
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient (Z)-selective semihydrogenation of alkynes catalyzed by air-stable imidazolyl amino molybdenum cluster sulfides†
María
Gutiérrez-Blanco
 a,
Eva
Guillamón
a,
Vicent S.
Safont
a,
Andrés G.
Algarra
b,
M. Jesús
Fernández-Trujillo
b,
Kathrin
Junge
c,
Manuel G.
Basallote
*b,
Rosa
Llusar
*a and
Matthias
Beller
*c
a,
Eva
Guillamón
a,
Vicent S.
Safont
a,
Andrés G.
Algarra
b,
M. Jesús
Fernández-Trujillo
b,
Kathrin
Junge
c,
Manuel G.
Basallote
*b,
Rosa
Llusar
*a and
Matthias
Beller
*c
aDepartament de Química Física i Analítica, Universitat Jaume I, Av. Sos Baynat s/n, 12071 Castelló, Spain. E-mail: rosa.llusar@uji.es
bDepartamento de Ciencia de los Materiales e Ingeniería Metalúrgica y Química Inorgánica, Instituto de Biomoléculas (INBIO), Facultad de Ciencias, Universidad de Cádiz, Apartado 40, Puerto Real, 11510 Cádiz, Spain
cLeibniz-Institute for Catalysis, Albert-Einstein-Street 29a, 18059 Rostock, Germany
First published on 16th February 2023
Abstract
Imidazolyl amino cuboidal Mo3(μ3-S)(μ-S)3 clusters have been investigated as catalysts for the semihydrogenation of alkynes. For that purpose, three new air-stable cluster salts [Mo3S4Cl3(ImNH2)3]BF4 ([1]BF4), [Mo3S4Cl3(ImNH(CH3))3]BF4 ([2]BF4) and [Mo3S4Cl3(ImN(CH3)2)3]BF4 ([3]BF4) have been isolated in moderate to high yields and fully characterized. Crystal structures of complexes [1]PF6 and [2]Cl confirm the formation of a single isomer in which the nitrogen atoms of the three imidazolyl groups of the ligands are located trans to the capping sulfur atom which leaves the three bridging sulfur centers on one side of the trimetallic plane while the amino groups lie on the opposite side. Kinetic studies show that the cluster bridging sulfurs react with diphenylacetylene (dpa) in a reversible equilibrium to form the corresponding dithiolene adduct. Formation of this adduct is postulated as the first step in the catalytic semihydrogenation of alkynes mediated by molybdenum sulfides. These complexes catalyze the (Z)-selective semihydrogenation of diphenylacetylene (dpa) under hydrogen in the absence of any additives. The catalytic activity lowers sequentially upon replacement of the hydrogen atoms of the N–H2 moiety in 1+ without reaching inhibition. Mechanistic experiments support a sulfur centered mechanism without participation of the amino groups. Different diphenylacetylene derivatives are selectively hydrogenated using complex 1+ to their corresponding Z-alkenes in excellent yields. Extension of this protocol to 3,7,11,15-tetramethylhexadec-1-yn-3-ol, an essential intermediate for the production of vitamin E, affords the semihydrogenation product in very good yield.
Introduction
The stereoselective reduction of internal alkynes to disubstituted alkenes by using hydrogen is a highly valuable reaction in the bulk and fine chemical industries.1 In particular, (Z)-alkenes are found in several bioactive compounds, pharmaceuticals and agrochemicals.2 The selective synthesis of these products has been traditionally achieved by heterogenous catalysis where the Pd-based Lindlar catalyst is still the most common choice.3 However, for several substrates drawbacks such as isomerization and overreduction are still unsolved. Although heterogeneous catalysis is frequently preferred by industry, homogenous catalysis offers some advantages such as a more rational tuning of the catalysts as well as its potential to be used as models capable to emulate the reactivity of its solid analogues. In this regard, homogenous systems can also provide valuable mechanistic information.Most of the reported homogeneous catalysts for the (Z)-stereoselective hydrogenation of alkynes to alkenes make use of alternative hydrogen donors such as boranes, silanes, alcohols, formic acid or water, while the number of systems which operate using molecular hydrogen as the reductant are surprisingly scarce.4 Due to economic and environmental reasons, there is a growing interest to replace noble metals by non-precious ones and to optimize the atom economy using hydrogen as reductant. Most examples include mononuclear 3d transition metal complexes based on Mn,5,6 Fe,7 Co,8,9 (Fig. 1) or cubane-type polynuclear Cu10 compounds functionalized with phosphine ligands. As a very recent example, Milstein and co-workers have evidenced the great potential of Mg pincer complexes as catalysts for the transformation of diphenylacetylene derivatives to the corresponding (Z)-alkenes with good to very good diastereoselectivities.11 Previously, our groups have also shown that low-valent Mo pincer complexes and cuboidal Mo3S4 clusters are also efficient homogeneous catalysts for this transformation.12,13 Notably, pincer complexes utilizing sophisticated phosphine ligands dominate this chemistry and more importantly, mechanistic investigations on these systems have revealed unique reaction pathways. Such studies combining control experiments with DFT calculations revealed that hydrogenation by PNHP and PNHS Mn and Mo pincer complexes proceeds via an outer sphere reaction mechanism utilizing the amino moiety in the ligand backbone for metal–ligand cooperativity.5,6,12
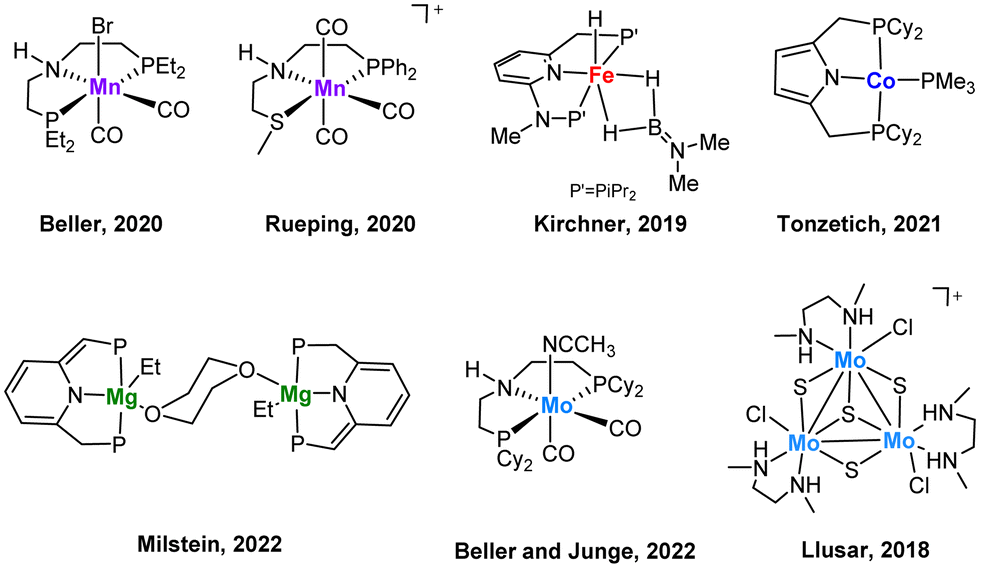 | ||
| Fig. 1 Selected examples of homogeneous non-noble metal (pre)catalysts for an efficient Z-selective semihydrogenation of internal alkynes. | ||
In these cases, a strong base seems to be needed to form the catalytically active species, likely an amido complex, generated by deprotonation of the amine of the ligand backbone. This amido complex presumably activates the hydrogen molecule with the corresponding formation of an R2(H)N–M–H hydrido moiety where the two hydrogen atoms are transferred to the alkyne to produce the (Z)-alkene without any direct interaction between the substrate and the metal. On the other hand, pincer PNP complexes of Fe, Co and Mg operate under base-free conditions and H2 activation generates catalytically active M–H hydrides, which react with the alkyne to form η2-olefin species.7,9 In the case of Mg, a metal–ligand cooperation mechanism (MLC) involving the aromatization/dearomatization of the pincer compound is proposed.11
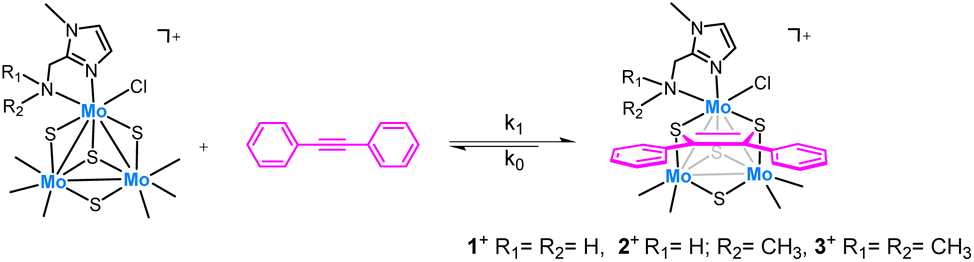
Interestingly, trinuclear Mo3S4 clusters containing inexpensive diamino ligands also catalyze the semihydrogenation of diphenylacetylene (dpa) with an acceptable (Z)-stereoselectivity (Z/E ca. 6/1) in the absence of a base, albeit harsh conditions were required (12% mol catalyst, 100 bars of H2 and 150 °C) to achieve moderate conversion (ca. 60%). This complex operates through an unusual cluster catalysis mechanism, depicted in Fig. 2, where only the three bridging sulfur atoms of the cluster act as the active sites for this transformation.13
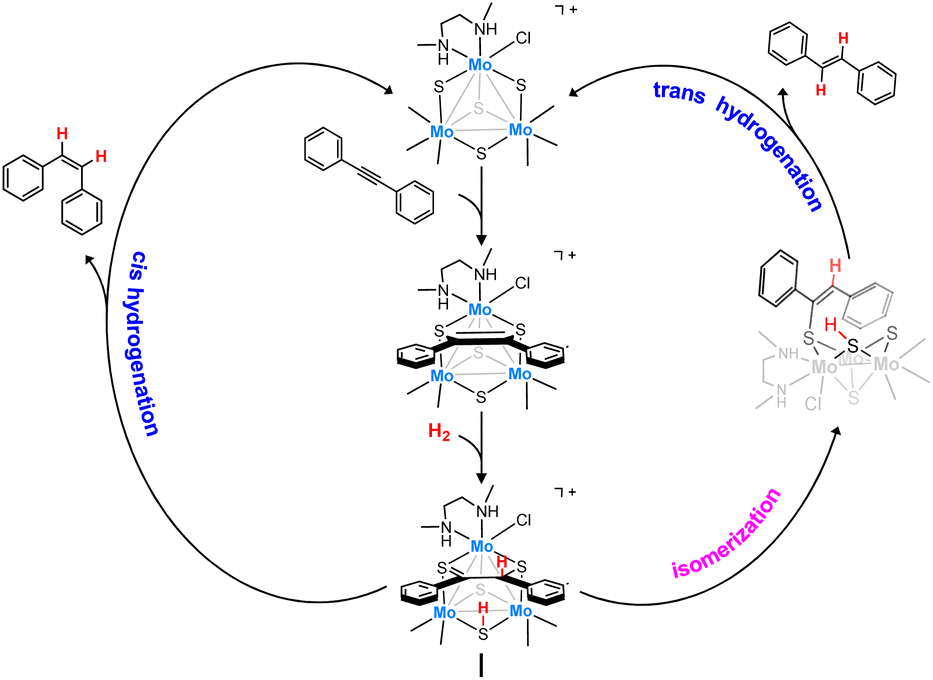 | ||
| Fig. 2 Simplified catalytic cycle for the semihydrogenation of dpa in the presence of Mo3S4 clusters. | ||
In a first step, the alkyne molecule reacts with two of the bridging sulfides to form a dithiolene adduct. Next, hydrogen activation takes place through the third bridging sulfur atom and one of the dithiolene carbon atoms. Finally, the half-hydrogenated intermediate undergoes a reductive elimination step to produce the desired (Z)-alkene or to evolve into an isomerized analogue which after elimination affords the (E)-alkene. The relative energy barrier of these two processes, cis-hydrogenation vs. trans isomerization, determines the stereoselectivity of the reaction.
The low cost and low toxicity of molybdenum also makes this metal an attractive substitute to noble metals as hydrogenation catalysts although this alternative has received little attention in comparison to first row transition metals.14,15 Motivated by our earlier results on alkyne semihydrogenation using Mo3S4 cluster catalysts, we decided to explore the effect of the outer bidentate ligands. Earlier studies in our group have shown that diphosphino and aminophosphino molybdenum complexes are not efficient catalysts for the direct hydrogenation of organic substrates such as nitroarenes.16 In this work, we turned our attention to bidentate nitrogen donor ligands containing imidazolyl moieties inspired by the excellent activity in hydrogenation processes that certain noble and non-noble metal complexes, i.e. Ru17,18 and Mn19 exhibit when decorated with such imidazolyl-based ligands.
Herein, we report a novel protocol for the selective (Z)-semihydrogenation of a series of alkynes using a Mo3S4 cluster catalyst decorated with imidazolyl amino ligands. Three new cluster complexes, which differ in the nature of the amino group: –NH2, –NH(CH3) and –N(CH3)2 have been prepared to investigate the influence of the Mo-amino functionality. Mechanistic experiments aimed to understand the reaction mechanism are also presented. The new [Mo3S4Cl3(ImNH2)3]+ (1+) complex has been applied to the catalytic semihydrogenation of several diphenylacetylene derivatives affording the (Z)-alkenes with excellent yields. Complex 1+ also catalyses the semihydrogenation of an industrially relevant terminal alkyne substrate.
Experimental
General remarks
Elemental analyses were carried out on a EuroEA3000 Eurovector analyser. Electrospray mass spectra were recorded with a QuattroLC (quadrupole–hexapole–quadrupole) mass spectrometer with an orthogonal Z-spray electrospray interface (Micromass, Manchester, UK). The cone voltage was set at 20 V unless otherwise stated using CH3CN as the mobile phase solvent. Sample solutions were infused using a syringe pump directly connected to the ESI source at a flow rate of 10 mL min−1 and a capillary voltage of 3.5 kV was used in the positive scan mode. Nitrogen was employed as drying and nebulizing gas. Isotope experimental patterns were compared with theoretical patterns obtained using the MassLynx 4.1 program.201H, 13C NMR and 1H–13C gradient HSQC spectra were recorded on a Bruker Avance III HD 400 MHz or 300 MHz spectrometers using CD3CN as solvent. Gas chromatography analyses were performed on an Agilent 7820A GC system equipped with a FID and a capillary column Agilent (HP-5, 30 m × 0.32 mm × 0.25 mm). Mass determination was carried out on a GC-Mass Agilent 5977E network equipped with a mass-selective detector. Kinetic experiments on the cycloaddition of diphenylacetylene to the clusters were carried out using a Cary 50 spectrophotometer provided with a thermostatted multicell holder. All experiments were conducted at 25.0 °C under pseudo-first order conditions of alkyne excess, and the spectral changes were fitted satisfactorily in all cases to a model with a single kinetic step.Catalyst preparation
All reactions were performed under a nitrogen atmosphere using standard Schlenk techniques. The trinuclear precursor Mo3S4Cl4(PPh3)3(H2O)2 was prepared according to the literature procedure.21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 50 mL), boiling hexane (50 mL) and diethyl ether to afford the green solid that was characterized as [Mo3S4Cl3(ImNH2)3]BF4 (112 mg, 67% yield). Elemental analysis (%) calcd for Mo3S4Cl3C15H27N9BF4: C 19.11, H 2.89, N 13.37; found: C 19.80, H 3.00, N 13.50; 1H NMR (CD3CN, 400 MHz): δ = 3.46 (br, 3H, NH, HA), 3.81 (s, 9H, N-CH3, HC), 3.95 (ddd, J = 16.3, 6.0 and 1.6 Hz, 3H, CH, HB), 4.00 (br, 3H, NH, HA), 4.44 (ddd, J = 16.3, 7.7 and 3.4 Hz, 3H, CH, HB), 7.48 (d, J = 1.6 Hz, 3H, CH, HD), 8.13 (d, J = 1.6 Hz, 3H, CH, HE); 13C {1H} NMR (CD3CN, 101 MHz): δ = 35.28 (s, CH2, CC), 42.50 (s, CH3, CB), 125.78 (s, CH, CD), 132.54 (s, CH, CE), 151.54 (s, C, CF); ESIMS (MeOH, 20 V): m/z: 856.0 [M+].
:1, 50 mL), boiling hexane (50 mL) and diethyl ether to yield an air-stable product characterized as [Mo3S4Cl3(ImNMe2)3]BF4 (70.3 mg, 94%). Elemental analysis (%) calcd for Mo3S4Cl3C15H27N9BF4: C 24.56, H 3.83, N 12.28; found: C 25.38, H 3.78, N 11.90; 1H NMR (CD3CN, 400 MHz): δ = 2.96 (s, 18H, CH3, HA), 3.91 (s, 9H, CH3, HC), 4.61 (s, 6H, CH2, HB), 7.53 (d, J = 2.0 Hz, 3H, CH, HD), 7.56 (d, J = 2.0 Hz, 3H, CH, HE); 13C {1H} NMR (CD3CN, 101 MHz): δ = 36.59 (s, CH3, CC), 44.85 (s, CH3, CA), 49.28 (s, CH2, CB), 122.45 (s, CH, CD), 127.02 (s, CH, CE), 136.33 (s, C, CF); ESIMS (CH3CN, 20 V): m/z: 940.0 [M+].
:1, 50 mL), boiling hexane (50 mL) and diethyl ether to afford the green solid that was characterized as [Mo3S4Cl3(ImNH2)3]BF4 (112 mg, 67% yield). Elemental analysis (%) calcd for Mo3S4Cl3C15H27N9BF4: C 19.11, H 2.89, N 13.37; found: C 19.80, H 3.00, N 13.50; 1H NMR (CD3CN, 400 MHz): δ = 3.46 (br, 3H, NH, HA), 3.81 (s, 9H, N-CH3, HC), 3.95 (ddd, J = 16.3, 6.0 and 1.6 Hz, 3H, CH, HB), 4.00 (br, 3H, NH, HA), 4.44 (ddd, J = 16.3, 7.7 and 3.4 Hz, 3H, CH, HB), 7.48 (d, J = 1.6 Hz, 3H, CH, HD), 8.13 (d, J = 1.6 Hz, 3H, CH, HE); 13C {1H} NMR (CD3CN, 101 MHz): δ = 35.28 (s, CH2, CC), 42.50 (s, CH3, CB), 125.78 (s, CH, CD), 132.54 (s, CH, CE), 151.54 (s, C, CF); ESIMS (MeOH, 20 V): m/z: 856.0 [M+].
:1, 50 mL), boiling hexane (50 mL) and diethyl ether to yield an air-stable product characterized as [Mo3S4Cl3(ImNMe2)3]BF4 (70.3 mg, 94%). Elemental analysis (%) calcd for Mo3S4Cl3C15H27N9BF4: C 24.56, H 3.83, N 12.28; found: C 25.38, H 3.78, N 11.90; 1H NMR (CD3CN, 400 MHz): δ = 2.96 (s, 18H, CH3, HA), 3.91 (s, 9H, CH3, HC), 4.61 (s, 6H, CH2, HB), 7.53 (d, J = 2.0 Hz, 3H, CH, HD), 7.56 (d, J = 2.0 Hz, 3H, CH, HE); 13C {1H} NMR (CD3CN, 101 MHz): δ = 36.59 (s, CH3, CC), 44.85 (s, CH3, CA), 49.28 (s, CH2, CB), 122.45 (s, CH, CD), 127.02 (s, CH, CE), 136.33 (s, C, CF); ESIMS (CH3CN, 20 V): m/z: 940.0 [M+].
X-ray data collection and structure refinement
Suitable crystals for X-ray studies of complex 1+ were obtained by slow evaporation of a sample of [1]BF4 in CH3CN the presence of an excess of NH4PF6. Crystals of 2+ were grown by slow evaporation of a sample solution of 2+ in CH3OH acidified with HCl. Diffraction data collection was performed at 200 K on an Agilent Supernova diffractometer equipped with an Atlas CCD detector using CuKα radiation (λ = 1.54184 Å). No instrument or crystal instabilities were observed during data collection. Absorption corrections based on the multiscan method were applied.22,23 The structure was solved by direct methods and refined by the full-matrix methods based on F2 with the program SHELXL-13 using the Olex2 software package.24,25 Graphics were performed with the Diamond visual crystal structure information system software.26![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.2357(3), b = 12.7929(3), c = 13.9167(4) Å; α = 68.076(3)°, β = 84.513(2)°, γ = 81.883(2)°; V = 1671.76(8) Å3; T = 200.00(14); Z = 2; μ(MoKα) = 14.717 mm−1; reflections collected/unique = 34375/6824 (Rint = 0.0664). The final refinement converged with R1 = 0.0400 and wR2 = 0.0881 for all reflections, GOF = 1.0340 a dmax./min residual electron density 0.96/−0.84 e Å−3.
, a = 9.67045(18), b = 12.8501(2), c = 13.6166(3) Å; α = 79.6676(15)°, β = 86.3570(15)°, γ = 88.6075(14)°; V = 1661.14 Å3; T = 200.00(14); Z = 2; μ(CuKα) = 1.710 mm−1; reflections collected/unique = 30528/6409 (Rint = 0.00272). The final refinement converged with R1 = 0.0302 and wR2 = 0.0684.
, a = 10.2357(3), b = 12.7929(3), c = 13.9167(4) Å; α = 68.076(3)°, β = 84.513(2)°, γ = 81.883(2)°; V = 1671.76(8) Å3; T = 200.00(14); Z = 2; μ(MoKα) = 14.717 mm−1; reflections collected/unique = 34375/6824 (Rint = 0.0664). The final refinement converged with R1 = 0.0400 and wR2 = 0.0881 for all reflections, GOF = 1.0340 a dmax./min residual electron density 0.96/−0.84 e Å−3.
, a = 9.67045(18), b = 12.8501(2), c = 13.6166(3) Å; α = 79.6676(15)°, β = 86.3570(15)°, γ = 88.6075(14)°; V = 1661.14 Å3; T = 200.00(14); Z = 2; μ(CuKα) = 1.710 mm−1; reflections collected/unique = 30528/6409 (Rint = 0.00272). The final refinement converged with R1 = 0.0302 and wR2 = 0.0684.
The structures of the complex salts [1]PF6·CH3CN and [2]Cl·CH3OH were refined in the triclinic P space group. The non-hydrogen atoms of the clusters and the counterions were refined anisotropically, whereas the positions of all hydrogen atoms in the ligands were generated geometrically, assigned isotropic thermal parameters, and allowed to ride on their respective parent carbon atoms. In both cases the solvent molecules in the crystal were refined anisotropically and their hydrogen atoms were included at their idealized positions.
CCDC 2210047 and 2210042† contains the supplementary crystallographic data for this paper.
Catalytic activity tests
Results and discussion
Synthesis and characterization
Three new trinuclear cluster salts of formula [Mo3S4Cl3(ImNH2)3]BF4 ([1]BF4), [Mo3S4Cl3(ImNH(CH3))3]BF4 ([2]BF4) and [Mo3S4Cl3(ImN(CH3)2)3]BF4 ([3]BF4) have been prepared by extending the procedure developed in our group and represented in Scheme 1.27 In this method the outer ligands of the Mo3S4Cl4(PPh3)3(H2O)2 precursor are partially replaced by imidazolylamino ligands. Acidification of the reaction media avoids formation of hydroxo species which result from the partial replacement of the outer chlorine ligands. As found for the aminophosphine M3S4 (Mo or W) derivatives, only one among all possible isomers is formed in moderate to high yields, 67% for [1]BF4, 64% for [2]BF4 and 94% for [3]BF4.28 All clusters salts were fully characterized by NMR, elemental analysis, and mass spectrometry.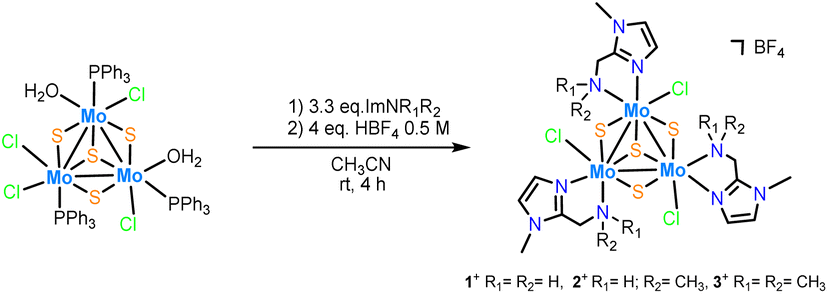 | ||
| Scheme 1 Synthesis of Mo3S4 cluster complexes decorated with imidazolylamino ligands. | ||
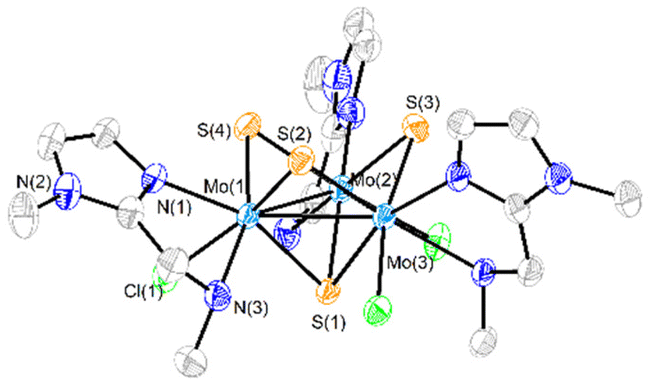
The crystal structures of [1]PF6 and [2]Cl have been determined by single crystal X-ray diffraction and they share structural features. Fig. 3 and 4 show ORTEP representations of the 1+ and 2+ cations together with relevant bond distances. Both cations contain an incomplete Mo3S4 cubane-type structure in which the molybdenum and sulfur atoms occupy adjacent vertices with a missing metal atom. In general, the metal–metal and metal–sulfur bond distances follow the same tendencies observed for other trinuclear Mo3S4 species.27,29 The three outer positions on each metal center are occupied by one chlorine atom and two nitrogen atoms from the imidazolyl amino ligand.
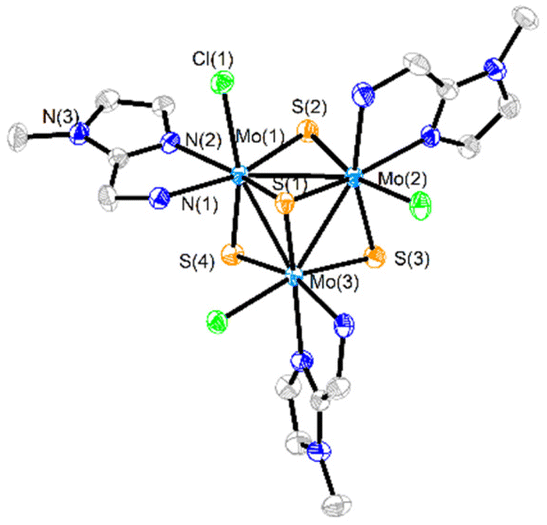 | ||
| Fig. 3 ORTEP representation (50% probability ellipsoids) of the cationic cluster [Mo3S4Cl3(ImNH2)3] + (1+). Hydrogen atoms have been omitted for clarity. Standard deviations of average distances are given in brackets. Average bond distances: Mo–Mo = 2.7378 [7], Mo-(μ3-S) = 2.328 [2], Mo-(μ-S)trans-Cl = 2.28 [1], Mo-(μ-S)trans-NH2 = 2.29 [1] Å. | ||
 | ||
| Fig. 4 ORTEP representation (50% probability ellipsoids) of the cationic cluster [Mo3S4Cl3(ImNHCH3)3]+ (2+). Standard deviations of average distances are given in brackets. Hydrogen atoms have been omitted for clarity. Average bond distances: Mo–Mo = 2.759 [3], Mo-(μ3-S) = 2.338 [4], Mo-(μ-S)trans-Cl = 2.288 [6], Mo-(μ-S)trans-NHMe = 2.289[6] Å. | ||
As previously mentioned, only one isomer is formed, wherein all three nitrogen atoms from the imidazolyl group are located trans to the capping sulfur while the ones from the amino group are placed trans to the bridging sulfur. Therefore, the capping sulfur, the chlorine atoms, and the amino groups of the ligand lie on the same side of the trimetallic plane.
The short Cl⋯H(–N) distances in 1+ (2.458 to 2.802 Å) and 2+ (2.359 to 2.951 Å) between the chlorine atom and the hydrogen atom of the amino groups of the adjacent metal center evidence the presence of hydrogen bonding interactions. Moreover, in the case of complex 1+ the above-mentioned values are comparable to the Cl⋯H(–N) distances of the chlorine atom and the amino group on the same metal center, with values ranging from 2.747 to 2.938 Å.
The solid-state structures of both cations, 1+ and 2+, are preserved in solution as evidenced by 13C NMR and 1H NMR spectroscopy. All attempts to obtain single crystals of 3+ were unfruitful but the product was fully characterized by spectrometric and spectroscopic techniques. The C3 symmetry of the three complexes has been confirmed in solution (Fig. SI1–SI6†). The 13C NMR spectra show five characteristic singlets, common for all three compounds, four signals of the imidazole ring and one to the methylene linker group. Nevertheless, the 1H NMR spectrum of 3+ reveals a slightly different solution behaviour compared to that of the other two clusters. In clusters 1+ and 2+, the signals for the methylene hydrogen atoms are clearly diastereotopic with resolved multiplets showing both the germinal coupling and the coupling to the amino hydrogen/s in agreement with their crystal structures. In contrast, this multiplet signal appears as a singlet in compound 3+, which suggests the existence of a weak Mo-NMe2 bond that allows for rapid dissociation–coordination and results in a time-averaged signal for the two methylene protons.
Reactivity versus alkynes and reaction kinetics
As mentioned in the Introduction, formation of dithiolene adducts upon interaction of alkynes with the bridging sulfur atoms of the cuboidal Mo3S4 unit is the first step of the mechanism for the semihydrogenation of alkynes catalyzed by these cluster complexes.13 It is well stablished that the [3 + 2] cycloaddition reaction between the alkynes and these clusters, depends on the alkyne substituents as well as on the nature of the cluster ligands.30,31 Formation of very stable Mo3S4 dithiolene adducts is detrimental for the catalytic semihydrogenation of alkynes. For example, the [Mo3S4Cl3(dmen)3]+ (dmen = N,N′-dimethylethylenediamine) cluster reacts with dimethyl acetylcarboxylate (dmad) to form a stable adduct and as a consequence it is not an active catalyst for alkyne semihydrogenation. On the other hand, the [Mo3S4Cl3(dmen)3]+ cluster does not react appreciably with diphenylacetylene (dpa), and thus it catalyzes its semihydrogenation although under harsh conditions. In our case, cluster complexes [1]BF4, [2]BF4 and [3]BF4 react with dpa to form the corresponding dithiolene adduct as evidenced by the appearance of a band in the UV-VIS spectra around 900 nm, characteristic of the formation of the [3 + 2] cycloaddition product (Fig. SI7†).32 Adduct formation is also evidenced by 1H NMR spectroscopy where new signals appear due to the loss of symmetry upon adduct formation (Fig. SI8†).21 However, these new signals coexist with those of 1+, 2+ and 3+ which indicates that formation of the addition dithiolene products occur under conditions of reversible equilibrium. This assumption is further confirmed by the electrospray ionization mass spectrometry (ESI-MS) of the reaction mixture (Fig. SI9†). The ESI-MS spectrum of 1+ in CH3CN shows a prominent peak centered at m/z = 856 associated to the 1+ cation according to its m/z value and calculated isotopic pattern. Upon reaction with an excess of dpa (20 eq., room temperature, CH3CN), the peak at m/z = 856 remains and a new peak of similar intensity emerges at m/z = 1034 identified as the [1 + dpa]+ cycloaddition product. Incidentally, no evidence exists regarding the addition of a second dithiolene molecule. A similar situation is observed when monitoring the reaction between [2]BF4 and [3]BF4 with dpa in CH3CN. That is, the cluster cations coexist with the cycloaddition dithiolene adduct even in the presence of a 20 molar excess of dpa.To obtain additional information on the process, the kinetics of reaction of [1]BF4, [2]BF4 and [3]BF4 with dpa were studied in acetonitrile solutions at 25 °C. The reaction was found to occur in a timescale suitable for monitoring the process with a conventional UV-VIS spectrophotometer. The spectral changes observed under pseudo-first order conditions of dpa excess (Fig. SI7†) clearly show the formation of the cycloaddition product with the previously commented band at 900 nm. These changes can be satisfactorily fitted using a kinetic model with a single kinetic step, and the values derived for the observed rate constant (kobs) show a linear dependence with the alkyne concentration that was fitted using eqn (1) (Fig. 5). The values so derived for the slope and the intercept are listed in Table 1.
| kobs = k0 + k1[dpa] | (1) |
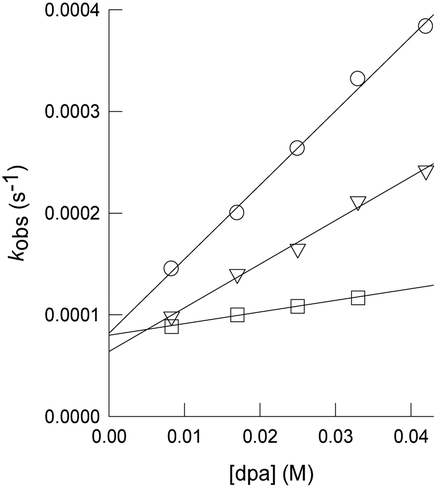 | ||
| Fig. 5 Plot of the dependence with the dpa concentration of the observed rate constants for the reaction of clusters [1]BF4 (circles), [2]BF4 (triangles), and [3]BF4 (squares), with an excess of dpa in CH3CN solutions at 25 °C. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Cluster | k 1 (M−1 s−1) | k 0 (s−1) | K e (M−1) |
| a Reaction conditions: 25 °C in CH3CN, pseudo-first order excess of dpa (0.008–0.042 M). | ||||
| 1 | 1+ | 7.3(3) × 10−3 | 8.2(7) × 10−5 | 89(8) |
| 2 | 2+ | 4.3(2) × 10−3 | 6.4(6) × 10−5 | 67(7) |
| 3 | 3+ | 1.14(4) × 10−3 | 8.0(1) × 10−5 | 14.0(5) |
In agreement with the NMR and ESI-MS results, the existence of a non-zero intercept in Fig. 5 suggests the possibility that the reaction occurs under conditions of reversible equilibrium, which is confirmed by an increase of the spectral change magnitude with the dpa concentration, so that k1 corresponds to the formation of the cycloaddition product, and k0 to the reverse process. While the rate of formation (k1) decreases gradually with the number of the amino methyl groups of the ligand, the rate of the reverse reaction (k0) shows smaller changes. Therefore, the equilibrium constant decreases upon methylation of the amino groups of the ligand. These results differ from those previously reported for the related diamino cluster, for which we were unable to observe formation of significant amounts of the cycloaddition product.13
Catalytic performance and mechanistic insights
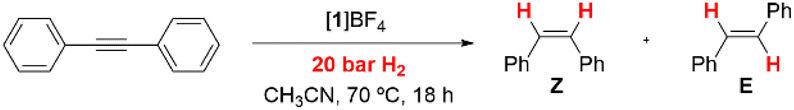
Evaluation of the catalytic activity of [1]BF4, [2]BF4 and [3]BF4 was performed on the semihydrogenation of diphenylacetylene as model reaction. Reaction conditions for the [1]BF4 cluster salt catalyst were optimized by varying the temperature and H2 pressure (Table SI1†). Full conversion and quantitative yield towards the formation of the (Z)-isomer was achieved under much milder conditions (70 °C and 20 bar) compared to previously known related molybdenum catalysts. For example, a conversion of 21% (18% of the (Z)-isomer and 3% of the (E)-isomer) was obtained using the diamino [Mo3S4Cl3(dmen)3]+ cluster catalyst (18 h at 150 °C, 100 bar H2 pressure and 5 mol% of the catalyst) in acetonitrile.13 Further attempts to lower the pressure and/or temperature led to a dramatic decrease of the catalytic activity. Incidentally, our optimized conditions are the typical conditions employed for the hydrogenation of nitroarenes mediated by the diamino [Mo3S4Cl3(dmen)3]+ catalyst.16 Then, the solvent influence was investigated. Similar activities are found for CH3CN and CH3OH (Table SI2†) while THF reduced the dpa conversion to 90%. No reaction is observed in 2-fluoroethanol or hexafluoroisopropanol. Hence at this point, we decided to continue the study in CH3CN.Cluster evolution during the catalytic semihydrogenation of dpa using [1]BF4 was monitored from batch experiments at different reaction times (Fig. SI10†). The molecular peak due to 1+ (m/z = 856) coexists with the [1 + dpa]+ (m/z = 1034) cycloaddition product. As expected, the intensity of the [1 + dpa]+ peak decreases with time. At the end of our catalytic protocol, the only peak registered, that of 1+, remains intact; therefore, catalysis occurs without degradation of 1+ which is indicative of a cluster catalysis process.
Next, we investigated the influence of the structure of the ligand on the catalytic activity for the semihydrogenation of diphenylacetylene at 70 °C and 20 bars of H2 (Table 2, entries 2–4). Substitution of one hydrogen of the N–H moiety in 1+, leads to a significant decrease in the catalytic activity (Table 2, entry 2). The total alkylation of the N–H moiety (Table 2, entries 3 and 4) with methyl substituents does not inhibit the catalytic conversion but drastically lowers the yield. The activity of 3+ is comparable with that of the diamino [Mo3S4Cl3(dmen)3]BF4 catalysts (Table 2, entry 5).13
|
|
||||
|---|---|---|---|---|
| Entry | Deviation | Conversionb [%] | Yieldb [%] | |
| Z | E | |||
| a Standard conditions: diphenylacetylene (0.1 mmol), H2 pressure (20 bar), 70 °C, catalyst (5 mol%), CH3CN, 18 h. b Determined by GC analysis using n-hexadecane as an internal standard. c Catalyst (12 mol%). d Catalyst (12 mol%) H2 pressure (50 bar), 150 °C. e Stoichiometric reaction, 0.01 mmol diphenylacetylene, 0.01 mmol cluster [1-d6]BF4, CD3CN. | ||||
| 1 | None | >99 | >99 | — |
| 2 | [2]BF4 | 40 | 40 | — |
| 3 | [3]BF4 | 7 | 7 | — |
| 4 | [3]BF4c |
23 | 23 | — |
| 5 | [Mo3S4Cl3(dmen)3]BF4d |
13 | 10 | 3 |
| 6 | [Mo3S4Cl3(ImND2)3]BF4e |
>99 | >99 | — |
| 7 | Addition of CuCl | 0 | — | — |
| 8 | Addition of Et3N (1 equiv.) | 30 | 29 | — |
| 9 | Addition of Et3N (2 equiv.) | 7 | 7 | — |
| 10 | Addition of Et3N (3 equiv.) | 7 | 7 | — |
An extra evidence of the passive role of the N–H moiety is that no deuterated (Z)-stilbene (Fig. SI11†) was formed by using the 1-d6+ cluster catalyst (Fig. SI12†) deuterated at the amino positions (Table 2, entry 6). These experiments rule out the involvement of the N–H moiety of the ligand backbone in the reaction. In contrast, there are strong evidences on metal–ligand cooperativity when low-valent molybdenum PNHP pincer complexes are employed as catalysts for this transformation which give support in this last case to an outer sphere reaction mechanism.12
Then, mechanistic evidence for a sulfur-based reaction mechanism were investigated. For that purpose, CuCl was added to the catalytic reaction mixture in order to block the three bridging sulfur atoms of the cluster. According to ESI-MS formation of the heterometallic [Mo3(CuCl)S4Cl3(ImNH2)3]+ (m/z = 956) and [Mo3(Cu(dpa))S4Cl3(ImNH2)3]+ (m/z = 1099) cluster cations took place on the basis of their m/z ratios and isotopic pattern (Fig. SI13†). Under these conditions, the catalytic process is inhibited (Table 2, entry 7) as previously observed for the semihydrogenation of alkynes and hydrogenation of diazobenzene mediated by the diamino [Mo3S4Cl3(dmen)3]+ catalysts, which was also attributed to the formation of Mo3S4Cu species.13,33
In the case of the catalytic diazobenzene hydrogenation, hydrogen activation occurs at the bridging sulfur atoms and results in the formation of a dithiol [Mo3S2(SH)2Cl3(dmen)3]+ adduct from which hydrogen is transferred to form first diphenylhydrazyne and next aniline through two interconnected cycles sharing a common reaction step which involves the above Mo3S4 dithiol adduct.33 Due to the acidic character of this proton S–H groups, addition of a base such as Et3N inhibits the hydrogenation process by providing an additional reaction pathway which capture these dithiol H atoms. In our case, the same tendency has been observed and addition of Et3N amine inhibits catalysis (Table 2, entries 8–10) suggesting a mechanism for hydrogen activation in which acidic protons are being generated.
The above control experiments indicate a sulfur-centered cluster catalysis mechanism without metal–ligand cooperativity. However, details on H2 activation by the bridging sulfur atoms of the cluster and their subsequent transfer to the alkyne molecule to selectively afford the (Z)-alkene remain unclear.
Substrate scope
Having in hand the optimal conditions for dpa semihydrogenation mediated by [1]BF4, dpa derivatives featuring different substituents were tested to analyze the tolerance of the system (Table 3).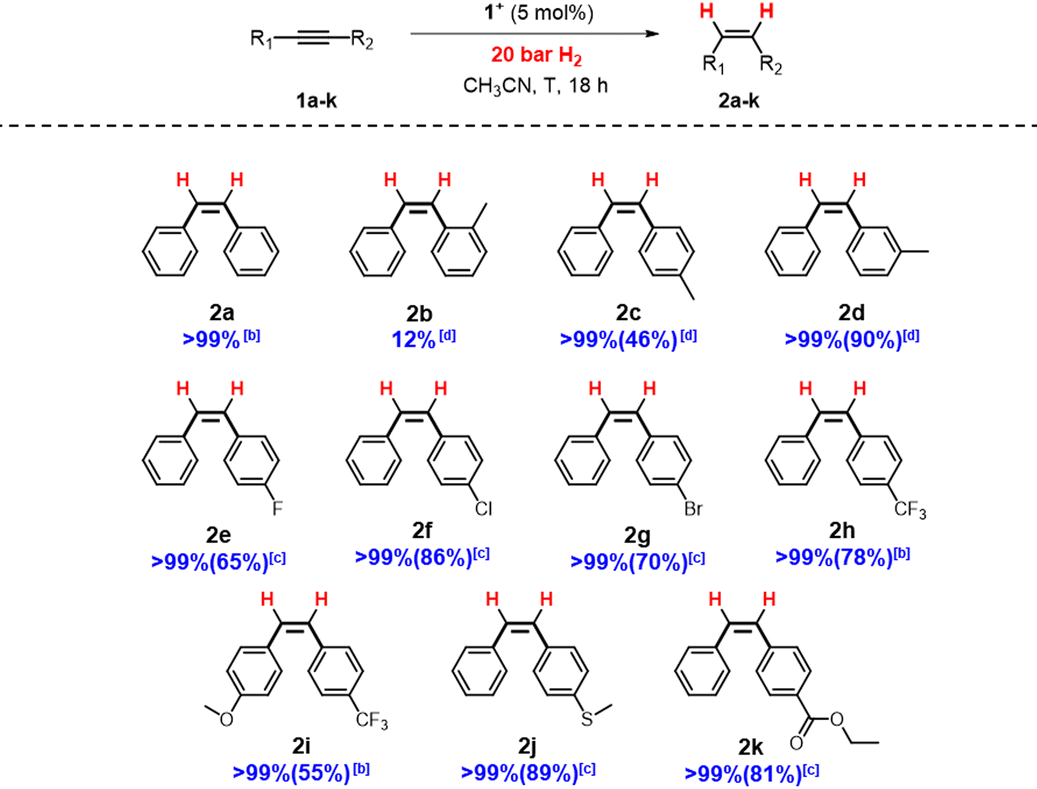
Compounds containing a methyl group in the para (2c) and meta (2b) positions were successfully reduced with excellent selectivity. However, in the case of the ortho derivative (2b) there was a drop of the conversion likely due to steric hindrance.12,34 Alkynes bearing halides as fluorine, chlorine, bromine or trifluoromethyl groups are smoothly transformed to the respective (Z)-alkenes. Remarkably, no dehalogenation products have been detected for any of the halogenated dpa derivatives. Also, substrates containing both trifluoromethyl and methoxy groups (2i) afforded good yields and stereoselectivity. Additionally, thioethers (2j) or ester groups (2k) on the alkynes are well tolerated.
Taking into consideration that [1]BF4 is an efficient catalyst for the semihydrogenation of internal alkynes, we decided to extend our study to terminal alkynes. The industrially relevant substrate chosen was 3,7,11,15-tetramethylhexadec-1-yn-3-ol, represented in Fig. 6, which is an essential intermediate for the production of vitamin E.35 Application of the optimized catalytic protocol developed for dpa semihydrogenation results in no conversion of the terminal alkyne substrate.
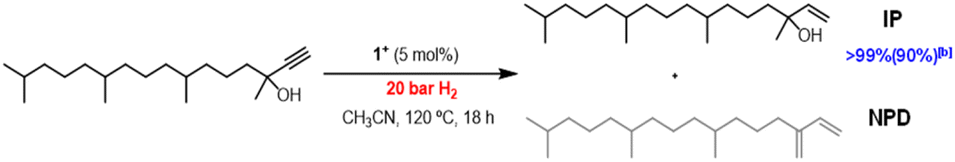 | ||
| Fig. 6 Semihydrogenation of DIP. Reaction conditions: 0.1 mmol alkyne, 5 mol% catalyst, 20 bar H2, 120 °C, CH3CN (2 mL), 18 h. Conversions and yields determined by GC analysis using n-hexadecane as an internal standard. | ||
Temperature and pressure were then optimized (Table SI3†) and quantitative conversions were achieved at 120 °C and 20 bar H2. In terms of chemoselectivity, the major product is the desired isophytol (IP). Notably, no over hydrogenation product was observed during the reaction and the only detected side product corresponds to the dehydration species, neophytadiene (NPD). Then, the influence of the reaction time was investigated to hamper this side-reaction (see Fig. SI32†). While the reaction time can be reduced from 18 h to 8 h maintaining conversion and yield, this has no effect on the final NPD concentration. Unfortunately, all attempts to avoid this side reaction were unfruitful. To the best of our knowledge, it is the first time that a homogenous molybdenum cluster performs this reaction. Further experiments are under investigation to achieve softer conditions.
Conclusions
In summary, we have synthesized and characterized three new Mo3S4 complexes featuring imidazolyl amino ligands with different degrees of methylation in the amino moiety. The bridging sulfur atoms of these Mo3(μ3-S)(μ-S) clusters interact with the alkyne, e.g. dpa to form a dithiolene Mo3(μ3-S)(μ-S)(μ3-SC(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Ph)S) adduct under conditions of reversible equilibrium. The [Mo3S4Cl3(ImNH2)]BF4 cluster salt is an efficient catalyst for the (Z)-selective semihydrogenation of dpa under much milder reaction conditions than those previously optimized for related diamino Mo3S4 cluster analogues. Mechanistic control experiments support a sulfur-based mechanism and rule out the operation of an outer sphere reaction mechanism with participation of the ligand NH group. Extension of our catalytic protocol to other internal alkynes revealed an excellent (Z)-selectivity. In addition, the catalytic protocol can also be adapted for the semihydrogenation of alkynols; in particular, to an essential vitamin E precursor. To the best of our knowledge, it is the first time that a homogenous molybdenum cluster performs this type of reaction.
C(Ph)S) adduct under conditions of reversible equilibrium. The [Mo3S4Cl3(ImNH2)]BF4 cluster salt is an efficient catalyst for the (Z)-selective semihydrogenation of dpa under much milder reaction conditions than those previously optimized for related diamino Mo3S4 cluster analogues. Mechanistic control experiments support a sulfur-based mechanism and rule out the operation of an outer sphere reaction mechanism with participation of the ligand NH group. Extension of our catalytic protocol to other internal alkynes revealed an excellent (Z)-selectivity. In addition, the catalytic protocol can also be adapted for the semihydrogenation of alkynols; in particular, to an essential vitamin E precursor. To the best of our knowledge, it is the first time that a homogenous molybdenum cluster performs this type of reaction.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The financial support of the Spanish Ministerio de Economía y Competitividad (grants PRE2019-088511, and PID2019-107006GB-C22), Generalitat Valenciana (grant CIAICO/2021/122) and Universitat Jaume I (grants UJI-B2019-30, UJI-B2021-29 and UJI-B2022-56) is gratefully acknowledged. The authors also thank the Serveis Centrals d'Instrumentació Científica (SCIC) of the Universitat Jaume I for providing us with materials characterization facilities. Furthermore, we thank Leibniz-Institute for Catalysis for the facilities.References
- M. Crespo-Quesada, F. Cárdenas-Lizana, A. L. Dessimoz and L. Kiwi-Minsker, Modern trends in catalyst and process design for alkyne hydrogenations, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS
.
- B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. Lansink Rotgerink, K. Möbus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, New developments in hydrogenation catalysis particularly in synthesis of fine and intermediate chemicals, Appl. Catal., A, 2005, 280, 17–46 CrossRef CAS
- H. Lindlar, Ein neuer Katalysator für selektive Hydrierungen, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS
- D. Decker, H. J. Drexler, D. Heller and T. Beweries, Homogeneous catalytic transfer semihydrogenation of alkynes-an overview of hydrogen sources, catalysts and reaction mechanisms, Catal. Sci. Technol., 2020, 10, 6449–6463 RSC
- M. Garbe, S. Budweg, V. Papa, Z. Wei, H. Hornke, S. Bachmann, M. Scalone, A. Spannenberg, H. Jiao, K. Junge and M. Beller, Chemoselective semihydrogenation of alkynes catalyzed by manganese(I)-PNP pincer complexes, Catal. Sci. Technol., 2020, 10, 3994–4001 RSC
- V. Zubar, J. Sklyaruk, A. Brzozowska and M. Rueping, Chemoselective Hydrogenation of Alkynes to (Z) -Alkenes Using an Air-Stable Base Metal Catalyst, Org. Lett., 2020, 22, 5423–5428 CrossRef CAS
- N. Gorgas, J. Brünig, B. Stöger, S. Vanicek, M. Tilset, L. F. Veiros and K. Kirchner, Efficient Z-Selective Semihydrogenation of Internal Alkynes Catalyzed by Cationic Iron(II) Hydride Complexes, J. Am. Chem. Soc., 2019, 141, 17452–17458 CrossRef CAS
- C. Chen, Y. Huang, Z. Zhang, X. Q. Dong and X. Zhang, Cobalt-catalyzed (Z)-selective semihydrogenation of alkynes with molecular hydrogen, Chem. Commun., 2017, 53, 4612–4615 RSC
- H. Alawisi, H. D. Arman and Z. J. Tonzetich, Catalytic Hydrogenation of Alkenes and Alkynes by a Cobalt Pincer Complex: Evidence of Roles for Both Co(I) and Co(II), Organometallics, 2021, 40, 1062–1070 CrossRef CAS
- K. Semba, R. Kameyama and Y. Nakao, Copper-Catalyzed Semihydrogenation of Alkynes to Z-Alkenes, Synlett, 2015, 26, 318–322 CrossRef CAS
- Y. Liang, U. K. Das, J. Luo, Y. Diskin-Posner, L. Avram and D. Milstein, Magnesium Pincer Complexes and Their Applications in Catalytic Semihydrogenation of Alkynes and Hydrogenation of Alkenes: Evidence for Metal–Ligand Cooperation, J. Am. Chem. Soc., 2022, 144, 19115–19126 CrossRef CAS
- N. F. Both, A. Spannenberg, K. Junge and M. Beller, Low-Valent Molybdenum PNP Pincer Complexes as Catalysts for the Semihydrogenation of Alkynes, Organometallics, 2022, 41, 1797–1805 CrossRef CAS
- A. G. Algarra, E. Guillamón, J. Andrés, M. J. Fernández-Trujillo, E. Pedrajas, J. Á. Pino-Chamorro, R. Llusar and M. G. Basallote, Cuboidal Mo3S4 Clusters as a Platform for Exploring Catalysis: A Three-Center Sulfur Mechanism for Alkyne Semihydrogenation, ACS Catal., 2018, 8, 7346–7350 CrossRef CAS
- M. V. Joannou, M. J. Bezdek and P. J. Chirik, Pyridine(diimine) Molybdenum-Catalyzed Hydrogenation of Arenes and Hindered Olefins: Insights into Precatalyst Activation and Deactivation Pathways, ACS Catal., 2018, 8, 5276–5285 CrossRef CAS
- T. Vielhaber, K. Faust and C. Topf, Group 6 Metal Carbonyl Complexes Supported by a Bidentate PN Ligand: Syntheses, Characterization, and Catalytic Hydrogenation Activity, Organometallics, 2020, 39, 4535–4543 CrossRef CAS
- E. Pedrajas, I. Sorribes, A. L. Gushchin, Y. A. Laricheva, K. Junge, M. Beller and R. Llusar, Chemoselective Hydrogenation of Nitroarenes Catalyzed by Molybdenum Sulphide Clusters, ChemCatChem, 2017, 9, 1128–1134 CrossRef CAS
- J. R. Cabrero-Antonino, E. Alberico, H.-J. Drexler, W. Baumann, K. Junge, H. Junge and M. Beller, Efficient Base-Free Hydrogenation of Amides to Alcohols and Amines Catalyzed by Well-Defined Pincer Imidazolyl–Ruthenium Complexes, ACS Catal., 2016, 6, 47–54 CrossRef CAS
- R. Adam, E. Alberico, W. Baumann, H. J. Drexler, R. Jackstell, H. Junge and M. Beller, NNP-Type Pincer Imidazolylphosphine Ruthenium Complexes: Efficient Base-Free Hydrogenation of Aromatic and Aliphatic Nitriles under Mild Conditions, Chem. – Eur. J., 2016, 22, 4991–5002 CrossRef CAS
- V. Papa, J. R. Cabrero-Antonino, E. Alberico, A. Spanneberg, K. Junge, H. Junge and M. Beller, Efficient and selective hydrogenation of amides to alcohols and amines using a well-defined manganese–PNN pincer complex, Chem. Sci., 2017, 8, 3576–3585 RSC
-
MassLynx, 4.1, Waters Corporation, Milford, MA, 2005 Search PubMed
- V. P. Fedin, M. N. Sokolov, Y. V. Mironov, B. A. Kolesov, S. V. Tkachev and V. Y. Fedorov, Triangular thiocomplexes of molybdenum: reactions with halogens, hydrohalogen acids and phosphines, Inorg. Chim. Acta, 1990, 167, 39–45 CrossRef CAS
-
CrysAlis, version 171.36.24, Agilent Technologies, Santa Clara, CA, 2012 Search PubMed
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
-
K. Brandenburg and H. Putz, DIAMOND, Crystal Impact GbR, Bonn, Germany, 2005 Search PubMed
- E. Pedrajas, I. Sorribes, K. Junge, M. Beller and R. Llusar, A Mild and Chemoselective Reduction of Nitro and Azo Compounds Catalyzed by a Well-Defined Mo3S4 Cluster Bearing Diamine Ligands, ChemCatChem, 2015, 7, 2675–2681 CrossRef CAS
- T. F. Beltrán, J. Á. Pino-Chamorro, M. J. Fernández-Trujillo, V. S. Safont, M. G. Basallote and R. Llusar, Synthesis and structure of trinuclear W3S4 Clusters bearing aminophosphine ligands and their reactivity toward halides and pseudohalides, Inorg. Chem., 2015, 54, 607–618 CrossRef
- A. L. Gushchin, Y. A. Laricheva, P. A. Abramov, A. V. Virovets, C. Vicent, M. N. Sokolov and R. Llusar, Homoleptic Molybdenum Cluster Sulfides Functionalized with Noninnocent Diimine Ligands: Synthesis, Structure, and Redox Behavior, Eur. J. Inorg. Chem., 2014, 4093–4100 CrossRef CAS
- J. A. Pino-Chamorro, Y. A. Laricheva, E. Guillamón, M. J. Fernández-Trujillo, E. Bustelo, A. L. Gushchin, N. Y. Shmelev, P. A. Abramov, M. N. Sokolov, R. Llusar, M. G. Basallote and A. G. Algarra, Cycloaddition of alkynes to diimino Mo3S4 cubane-type clusters: a combined experimental and theoretical approach, New J. Chem., 2016, 40, 7872–7880 RSC
-
A. G. Algarra and M. G. Basallote, in Advances in Inorganic Chemistry, ed. C. D. Hubbard and R. van Eldik, Academic Press, 2017, vol. 8, pp. 311–342 Search PubMed
- Y. Ide, M. Sasaki, M. Maeyama and T. Shibahara, Adducts of Acetylene and Dimethylacetylenedicarboxylate at Sulfurs in Sulfur-Bridged Incomplete Cubane-Type Tungsten Clusters, Inorg. Chem., 2004, 43, 602–612 CrossRef CAS
- E. Guillamón, M. Oliva, J. Andrés, R. Llusar, E. Pedrajas, V. S. Safont, A. G. Algarra and M. G. Basallote, Catalytic Hydrogenation
of Azobenzene in the Presence of a Cuboidal Mo3S4 Cluster via an Uncommon Sulfur-Based H2 Activation Mechanism, ACS Catal., 2021, 11, 608–614 CrossRef
- R. Kusy, M. Lindner, J. Wagner and K. Grela, Ligand-to-metal ratio controls stereoselectivity: Highly functional-group-tolerant, iridium-based, (E)-selective alkyne transfer semihydrogenation, Chem. Catal., 2022, 2, 1346–1361 CrossRef
-
K. Baldenius, L. von dem Bussche-Hünnefeld, E. Hilgemann, P. Hoppe and R. Stürmer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2021, pp. 1–21 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the synthesized compounds, UV-VIS and ESI-MS analyses. CCDC 2210047 [1]PF6·CH3CN and 2210042 [2]Cl·CH3OH. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02755k |
| This journal is © the Partner Organisations 2023 |