they remained low at approx. 10−7 S cm−1 at 70 °C. The further addition of NaFSI salt to the polymer blend (poly-NaMTFSI and poly-P111i4MTFSI) enhanced the ionic conductivity by up to four orders of magnitude; however, it re-coupled the ion transport to the Tg, suggesting a change in the ion transport mechanism. The Tg was not significantly affected after the first 10 mol% NaFSI addition, which contrasts with the polyelectrolytes with the polycationic backbone and where the anion is the counterion. Disappointingly, the sodium cation transference number was low, 0.3 at 70 °C, suggesting that the FSI anion may be the dominant charge carrier. Nevertheless, the work here has shown that blending of polyelectrolytes may be a facile method to create new polymer electrolyte materials with decoupled ion transport, and at the fundamental level, it points to the distinct conduction processes that can eventuate. A deeper understanding of the structure–property relationships in such materials and the transport mechanisms would further advance the field of solid polymer electrolytes for advanced batteries. The in-depth studies to be considered for further understanding of these materials include ion diffusion such as NMR, dielectric spectroscopy and FTIR measurements and detailed electrochemical analysis of long term charge–discharge cycling.Experimental
Materials
3-(Chlorosulfonyl)propyl methacrylate (100 g), anhydrous THF ≥ 99.9%, inhibitor free anhydrous DMF 99.8%, dichloromethane, trifluoromethanesulfonamide (NH2SO2CF3), triethylamine ≥99.5%, anhydrous magnesium sulfate ≥99.5% (500 g), sodium hydride (NaH), 10 g, dry 90%, 4,4′-azobis(4-cyanovaleric acid) and azobisisobutyronitrile (AIBN) were all purchased from Sigma-Aldrich Pvt. Ltd. Isobutyltrimethylphosphonium bromide (P111i4Br) ≥ 99% was purchased from Boron Molecular, Australia (Scheme 2).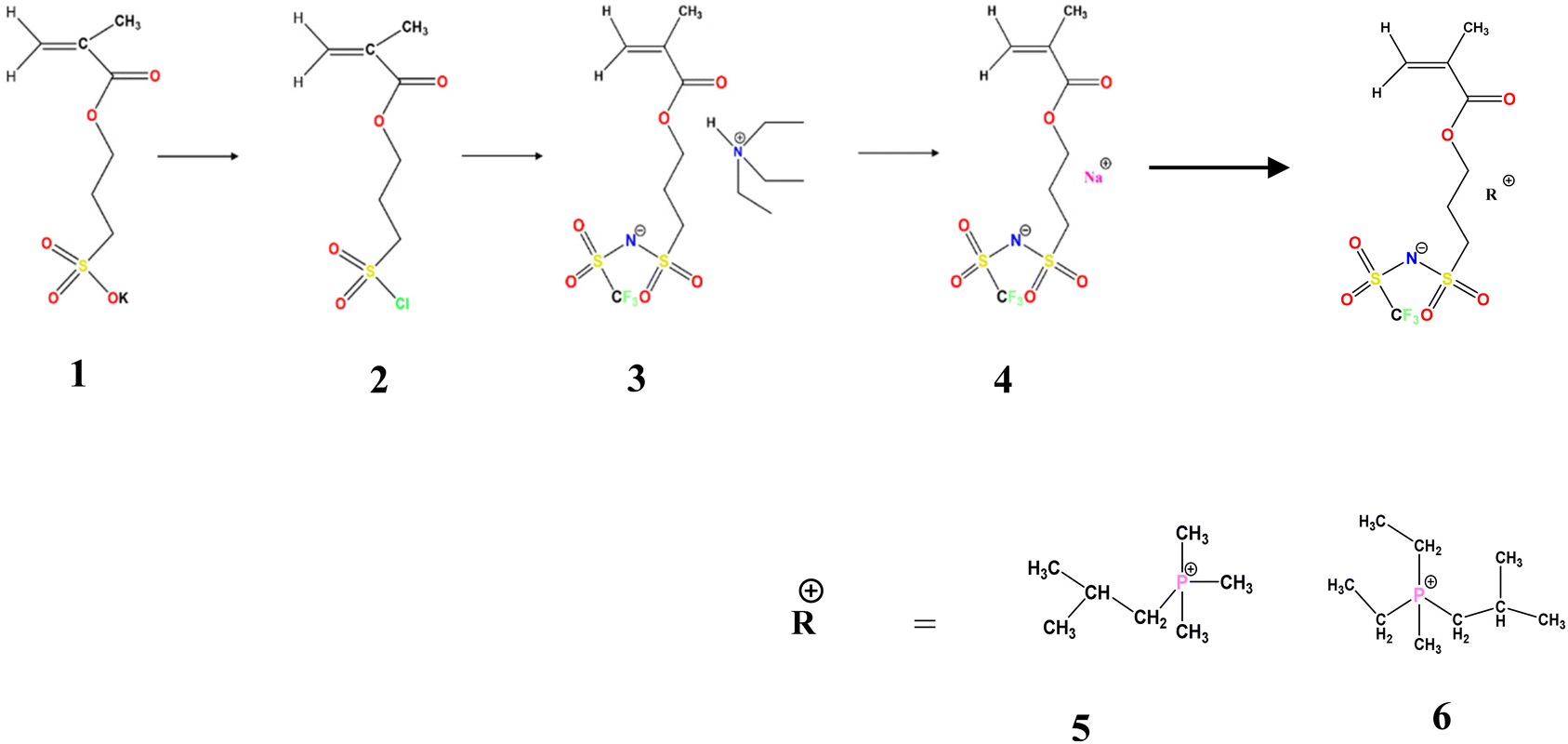 | ||
| Scheme 2 Synthetic route for the preparation of monomers 4, 5 and 6. | ||
Monomer synthesis: three different monomer molecules 4, 5, 6 were synthesized using a three step procedure described below.
Step 1: for the synthesis of 3-(chlorosulfonyl)propyl methacrylate (2), 15 g of potassium 3-(methacryloyloxy) propane-1-sulfonate/3-sulfopropyl methacrylate potassium salt (KSPM) was placed in a round bottomed flask filled with argon. 25 ml of anhydrous dry THF was added to the flask; to this solution, 1.7 ml of dry DMF (acts as a catalyst) was further added via a syringe. This whole reaction flask was maintained at 0 °C in an ice bath. Then, 39.9 g, 0.335 mol of thionyl chloride was added dropwise and the mixture was left stirring overnight. The obtained suspension was carefully added to ice water (approx. 200 mL, which was prepared by adding liquid N2 to water in a clean flask); once the suspension settled down, the upper aqueous layer was decanted. The oily organic layer was diluted with 80 ml of dichloromethane (CH2Cl2), and the CH2Cl2 layer was then washed with brine water (6 × 25 mL) in a separating flask and dried with MgSO4 with stirring for 1 hour. The MgSO4 was filtered off under vacuum on a Schlenk line to obtain the product (2). Analysis: C7H11ClO4S (226.68): 1H NMR (400.07 MHz, CDCl3): δ = 6.09(s, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)–), 6.05 (s, 1H, CH2C(CH3)–), 4.26(t, 2H, CO–O–CH2–), 3.72(m, 2H, –CH2–SO2Cl), 2.07(m, 2H, CO–O–CH2–CH2–), 1.89(s, 3H, CH2C(CH3)–)
C(CH3)–), 6.05 (s, 1H, CH2C(CH3)–), 4.26(t, 2H, CO–O–CH2–), 3.72(m, 2H, –CH2–SO2Cl), 2.07(m, 2H, CO–O–CH2–CH2–), 1.89(s, 3H, CH2C(CH3)–)
Step 2: for the synthesis of triethyl ammonium 1-[3-(methacryloxy)propylsulfonyl]-1-(trifluoromethane-sulfonyl) imide 3, trifluoromethanesulfonamide NH2SO2CF3 (7.3 g, 0.049 mol) was placed in a round bottomed flask by flushing Ar, and anhydrous N(C2H5)3 (10.9 g, 0.107 mol) was added under stirring. 40 ml of anhydrous THF was further added to the mixture and the resultant solution was cooled at 0 °C in an ice bath. 3-Chlorosulfonyl)propyl methacrylate (product 2) (11.1 g, 0.049 mol) was dissolved in 15 mL of anhydrous THF. This solution was added dropwise to the solution of THF with NH2SO2CF3 and N(C2H5)3 under an inert atmosphere and the reaction was left stirring at 0 °C for 1 hour and at room temperature. The obtained precipitate was filtered off and the remaining THF was removed by using a rotavap and a slightly yellow oily residue was obtained, which was dissolved in 90 mL of CH2Cl2. The dichloromethane was washed with brine water (4 × 35 mL) and dried with MgSO4. It was further filtered, and the excess dichloromethane was removed by a rotavap followed by drying under vacuum on a Schlenk line and stored in a refrigerator at 3.8 °C. Analysis: C14H27F3N2O6S2 (440.50): 1H NMR (400.07 MHz, CDCl3) δ = 6.02 (s, 1H, CH2C(CH3)–), 5.69 (t, 1H, CH2C(CH3)), 4.18 (t, 2H, CO–O–CH2–), 3.16–2.99 (m, 8H, H–N (CH2CH3)3 and CH2–SO2–N–), 2.05–1.97 (m, 2H, O–CH2–CH2–CH2–), 1.89(s, 3H, CH2C(CH3)–), 1.14 (t, 9H, H–N(CH2CH3)3).
Step 3: for the synthesis of the Na monomer (NaMTFSI), 4, 1.5 eq. of NaH (0.8 g, 0.033 mol) (purchased from Sigma Aldrich) was dissolved in 20 mL of anhydrous THF in an addition flask. In a round bottomed flask, 9.8 g of product 3 was dissolved in 30 mL of anhydrous THF in an ice bath (0 °C) under an inert atmosphere. The NaH solution was added dropwise to the product 3 solution. The reaction was left stirring for 2 hours at room temperature, and then unreacted NaH was removed by filtration, and THF was removed by a rotavap. The concentrated product 4 was washed with cyclohexane under vigorous stirring (3 × 20 mL) and the excess hexane was decanted. We obtained a yellow oily residual product 4, to which dry dichloromethane was added until the oil residue dissolved. This sample was stored in a refrigerator overnight below 3.8 °C to form white crystals of a NaMTFSI monomer with a product yield of 48%. The white crystals were filtered off the next day, washed with diethyl ether and dried on a Schlenk line. Analysis: C8H11F3NO6S2 (360.59) 1H NMR (400.07 MHz, CDCl3) δ = 6.02 (s, 1H, CH2C(CH3)–), 5.69 (s, 1H, CH2C(CH3)), 4.18(t, 2H, CO–O–CH2–), 3.16 (t, CH2–SO2–N–), 2.05–1.97(m, 2H, O–CH2–CH2–CH2–), 1.89(s, 3H, CH2C(CH3)–).
P111i4MTFSI monomer 5 was synthesised in a one-step procedure. Product 4 NaMTFSI (2.34 g, 0.0064 mol) was dissolved in 15 mL of Milli-Q water in a beaker. In a separate beaker, 1.5 eq. of P111i4Br (2.07 g, 0.0097 mol) was dissolved in 10 mL of Milli-Q water. The P111i4Br solution was added dropwise to a solution containing NaMTFSI using an addition flask and the reaction was kept stirring for 30–40 min at room temperature. While the reaction was going on, 15 mL of CH2Cl2 was added and the reaction was left stirring for an additional 1 hour at room temperature. The upper aqueous layer was decanted. The oily residue was washed with brine water (3 × 15 mL) to remove excess CH2Cl2 and then it was further dried with MgSO4 and filtered. The obtained viscous oily layer was further subjected to rotavap drying to remove any residue of CH2Cl2; 0.01 M hydroquinone inhibitor was added before rotavap drying to avoid self-polymerisation. Product 5 finally underwent an additional drying step under vacuum on a Schlenk line and finally stored in the refrigerator at <5 °C. Analysis: C16H32F3NO6S2 (471.25) 1H NMR (400.13 MHz, CDCl3) 6.02 (s, 1H, CH2C(CH3)–), 5.49 (s, 1H, CH2C(CH3)), 4.18 (t, 2H, CO–O–CH2–), 3.16 (m, CH2–SO2–N–), 2.19–2.05 ((m, 2H, O–CH2–CH2–CH2–)+(2H–CH–CH2)), 2.01–1.939 (m, CH3–CH–CH3), 1.891 (s, 3H, CH2C(CH3)–), 1.87–1.84(s, 3H, CH3–P–, CH3–P–, CH3–P–), 1.05 (s, 3H CH3–CH–), 1.02 (s, 3H CH3–CH–).
P122i4MTFSI monomer 6 was synthesised in a one-step procedure. Product 4 NaMTFSI (2.0 g, 0.0055 mol) was dissolved in 15 mL of Milli-Q water in a beaker, while in a separate beaker, 1.5 eq. of P122i4TSO (2.75 g, 0.0083 mol) was dissolved in 10 mL of Milli-Q water. The P122i4TSO solution was added dropwise to a solution containing NaMTFSI through an addition flask and the reaction was kept stirring for 30–40 min at room temperature. While the reaction was still going on, 15 mL of CH2Cl2 was added and the reaction mixture was left stirring for an additional 1 hour at room temperature. The upper aqueous layer was subsequently decanted and the oily residue was washed with brine water (3 × 15 mL) to remove excess CH2Cl2, and then it was further dried with MgSO4 and filtered off. The obtained viscous oily layer was further subjected to rotavap drying to remove any residue of CH2Cl2, and then 0.01 M hydroquinone inhibitor was added before rotavap drying to avoid self-polymerisation. Product 6 was finally extra dried under vacuum on a Schlenk line and stored in the refrigerator. Analysis: C17H33F3NO6PS2 (499.54) 1H NMR (400.13 MHz, CDCl3) δ = 6.02 (s, 1H, CH2C(CH3)–), 5.69 (s, 1H, CH2C(CH3)), 4.18 (t, 2H, CO–O–CH2–), 3.16 (m, CH2–SO2–N–), 2.29–2.15 (m, 4H, CH3–CH2–P–CH2–CH3), 2.08 (s, 2H CH2–C–CH3), 2.00 (m, 2H, O–CH2–CH2–CH2–), 1.87 (s, 3H, CH3–P–CH2), 1.26–1.10 (m, CH3–CH2–P–CH2–CH3), 1.05 (s, CH2C(CH3)), 1.04–1.02(s, 6H CH3–C–CH3), 0.8(s, 2H, P–CH2–C–CH3).
Polymer synthesis routes
The polymerisation of NaMTFSI 7 was done in water as a solvent in a 1:1 ratio as follows: 2.0 g of NaMTFSI was dissolved in 2 mL of Milli-Q water and to this solution, 0.02 g of 4,4′-azobis(4-cyanovaleric acid) initiator was added. The solution was mixed well and degassed with Ar gas for 1 hour, and the reaction flask was covered with Al foil to avoid light as the initiator is light sensitive. After degassing, the reaction flask was immersed in an oil bath at 80 °C and left overnight. After the polymerisation was completed, the water was removed with a rotavap and excess water was removed using a lyophilizer before drying on the Schlenk line. Finally the white product 7 with 35% yield was obtained and stored in an argon-filled glove box. Analysis: C8H11F3NO6S2 (360.59) 1H NMR (400.07 MHz, DMSO) δ = 3.06 (m, 2H, O–CH2–CH2–CH2–), 1.956 (t, CH2–SO2–N–), 1.17 (t, 2H, CO–O–CH2–), 1.08 (s, 1H, CH–C(CH), 1 g of product 6 was dissolved in 1 mL of DMF; to this solution, 0.01 g of AIBN was added as the initiator, and then the solution was mixed well and degassed with Ar gas for 1 hour before the reaction flask was immersed in an oil bath at 90 °C and left overnight. After polymerisation the obtained polymer was washed with diethyl ether (2 × 40 mL); the final product 8 was further dried under vacuum on a Schlenk line and stored in an argon-filled glove box. Analysis: C16H32F3NO6S2 (471.25) 1H NMR (400.07 MHz, DMSO) δ = 3.06(m, 2H, O–CH2–CH2–CH2–), 2.05 (t, 2H–CH–CH2)), 2.01–1.939 (m, CH3–CH–CH3), 1.956(t, CH2–SO2–N–), 1.891 (s, 3H, CH2C(CH3)–), 1.87–1.84(s, 3H, CH3–P–, CH3–P–, CH3–P–), 1.17(t, 2H, CO–O–CH2–), 1.08 (s, 1H, CH–C(CH)), 1.05 (s, 3H CH3–CH–), 1.02 (s, 3H CH3–CH–).
Polymer 7 was similarly prepared from monomer 5 with a 35% yield and dried as above, before being stored in an argon filled glove box. Analysis: C17H33F3NO6PS2 (499.54) 1H NMR (400.13 MHz, DMSO) δ = 3.06 (m, 2H, O–CH2–CH2–CH2–), 2.29–2.15 (m, 4H, CH3–CH2–P–CH2–CH3), 2.08 (s, 2H CH2–C–CH3), 2.00 (m, 2H, O–CH2–CH2–CH2–), 1.956 (t, CH2–SO2–N–), 1.87 (s, 3H, CH3–P–CH2), 1.26 (m, CH3–CH2–P–CH2–CH3), 1.17 (t, 2H, CO–O–CH2–), 1.08 (s, 1H, CH–C(CH)), 1.04–1.02 (s, 6H CH3–C–CH3), 0.8 (s, 2H, P–CH2–C–CH3). The formation of polymers 7, 8 and 9 was analysed with the help of 1H NMR as shown in Fig. S3.† All the three polyelectrolytes were amorphous in nature.
Electrolyte membrane preparation
The electrolyte membrane was prepared by mixing poly NaMTFSI (7) with poly-P111i4MTSFI (8) and poly-P122i4MTSFI (9) in acetonitrile separately. The different ratios used are summarised in Table 1. Once the homogeneous mixture was formed, the solvent was removed completely under vacuum under an Ar atmosphere and dried. The dried powder was then hot pressed at different temperatures ranging from 70 to 100 °C to obtain the electrolyte membrane. Once the membrane was formed, it was further dried under vacuum to remove any residual solvents and stored in an Ar filled glove box.Instrumentation
:50 mol% polymer blends of poly-NaMTFS: poly-P111i4MTFSI and poly-NaMTFSI: poly-P122i4MTFSI. The sample was studied in the temperature range of 30 to 150 °C. The sample was first cooled and then heated at 2 K min−1. The sample was cut into 1.5 × 5 mm. The storage modulus and tan(δ) were determined using tensile mode with a frequency of 1 Hz and an elongation of 0.02 mm.
Electrochemical characterisation of 30 mol% NaFSI salt as a plasticizer in a 50:50 mol% polymer blend of poly-P111i4MTFSI and poly-NaMTFSI
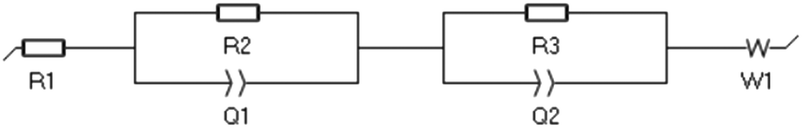
where R1 is the bulk resistance of the electrolyte. R2 and R3 represent the resistance related with two electrode surfaces. The EC-Lab V11.27 software was used to analyse the data and the fitting method is Randomize + Simplex.
Author contributions
Sneha Malunavar: experiments, data analysis, and writing – original draft. Luca Porcarelli: methodology design and supervision. Patrick Howlett: project conceptualization, manuscript editing and supervision, David Mecerreyes: conceptualization of polymer synthesis and contribution to manuscript editing, and Maria Forsyth: project conceptualization, analysis and interpretation of data, and manuscript preparation.Conflicts of interest
There are no conflicts of interest.Acknowledgements
S. M. acknowledges Deakin University for support via the Deakin University postgraduate research scholarship, L. P. and D. M. acknowledge the IONBIKE RISE for financial support through European Union's Horizon 2020 research and innovation programme under Marie Skłodowska-Curie Grant Agreement No. 823989, M. F. acknowledges the Australian Research Council (ARC) for funding through Discovery Programme DP160101178 and the ARC Industry Transformation Training Centre for Future Energy Technologies (storEnergy), M. F. and D. M. also thank the Ikerbasque Foundation for Science for their support, And M. G. and M. A. acknowledge the Ministerio de Ciencia e Innovación for the financial support through the PID2019-107468RB-C22 and PLEC2021-007929 projects.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- X. Chen, Z. Guan, F. Chu, Z. Xue, F. Wu and Y. Yu, InfoMat, 2022, 4(1), e12248 CrossRef CAS.
- C. Zhao, J. Guo, Z. Gu, X. Wang, X. Zhao, W. Li, H. Yu and X. Wu, Nano Res., 2022, 15(2), 925–932 CrossRef CAS.
- M. Yu, Z. Gu, J. Guo, C. Wang and X. Wu, Chem. Commun., 2022, 58, 6813–6816 RSC.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- W. Gorecki, M. Jeannin, E. Belorizky, C. Roux and M. Armand, J. Phys.: Condens.Matter, 1995, 7, 6823–6832 CrossRef CAS.
- V. Di Noto, S. Lavina, G. A. Giffin, E. Negro and B. Scrosati, Electrochim. Acta, 2011, 57, 4–13 CrossRef CAS.
- C. Li, H. Zhang, L. Otaegui, G. Singh, M. Armand and L. M. Rodriguez-Martinez, J. Power Sources, 2016, 326, 1–5 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- A. M. Stephan, Eur. Polym. J., 2006, 42, 21–42 CrossRef.
- J. W. Fergus, J. Power Sources, 2010, 195, 4554–4569 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Electrochim. Acta, 2015, 175, 18–34 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- X. G. Sun, J. B. Kerr, C. L. Reeder, G. Liu and Y. Han, Macromolecules, 2004, 37, 5133–5135 CrossRef CAS.
- M. Doyle, T. F. Fuller and J. Newman, Electrochim. Acta, 1994, 39, 2073–2081 CrossRef CAS.
- D. J. Bannister, G. R. Davies, I. M. Ward and J. E. McIntyre, Polymer, 1984, 25, 1291–1296 CrossRef CAS.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- S. Zheng, Q. Liu and L. Yang, Polymer, 1994, 35, 3740–3744 CrossRef.
- S. A. Mohd Noor, D. Gunzelmann, J. Sun, D. R. MacFarlane and M. Forsyth, J. Mater. Chem. A, 2014, 2, 365–374 RSC.
- S. A. M. Noor, J. Sun, D. R. Macfarlane, M. Armand, D. Gunzelmann and M. Forsyth, J. Mater. Chem. A, 2014, 2, 17934–17943 RSC.
- C. R. Pope, K. Romanenko, D. R. MacFarlane, M. Forsyth and L. A. O'Dell, Electrochim. Acta, 2015, 175, 62–67 CrossRef CAS.
- Y. V. Oza, D. R. Macfarlane, M. Forsyth and L. A. O'Dell, Electrochim. Acta, 2015, 175, 80–86 CrossRef CAS.
- A. S. Shaplov, P. S. Vlasov, M. Armand, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. M. Pavlov, C. Wandrey, I. A. Godovikov and Y. S. Vygodskii, Polym. Chem., 2011, 2, 2609–2618 RSC.
- L. Porcarelli, A. S. Shaplov, M. Salsamendi, J. R. Nair, Y. S. Vygodskii, D. Mecerreyes and C. Gerbaldi, ACS Appl. Mater. Interfaces, 2016, 8, 10350–10359 CrossRef CAS PubMed.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- G. J. Tudryn, W. Liu, S. W. Wang and R. H. Colby, Macromolecules, 2011, 44, 3572–3582 CrossRef CAS.
- W. Wang, G. J. Tudryn, R. H. Colby and K. I. Winey, J. Am. Chem. Soc., 2011, 133, 10826–10831 CrossRef CAS PubMed.
- Y. Wang, A. L. Agapov, F. Fan, K. Hong, X. Yu, J. Mays and A. P. Sokolov, Phys. Rev. Lett., 2012, 108, 1–5 Search PubMed.
- G. Huang, L. Porcarelli, Y. Liang, M. Forsyth and H. Zhu, ACS Appl. Energy Mater., 2021, 4, 10593–10602 CrossRef CAS.
- O. Danyliv and A. Martinelli, J. Phys. Chem. C, 2019, 123, 14813–14824 CrossRef CAS.
- S. A. Mohd Noor, D. Gunzelmann, J. Sun, D. R. MacFarlane and M. Forsyth, J. Mater. Chem. A, 2014, 2, 365–374 RSC.
- S. Malunavar, A. Gallastegui, X. Wang, F. Makhlooghiazad, D. Mecerreyes, M. Armand, M. Galceran, P. C. Howlett and M. Forsyth, ACS Appl. Polym. Mater., 2022, 4(12), 8977–8986 CrossRef CAS.
- P. G. Bruce, M. T. Hardgrave and C. A. Vincent, Solid State Ionics, 1992, 53–56, 1087–1094 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00245d |
| This journal is © The Royal Society of Chemistry 2024 |