
Iodide/triiodide redox shuttle-based additives for high-performance perovskite solar cells by simultaneously passivating the cation and anion defects†
Huimin
Xiang
a,
Jingsheng
He
a,
Ran
Ran
a,
Wei
Zhou
a,
Wei
Wang
*a and
Zongping
Shao
 *ab
*ab
aState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing 210009, China. E-mail: wangwei@njtech.edu.cn; shaozp@njtech.edu.cn
bWA School of Mines: Minerals, Energy and Chemical Engineering, Curtin University, Perth, WA 6845, Australia
First published on 31st January 2023
Abstract
Halide perovskite solar cells (PSCs) have received remarkably increasing interests due to their facile fabrication procedures, use of cost-effective raw materials, and high power conversion efficiencies (PCEs) during the past 10 years. Nevertheless, the state-of-the-art organic–inorganic PSCs suffer from high defect concentration and inferior humid/thermal stability, significantly restricting the widespread applications of PSCs. More specifically, point defects including metallic lead (Pb0) and halide iodine (I0) are easily generated in Pb/I-based PSCs during fabrication processes and operational conditions due to the inferior interaction between the anions and cations in halide perovskites and promote detrimental carrier recombination and ion migration, leading to inferior PCEs and durability of the PSCs. Herein, to tackle the above-mentioned issues, iodide/triiodide (I−/I3−) redox shuttles as a new additive were introduced to simultaneously passivate the cation and anion defects of methylammonium lead iodide (MAPbI3)-based PSCs. In particular, I−/I3− redox shuttles play a vital role in regenerating the cation (Pb0) and anion (I0) defects through the redox cycles of Pb0/Pb2+ and I0/I−. Consequently, the cell with an optimized amount of I−/I3− additive generated a superior PCE of 20.4%, which was 12% higher than the pristine device (18.2%). Furthermore, the introduction of the I−/I3− additive remarkably improved the humid and thermal stability of MAPbI3-based PSCs. This work manifests the importance of the design of redox shuttle-based additives to boost the efficiency and durability of organic–inorganic PSCs.
1. Introduction
Developing photovoltaic cells is regarded as one of the most effective approaches to conquer the challenges associated with global energy demands and environmental pollution by efficiently utilizing the abundant, renewable, and clean solar energy.1 Among various photovoltaic cells (solar cells), organic–inorganic halide perovskite solar cells (PSCs) have received significant attention because of the ultrafast boost in the power conversion efficiencies (PCEs) from 3.8% to 25.7% in the past 13 years.2,3 More specifically, the high PCEs of organic–inorganic PSCs mainly originate from the unique optical/electronic properties of Pb/I-based organic–inorganic perovskites as light absorbers, such as their tunable band gaps, large light-absorption coefficient, long carrier-diffusion length, and excellent carrier mobility.4–8 In particular, Pb/I-based organic–inorganic perovskites with CH3NH3+ (MA+) cation, CH3(NH2)2+ (FA+) cation, and mixed cations at the A-site have recently exhibited high PCEs of 22–25%.9 However, the estimated lifetime of encapsulated PSCs is far away from commercial silicon-based solar cells, which is mainly attributed to the weak interaction between cations and anions in halide perovskites, leading to inferior stability under various conditions, such as humidity, oxygen, ultraviolet (UV) light, and high temperatures.10–15Several strategies such as encapsulation, adding a UV filter, and surface modification have been reported to alleviate the performance degradation of PSCs by temporarily excluding external environmental factors.16–21 However, some unavoidable factors such as light illumination, heat, and electric field under the operation conditions of PSCs are also critical elements in the degradation of Pb/I-based organic–inorganic perovskites induced by the breaking of the lattice bonding of I− and Pb2+ ions.22,23 It has been reported that I− and Pb2+ are easily transformed to I0 and Pb0 (point defects) under high-temperature or illumination conditions, which not only act as carrier-recombination centers but can also accelerate the decomposition of Pb/I-based halide perovskites, leading to inferior PCEs and device lifetime.24,25 Point defects are considered as deep-level defects in PSCs and can capture and annihilate photoinduced electrons, leading to nonradiative carrier recombination, while most of the energy generated in the carrier annihilation is released into the halide perovskite lattice in the form of phonons. Such nonradiative carrier recombination will reduce the steady-state density, diffusion length, and lifetime of the photoexcited carriers, leading to serious performance degradation and the hysteresis of PSCs.26,27 To tackle the crucial issues of point defects in organic–inorganic Pb/I-based PSCs, tremendous attempts have been made recently such as additive engineering and surface passivation.28–40 Additives with functional groups such as O-, N-, or S-donors introduced into the perovskite precursor solution can coordinate with unpaired Pb2+ and stabilize Pb atoms in the perovskite lattice.41 In particular, metal/organic halide salt additives such as NaX, CsX, KX, RbX, MAX, and FAX (where X = F, Cl, Br) have been added in to the precursor solution and/or the lattice of halide perovskites, showing two main functions: (1) cations with a suitable radius can occupy the A-site to stabilize the perovskite structure and adjust its band gaps and photoelectric properties and (2) anions anchor Pb2+ through hydrogen bonds to inhibit the movement and immigration of Pb2+ cations.42 On the other hand, the surface modification of perovskite films aims to passivate grain boundaries and/or surface defects, forming a protective layer and constructing well-matched energy levels for PSCs.43,44 It should be noted that most of the used additives and surface modifiers are sacrificial agents nowadays, which are not able to continuously passivate the newly formed defects during the operation of PSCs. Thus, the continuous and efficient elimination of newly generated defects is highly crucial for extending the lifetime of PSCs.
Several sustainably utilized agents have attracted significant attention for removing the regenerated defects, which would be beneficial for promoting the lifetimes of PSCs.42,45,46 For instance, Shi et al. introduced spiropyran molecules as a capping layer on the perovskite film, which incessantly passivated the renewable Pb vacancies in FAPbI3-based PSCs, leading to much improved PCEs and long-term stability under sunlight illumination.42 In another work, Wang et al. reported that the Eu3+/Eu2+ redox pair could continuously oxidize Pb0 and reduce I0 defects circularly in FAPbI3-based PSCs to achieve enhanced PCEs and operational durability.41 Similarly, the introduction of tributyl phosphine (TBUP)/tributyl phosphine oxide (TBPO) redox couples could efficiently reduce the concentrations of Pb0 and I0 defects in the fabrication processes and the operational conditions of FAPbI3-based PSCs, significantly improving the thermal stability.46 In addition, triiodide (I3−) species have also been reported to decrease the iodide vacancies in FAPbI3-based PSCs to achieve a high PCE of 22.1%.47 In this work, I−/I3− redox shuttles as a new additive were introduced into a perovskite precursor solution of MAPbI3-based PSCs, simultaneously passivating the cation and anion defects by regenerating the Pb0 and I0 defects through the redox cycles of Pb0/Pb2+ and I0/I−. As a result, after optimizing the amount of I−/I3− additive, the MAPbI3-based cell generated a much better PCE of 20.4% than that of the pristine cell (18.2%). Furthermore, the moisture and high-temperature stability of the MAPbI3-based PSCs were also significantly enhanced after introducing the I−/I3− redox couple-based additive. The current study can provide some valuable insights for the rational design of redox couple-based additives for achieving high-performance PSCs.
2. Experimental
2.1 Cell fabrication
The F−-doped SnO2 (FTO) glasses (2.2 mm-thick, ∼7 Ω sq−1) were sequentially cleaned by acetone, isopropanol, deionized water, and ethanol for half an hour, respectively, then dried by an inert gas and further treated in oxygen plasma for 5 min. A compact TiO2 (c-TiO2) layer was fabricated on the cleaned FTO substrate by spin-coating the titanium diisopropoxide bis(acetylacetonate)/n-butanol solution, which was further annealed at 500 °C for half an hour in air. A mesoporous TiO2 (m-TiO2) layer was subsequently fabricated on the c-TiO2 layer by spin-coating the TiO2 paste (30NR-D) dispersed in ethanol, which was subsequently annealed at 500 °C for half an hour in air. Next, 0.6 M I−/I3− solution was prepared by a mixed solid iodine (I2) and 1-butyl-3-methylimidazolium iodide in mixed dimethyl sulfoxide (DMSO)/N,N-dimethylformamide (DMF) solution. The MAPbI3 precursor solution was prepared by mixing MAI and PbI2 in DMF/DMSO (7/3, v/v) solution. Then, I−/I3− solutions at fixed volume ratios of 1%, 2%, and 3% were added in to the perovskite solution, and the corresponding perovskite films and PSCs were denoted as 1%-redox, 2%-redox, and 3%-redox, respectively. For the preparation of the hole-transporting layer (HTL), 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD), FK209, 4-tert-butylpyridine, and Li-TFSI were dissolved in chlorobenzene at fixed proportions.29 The fabrications of the perovskite layer and HTL were sequentially conducted in the glove box.29 Last, the Ag electrode with a thickness of 100 nm and an active area of 0.0625 cm2 was thermally evaporated on the HTL surface. Carbon-based HTL-free devices were fabricated by doctor blading under ambient conditions for the thermal stability test.2.2 Characterizations
A source meter (Keithley 2400) combined with a solar simulator (Zolix, SS150A) was used to obtain the photocurrent density–voltage (J–V) curves under the condition of AM 1.5G irradiation (100 mW cm−2). X-ray diffraction (XRD) patterns of the various films were tested by X-ray diffractometry (Cu Kα radiation, Rigaku Co., Ltd). The surface and cross-sectional microstructures (morphology and roughness) of various films were tested by scanning electron microscopy (SEM, Hitachi S-4800) and atomic force microscopy (AFM, SmartSPM, Horiba), respectively. The surface elemental states of various perovskite films were studied by X-ray photoelectron spectroscopy (XPS, Al Kα X-ray source, PHI5000 Versa Probe). The time-resolved photoluminescence (TRPL) and photoluminescence (PL) spectra were measured by fluorescence spectrophotometry (Edinburgh Instruments, FLS980, and PerkinElmer, FL6500, respectively). UV-Vis-NIR spectrophotometry (PerkinElmer, Lambda 750S) was used to obtain the UV-vis spectra of various films and cells. The external quantum efficiency (EQE) profiles were tested using a Solar Cell Scan 100 instrument (Zolix). The electrochemical impedance spectroscopy (EIS) profiles were measured on an electrochemical station (CHI760) under dark conditions with a frequency range of 106–0.1 Hz.3. Results and discussion
A traditional I−/I3− redox shuttle has been extensively utilized in dye-sensitized solar cells to efficiently regenerate the dye.48,49 It has been reported that I− anion-based additives, such as CsI, KI, and PbI2, can reduce the amounts of Pb0 and I0 defects.50–52 However, these sacrificial additives are not able to eliminate renewable (newly formed) point defects of PSCs under operational conditions. On this basis, I−/I3−-based redox shuttles were introduced here as additives in perovskite films to continuously passivate the Pb0 and I0 defects. To prepare the I−/I3−-based solution, I2 and 1-butyl-3-methylimidazolium iodide were dissolved in DMF/DMSO solution with the formation of I3− based on the equation: I2 + I− ⇌ I3−. Different volume ratios of I−/I3− redox shuttles (1%, 2%, and 3%) were added into perovskite precursor solution, with the typical digital photographs shown in Fig. S1.† Compared with the perovskite solution without adding the redox additive, the color of the precursor solution became deeper yellow after the addition of I−/I3− additives. A typical cross-sectional SEM image of the fabricated PSCs in this study with the configuration of FTO/c-TiO2/m-TiO2/perovskite/spiro-OMeTAD/Ag is shown in Fig. S2† and the thickness of the perovskite layer was around 400 nm. The photocurrent–voltage (J–V) curves of the best-efficiency devices fabricated without and with the redox additives are shown in Fig. 1a and the corresponding photovoltaic parameters are displayed in Table S1 and Fig. S3.† It was found that the PCEs of the MAPbI3-based cells first increased then decreased with the increasing amount of redox additives (Fig. 1b and c). The best-performing 2%-redox cell produced a superior PCE of 20.4% with an open-circuit voltage (Voc) of 1.11 V, a short-circuit current (Jsc) of 23.8 mA cm−2, and a fill factor (FF) of 0.770, while the pristine device generated a PCE of 18.2% with a Voc of 1.08 V, a Jsc of 22.8 mA cm−2, and an FF of 0.744. It should be noted that a slight decrease in the cell performance was observed in the 3%-redox cell, which may be attributed to the reaction between the excessive redox additives and mobile ions in perovskite films with the regeneration of I2.24Fig. 1d displays the EQE curves of the pristine and 2%-redox cells, whereby the 2%-redox device exhibited higher EQE values than those of the pristine cell over the whole visible light range (380–780 nm), indicating the improved light-absorption capability of the 2%-redox cell.53 In addition, the 2%-redox cell displayed a superior integrated Jsc value of 23.4 mA cm−2 compared to that of the pristine cell (21.8 mA cm−2), agreeing well with the J–V results.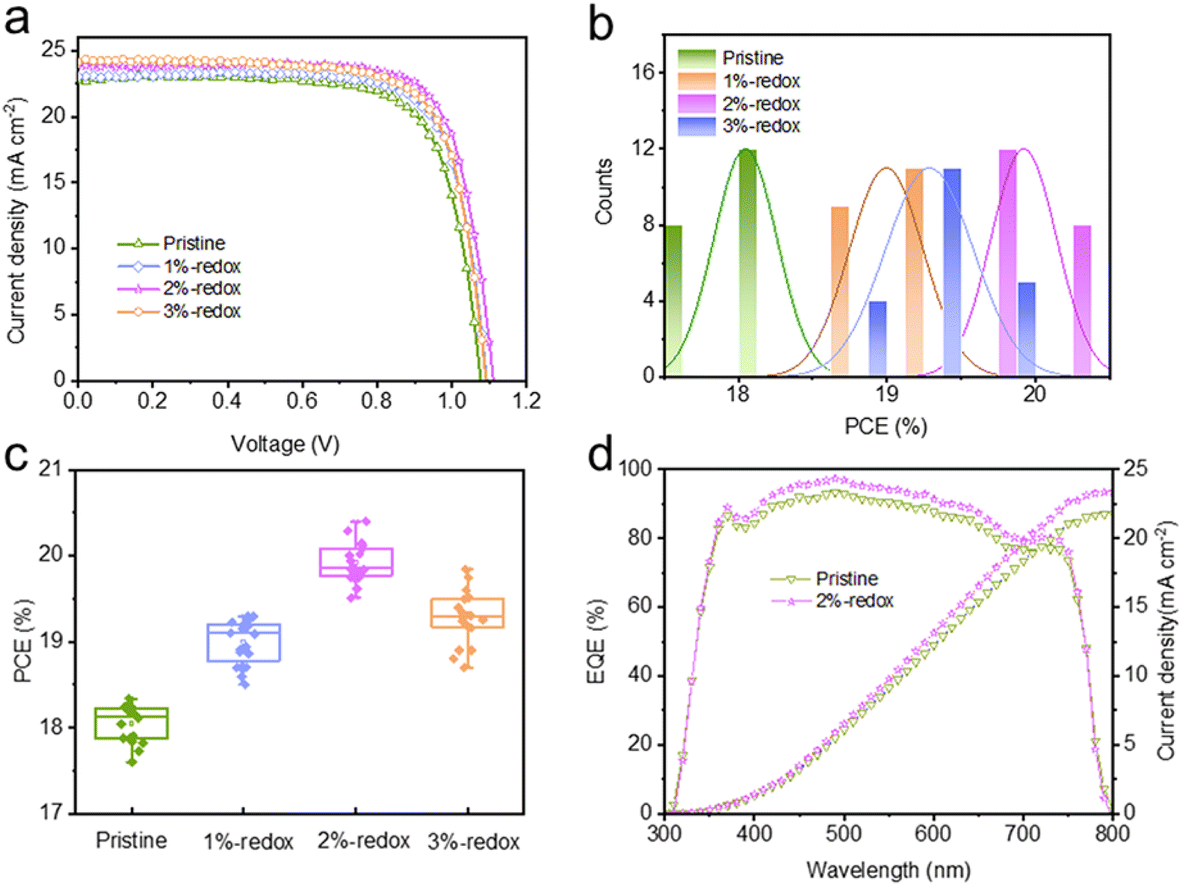 | ||
| Fig. 1 (a) J–V curves of the cells without and with redox additives at different volume ratios. PCE distributions of 20 devices without and with redox additives at different volume ratios: (b) histogram and (c) box chart. (d) EQE spectra and integrated current densities of the pristine and 2%-redox cells. | ||
The influence of the redox additives on the crystal structure and crystallinity of the perovskite films was investigated by XRD. As depicted in Fig. 2a, the major characteristic peaks of MAPbI3 perovskites at 14.29° and 28.38° were assigned to the (110) and (220) planes and exhibited no obvious shifts, indicating the introduction of redox additives showed no obvious influence on the crystal structure of the MAPbI3 perovskites.54,55 Furthermore, the redox additive-modified perovskite films displayed much higher intensities for the (110) and (220) peaks in the XRD patterns, suggesting that the redox additive enhanced the crystallinity of the perovskite film. We further compared the full width at half maximum (FWHM) values of the (110) diffraction peak of the pristine and various redox additive-modified perovskite films, and the results are shown in Fig. 2b. It was found that the introduction of redox additives effectively reduced the FWHM value of the (110) peak, suggesting the much improved crystallinity of the perovskite film induced by the redox additive. In addition, based on the calculated crystallite sizes of various perovskite films by Scherrer's equation in Fig. 2b,56,57 the crystallite sizes of the perovskite films first increased and then decreased with the increasing amount of redox additive. In particular, the 2%-redox film exhibited the smallest FWHM and largest crystallite sizes, demonstrating a much improved perovskite film quality induced by the proper concentration of redox additive. SEM and AFM were employed to investigate the impacts of the redox additives on the morphology and surface roughness of the perovskite films. As displayed in Fig. 2c–f, the top-view SEM images revealed that the redox additive with a proper concentration effectively increased the grain sizes, and reduced the amounts of pinholes, cracks, and grain boundaries of the pristine MAPbI3 film. As displayed in Fig. 2g–j and S4a–d,† the pristine, 1%-redox, 2%-redox, and 3%-redox perovskite films exhibited root-mean-square (RMS) roughness values of 51.5, 42.5, 36.7, and 52.6 nm, respectively. The remarkably reduced RMS value of the 2%-redox film indicated a compact film surface with less traps and pinholes, providing better contact between the perovskite film and HTL to promote the interfacial carrier transport.28,30
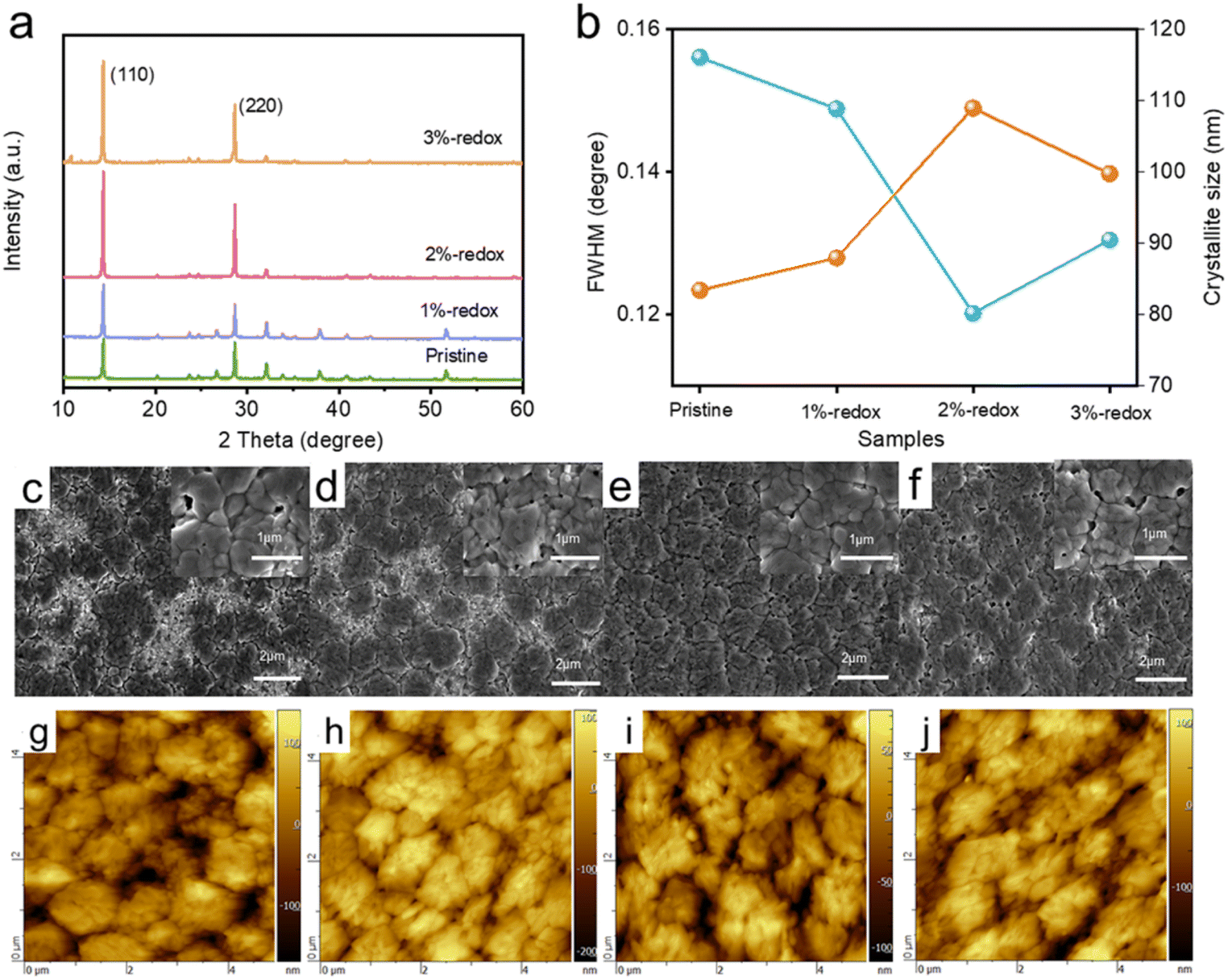 | ||
| Fig. 2 (a) XRD patterns of perovskite films without and with redox additives. (b) FWHM values of the (110) diffraction peaks and crystalline sizes of perovskite films without and with redox additives. Top-view SEM and AFM images of (c and g) pristine, (d and h) 1%-redox, (e and i) 2%-redox, and (f and j) 3%-redox films. | ||
XPS was used to explore the influences of the I−/I3−-based redox additives on the surface elemental states of various ions of perovskite films (Fig. S5†). As depicted in Fig. 3a, Pb2+ 4f5/2 and Pb2+ 4f7/2 were located at binding energies (BEs) of 142.8 and 137.9 eV, respectively, while the peaks at BEs of 141.1 and 136.2 eV were assigned to Pb0 species.58,59 It was obvious that a considerable amount of Pb0 defects was distributed on the surface of pristine perovskite film, while the Pb0 content of the 2%-redox film was significantly reduced. The intensity ratio of Pb0/(Pb0 + Pb2+) was further calculated to investigate the Pb0 content in the pristine and 2%-redox films. As shown in Fig. 3b, the 2%-redox film exhibited a much decreased Pb0/(Pb0 + Pb2+) ratio of 3.28% compared with the pristine perovskite film (8.00%) due to the effective oxidation of Pb0 defects by I3−. As for the I0 species, the exact I0/(I0 + I−) ratio was very difficult to obtain via peak fitting due to the volatility of I0 species during the annealing process. Thus, we adopted the I/Pb ratios and BE shifts of I 3d5/2 to reflect the concentration evolution of I0 species in the pristine and 2%-redox films. As depicted in Fig. 3c, the intensity ratios of I/Pb were 2.77 and 2.90 for the pristine and 2%-redox films, respectively, indicating the smaller amount of I0 species in the 2%-redox film.44,45 Moreover, the shifts of the I 3d5/2 XPS peaks to lower BEs in the 2%-redox film also suggested the reduced amount of I0 species.44 Accordingly, we proposed the oxidation–reduction mechanism of the I−/I3− redox shuttle in the MAPbI3 film based on the equations: Pb0 + I3− → PbI2 + I− and 2I0 + I− → I3−, as depicted in Fig. 3d. It could be concluded that the introduction of redox additives effectively reduced the amounts of Pb0 and I0 defects through continuous regeneration and redox reactions.
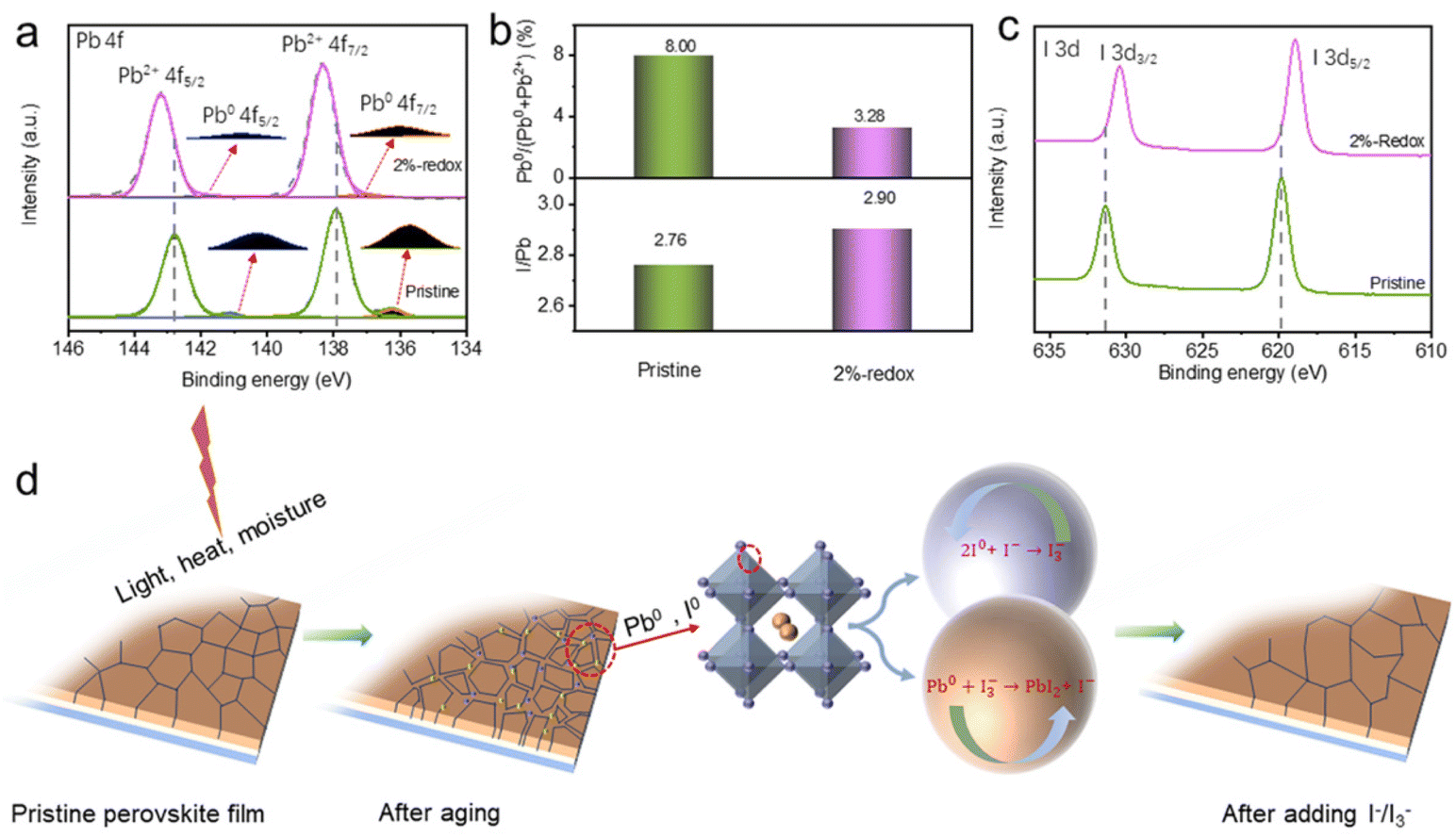 | ||
| Fig. 3 (a) Pb 4f XPS spectra, (b) Pb0/(Pb0 + Pb2+) and I/Pb ratios, (c) I 3d XPS spectra of the pristine and 2%-redox films. (d) Proposed mechanism of the I−/I3−-based redox additive on passivating the Pb0 and I0 defects via continuous regeneration and redox reactions. | ||
To examine the impacts of the I−/I3− redox shuttle-based additives on the light-absorption capacity and band gaps of MAPbI3 films, the UV-vis spectra of various MAPbI3 films were tested. As depicted in Fig. 4a, the addition of redox additives enhanced the light-absorption capacity of the perovskite films, while the optimized 2%-redox film exhibited the highest light-absorption intensity without obviously changing the band gap values (1.583 and 1.577 eV for the pristine and 2%-redox films, as shown in Fig. S6†), which was attributed to the much enhanced quality and morphology of the 2%-redox film, as evidenced by the SEM results. Based on the steady-state PL spectra of various MAPbI3 films deposited on an insulating substrate in Fig. 4b, compared with the PL spectra of the pristine MAPbI3 film, the 1%-redox film showed a blue-shift in the PL peak, while the 2%-redox film exhibited a red-shift in the PL peak, which may have contributed to the increased Jsc value of the corresponding 2%-redox device.60,61 The higher PL peak intensity of the 2%-redox film indicated that the introduction of redox additives effectively suppressed the amount of nonradiative recombination centers and slightly extended the light-absorption range of the perovskite films, which would be beneficial for the performance enhancement of the corresponding PSCs.62 In addition, the TRPL technique was conducted to investigate the carrier-transport capability of the perovskite films after introducing the redox additive. As shown in Fig. 4c, the average carrier lifetime values (τ) of the pristine and 2%-redox films obtained from biexponential decay fitting (Table S2†) were 49.0 and 68.0 ns, respectively.63 This indicated that the redox additive remarkably inhibited the nonradiative carrier recombination by eliminating the deep-level defects, such as Pb0 and I0 species, which also contributed to the higher Voc value of the 2%-redox cell.64,65 In order to explore the interfacial charge-transfer dynamics, EIS was conducted at 0.9 V under dark conditions. As depicted in Fig. 4d, the fitted Nyquist plots displayed a low-frequency arc, which was inversely proportional to the interfacial carrier-recombination degree.66 As could be seen, the 2%-redox cell displayed a higher recombination resistance (Rrec) value of 7776 Ω than those of the pristine (2031 Ω), 1%-redox (3694 Ω), and 3%-redox (6704 Ω) cells, suggesting that the interfacial carrier-recombination process was remarkably inhibited by introducing a proper amount of the redox additive, which was beneficial for the photovoltaic performance.
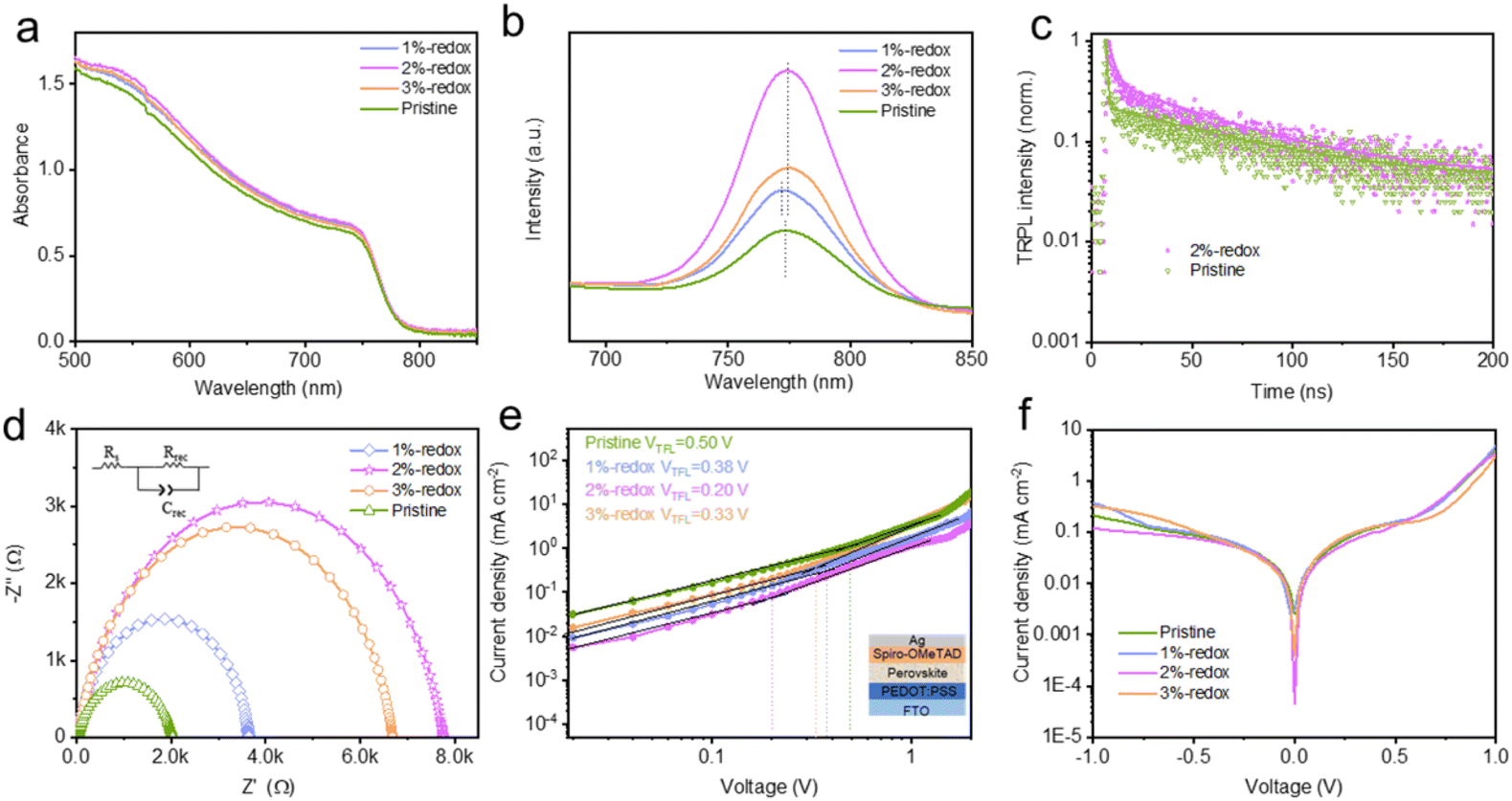 | ||
| Fig. 4 (a) UV-vis absorption spectra of various perovskite films with and without redox additives. (b) PL and (c) TRPL spectra of different perovskite films with and without redox additives deposited on non-conductive substrates. (d) EIS spectra of devices with and without redox additives at 0.9 V in the dark. (e) Dark J–V curves of the hole-only devices with and without redox additives. (f) Dark J–V curves of the pristine and redox additive-modified cells. | ||
The hole-defect densities (Nd) of the pristine and various redox additive-modified films were calculated by using the space-charge-limited current mode in hole-only PSCs with a configuration of FTO/PEDOT:PSS/perovskite/spiro-OMeTAD/Ag under dark conditions. The Nd values could be obtained from the equation: 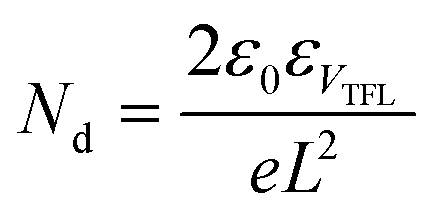 , where ε0 and ε are the vacuum dielectric constant and relative permittivity, VTFL is the trap-filled limit voltage, e is the elementary charge of electron, and L is the thickness of the perovskite film (∼400 nm).67
, where ε0 and ε are the vacuum dielectric constant and relative permittivity, VTFL is the trap-filled limit voltage, e is the elementary charge of electron, and L is the thickness of the perovskite film (∼400 nm).67
As depicted from Fig. 4e, the VTFL values of the pristine, 1%-redox, 2%-redox, and 3%-redox cells were 0.50, 0.38, 0.20, and 0.33 V, corresponding to Nd values of 6.36, 4.84, 2.54, and 4.20 × 1015 cm−3, respectively, further verifying the vital role of the redox additives on reducing the defects of the perovskite film. Based on the dark J–V curves of various PSCs in Fig. 4f, the 2%-redox cell exhibited the lowest dark current among the four cells, indicating a much reduced current loss due to the improved interfacial charge transfer induced by the suppressed defect concentration.30
As mentioned above, the Pb0 and I0 defects derived from the perovskite structure acted as nonradiative recombination centers to promote the detrimental ion migration, leading to severe performance degradation under humid and high-temperature conditions.24 The moisture and thermal stability of the pristine and 2%-redox cells are displayed in Fig. 5a and b, respectively. As can be seen in Fig. 5a, the 2%-redox cell exhibited a superior PCE retention ratio of ∼78% after 864 h storage in ambient conditions at 25–30 °C and a relative humidity (RH) of ∼25%, which was much better than that of the pristine cell (PCE retention ratio of ∼25%) under the same conditions. HTL-free carbon-based PSCs were utilized to study the impacts of the redox additive on the thermal stability due to the unstable organic HTL. The thermal stability of the HTL-free carbon-based devices is depicted in Fig. 5b, where it can be seen that the 2%-redox cell retained 46% of the primary PCE after aging at 85 °C in humid air (RH: 20%) for 720 h, while the PCE of the pristine cell declined to 24% under the same conditions, which was mainly attributed to the rapidly degraded FF. As shown in Fig. S7 and Table S3,† the HTL-free 2%-redox cell delivered a higher PCE of 10.3% than that of the unmodified cell (9.6%), further indicating the superiority of the redox additive in boosting the cell performance. The above results suggested that the redox additives played a crucial role in improving the moisture and high-temperature durability of MAPbI3-based PSCs.
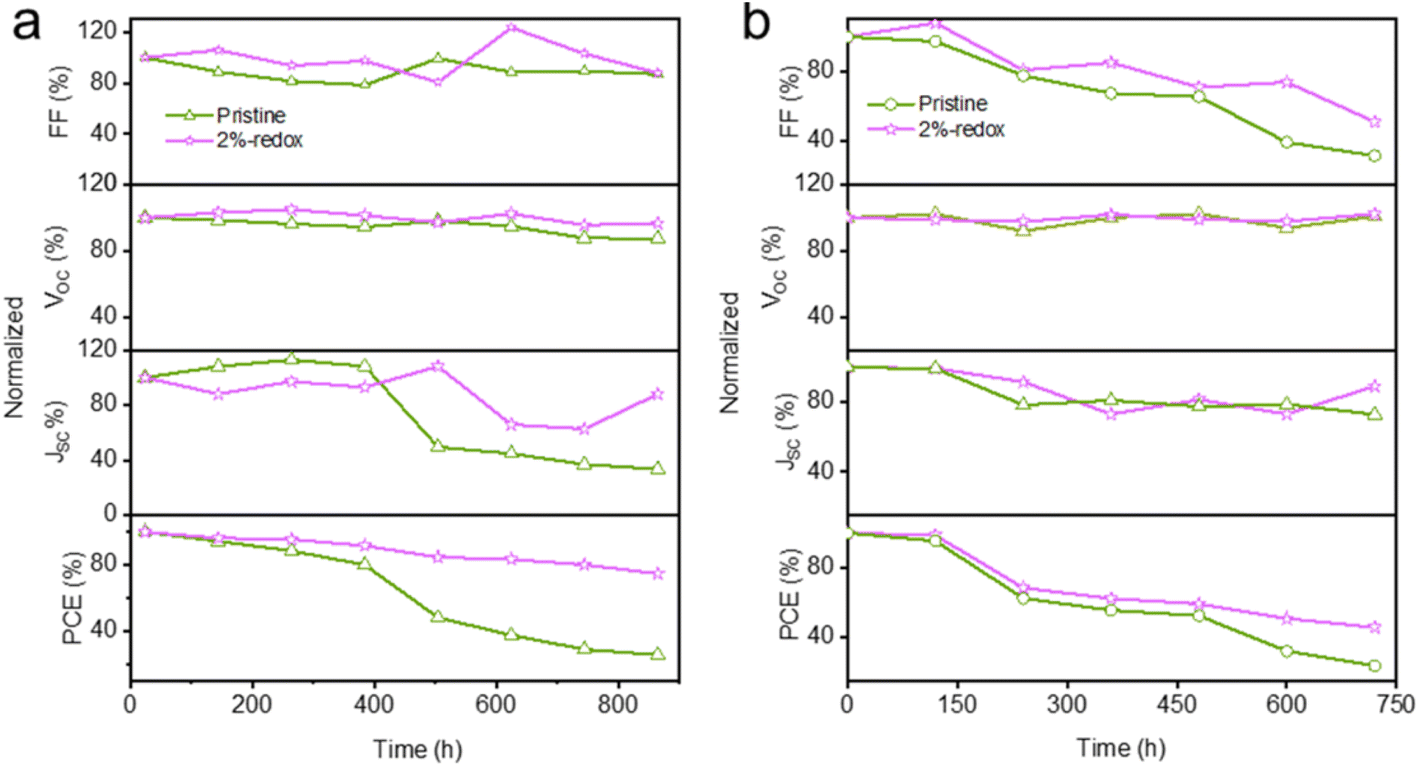 | ||
| Fig. 5 Normalized photovoltaic parameters of the pristine and 2%-redox cells stored in humid air at (a) 25–30 °C (with spiro-OMeTAD HTL) and (b) 85 °C (HTL-free). | ||
4. Conclusions
In summary, iodide/triiodide redox shuttles were employed as effective additives to simultaneously passivate the deep-level cation and anion defects (e.g., Pb0 and I0) by constructing redox cycles of Pb0/Pb2+ and I0/I− for the continuous regeneration of Pb0 and I0 defects, which effectively reduced the nonradiative carrier recombination and facilitated the interfacial carrier transport. In addition, the introduction of redox additives also enhanced the perovskite film quality by enlarging the grain sizes, increasing the crystallinity, reducing the amount of pinholes, and improving the light-absorption capability. As a result, the device with the optimized amount of redox additive generated a higher PCE of 20.4% than that of the pristine cell (18.2%) due to the remarkable enhancement in Voc and FF values induced by the effective passivation of the deep-level Pb0 and I0 defects. Moreover, the redox additive-modified cell also showed much superior moisture/thermal durability to that of the unmodified cell and stability due to the improved perovskite film quality and suppressed defect concentration. This study provides a new and effective redox shuttle-based additive to eliminate the deep-level defects in perovskite films, simultaneously improving the PCEs and durability of MAPbI3-based PSCs, which can promote the widespread application of perovskite photovoltaics.Author contributions
Huimin Xiang: investigation, formal analysis, visualization, writing – original draft, data curation; Jingsheng He: formal analysis; Ran Ran: writing – review & editing; Wei Zhou: funding acquisition, writing – review & editing; Wei Wang: supervision, conceptualization, writing – review & editing, funding acquisition, validation; Zongping Shao: supervision, conceptualization, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by National Natural Science Foundation of China (no. 22279057). The Postgraduate Research & Practice Innovation Program of Jiangsu Province (no. KYCX22_1352).References
- W. Wang, M. O. Tade and Z. Shao, Chem. Soc. Rev., 2015, 44, 5371–5408 RSC
.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
- NREL, Best Research Cell Efficiencies, https://www.nrel.gov/pv/cell-efficiency.html, accessed November 2022.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano and J.-P. Correa-Baena, Sci. Adv., 2016, 2, e1501170 CrossRef PubMed
- T. Wang, B. Daiber, J. M. Frost, S. A. Mann, E. C. Garnett, A. Walsh and B. Ehrler, Energy Environ. Sci., 2017, 10, 509–515 RSC
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed
- D. J. Keeble, J. Wiktor, S. K. Pathak, L. J. Phillips, M. Dickmann, K. Durose, H. J. Snaith and W. Egger, Nat. Commun., 2021, 12, 5566 CrossRef CAS PubMed
- D. C. Jordan and S. R. Kurtz, Prog. Photovolt.: Res. Appl., 2013, 21, 12–29 CrossRef
- F. Qi, K. Jiang, F. Lin, Z. Wu, H. Zhang, W. Gao, Y. Li, Z. Cai, H. Y. Woo, Z. Zhu and A. K. Y. Jen, ACS Energy Lett., 2021, 6, 9–15 CrossRef CAS
- J. S. Yun, J. Kim, T. Young, R. J. Patterson, D. Kim, J. Seidel, S. Lim, M. A. Green, S. Huang and A. Ho-Baillie, Adv. Funct. Mater., 2018, 28, 1705363 CrossRef
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed
- W. Li, W. Zhang, S. Van Reenen, R. J. Sutton, J. Fan, A. A. Haghighirad, M. B. Johnston, L. Wang and H. J. Snaith, Energy Environ. Sci., 2016, 9, 490–498 RSC
- Y. Bai, X. Meng and S. Yang, Adv. Energy Mater., 2018, 8, 1701883 CrossRef
- Z. Liu, B. Sun, T. Shi, Z. Tang and G. Liao, J. Mater. Chem. A, 2016, 4, 10700–10709 RSC
- L. Shi, M. P. Bucknall, T. L. Young, M. Zhang, L. Hu, J. Bing, D. S. Lee, J. Kim, T. Wu, N. Takamure, D. R. McKenzie, S. Huang, M. A. Green and A. W. Y. Ho-Baillie, Science, 2020, 368, eaba2412 CrossRef CAS PubMed
- C. Kan, Z. Tang, Y. Yao, P. Hang, B. Li, Y. Wang, X. Sun, M. Lei, D. Yang and X. Yu, ACS Energy Lett., 2021, 6, 3864–3872 CrossRef CAS
- W. Chen, J. Zhang, G. Xu, R. Xue, Y. Li, Y. Zhou, J. Hou and Y. Li, Adv. Mater., 2018, 30, 1800855 CrossRef PubMed
- M. Karimipour, S. Khazraei, B. J. Kim, G. Boschloo and E. M. J. Johansson, Nano Energy, 2022, 95, 107044 CrossRef CAS
- H. Zhang, Z. Shi, L. Hu, Y.-Y. Tang, Z. Qin, W.-Q. Liao, Z. S. Wang, J. Qin, X. Li, H. Wang, M. Gusain, F. Liu, Y. Pan, M. Xu, J. Wang, R. Liu, C. Zhang, R.-G. Xiong, W. E. I. Sha and Y. Zhan, Adv. Funct. Mater., 2021, 31, 2100205 CrossRef CAS
- R. K. Gunasekaran, D. Chinnadurai, A. R. Selvaraj, R. Rajendiran, K. Senthil and K. Prabakar, ChemPhysChem, 2018, 19, 1507–1513 CrossRef CAS PubMed
- G. Tumen-Ulzii, C. Qin, D. Klotz, M. R. Leyden, P. Wang, M. Auffray, T. Fujihara, T. Matsushima, J.-W. Lee, S.-J. Lee, Y. Yang and C. Adachi, Adv. Mater., 2020, 32, 1905035 CrossRef CAS PubMed
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Nat. Energy, 2016, 2, 16195 CrossRef
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung, S. Yoo, S. H. Im, R. H. Friend and T.-W. Lee, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed
- P. Liu, W. Wang, S. Liu, H. Yang and Z. Shao, Adv. Energy Mater., 2019, 9, 1803017 CrossRef
- S. Yuan, J. Wang, K. Yang, P. Wang, X. Zhang, Y. Zhan and L. Zheng, Nanoscale, 2018, 10, 18909–18914 RSC
- P. Liu, Y. Chen, H. Xiang, X. Yang, W. Wang, R. Ran, W. Zhou and Z. Shao, Small, 2021, 17, 2102186 CrossRef CAS PubMed
- Y. Chen, X. Yang, P. Liu, W. Wang, R. Ran, W. Zhou and Z. Shao, Sol. RRL, 2021, 5, 2000621 CrossRef CAS
- X. Yang, A. Xie, H. Xiang, W. Wang, R. Ran, W. Zhou and Z. Shao, Appl. Phys. Rev., 2021, 8, 041402 CAS
- Q. Wang, W. Zhang, Z. Zhang, S. Liu, J. Wu, Y. Guan, A. Mei, Y. Rong, Y. Hu and H. Han, Adv. Energy Mater., 2020, 10, 1903092 CrossRef CAS
- Y. Chen, N. Li, L. Wang, L. Li, Z. Xu, H. Jiao, P. Liu, C. Zhu, H. Zai, M. Sun, W. Zou, S. Zhang, G. Xing, X. Liu, J. Wang, D. Li, B. Huang, Q. Chen and H. Zhou, Nat. Commun., 2019, 10, 1112 CrossRef PubMed
- X. Li, X. Wu, B. Li, Z. Cen, Y. Shang, W. Lian, R. Cao, L. Jia, Z. Li, D. Gao, X. Jiang, T. Chen, Y. Lu, Z. Zhu and S. Yang, Energy Environ. Sci., 2022, 25, 4813–4822 RSC
- H. Xiang, P. Liu, W. Wang, R. Ran, W. Zhou and Z. Shao, J. Mater. Sci. Technol., 2022, 113, 138–146 CrossRef
- T. Wang, Y. Li, Q. Cao, J. Yang, B. Yang, X. Pu, Y. Zhang, J. Zhao, Y. Zhang, H. Chen, A. Hagfeldt and X. Li, Energy Environ. Sci., 2022, 15, 4414–4424 RSC
- B. Wang, H. Li, Q. Dai, M. Zhang, Z. Zou, J.-L. Brédas and Z. Lin, Angew. Chem., Int. Ed., 2021, 60, 17664–17670 CrossRef CAS PubMed
- J. Zhang, H. Luo, W. Xie, X. Lin, X. Hou, J. Zhou, S. Huang, W. Ou-Yang, Z. Sun and X. Chen, Nanoscale, 2018, 10, 5617–5625 RSC
- P. Zhang, Y. Chen, S. Wu, X. Li, M. Liu and S. Li, Nanoscale, 2022, 14, 35–41 RSC
- X. Yang, Y. Ni, Y. Zhang, Y. Wang, W. Yang, D. Luo, Y. Tu, Q. Gong, H. Yu and R. Zhu, ACS Energy Lett., 2021, 6, 2404–2412 CrossRef CAS
- D. Huang, H. Xiang, R. Ran, W. Wang, W. Zhou and Z. Shao, Nanomaterials, 2022, 12, 2592 CrossRef CAS PubMed
- L. Fu, H. Li, L. Wang, R. Yin, B. Li and L. Yin, Energy Environ. Sci., 2020, 13, 4017–4056 RSC
- Y.-R. Shi, K.-L. Wang, Y.-H. Lou, G.-L. Liu, C.-H. Chen, J. Chen, L. Zhang and Z.-K. Wang, Adv. Mater., 2022, 34, 2205338 CrossRef CAS PubMed
- H. Xiang, P. Liu, W. Wang, R. Ran, W. Zhou and Z. Shao, Chem. Eng. J., 2020, 420, 127599 CrossRef
- X. Yang, W. Wang, R. Ran, W. Zhou and Z. Shao, Energy Fuels, 2020, 34, 10513–10528 CrossRef CAS
- L. Wang, H. Zhou, J. Hu, B. Huang, M. Sun, B. Dong, G. Zheng, Y. Huang, Y. Chen, L. Li, Z. Xu, N. Li, Z. Liu, Q. Chen, L.-D. Sun and C.-H. Yan, Science, 2019, 363, 265–270 CrossRef CAS PubMed
- Z. Wu, M. Zhang, Y. Liu, Y. Dou, Y. Kong, L. Gao, W. Han, G. Liang, X. L. Zhang, F. Huang, Y.-B. Cheng and J. Zhong, J. Energy Chem., 2021, 54, 23–29 CrossRef CAS
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed
- A. Abbotto and N. Manfredi, Dalton Trans., 2011, 40, 12421–12438 RSC
- Z. She, Y. Cheng, L. Zhang, X. Li, D. Wu, Q. Guo, J. Lan, R. Wang and J. You, ACS Appl. Mater. Interfaces, 2015, 7, 27831–27837 CrossRef CAS PubMed
- A. Kogo and M. Chikamatsu, Nanoscale, 2020, 12, 21605–21609 RSC
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 7232–7238 CrossRef CAS PubMed
- H. Wang, Z. Wang, Z. Yang, Y. Xu, Y. Ding, L. Tan, C. Yi, Z. Zhang, K. Meng, G. Chen, Y. Zhao, Y. Luo, X. Zhang, A. Hagfeldt and J. Luo, Adv. Mater., 2020, 32, 2000865 CrossRef CAS PubMed
- H. Xiang, P. Liu, R. Ran, W. Wang, W. Zhou and Z. Shao, Renewable Sustainable Energy Rev., 2022, 166, 112614 CrossRef CAS
- A. Shit and A. K. Nandi, Phys. Chem. Chem. Phys., 2016, 18, 10182–10190 RSC
- W. Zhou, D. Li, Z. Xiao, Z. Wen, M. Zhang, W. Hu, X. Wu, M. Wang, W.-H. Zhang, Y. Lu, S. Yang and S. Yang, Adv. Funct. Mater., 2019, 29, 1901026 CrossRef
- W. Xu, G. Lei, C. Tao, J. Zhang, X. Liu, X. Xu, W.-Y. Lai, F. Gao and W. Huang, Adv. Funct. Mater., 2018, 28, 1802320 CrossRef
- H. Kim, J. W. Lee, G. R. Han, S. K. Kim and J. H. Oh, Adv. Funct. Mater., 2021, 31, 2008801 CrossRef CAS
- B. Conings, L. Baeten, C. De Dobbelaere, J. D'Haen, J. Manca and H. G. Boyen, Adv. Mater., 2014, 26, 2041–2046 CrossRef CAS PubMed
- R. Lindblad, D. Bi, B.-w. Park, J. Oscarsson, M. Gorgoi, H. Siegbahn, M. Odelius, E. M. Johansson and H. k. Rensmo, J. Phys. Chem. Lett., 2014, 5, 648–653 CrossRef CAS PubMed
- J. N. Fru, N. Nombona and M. Diale, Vacuum, 2020, 182, 109727 CrossRef CAS
- A. Y. Alsalloum, B. Turedi, K. Almasabi, X. Zheng, R. Naphade, S. D. Stranks, O. F. Mohammed and O. M. Bakr, Energy Environ. Sci., 2021, 14, 2263–2268 RSC
- Z. Liu, F. Cao, M. Wang, M. Wang and L. Li, Angew. Chem., Int. Ed., 2020, 59, 4161–4167 CrossRef CAS PubMed
- Y. Yao, M. Wang, Q. Cai and D. Wei, Nanoscale, 2022, 14, 7203–7210 RSC
- S. Yang, H. Zhao, Y. Han, C. Duan, Z. Liu and S. Liu, Small, 2019, 15, 1904387 CrossRef CAS PubMed
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS
- X. Yang, H. Xiang, J. Huang, C. Zhou, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Colloid Interface Sci., 2022, 628, 476–485 CrossRef CAS PubMed
- X. Yang, Y. Chen, P. Liu, H. Xiang, W. Wang, R. Ran, W. Zhou and Z. Shao, Adv. Funct. Mater., 2020, 30, 2001557 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06710b |
| This journal is © The Royal Society of Chemistry 2023 |