
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inherent limitations of the hydrogen-bonding UPy motif as self-healing functionality for polymer electrolytes†
Cuc
Thu Mai
 ,
Harish
Gudla
,
Guiomar
Hernández
,
Kristina
Edström
and
Jonas
Mindemark
*
,
Harish
Gudla
,
Guiomar
Hernández
,
Kristina
Edström
and
Jonas
Mindemark
*
Department of Chemistry – Ångström Laboratory, Uppsala University, Box 531, SE-751 21 Uppsala, Sweden. E-mail: Jonas.Mindemark@kemi.uu.se.
First published on 22nd March 2024
Abstract
The development of advanced materials displaying reversible functionalities, such as self-healing is particularly desirable for energy storage devices, since the cycle life of many rechargeable batteries is limited due to the irreversible mechanical damages over the cycling processes. Hydrogen-bonding self-healing polymers functionalized with ureido pyrimidinone (UPy) has received great interest for energy storage applications, particularly for polymer electrolytes. Herein, we design a star-branched poly(ε-caprolactone-co-trimethylene carbonate) end-capped with UPy groups for both reinforced mechanical and desired self-healing properties in the polymer electrolytes. Despite the versatile implementation and strong bonding association, the benefits of hydrogen-bonding UPy functionalities are diminished after the dissolution of LiTFSI salt in the self-healing polymer matrix. Experimental analysis and molecular dynamics simulations were performed to gain insight into the dynamics of the self-healing polymer electrolyte system. FTIR shows a dramatic decrease in the intensities of the hydrogen-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signals belonging to UPy motifs after adding LiTFSI salt, indicative of a significant reduction in the total number of hydrogen-bonding and more loosened cross-linked polymer network. This is also noticed as a simultaneous deterioration of the mechanical properties. Molecular dynamics simulations reveal that the complex interplay of CO--Li+ coordination bonds and hydrogen bonding between TFSI anions and UPy motifs are responsible for the mechanical deterioration of the self-healing polymer electrolytes.
O signals belonging to UPy motifs after adding LiTFSI salt, indicative of a significant reduction in the total number of hydrogen-bonding and more loosened cross-linked polymer network. This is also noticed as a simultaneous deterioration of the mechanical properties. Molecular dynamics simulations reveal that the complex interplay of CO--Li+ coordination bonds and hydrogen bonding between TFSI anions and UPy motifs are responsible for the mechanical deterioration of the self-healing polymer electrolytes.
1. Introduction
Solid-state electrolytes have been thrust into the spotlight for the revival of high-energy lithium metal batteries (LMBs) given the anticipated advantages in safety, working temperature range, and improved energy density as compared to conventional liquid electrolytes.1 Among these, solid polymer electrolytes (SPEs) offer outstanding advantages over their inorganic counterparts in terms of tunability and easy processability of the polymer matrices.2–6 Abundant organic chemistry tools pave the way for more complex polymer engineering, enabling the integration of specific functionalities in the polymer scaffolds. Despite these merits, the practical application of SPEs is set back by the canonical trade-off between mechanical performance and ionic conductivity.3,7–11 Highly elastic, potentially reinforceable, and healable polymer electrolytes are highly sought to tackle the dendrite growth in lithium metal batteries. Mechanically strengthened polymer electrolytes, on the other hand, tend to exhibit poor ionic conductivity and inferior electrochemical performance. To date, no single material exists that can satisfy all the aforementioned challenges.Numerous efforts have been dedicated to further improve the performance of SPEs through various structural modification strategies. In particular, SPEs integrated with self-healing functionalities have been synthesized and intensively investigated. The built-in self-healing ability allows network repair of physical damages during fabrication and cycling, hence prolonging the lifespan and improving the safety of the solid-state batteries.12,13 Self-healing properties of SPEs can be realized through the reversibility of dynamic covalent and non-covalent bonds such as hydrogen bonds (H-bonds), disulfides, boronic esters, etc.8 This dynamic bonding facilitates inter- and intramolecular interactions, which reinforces the polymer network and enhances the mechanical strength. If dendrite growth penetrates the electrolyte and creates cracks, the cracks can self-heal under the action of dynamic bonding. H-bonds have been widely used to prepare dynamic and self-healing polymers, which can heal under ambient conditions.14 Multi-hydrogen bond arrays have particularly received extensive attention because of the simple design, selectivity, high strength, and directionality. Ureido pyrimidinone (UPy) developed by Meijer and colleagues15 is one of the most accessible and strong H-bond motifs with high association constants that can exceed 106 M−1.14 UPy motifs self-assemble into dimers via complementary quadruple H-bonding. During the assembly process, the UPy dimers further associate into larger domains with high crystallinity, enabling the formation of a 3D network with high crosslinking density through the connection of UPy dimers.16 Regarding polymer electrolyte engineering, the high bonding association constant and directional self-assembly of UPy render the polymer excellent mechanical properties that could otherwise only be achieved in covalently cross-linked polymers, whereas the reversibility at elevated operating temperature promotes the self-healing ability.17–19
Recent studies have proposed paradigms of polymer electrolytes utilizing UPy self-healing motifs to enable self-repairing properties and mechanical enhancement.6,8,18,20–23 Xue et al. synthesized a highly stretchable and self-healing polymer electrolyte by tethering UPy motifs to a PEGMA polymer.20 The resulting polymer electrolyte not only displayed excellent thermal and electrochemical properties but also exhibited a stable cycling performance even after cutting and healing. In other study, the same group reported the successful fabrication of self-healing polymer composite electrolyte using UPy-functionalized SiO2 and a PEG polymer matrix containing UPy.24 The UPy H-bonding between silica nanoparticles and polymer chains greatly promoted the uniform dispersity of nanoparticles in the polymer matrix. This, as a result, improves the interface between SiO2 and the polymer and provides fast conduction channel for Li ion transportation. Despite promising results, a deterioration in the elasticity of UPy-functionalized polymers after adding LiTFSI was reported while little explanation has been provided.4,25 Cui and Bao et al. attributed the weakened mechanical effect in their PEG-UPy electrolytes to the steric hindrance of large TFSI anions with the dimerization and chain packing of the UPy motifs.4 On the contrary, Binder et al. believed that the coordination bonding between the carbonyl (CO) group in UPy motifs and Li+ ions to be the contributing factor.25 As with most types of noncovalent interactions, the chemistry of polymer backbones and the environments dictates the behaviour of H-bonded supramolecular polymers. For instance, polar polymer backbones can introduce competitive H-bonding, which can disrupt the association of supramolecular groups. The presence of ether linkages, hydroxyl groups, and other polar functionalities has also been shown to interfere with the association of UPy motifs.26 For this reason, it is vital to understand the interactions between UPy motifs with electrolyte ions when it comes to designing novel polymer electrolytes with intelligent and tuneable properties.
In this study, we aim to provide a better understanding on the effect of electrolyte salts (LiTFSI, LiPF6, and NaTFSI) on the H-bond structures and dynamics of UPy motifs in SPEs. A star-branched poly(ε-caprolactone-co-trimethylene carbonate) PCL-PTMC copolymer with UPy motifs coupled to the chain ends was synthesized and its electrochemical, physical and mechanical properties were examined. Compared to pure PCL-PTMC, the self-healing PCL-PTMC (shPCL-PTMC) features outstanding mechanical properties which, however, deteriorate considerably after adding electrolyte salts. More specifically, LiTFSI leads to the largest decrease in elasticity, followed by LiPF6 and NaTFSI. This weakening effect in mechanical properties becomes even more pronounced at elevated temperatures, highlighting an unresolved challenge for practical application of SPEs in lithium batteries. In a good correlation to that, FTIR results reveal clear evidence for the decreased intensities of UPy H-bonding in the self-healing SPEs. Molecular dynamics simulations further demonstrate how the coordination bonds between Li+ ions with UPy and H-bonding between TFSI anions with UPy are accountable for the dissociation of the quadruple UPy H-bonding.
2. Experimental
2.1 Materials
ε-Caprolactone (εCL), 1,6-hexamethylenediisocyanate, 2-amino-4-hydroxy-6-methylpyrimidine, dibutyltindilaurate (DBTDL), and 2-hydroxymethyl-1,3-propanediol were obtained from Sigma-Aldrich and trimethylene carbonate (TMC) was from Richman Chemical. ε-CL was dried with CaH2 overnight and distilled under vacuum prior to transferring into glovebox. Lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) (Sigma-Aldrich) was dried at 120 °C for 48 h under vacuum prior to use.2.2 Materials synthesis
), 10.17 ppm (1H, s, –CH2NHCO–), 11.85 ppm (1H, s, –CH2NHCONH–), 13.10 ppm (1H, s, –NHC(CH3)). 13C NMR (CDCl3): δ = 19.02, 26.24, 26.31, 29.38, 31.27, 39.86, 42.96, 106.79, 141.10, 154.79, 156.68, 173.16. FTIR ν(cm−1) = 2277 (–NCO stretch), 1698 (NH–CO–NH–), 1662 (CO), 1570 (CN), 1521 (CC), 1251 (C–N).
O), 1195 (–C–O–C).
), 10.12 ppm (1H, s, –CH2NHCO–), 11.85 ppm (s, –CH2NHCONH–), 13.12 ppm (s, –NHC(CH3)). FTIR ν(cm−1) = 2946 (C–H vibration), 1730 (CO), 1700, 1669, 1585 & 1525 (UPy).
2.3 Materials characterisation
FTIR analysis was performed on an IRTracer-100 spectrometer, Shimadzu. 1H NMR spectra were recorded in CDCl3 on a JEOL ECZ 400S 400 MHz NMR spectrometer. Differential scanning calorimetry was performed using a METTLER TOLEDO DSC 3+, STARe System. The samples were weighed and hermetically sealed in aluminium pans. The samples were first rapidly cooled down to −80 °C, then heated at 10 °C min−1 to 150 °C, cooled at 5 °C min−1 to −80 °C and lastly heated to 150 °C at 10 °C min−1. The second heating ramp was used for measurement.2.4 Electrolyte preparation
The polymers and electrolyte salt were dissolved in dry THF (ca. 100 mg of polymer and 1.5 mL of THF with a corresponding amount of electrolyte salt). The films were cast in PTFE moulds (20 mm in diameter), and the solvent was removed via controlled evaporation in a vacuum oven; the pressure was reduced to full vacuum (<1 mbar) over 20 h before heating at 60 °C at full vacuum for 40 h. The resulting films were ca. 0.2 mm in thickness. All procedures were performed in an argon-filled glovebox.2.5 Rheological characterisation
Rheological studies were performed on a stress-controlled TA Discovery HR-1 rheometer, equipped with a Peltier temperature controller. A parallel plate geometry (∅ = 8 mm) was used. Frequency sweeps (0.01–10 Hz) at constant strain amplitude (1%) were performed to determine the elastic (G′) and viscous (G′′) moduli. All measurements were performed at 25 and 40 °C (10 K increment) with an isothermal hold for 10 min prior to each measurement.2.6 Electrochemical impedance spectroscopy (EIS)
The ionic conductivity was measured using impedance spectroscopy with an SI 1260 Impedance Gain-Phase Analyzer (Schlumberger). The polymer electrolyte films were sandwiched between two stainless steel blocking electrodes in a CR2025 coin cell. The samples were equilibrated at every temperature for ca. 30 min before a new recording was performed. The measurements were done in the frequency range of 1 Hz to 10 MHz with an amplitude of 10–50 mV during heating from 20 °C up to 90 °C and during cooling back down to 20 °C. The data were fitted to a Debye equivalent circuit to obtain the electrolyte resistance during heating, from which the ionic conductivity was calculated.2.7 Molecular dynamics simulations
Four MD simulation boxes are constructed for PCL-PTMC and shPCL-PTMC polymer electrolyte with LiTFSI, LiPF6, and NaTFSI concentration of [cation]/[CO] = 0.125. To keep the total number of repeating units constant, the number of polymers in the simulation boxes was 30 and 6 for a polymer chain length of 20 and 100 repeating units, respectively. All simulations were carried out in GROMACS 202129 and General AMBER force field (GAFF)30 parameters were used for describing bonding and non-bonding interactions. The velocity Verlet algorithm was used for integrating Newton's equations of motion with a time step of 1 fs. More details regarding the MD options and force field parameters can be found elsewhere.31 All the H-bonds in the system were constrained using the LINCS algorithm. The protocol for the equilibrating step was 5 ns NVT (constant number, volume, and temperature) simulation at 400 K followed by 10 ns NPT (constant number, pressure, and temperature) simulation at 1 bar and with varying temperatures from 400–1000–400 K to ensure the randomly equilibrated structure. Bussi–Donadio–Parrinello thermostat32 and the Parrinello–Rahman barostat33 with time constant of 1 ps were used for NVT and NPT simulations. Then finally a 400 ns NPT simulation for the production run where thermodynamic properties and trajectory were saved every 0.5 ps and 5 ps, respectively.
3. Results and discussion
3.1 Materials design and polymer synthesis
Self-healing polymer electrolytes were prepared based on a PCL-PTMC backbone, which serves as a core unit for the formation of a cross-linked polymer network. With a glass transition temperature that is lower for PCL (−60 °C) than for PTMC (−15 °C), the chain flexibility can be increased by means of the caprolactone comonomer, thereby achieving the effect of increasing the ionic conductivity. In particular, the PCL-PTMC copolymer with a monomer ratio of ε-caprolactone (εCL) to trimethylene carbonate (TMC) of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 has been demonstrated to exhibit a combination of high ionic conductivity and high Li+ transference number (t+>0.5).34 The PCL-PTMC copolymer was synthesized by ring-opening polymerization of εCL and TMC with a multifunctional initiator, 2-ethyl-2-(methylhydroxy)propane-1,3-diol illustrated in Scheme 1. In order to create a densely cross-linked network, the macromolecular star-branched PCL-PTMC was synthesised using a feed ratio of [εCL]:[TMC]:[initiator] of 40:10:1 (for DP = 50, Mn, theoretical = 5586 g mol−1). Full conversion was evidenced by 1H NMR (Fig. S1†); hence, the crude PCL-PTMC was used without further purification.
:20 has been demonstrated to exhibit a combination of high ionic conductivity and high Li+ transference number (t+>0.5).34 The PCL-PTMC copolymer was synthesized by ring-opening polymerization of εCL and TMC with a multifunctional initiator, 2-ethyl-2-(methylhydroxy)propane-1,3-diol illustrated in Scheme 1. In order to create a densely cross-linked network, the macromolecular star-branched PCL-PTMC was synthesised using a feed ratio of [εCL]:[TMC]:[initiator] of 40:10:1 (for DP = 50, Mn, theoretical = 5586 g mol−1). Full conversion was evidenced by 1H NMR (Fig. S1†); hence, the crude PCL-PTMC was used without further purification.
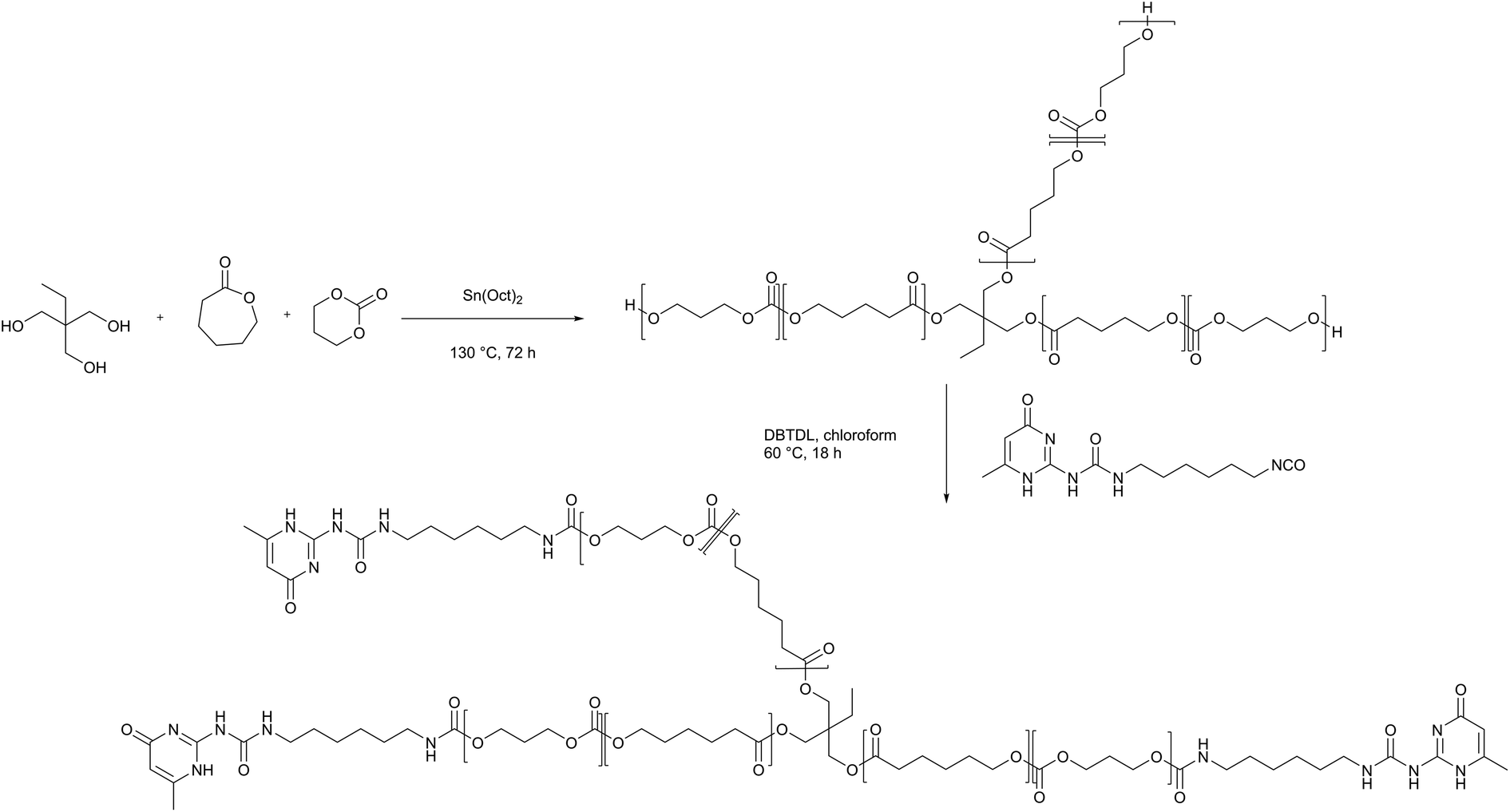 | ||
| Scheme 1 Synthesis of self-healing star-branched PCL-PTMC (shPCL-PTMC). | ||
An isocyanate UPy synthon (UPy-hexyl isocyanate) was prepared as previously reported28 and ultimately incorporated to the polymer end groups through an amidation reaction between the isocyanate synthon and the hydroxyl end groups (Scheme 1).4 The self-healing PCL-PTMC (shPCL-PTMC) was collected and purified by precipitating into cold diethyl ether. The chemical structure of shPCL-PTMC was verified by 1H NMR (Fig. S2†). Three singlet peaks originating from UPy quadruple H-bonding in chloroform appear at δ = 10.12, 10.85, and 13.12 ppm.35–37 The resonance of the two methylene –CH2– adjacent to the NHCO carbamate group was clearly visible at around 3.2 ppm, confirming the successful functionalization of UPy to PCL-PTMC. The composition of the resulting self-healing polymer can be determined based on the integral area ratio of methylene protons (H-h) to methyl protons belonging to TMC monomer (H-m). The characteristic main signals of the TMC, CL units, and UPy exhibit relative intensities in close concordance with the targeted molar ratios between the monomers (TMC:εCL:UPy = 40:10:3). This demonstrates that the end-groups in the starting PCL-PTMC were retained and fully functionalised.
3.2 Characterisation of shPCL-PTMC polymer electrolytes
In the forthcoming discussion, we will firstly study the rheological effects of the electrolyte salt ions on the dynamics of quadruple H-bonds and the mechanical performance of shPCL-PTMC electrolytes in comparison to the non-self-healing PCL-PTMC polymer electrolyte. We then probe the change in UPy quadruple H-bonding association using FTIR analysis and molecular dynamics simulations to understand the effects of the electrolyte salts. Three samples of SPEs containing different electrolyte salts – LiTFSI, LiPF6, and NaTFSI – were prepared at a fixed ratio of Li+/Na+ ions to CO groups of 1:8. With this selection of salts, both effects of different cations and different anions on the electrolyte properties can be distinguished. For comparison purposes, a fixed salt concentration was selected in a range where optimal ionic conductivity is typically reported for a wide range of SPEs.
000 g mol−1) was used instead as a reference material for comparison.
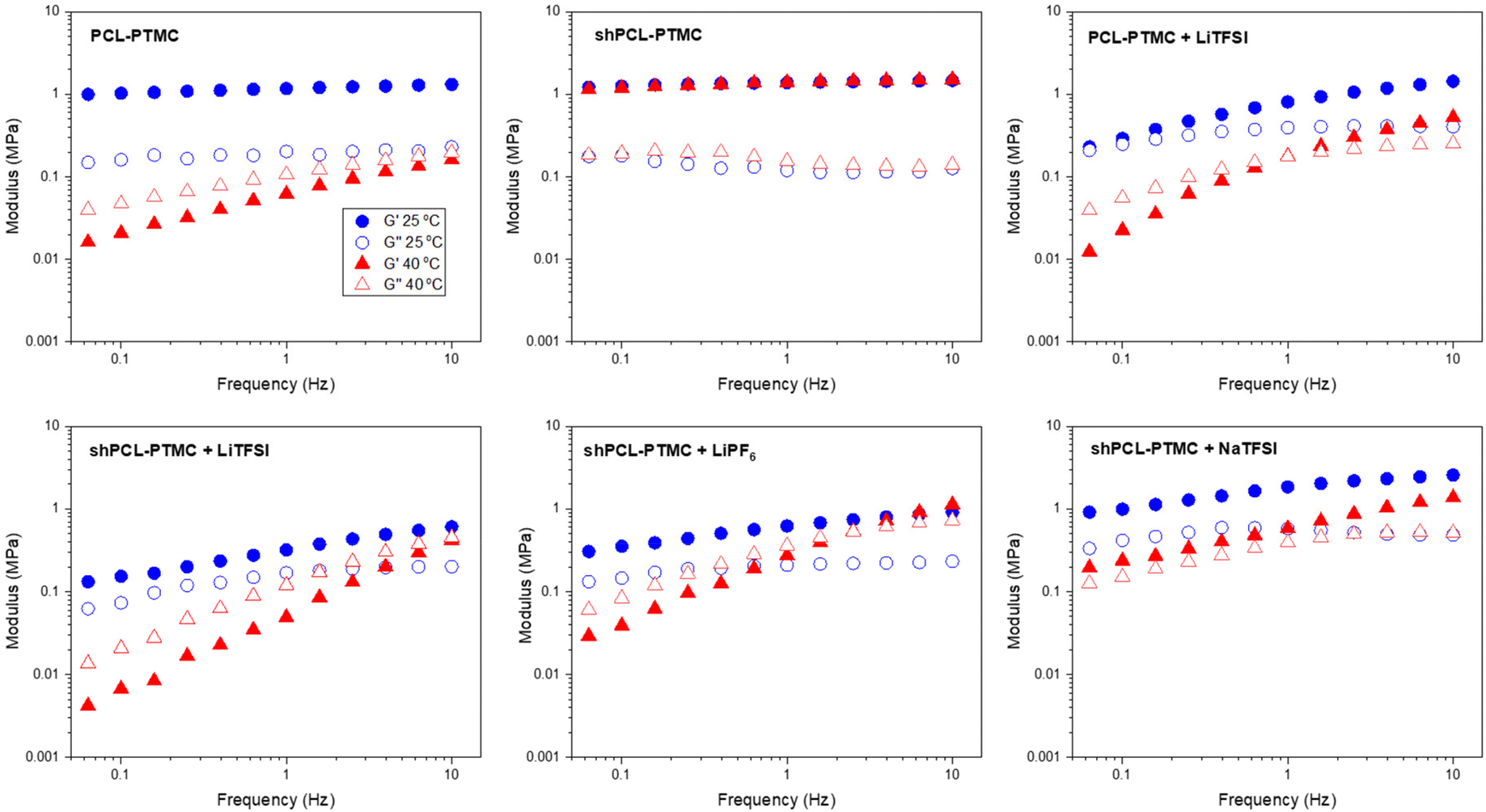 | ||
| Fig. 1 Oscillatory sweep of HMW PCL-PTMC, shPCL-PTMC, and the polymer electrolytes containing LiTFSI, LiPF6, and NaTFSI at 25 °C and 40 °C. | ||
Although HMW PCL-PTMC is typically mostly amorphous, at 25 °C there is still some crystallinity in the rheology sample, which could be noticed from the sample being partly opaque. Even this limited amount of crystallites was sufficient to mechanically stabilize the material, as seen from the consistently elastic behavior (G′ ≫ G′′) in Fig. 1. Either adding LiTFSI salt or heating the sample to 40 °C completely disrupts this crystallinity, leading to a deterioration of the mechanical stability and the PCL-PTMC samples instead show predominantly viscous behavior (G′ < G′′).
In comparison, the effect of loss of crystallinity can be counteracted by the UPy motifs, as seen from the significant mechanical enhancement for shPCL-PTMC, which shows elastic behavior at 40 °C, compared to the predominantly viscous behavior in PCL-PTMC at the same temperature. For shPCL-PTMC, a plateau region for the storage modulus can be seen at intermediate frequencies indicating physically cross-linked network structures in shPCL-PTMC resulting from the long lifetime of UPy dimers.28 The moduli of the shPCL-PTMC sample also remain almost unaffected at 40 °C despite the increasing likelihood of H-bonding dissociation at elevated temperatures. This suggests that the shPCL-PTMC can withstand moderate heating. Furthermore, according to the time–temperature superposition principle, as an increase in temperature is equivalent to a decrease in frequency, this indicates that the pure shPCL-PTMC should also show mechanical stability at low rates of deformation or static loads which are the relevant conditions during battery cycling.
When an electrolyte salt is added, however, this picture completely changes. At ambient temperature, all SPE samples exhibit a predominantly elastic behaviour in the frequency range studied although for the lithium salt-containing SPEs (i.e., LiTFSI and LiPF6), the storage modulus G′ shows a noticeable decrease in magnitude as well as a greater frequency dependence, suggesting that the lithium salts mechanically weaken the self-healing polymer network more than NaTFSI.
After heating to 40 °C, the mechanical properties of the self-healing SPEs worsen considerably, and a crossover of G′ and G′′ appears where the mechanical properties transition from elastic to viscous. Applying the time–temperature superposition principle, this indicates mechanical deterioration at low frequencies also at lower temperatures and an instability under static loads. In contrast, the NaTFSI-containing electrolyte shows only a marginal reduction in the storage modulus G′, and its elastic behavior is retained throughout the measured frequency interval.
The striking discrepancy in mechanical behavior between the pristine self-healing polymer and the self-healing SPEs evidently demonstrate that the salts not only exert negative effects on the quadruple H-bonding association but also amplifies the H-bonding dissociation in response to elevated temperatures, which affects the lithium salt-containing SPEs the most. While the LiPF6-containing electrolyte shows better mechanical stability than its LiTFSI-containing counterpart, of all three self-healing polymer electrolytes the solid-like behavior is only retained for shPCL-PTMC with NaTFSI.
The variation in rheological behaviors of the self-healing SPEs demonstrates that there are likely dynamic interactions between the salt ions and UPy motifs, with both anionic and cationic effects contributing to the mechanical weakening observed in the self-healing SPEs. The interfering effect differs from ion to ion, largely depending on the sizes and the structures of ions. As for UPy–anion interaction, the PF6− ion should be considered as having less potential for H-bonding compared to TFSI ions which consist of several H-bond acceptors such as SO groups. It is reasonable to expect the disruptive effect from LiPF6 to be less than that from LiTFSI, which would also explain the observations in Fig. 1.
While the rheological analysis does not clearly reveal the culprit for the mechanical deterioration in self-healing SPEs, it does point out that both anions and cations appear to adversely affect the UPy quadruple H-bonding association. Since it is vital to have the SPE function as a mechanical separator throughout the battery operation, this phenomenon raises the question as to whether the self-healing UPy units are practically useful for long-term cycling of batteries under low-frequency or static mechanical pressure, and/or at conventionally elevated operating temperatures.
O.39 As illustrated in Fig. 2a, the Tg of the polymer electrolytes increases in the order of LiTFSI < NaTFSI < LiPF6. It was reported previously that Na+ cations form stronger coordination bonding to carbonyl groups in polymers and higher coordination number compared to Li+ cations.40 This would, as a result, lead to a stronger physically cross-linked network for sodium SPEs (as also indicated in Fig. 1). The difference in Tg between SPEs containing LiTFSI and LiPF6 indicates that shPCL-PTMC with LiPF6 possesses a more densely cross-linked network than the former. This result is in good agreement with the rheological data, which shows that LiPF6-containing electrolyte films were more mechanically stable with a higher elastic modulus than shPCL-PTMC with LiTFSI. On the basis of rheological analysis and thermal properties, it can be concluded that TFSI ions appear to exert more negative effects on the associations of UPy motifs than PF6− ions.
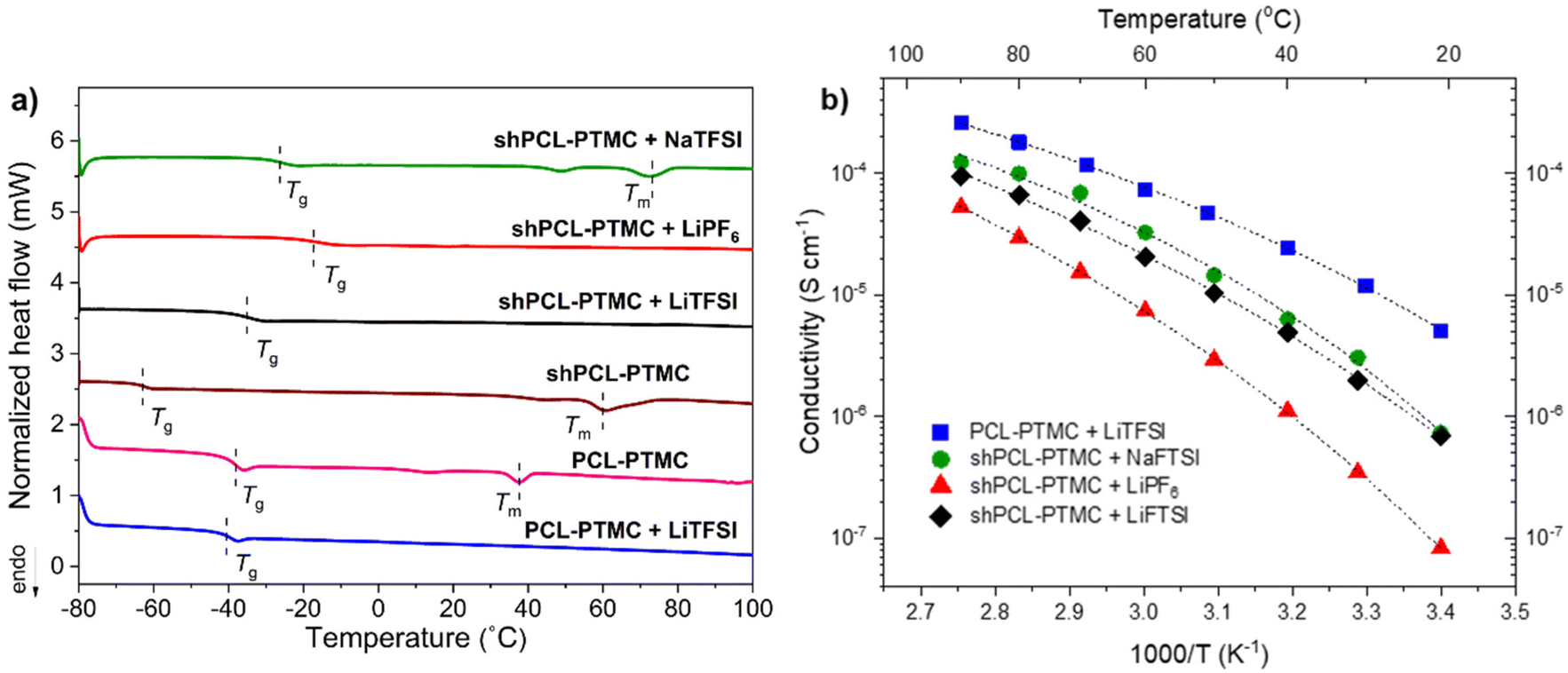 | ||
| Fig. 2 (a) DSC analysis of shPCL-PTMC and self-healing polymer electrolytes; (b) Ionic conductivity of PCL-PTMC, shPCL-PTMC with LiTFSI, shPCL-PTMC with NaTFSI, and shPCL-PTMC with LiPF6. The dashed lines in (b) represent VFT fits. | ||
As for the ionic conductivity of the electrolytes, VFT-type behavior is observed throughout the investigated temperature range (Fig. 2b). The self-healing SPEs exhibit a lower ionic conductivity compared to the unmodified PCL-PTMC polymer primarily due to the restricted polymer chains existing in the self-healing cross-linked network. shPCL-PTMC with LiTFSI exhibits an order of magnitude higher ionic conductivity than that of shPCL-PTMC with LiPF6 which correlates well with the rheology and DSC data. The ionic conductivity of both lithium SPEs is, however, still in the low range around 10−5 S cm−1 at 40 °C.
O acceptor in UPy dimers. To evaluate the individual and combined impacts of UPy–anion and UPy–cation interactions, we examine in detail the infrared absorption discrepancy of these groups in free and bonded states. The IR spectrum of pristine shPCL-PTMC displays a strong absorption at 1730 cm−1 for CO groups in the polymer backbone (Fig. 3a). The characteristic absorptions of UPy quadruple H-bonding appear at 1700, 1669, 1585, and 1525 cm−1, corresponding to urea CO, and amide CO, CN, and CC, respectively.25,35
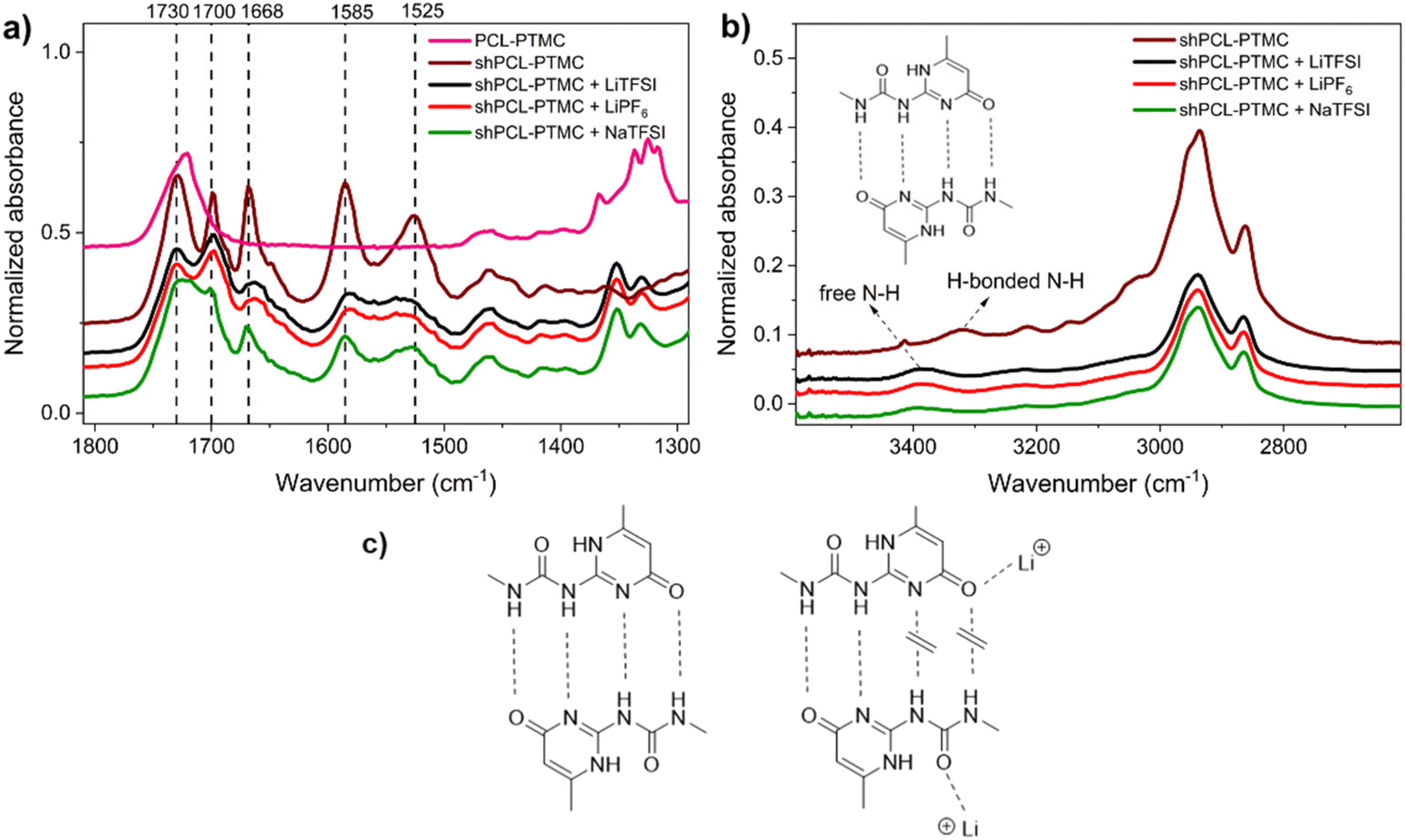 | ||
| Fig. 3 FTIR spectra of (a) pure shPCL-PTMC and self-healing polymer electrolytes with different salts LiTFSI, NaTFSI, and LiPF6; (b) in the UPy stretching region; (c) Proposed interactions of UPy-Li+ and UPy-TFSI. | ||
For the self-healing polymer electrolytes, a sharp drop in intensities of these infrared absorptions can be seen as indicative of weakened UPy quadruple H-bonding or a reduction in the number of H-bonds. Furthermore, a strong shift from 3323 cm−1 to 3387 cm−1 was observed for the urea N–H stretching, implying a transition from hydrogen-bonded N–H to non-hydrogen-bonded N–H (Fig. 3b).25 This is presumably due to the elimination of anchoring restrictions from hydrogen-bonded N–H bonds leading to a decrease in vibration frequency. SPEs containing lithium salts show very similar IR spectra with respect to absorbances and intensities, particularly in the region between 1800 and 1500 cm−1. In particular, the amide and urea CO stretching bands at 1700 and 1669 cm−1 appear noticeably broadened and shift to lower wavenumbers, indicative of bonded CO states other than H-bonded CO (Fig. 3c).
:TMC of 4:1 were simulated with LiTFSI, LiPF6, and NaTFSI. The number of polymers in the simulation boxes were 30 and the salt concentration was fixed at a ratio of [cation]/[COPCL-TMC] = 1:8. The molecular structures of the monomer units, UPy and anions are shown in Fig. 4a.
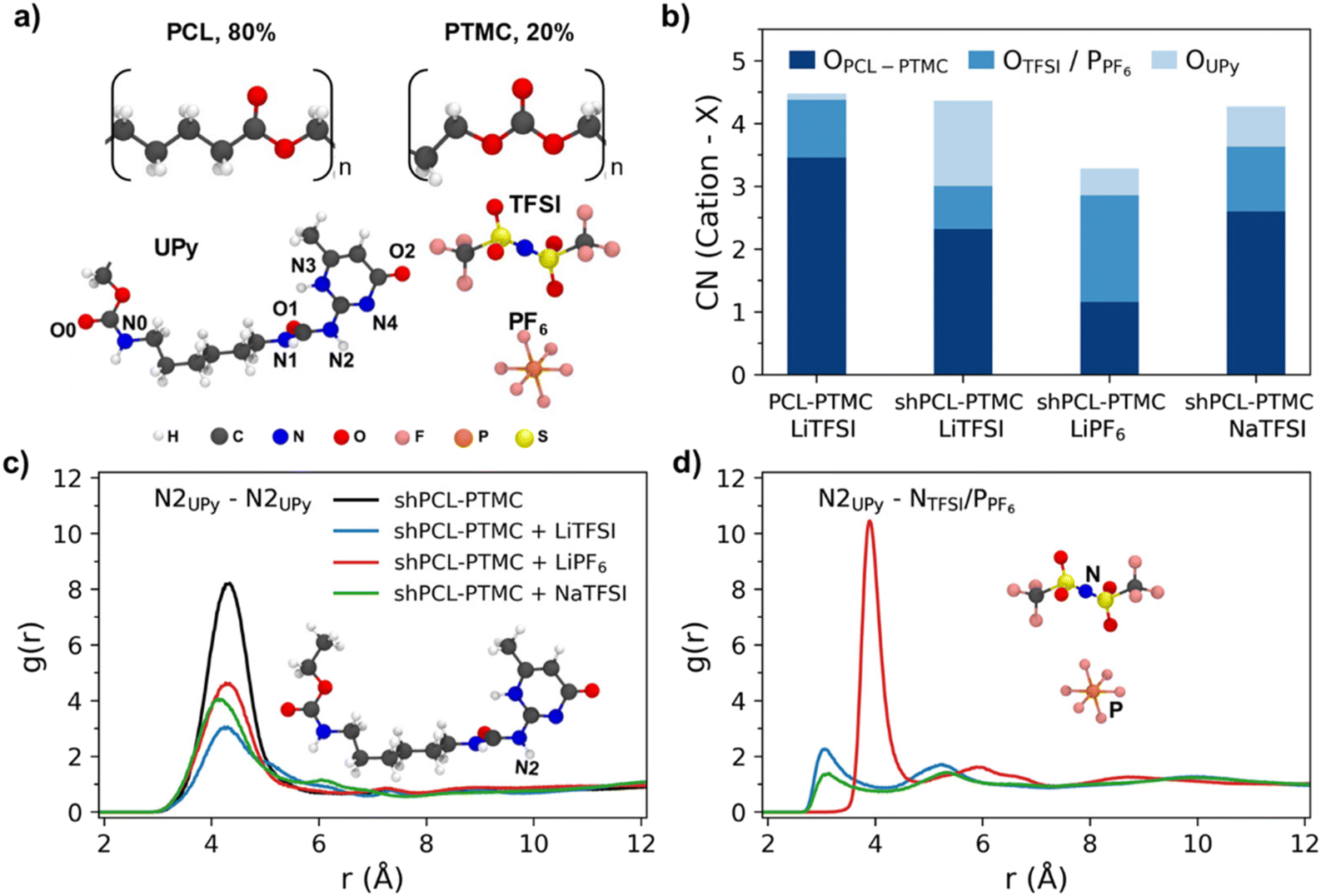 | ||
| Fig. 4 (a) The molecular structure of the repeating unit of PCL, PTMC, UPy, TFSI, and PF6 anions. (b) Coordination number (CN) of Li+ or Na+ with carbonyl O from PCL-PTMC (OPCL-PTMC), O from TFSI (OTFSI) or P from PF6− (PPF6), O from PCL-PTMC end group (OH), and O from UPy. (OUPy) (c) The radial distribution functions (g(r)) between the N2 atom of UPy to the N2 atom of UPy, and (d) the N2 atom of UPy to the central N atom of TFSI or central P atom of PF6−. | ||
To study the cation local coordination environments, the coordination number (CN) of Li+/Na+ with carbonyl COPCL-PTMC, O from TFSI or P from PF6−, and O atoms from end groups i.e., OH for PCL-PTMC and UPy for shPCL-PTMC, were calculated from their radial distribution functions g(r) (Fig. S3†).
From Fig. 4b, it is evident that there is a drastic increase in CN for Li+–OUPy accompanied by a drop in CN for Li+–OPCL-PTMC for self-healing SPEs compared to the PCL-PTMC electrolyte, indicating a shift in cation interactions from coordination by the polymer backbone to coordination by the UPy end group. This undesirable interaction between cations and the UPy group could lead to UPy H-bonding dissociation, which can be seen from the decrease in g(r) peaks in Fig. 4c. For the NaTFSI salt, the CN of Na+–OPCL-PTMC remains same but the CN of Na+–OUPy was less than that of Li+–OUPy in the case of LiTFSI, which is in good agreement with the FTIR results. To understand the role of anions, g(r) of UPy with anions was plotted in Fig. 4d. The presence of peaks confirms the interactions between anions and UPy groups. The results from radial distribution function (RDF) calculations suggest that there are interactions between the salt ions and the UPy groups from a statistical perspective but does not provide any information on specifics of the dynamical H-bonds.
To also study the dynamical properties, the number of H-bonds and their lifetimes were investigated for UPy–UPy and UPy–TFSI interactions. In the simulations, an H-bond is determined by a geometrical criterion, where the distance between the donor and acceptor is less than 3.5 Å and the angle between hydrogen donor and acceptor is less than 30°. The O–H and N–H groups are regarded as donors, and O and N are regarded as acceptors. The lifetime of the H-bonds is calculated from the average over all autocorrelation functions of the existence functions (either 0 or 1) of all H-bonds. As salt is added to shPCL-PTMC, both the number and lifetimes of H-bonds drop dramatically (Fig. 5), the lifetime of H-bonds between the UPy–TFSI being a few orders of magnitude lower than the UPy–UPy bonds. This both shows that the interactions with the salt ions disrupts the H-bonding of the UPy moieties and that the newly-formed H-bonds between the UPy groups and TFSI are highly dynamic. We also note that the number of UPy–UPy H-bonds (Fig. 5a) furthermore appears to correlate well with the rheological data in Fig. 1, showing both a higher number of H-bonds and a higher mechanical stability for shPCL-PTMC with NaTFSI than for either of the lithium varieties. From Fig. S4,† the opposite trends in the progression of the number of H-bonds of UPy–UPy and UPy–TFSI as a function of simulation time can also be observed. This further confirms that the UPy quadruple H-bonding as visualized in Fig. 6a is disrupted by the interactions between the UPy group and salt ions (Fig. 6b). The results strongly support our hypothesis about the undesirable interactions between electrolyte salt ions and the self-healing functionalities interfering with the dimerization of UPy motifs which consequently disrupts the dynamic H-bonding network.
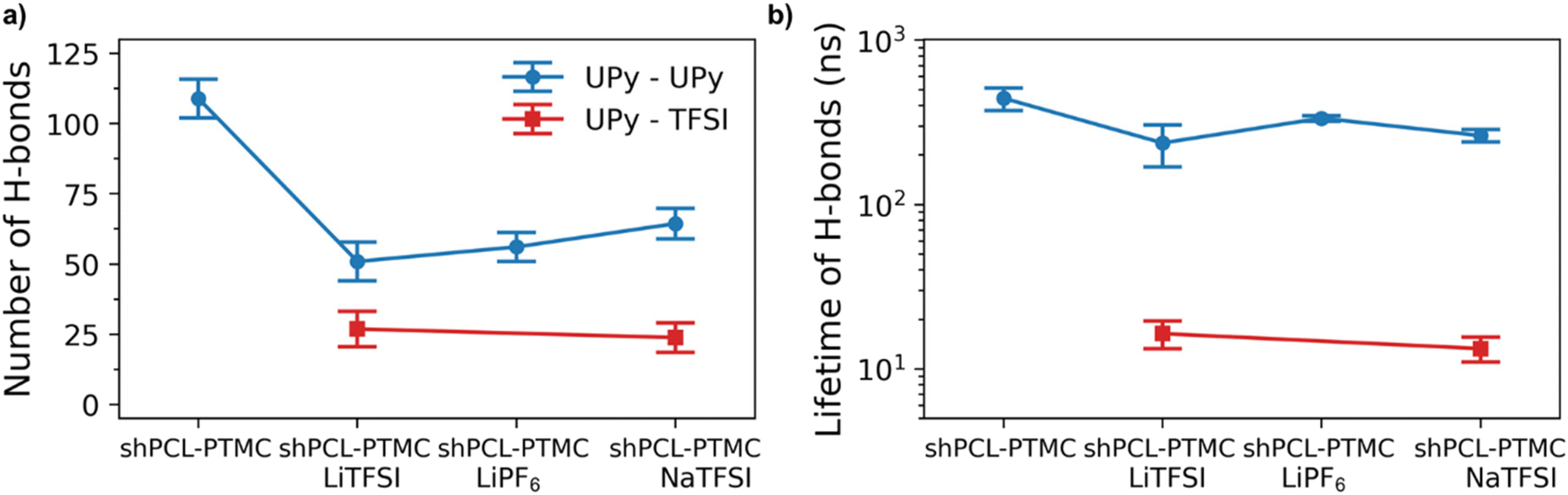 | ||
| Fig. 5 The average number of H-bonds (a) and their lifetimes (b) between the self-healing motifs (UPy–UPy) and between self-healing motif and TFSI (UPy–TFSI) for different polymer electrolyte systems, i.e., shPCL-PTMC with LiTFSI (red) and NaTFSI and LiPF6 (blue). | ||
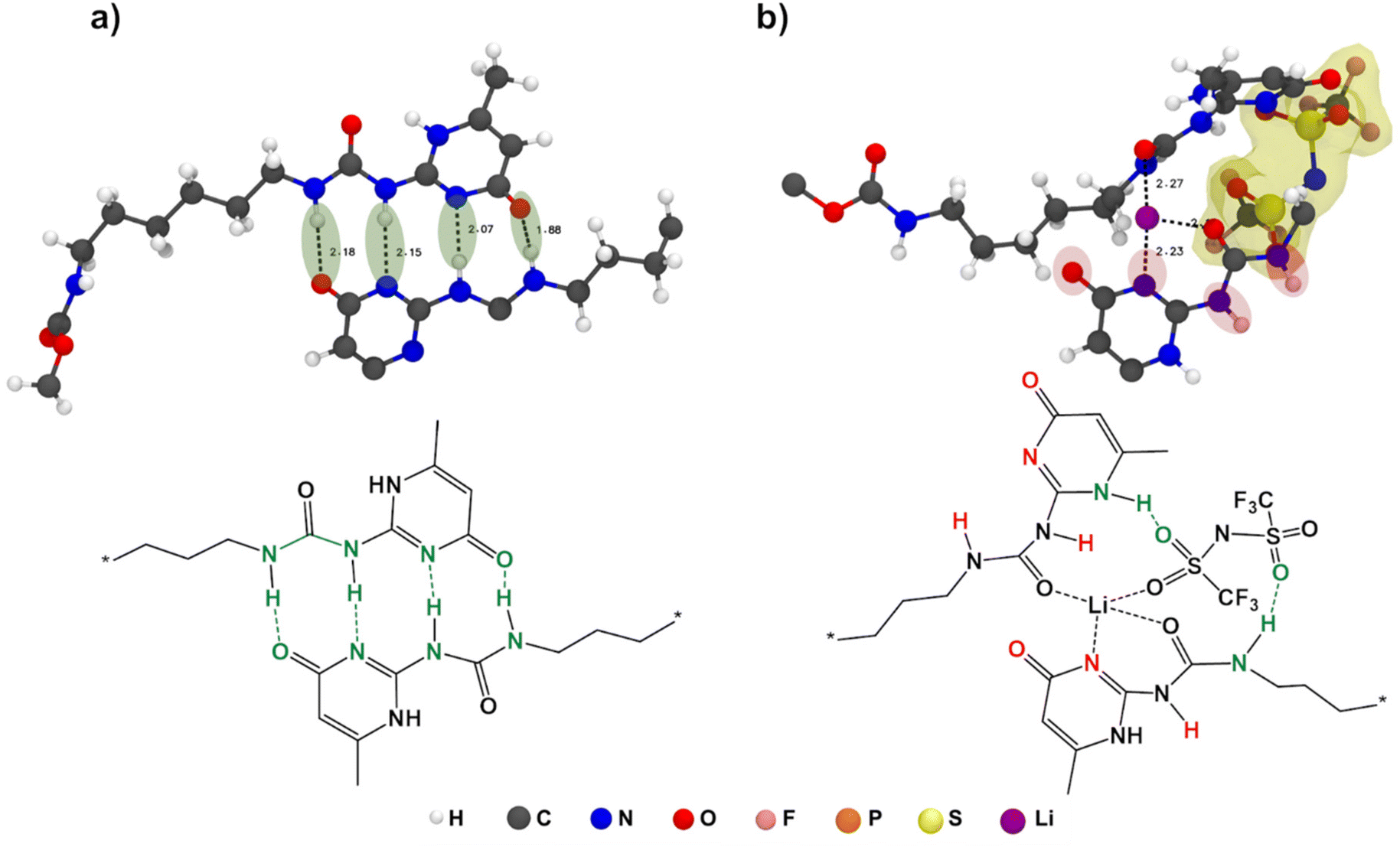 | ||
| Fig. 6 Visualization of H-bonding and coordination interactions between (a) UPy–UPy and (b) UPy–LiTFSI with the TFSI anion highlighted in yellow. | ||
4. Conclusions
The integration of smart functions into polymeric materials in batteries opens up avenues for the development of high-performance polymeric materials for batteries in general. Despite the merits of quadruple H-bonding UPy motifs, it is evident that the UPy-functionalized polymer electrolyte system presents its own unique set of challenges. In particular, the self-healing polymer, once combined with electrolyte salt, loses its mechanical stability, proving that UPy motifs are unfit to generate polymer electrolyte systems with long-term mechanical stability. Interactions of UPy with anions and cations from the salt are accountable for the dissociation of UPy quadruple H-bonds, leading to the weakening in mechanical strength of the self-healing polymers, effects that are highly salt-dependent. Some improvements in mechanical robustness may be achieved by adding additional grafted H-bonding groups along the polymer main chain instead of the end group functionalities used here, although that is likely to further stiffen the system and inhibit ion transport. More appropriate steps to take to counter these negative effects could be to combine dynamic non-covalent bonds with dynamic covalent bonds (e.g., disulfide bonds, Diels–Alder-formed bonds and reversible N–O bonds) to provide self-healing functionalities that are not affected by the high ionic strength of the electrolyte environment. The findings highlight the importance of a comprehensive understanding of underlying interactions between battery system components and polymer functionalities and point out the need for chemistry invention for the future development of high-performance polymeric materials for battery applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Knut and Alice Wallenberg Foundation (INTELiSTORE 139501042) and STandUP for Energy are acknowledged for financial support.References
- H. T. Zhang, H. Wu, L. Wang, H. Xu and X. M. He, J. Power Sources, 2021, 492, 229661 CrossRef CAS.
- D. Liu, Z. Tang, L. Luo, W. Yang, Y. Liu, Z. Shen and X.-H. Fan, ACS Appl. Mater. Interfaces, 2021, 13, 36320–36329 CrossRef CAS PubMed.
- A. Bergfelt, G. Hernández, R. Mogensen, M. J. Lacey, J. Mindemark, D. Brandell and T. M. Bowden, ACS Appl. Polym. Mater., 2020, 2, 939–948 CrossRef CAS.
- D. G. Mackanic, X. Yan, Q. Zhang, N. Matsuhisa, Z. Yu, Y. Jiang, T. Manika, J. Lopez, H. Yan, K. Liu, X. Chen, Y. Cui and Z. Bao, Nat. Commun., 2019, 10, 5384 CrossRef PubMed.
- P. Jaumaux, Q. Liu, D. Zhou, X. Xu, T. Wang, Y. Wang, F. Kang, B. Li and G. Wang, Angew. Chem., Int. Ed., 2020, 59, 9134–9142 CrossRef CAS PubMed.
- L. Zhang, P. Zhang, C. Chang, W. Guo, Z. H. Guo and X. Pu, ACS Appl. Mater. Interfaces, 2021, 13, 46794–46802 CrossRef CAS PubMed.
- K. R. Deng, S. P. Zhou, Z. L. Xu, M. Xiao and Y. Z. Meng, Chem. Eng. J., 2022, 428, 131224 CrossRef CAS.
- P. Guo, A. Su, Y. Wei, X. Liu, Y. Li, F. Guo, J. Li, Z. Hu and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 19413–19420 CrossRef CAS PubMed.
- N. Boaretto, T. Horn, M. Popall and G. Sextl, Electrochim. Acta, 2017, 241, 477–486 CrossRef CAS.
- A. Bergfelt, M. J. Lacey, J. Hedman, C. Sångeland, D. Brandell and T. Bowden, RSC Adv., 2018, 8, 16716–16725 RSC.
- Y. An, X. Han, Y. Liu, A. Azhar, J. Na, A. K. Nanjundan, S. Wang, J. Yu and Y. Yamauchi, Small, 2022, 18, 2103617 CrossRef CAS.
- W. Mai, Q. Yu, C. Han, F. Kang and B. Li, Adv. Funct. Mater., 2020, 1909912, DOI:10.1002/adfm.201909912.
- J. Chen, Y. Zhu, X. Chang, D. Pan, G. Song, Z. Guo and N. Naik, Adv. Funct. Mater., 2021, 31, 2104686 CrossRef CAS.
- S. C. Cummings, O. J. Dodo, A. C. Hull, B. Zhang, C. P. Myers, J. L. Sparks and D. Konkolewicz, ACS Appl. Polym. Mater., 2020, 2, 1108–1113 CrossRef CAS.
- J. Xu, X. Wang, H. Ruan, X. Zhang, Y. Zhang, Z. Yang, Q. Wang and T. Wang, Polym. Chem., 2022, 13, 2420–2441 RSC.
- M. Liu, P. Liu, G. Lu, Z. Xu and X. Yao, Angew. Chem., Int. Ed., 2018, 57, 11242–11246 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- Y. H. Jo, S. Q. Li, C. Zuo, Y. Zhang, H. H. Gan, S. B. Li, L. P. Yu, D. He, X. L. Xie and Z. G. Xue, Macromolecules, 2020, 53, 1024–1032 CrossRef CAS.
- E. Calabrese, L. Guadagno, M. Raimondo, A. Sorrentino, S. Russo, P. Longo and A. Mariconda, Macromol. Mater. Eng., 2023, 308, 2200500 CrossRef CAS.
- B. Zhou, D. He, J. Hu, Y. Ye, H. Peng, X. Zhou, X. Xie and Z. Xue, J. Mater. Chem. A, 2018, 6, 11725–11733 RSC.
- Y. Ren, Z. Cui, A. Bhargav, J. He and A. Manthiram, Adv. Funct. Mater., 2022, 32, 2106680 CrossRef CAS.
- L. Mezzomo, C. Ferrara, G. Brugnetti, D. Callegari, E. Quartarone, P. Mustarelli and R. Ruffo, Adv. Energy Mater., 2020, 10, 2002815 CrossRef CAS.
- E. R. Ezeigwe, L. Dong, R. Manjunatha, M. Tan, W. Yan and J. Zhang, Nano Energy, 2021, 84, 105907 CrossRef CAS.
- B. Zhou, Y. H. Jo, R. Wang, D. He, X. Zhou, X. Xie and Z. Xue, J. Mater. Chem. A, 2019, 7, 10354–10362 RSC.
- H. Rupp, R. Bhandary, A. Kulkarni and W. Binder, Adv. Mater. Technol., 2022, 7, 2200088 CrossRef CAS.
- C. B. Thompson and L. T. J. Korley, ACS Macro Lett., 2020, 9, 1198–1216 CrossRef CAS PubMed.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- P. Y. W. Dankers, Z. Zhang, E. Wisse, D. W. Grijpma, R. P. Sijbesma, J. Feijen and E. W. Meijer, Macromolecules, 2006, 39, 8763–8771 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- K. G. Sprenger, V. W. Jaeger and J. Pfaendtner, J. Phys. Chem. B, 2015, 119, 5882–5895 CrossRef CAS PubMed.
- T. Eriksson, H. Gudla, Y. Manabe, T. Yoneda, D. Friesen, C. Zhang, Y. Inokuma, D. Brandell and J. Mindemark, Macromolecules, 2022, 55, 10940–10949 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Eriksson, A. Mace, J. Mindemark and D. Brandell, Phys. Chem. Chem. Phys., 2021, 23, 25550–25557 RSC.
- M. Sadat-Shojai and S. Ghadiri-Ghalenazeri, New J. Chem., 2020, 44, 20155–20166 RSC.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- S. Bobade, Y. Wang, J. Mays and D. Baskaran, Macromolecules, 2014, 47, 5040–5050 CrossRef CAS.
- K. E. Feldman, M. J. Kade, T. F. A. de Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694–4700 CrossRef CAS.
- J. Mindemark, A. Sobkowiak, G. Oltean, D. Brandell and T. Gustafsson, Electrochim. Acta, 2017, 230, 189–195 CrossRef CAS.
- C. Sångeland, R. Younesi, J. Mindemark and D. Brandell, Energy Storage Mater., 2019, 19, 31–38 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00017j |
| This journal is © The Royal Society of Chemistry 2024 |