
The self-assembly of a pair of low-symmetry tetracarboxylic acid molecules and their co-assembly with bridging molecules at the liquid–solid interface†
Siqi
Zhang
ab,
Jianqiao
Li
b,
Linlin
Gan
b,
Lin
Ma
b,
Wei
Ma
d,
Min
Zhang
*a,
Faliang
Cheng
*a,
Ke
Deng
 *b and
Qingdao
Zeng
*bc
*b and
Qingdao
Zeng
*bc
aGuangdong Engineering and Technology Research Center for Advanced Nanomaterials, School of Environment and Civil Engineering, Dongguan University of Technology, Dongguan 523808, China. E-mail: mindear@dgut.edu.cn; chengfl@dgut.edu.cn
bCAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology (NCNST), Beijing 100190, China. E-mail: kdeng@nanoctr.cn; zenqd@nanoctr.cn
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
dState Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an 710049, China
First published on 30th January 2023
Abstract
The supramolecular self-assembly behavior of a pair of low-symmetry tetracarboxylic acid molecules (H4OBDB and H4ADDI) and their co-assembly behavior with TMA as a bridging molecule were studied at the liquid–solid interface. Scanning tunneling microscope (STM) observations revealed that H4OBDB and H4ADDI molecules both tend to form O-shaped dimers but end up forming different types of self-assembly structures. We also investigated the construction of two-component co-assembly structures by mixing H4OBDB or H4ADDI molecules with bridging molecules such as TMA. The two formed co-assembly structures are similar. Based on the analysis of the STM results and the density functional theory (DFT) calculations, the formation mechanism of the assembled structures was revealed.
Introduction
Supramolecular self-assembly is an impeccable system in the natural world for spontaneously forming well-organized materials, such as cellular membranes.1,2 Over the past few decades, many researchers have learned from nature and devoted themselves to the design and construction of complex functional supramolecular structures using simple molecular building blocks.3–7 The systematic study of molecular self-assembly is beneficial for simulating its properties in the designation of functional nanomaterials and devices. The key to obtaining a deep understanding of self-assembly is molecular-level research of the specific assembly system. Scanning tunneling microscopy (STM) is a powerful tool to characterize two-dimensional supramolecular assemblies with submolecular resolution.8–10 The liquid–solid interface between the molecular solution and the solid surface of highly oriented pyrolitic graphite (HOPG) provides an ideal planar solid substrate for supramolecular self-assembly and an environment for studying the dynamic process of molecular self-assembly under the synergistic effects of various driving forces in the system. Extensive STM investigations have been conducted at the liquid–solid interface in recent years.11–19Among the intermolecular noncovalent driving forces of molecular self-assembly, hydrogen bonding is the strongest and therefore one of the most important methodologies for preparing supramolecular structures. In addition, the selectivity and directivity of hydrogen bonds are noteworthy aspects concerning molecular self-assembly, and the self-assembly structures driven by the hydrogen bonds are more predictable and controllable.20–26 In particular, cyclic dimeric O–H⋯O hydrogen bonds between two carboxyl groups have been widely used in the construction of particular 2D supramolecular architectures at the liquid–solid interface.27–31 For instance, with the dimeric hydrogen bonds between carboxyl groups as the main driving force of self-assembly, trimesic acid (TMA) molecules can form a typical honeycomb network structure;32–34 tetracarboxylic acid molecules such as NN4A and H4ETTC can form a Kagomé or a quadrilateral network structure, respectively;35,36 flexible carboxylic porphyrin derivative IPETPP containing eight carboxyl groups can form both Kagomé and quadrilateral network structures.37
The position of the carboxyl groups at the molecule core determines whether long-range ordered structures can be observed.38,39 A bonding angle of 180° between carboxylic groups of adjacent molecules is ideal, which enables the molecules to be arranged into a relatively strong and highly predictable network structure.40 In general, structures stabilized by dimeric hydrogen bonds between carboxyl groups, which have well-ordered symmetrical geometries, were constructed using molecular building blocks that have a symmetrical distribution of carboxyl groups. In this case, C3-symmetric aromatic tricarboxylic/hexacarboxylic acids and D2h-symmetric aromatic tetracarboxylic acids are common building block choices. To enrich the topology of the assembled structure, it is of considerable interest to impose structural variations on a well-studied molecule type such as C3-symmetric tricarboxylic acid. For example, with a larger core size than TMA, C3-symmetric 1,3,5-benzenetribenzoic acid (BTB) tends to form a densely packed structure with a rectangular cavity rather than a hexagonal honeycomb structure that comprises weaker C–H⋯O bonds but increases molecular packing.41 When the symmetry of the building block is reduced, the assembled structure becomes less predictable, yet it opens up an opportunity to access a variety of possible structures. Although systematic studies are lacking, some self-assembly studies of low-symmetry building blocks have been carried out in recent years.42–44 Morrison et al. reduced the symmetry of BTB by changing the sites of carboxyl groups and the assembly structure was significantly diversified.45 Wang et al. observed two kinds of self-assembly of C2v-symmetric tetracarboxylic acid (TPTA).46 In our previous study, a low-symmetry pentacarboxylic acid (H5BHB) that participated in co-assembly with pyridine molecules using its C2-symmetric dimer as the basic building block was reported.47
In this article, we report the self-assembly of a pair of low-symmetry tetracarboxylic acid molecules (H4OBDB and H4ADDI) at the heptanoic acid solvent solution–HOPG liquid–solid interface. As shown in Scheme 1, H4OBDB and H4ADDI are similar in structure, both having a semicircular bent skeleton and an isophthalic acid group at two ends of the skeleton. The difference is that H4OBDB incorporates a diphenyl ether group, whereas H4ADDI contains an acridine group. Due to the rotation of C–O bonds, H4OBDB will be more flexible than the rigid H4ADDI molecule. Their synthesis procedures have been reported in the previous literature.48,49 Comparing the similarities and differences of their self-assembly behaviors will provide an understanding of how the different assembly factors are related to each other, with the key point being developing a diverse topology of assembly structures and greater sophistication in predicting self-assembly architectures using low-symmetry building blocks. We also investigated the construction of two-component co-assembly structures by mixing H4OBDB or H4ADDI molecules with bridging molecules such as TMA. Through STM observations and density functional theory (DFT) calculations, the formation mechanism of the assembled structures was revealed.
 | ||
| Scheme 1 Chemical structures of 4′,4′′′-oxybis[1,1′-biphenyl]-3,5-dicarboxylic acid (H4OBDB), 5,5′-(acridine-2,7-diyl)diisophthalic acid (H4ADDI), and trimesic acid (TMA). | ||
Results and discussion
Self-assembly of H4OBDB at the liquid–solid interface
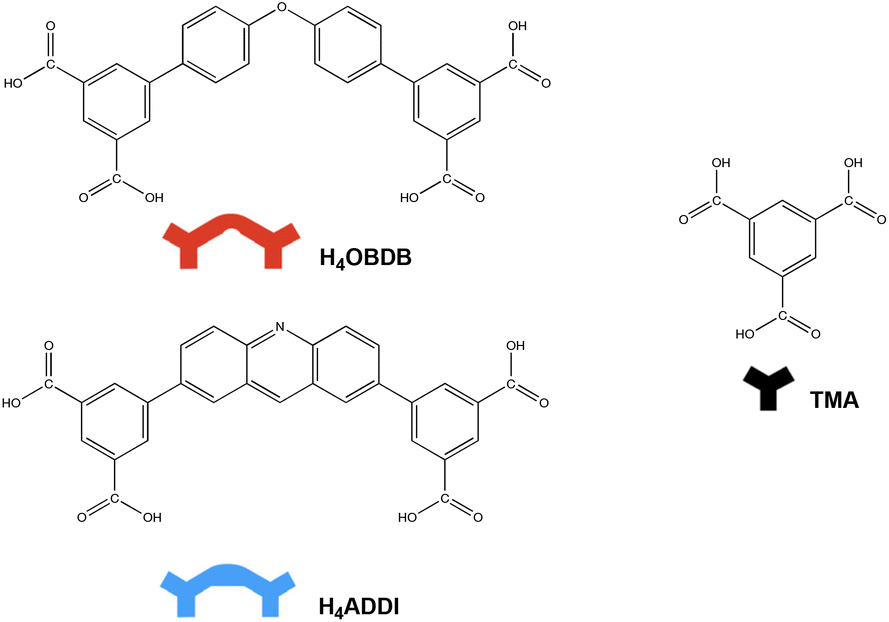
4′,4′′′-Oxybis[1,1′-biphenyl]-3,5-dicarboxylic acid (H4OBDB) is a semi-rigid bent-shaped molecule with a diphenyl ether group in the middle of the skeleton and an isophthalic acid group at each terminal. After depositing a droplet of H4OBDB heptanoic acid solvent solution on a freshly cleaved HOPG surface, a dense and well-ordered monolayer was formed immediately at the liquid–solid interface at room temperature. It can be seen from the STM image in Fig. 1a and b that the H4OBDB molecule assembled into two different types of patterns, which were marked by stru1 and stru2, respectively. In type stru1, the H4OBDB self-assembly structure is densely packed and exhibits a linear motif (denoted as H4OBDB_stru1), while in type stru2, the H4OBDB self-assembly structure is loosely arranged and exhibits a hexagonal symmetric motif (denoted as H4OBDB_stru2). The small-scale STM images of H4OBDB_stru1 and H4OBDB_stru2 with distinct molecular resolutions are shown in Fig. 1c and d, respectively. A careful analysis of these two assembled structures provides us with a preliminary understanding of the self-assembly behavior of the H4OBDB molecule. In Fig. 1c, the X-shaped highlighted contour is consistent with the skeleton of two neighboring H4OBDB molecules arranged “back to back”, whereas in Fig. 1d, the O-shaped highlighted contour agrees with the skeleton of two neighboring H4OBDB molecules arranged “face to face”. From the location of the hydrogen bond donor and acceptor moieties on its semicircular skeleton, it is not difficult to infer that two H4OBDB molecules can form double cyclic dimeric O–H⋯O hydrogen bonds between their carboxyl groups, thus forming an O-shaped dimer. At the same time, the central oxygen atom of the H4OBDB molecule can also form an O⋯H–C hydrogen bond with the hydrogen atom on its skeleton, allowing two H4OBDB molecules to appear as an X-shaped dimeric contour. Fig. 1e and f show the corresponding molecular models optimized by the DFT method based on the STM observation of H4OBDB_stru1 and H4OBDB_stru2, respectively. The molecular model is consistent with the STM images in terms of adsorbate geometry and unit cell parameters. It can clearly be seen that, in H4OBDB_stru1, H4OBDB molecules can form O-shaped dimers and are simultaneously attached by hydrogen bonds between diphenyl ether groups; in this case, the O-shaped dimers are stacked parallel to each other in columns. Three of the four carboxyl groups of the H4OBDB molecule bond with carboxyl groups of adjacent H4OBDB molecules, and the other one can also form an O⋯H–C hydrogen bond with the hydrogen atom on the benzene ring. In H4OBDB_stru2, only O-shaped dimers of the H4OBDB molecule appear and they are connected by hydrogen bonds between carboxyl groups to form a porous network structure. Further analysis of the thermodynamic stability of these two self-assembly structures will be conducted in combination with DFT-calculated data.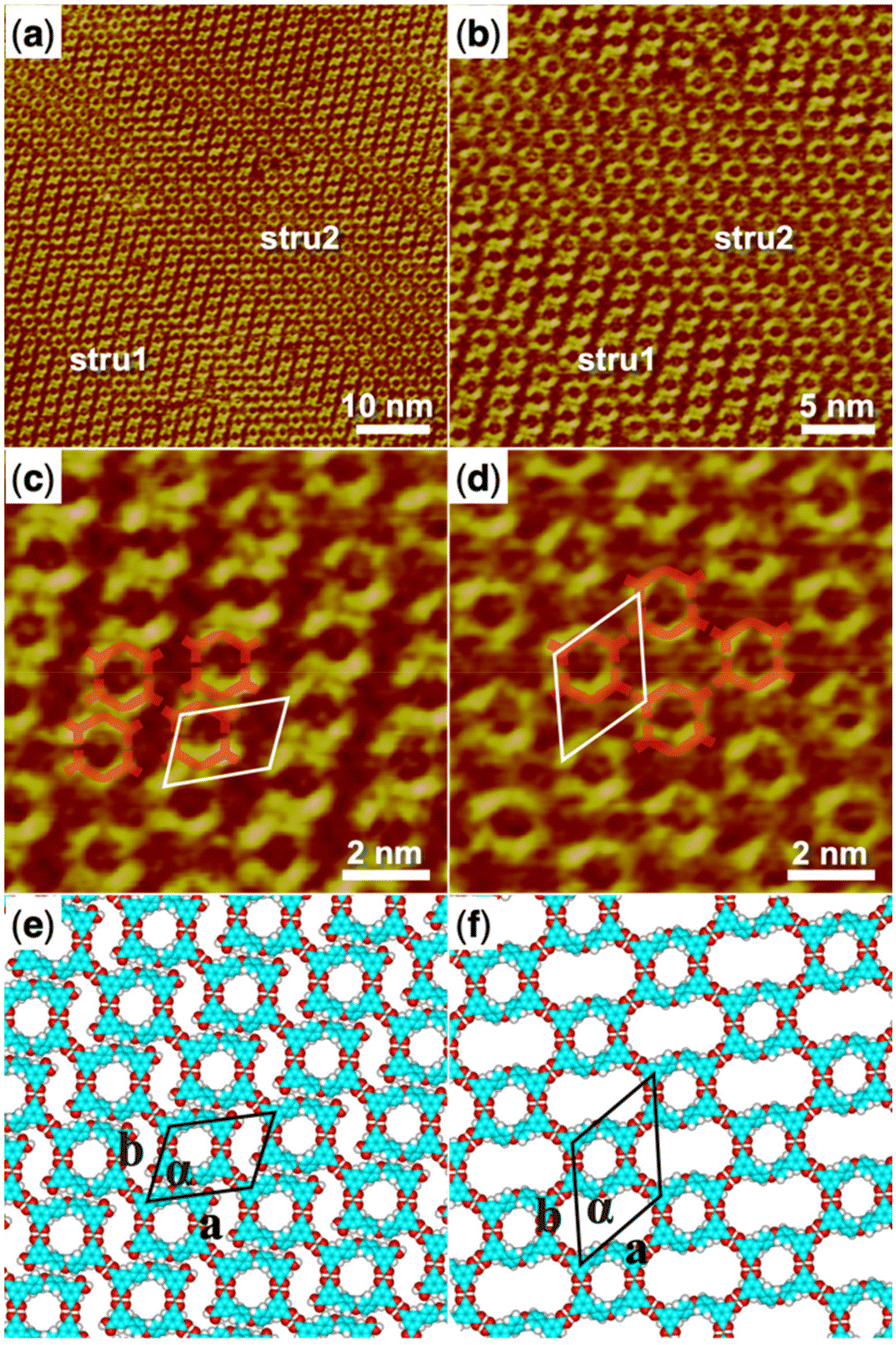 | ||
| Fig. 1 (a) STM image (60 nm × 60 nm) of H4OBDB self-assembly at the heptanoic acid–HOPG liquid–solid interface, Iset = 299.1 pA, Vbias = 699.8 mV. (b) STM image (30 nm × 30 nm) of H4OBDB self-assembly, Iset = 299.1 pA, Vbias = 699.8 mV. (c) High-resolution STM image (10 nm × 10 nm) of H4OBDB_stru1, Iset = 299.1 pA, Vbias = 699.8 mV. (d) High-resolution STM image (10 nm × 10 nm) of H4OBDB_stru2, Iset = 299.1 pA, Vbias = 699.8 mV. (e) Suggested molecular model for H4OBDB_stru1. (f) Suggested molecular model for H4OBDB_stru2. Unit cells were imposed on the STM image and its molecular model. Their measured and calculated parameters are shown in Table 1. | ||
Self-assembly of H4ADDI at the liquid–solid interface
5,5′-(Acridine-2,7-diyl)diisophthalic acid (H4ADDI) is also a bent-shaped tetracarboxylic molecule similar to H4OBDB, but with an acridine group in the middle of the skeleton. Two types of well-ordered self-assembly monolayers were formed as observed in large-scale STM images of Fig. 2a and b after depositing a droplet of heptanoic acid solution containing H4ADDI on the surface of the HOPG substrate, which was marked by stru1 and stru2. In type stru1, an alignment pattern of distinct O-shaped dimers was observed (denoted as H4ADDI_stru1). Compared with type stru1, type stru2 is arranged in a hexagonal symmetric pattern (denoted as H4ADDI_stru2) and has a lower proportion on the substrate. High-resolution STM images of H4ADDI_stru1 and H4ADDI_stru2 are shown in Fig. 2c and d, respectively. Although H4ADDI has a similar structure to H4OBDB, its assembly strategy is different from that of H4OBDB. Undoubtedly, the O-shaped contour in Fig. 2c corresponds to the dimer of H4ADDI formed by the double cyclic dimeric O–H⋯O hydrogen bonds between terminal carboxyl groups. As illustrated by the imposed blue-coloured symbol representing the H4ADDI molecule, the X-shaped dimer of H4ADDI does not occur, but a vacancy terminal carboxyl group of the H4ADDI molecule is observed to be close to the middle moiety of an adjacent H4ADDI molecule (as shown in the red dashed circles in Fig. 2c), forming a misplaced X-type linkage, and then the O-shaped dimers are assembled into H4ADDI_stru1. In Fig. 2d, the middle moiety of the H4ADDI molecule is attached to carboxyl groups of two adjacent molecules (as shown in the red dashed circles in Fig. 2d), which increases the molecular packing of the assembled H4ADDI_stru2. Fig. 2e and f show the corresponding molecular models optimized by the DFT method based on the STM observation of H4ADDI_stru1 and H4ADDI_stru2, respectively. As shown in the molecular models, N⋯H–O and O⋯H–C hydrogen bonds could be formed between the acridine group and the terminal carboxyl group of neighboring H4ADDI molecules. In H4ADDI_stru1, an acridine group is connected to a carboxyl group by a N⋯H–O hydrogen bond, while in H4ADDI_stru2, an acridine group is connected to a carboxyl group by a N⋯H–O hydrogen bond and to a second carboxyl group by a C–H⋯O hydrogen bond. The molecular models are in good agreement with the STM images. Further analysis of the thermodynamic stability of these two self-assembly structures will be conducted in combination with the DFT data in the following section.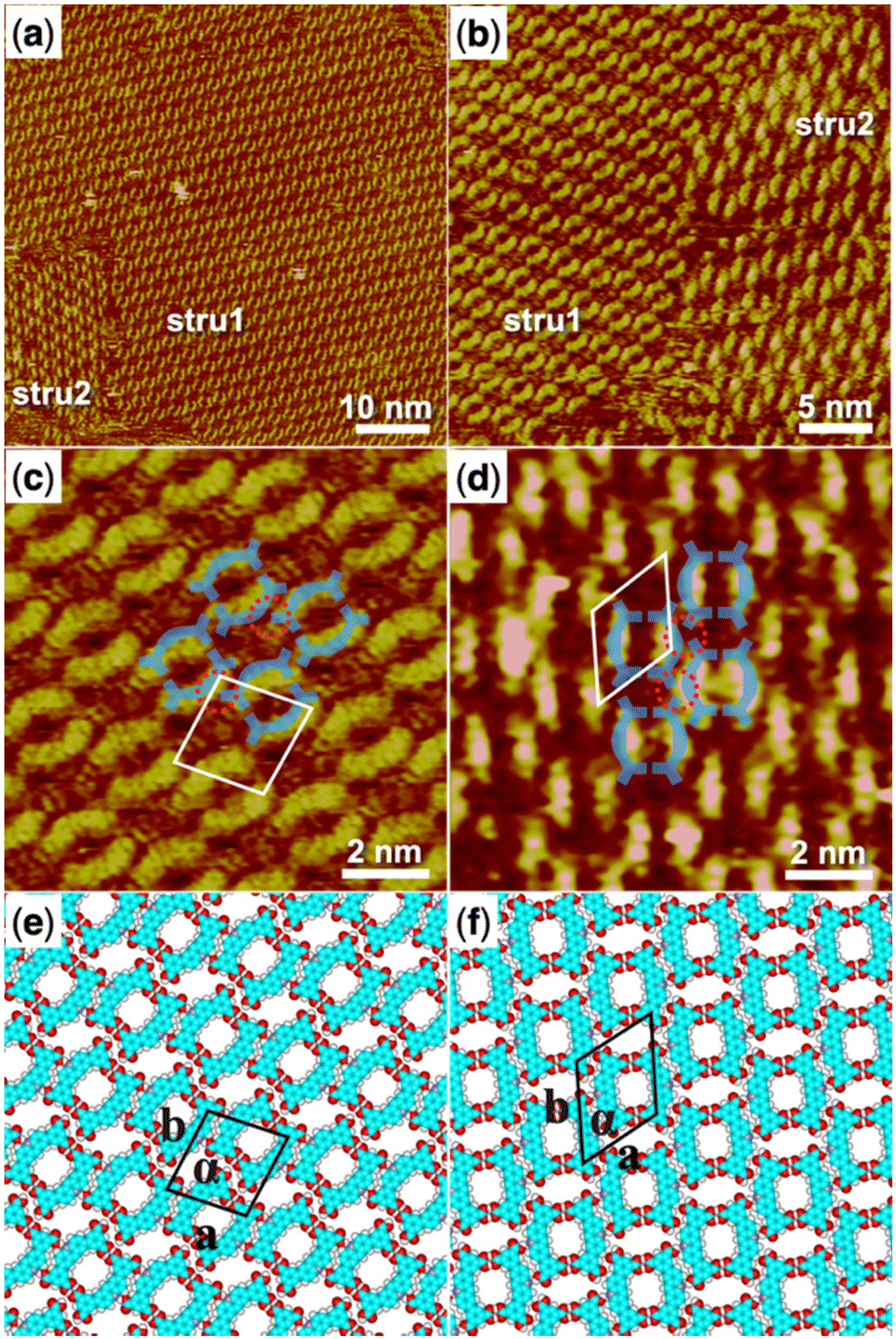 | ||
| Fig. 2 (a) STM image (60 nm × 60 nm) of H4ADDI self-assembly at the heptanoic acid–HOPG liquid–solid interface, Iset = 140.4 pA, Vbias = 923.5 mV. (b) STM image (30 nm × 30 nm) of H4ADDI self-assembly, Iset = 283.8 pA, Vbias = 661.0 mV. (c) High-resolution STM image (10 nm × 10 nm) of H4ADDI_stru1, Iset = 283.8 pA, Vbias = 661.0 mV. (d) High-resolution STM image (10 nm × 10 nm) of H4ADDI_stru2, Iset = 140.4 pA, Vbias = 923.5 mV. (e) Suggested molecular model for H4ADDI_stru1. (f) Suggested molecular model for H4ADDI_stru2. Unit cells were imposed on the STM image and its molecular model. Their measured and calculated parameters are shown in Table 1. | ||
Co-assembly of H4OBDB and TMA at the liquid–solid interface
We then started to investigate the trimesic acid (TMA) molecule as a bridging molecule to expand the self-assembly network of the H4OBDB molecule. When a droplet of heptanoic acid solution containing H4OBDB and TMA was added to the surface of HOPG, we observed that the assembled monolayer structure (denoted as H4OBDB–TMA) covers the entire surface immediately, as shown in the large-scale STM image in Fig. 3a and b. In the high-resolution STM image of Fig. 3c, it can be clearly seen that the dot-shaped structure representing the TMA molecule and the long bar-shaped structure representing the H4OBDB molecule mixed to form a porous hexagonal co-assembly pattern. Fig. 3d shows the corresponding DFT-optimized molecular model of H4OBDB–TMA. The unit cell parameters of the DFT model were in good agreement with those measured in the STM image, indicating that the model is reasonable. Two of the three carboxyl groups of each TMA molecule form cyclic dimeric O–H⋯O hydrogen bonds with two adjacent H4OBDB molecules and the other one can form two O–H⋯O hydrogen bonds with the other two adjacent H4OBDB molecules. Two H4OBDB molecules and two TMA molecules form the minimal repeated building block of the H4OBDB–TMA co-assembly structure, which is then spliced into a large range of an ordered pattern. The ratio of the H4OBDB molecule to the TMA molecule is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the co-assembly structure. With a periodic arrangement of nano-cavities of different sizes, the co-assembly structure can be further studied in the field of selective molecular recognition.
:1 in the co-assembly structure. With a periodic arrangement of nano-cavities of different sizes, the co-assembly structure can be further studied in the field of selective molecular recognition.
 | ||
| Fig. 3 (a) STM image (60 nm × 60 nm) of H4OBDB–TMA co-assembly at the heptanoic acid–HOPG liquid–solid interface, Iset = 228.9 pA, Vbias = 693.1 mV. (b) STM image (30 nm × 30 nm) of H4OBDB–TMA co-assembly, Iset = 296.0 pA, Vbias = 698.9 mV. (c) High-resolution STM image (10 nm × 10 nm) of H4OBDB–TMA co-assembly, Iset = 228.9 pA, Vbias = 693.1 mV. (d) Suggested molecular model for H4OBDB–TMA co-assembly. Unit cells were imposed on the STM image and its molecular model. Their measured and calculated parameters are shown in Table 1. | ||
Co-assembly of H4ADDI and TMA at the liquid–solid interface
The co-assembly of the H4ADDI molecule and the TMA molecule was also carried out and the large-scale STM image of the obtained H4ADDI–TMA co-assembly monolayer structure is shown in Fig. 4a and b. It can be observed that the H4ADDI–TMA structure is similar to the H4OBDB–TMA structure. In the high-resolution STM image in Fig. 4b, we can see that the TMA molecule appears as a dot-shaped structure and the H4ADDI molecule appears as a bar-shaped structure. Fig. 4d shows the corresponding molecular model of H4ADDI–TMA, which was based on the STM observation and optimized by the DFT method. The interactions between the molecules are quite clear, and the molecular model matches the STM image well. A TMA molecule also forms two cyclic dimeric O–H⋯O hydrogen bonds with two adjacent H4ADDI molecules and two O–H⋯O hydrogen bonds with the other two adjacent H4OBDB molecules. The ratio of the H4 ADDI molecule to the TMA molecule is also 1:1 in the co-assembly structure.
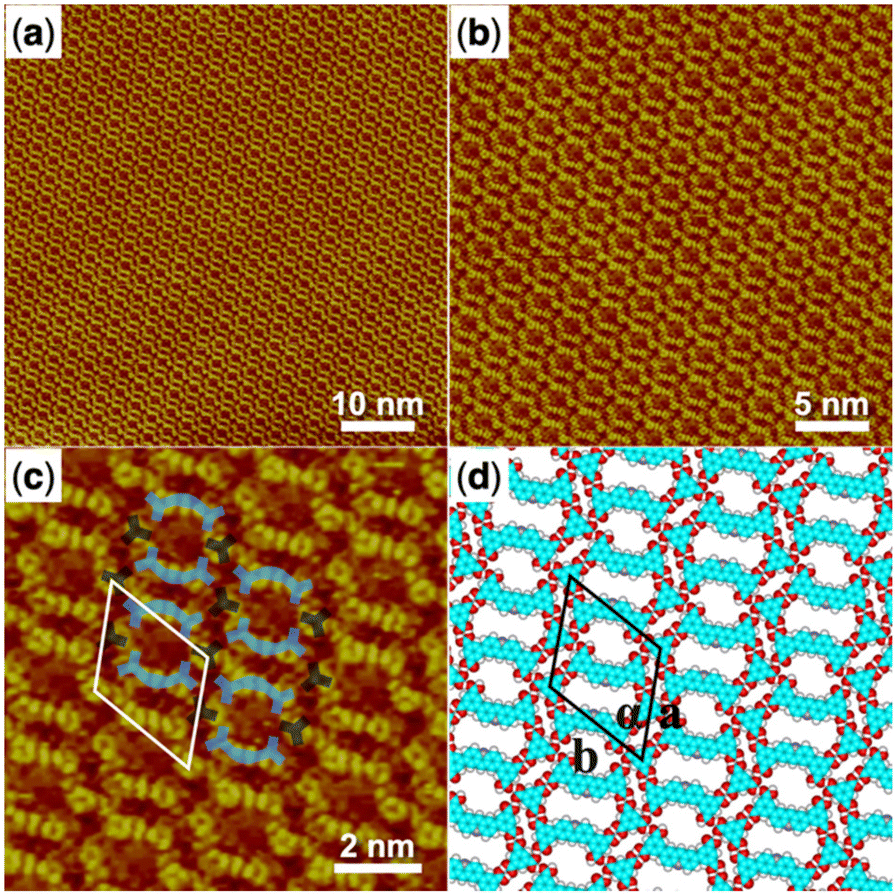 | ||
| Fig. 4 (a) STM image (60 nm × 60 nm) of H4ADDI–TMA co-assembly at the heptanoic acid–HOPG liquid–solid interface, Iset = 289.9 pA, Vbias = 727.5 mV. (b) STM image (30 nm × 30 nm) of H4ADDI–TMA co-assembly, Iset = 289.9 pA, Vbias = 727.5 mV. (c) High-resolution STM image (10 nm × 10 nm) of H4ADDI–TMA co-assembly, Iset = 289.9 pA, Vbias = 727.5 mV. (d) Suggested molecular model for H4ADDI–TMA co-assembly. Unit cells are imposed on the STM image and its molecular model. Their measured and calculated parameters are shown in Table 1. | ||
In conclusion, the self-assembly behavior of a pair of low-symmetry carboxylic acid molecules (H4OBDB and H4ADDI) and the co-assembly behavior of bridging molecules such as TMA were studied. H4OBDB and H4ADDI molecules have similar semicircular bent-shaped structures and both tend to form an O-shaped dimer via strong intermolecular double cyclic dimeric O–H⋯O hydrogen bonds between terminal carboxyl groups, but end up forming different monolayer self-assembly structures at the liquid–solid interface. The configurations of the self-assembled structures are determined by the connections between the O-shaped dimers. In addition to the cyclic dimeric O–H⋯O hydrogen bonds between carboxyl groups, for the H4OBDB molecule, the double O⋯H–C hydrogen bonds between diphenyl ether groups are also considerable driving forces for its self-assembly. For the H4ADDI molecule, its acridine group can connect to adjacent vacant carboxyl groups by a N⋯H–O hydrogen bond and a C–H⋯O hydrogen bond, which has an important impact on its self-assembly. After mixing with the TMA molecule, both H4OBDB and H4ADDI can form a precise arrangement of co-assembly with TMA through hydrogen bonding. The two co-assembly structures are similar because four terminal carboxyl groups of the tetracarboxylic molecule are all connected to the TMA bridging molecule by O–H⋯O hydrogen bonds, and thus the influence of different skeleton moieties on the co-assembly structure is limited.
Density functional theory (DFT) calculation results
To further reveal the formation mechanism of each assembled orderly structure at the liquid–solid interface from a theoretical perspective, we performed DFT calculations based on the above observed STM experimental results.The measured and calculated unit cell parameters for all assembled structures are summarized in Table 1. The calculated parameters agree well with the experimental data, indicating that our DFT results are reasonable. In the surface assembly system, the interaction between adsorbates and the substrate plays an important role. Therefore, we present the total energy (including the interaction energy between adsorbates and the interaction energy between adsorbates and the substrate) in Table 2. Furthermore, a reasonable way to compare the thermodynamic stability of differently assembled structures should be the total energy per unit area. Hence, we also present the total energy per unit areas of the assembled structures in Table 2.
| Unit cell parameters | ||||
|---|---|---|---|---|
| a (nm) | b (nm) | α (°) | ||
| H4OBDB_stru1 | Expt. | 2.5 ± 0.1 | 1.9 ± 0.1 | 67 ± 1 |
| Cal. | 2.50 | 1.90 | 66.7 | |
| H4OBDB_stru2 | Expt. | 2.4 ± 0.1 | 2.8 ± 0.1 | 55 ± 1 |
| Cal. | 2.45 | 2.90 | 55.0 | |
| H4ADDI_stru1 | Expt. | 2.1 ± 0.1 | 2.2 ± 0.1 | 79 ± 1 |
| Cal. | 2.05 | 2.15 | 79.5 | |
| H4ADDI_stru2 | Expt. | 2.0 ± 0.1 | 2.4 ± 0.1 | 60 ± 1 |
| Cal. | 2.10 | 2.40 | 60.0 | |
| H4OBDB–TMA | Expt. | 2.6 ± 0.1 | 2.7 ± 0.1 | 63 ± 1 |
| Cal. | 2.60 | 2.75 | 63.0 | |
| H4ADDI–TMA | Expt. | 2.6 ± 0.1 | 2.7 ± 0.1 | 61 ± 1 |
| Cal. | 2.65 | 2.75 | 61.0 | |
| Interactions between molecules (kcal mol−1) | Interactions between molecules and substrate (kcal mol−1) | Total energy (kcal mol−1) | Energy per unit area (kcal mol−1 Å−2) | |
|---|---|---|---|---|
| H4OBDB_stru1 | −95.030 | −73.116 | −168.146 | −0.385 |
| H4OBDB_stru2 | −116.215 | −76.067 | −192.282 | −0.330 |
| H4ADDI_stru1 | −82.396 | −103.266 | −185.662 | −0.428 |
| H4ADDI_stru2 | −81.366 | −104.063 | −185.429 | −0.425 |
| H4OBDB–TMA | −141.138 | −115.554 | −256.692 | −0.403 |
| H4ADDI–TMA | −127.224 | −154.624 | −281.848 | −0.442 |
In Table 2, we noticed that H4OBDB_stru1 had a lower total energy per unit area (−0.385 kcal mol−1 Å−2) than H4OBDB_stru2 (−0.330 kcal mol−1 Å−2). This means that H4OBDB_stru1 has higher thermodynamic stability than H4OBDB_stru2. This is due to the high molecular density of H4OBDB_stru1 and the strong hydrogen bond interactions between diphenyl ether groups of adjacent H4OBDB molecules. In the experiment, it was observed that H4OBDB_stru1 occupies a large domain area on the substrate and H4OBDB_stru2 only appears at the domain boundary, which is consistent with the thermodynamic theoretical results. As for H4ADDI_stru1 and H4ADDI_stru2, the values for total energy per unit area are −0.428 kcal mol−1 Å−2 and −0.425 kcal mol−1 Å−2, respectively. The theoretical result suggests that H4ADDI_stru1 and H4ADDI_stru2 have similar thermodynamic stabilities. In the experiment, it was also observed that H4ADDI_stru1 has a higher proportion on the substrate, meaning that there is competition between kinetic factors occurring in the self-assembly of the H4ADDI molecule, and the formation of H4ADDI_stru1 is a kinetic priority. According to the DFT-simulated single molecular models in Fig. S1 in the ESI,† the H4ADDI molecule has better planarity than H4OBDB on the graphite substrate and therefore has a stronger π–π stacking interaction with the substrate, which is reflected by the corresponding data in Table 2.
It is noteworthy that the values of the total energy per unit area of H4OBDB–TMA and H4ADDI–TMA co-assembly structures are lower than those of the H4OBDB and H4ADDI self-assembly structures, revealing that the two co-assembly structures are more thermodynamically stable than the self-assembly structures. This means that, from a thermodynamics perspective, the structural transformation from the self-assembly structure to two-component co-assembly structures could occur by adding TMA molecules, which is consistent with the experimental results. In general, all the DFT calculation results agreed well with the STM observations and were self-consistent. The STM observation combined with DFT calculations illustrated how different assembly driving forces, such as molecule–molecule interactions and molecule–substrate interactions, synergistically influence the formation process and structure of the assembled monolayers on the surface. In this study, the molecule–molecule interactions mainly refer to different hydrogen bonds, and the spatial shape of the molecules and the position of the hydrogen bond groups on the molecular skeleton determine the assembled structures in the long range.
Experimental methods
STM investigation
All commercial reagents were used as received without further purification. Heptanoic acid solvent was purchased from Aldrich. H4OBDB, H4ADDI, and TMA molecules were purchased from Jilin Chinese Academy of Sciences—Yanshen Technology Co., Ltd (chemical structures are shown in Scheme 1). The solutions for the STM experiment were obtained by dissolving the molecules in heptanoic acid solvent. Unless otherwise noted, all solutions had a concentration of around 1.0 × 10−4 M. Specifically, all solutions were diluted to 10% of the saturated concentration because their typical saturated concentrations were generally 1.0 × 10−3 M. Highly oriented pyrolytic graphite (HOPG, grade ZYB, NTMDT, Russia) was used as the substrate and was cleaved using adhesive tape. The STM samples were prepared by depositing a droplet (0.4 μL) of solution onto the bare surface of the freshly cleaved HOPG substrate. After the treatments, STM experiments were performed with a Nanoscope III scanning probe microscope system (Bruker, USA) operating in a constant current mode under ambient conditions. An STM probe tip was prepared by mechanically cutting Pt/Ir wire (80/20) and immersed in the deposited solution during imaging. The provided STM images are raw data without any treatment except for the flattening process. The detailed tunneling conditions are given in the corresponding figure captions.Computational details
The theoretical calculations were carried out using density functional theory (DFT) provided by the DMol3 code.50 We used the periodic boundary conditions (PBC) to describe the 2D periodic structure on the graphite in this work. The Perdew and Wang parameterization of the local exchange–correlation energy was applied in the local spin density approximation (LSDA) to describe exchange and correlation.51,52 All-electron spin-unrestricted Kohn–Sham wave functions were expanded on a local atomic orbital basis. A numerical basis set was applied for the large system. The calculations were equipped with the medium mesh and were all-electron ones. The self-consistent field procedure was performed with a convergence criterion of 10−5 a.u. on the energy and electron density. Combined with the experimental data, we optimized the unit cell parameters and the geometry of the adsorbates in the unit cell. When the energy and density convergence criteria reached the desired degree, we could obtain the optimized parameters and the interaction energy between adsorbates.The model system shows the interactions between the adsorbates and HOPG. In this investigation, the adsorption of adsorbates with a π-conjugated benzene-ring on graphite is similar to that of graphene, which helps us to perform calculations on infinite graphene monolayers using PBC. Graphene layers were separated by 40 Å in the normal direction. Graphene supercells were used and the Brillouin zone was sampled using a gamma point mesh when adsorbates were modeled on graphene. The interaction energy (Einter) of adsorbates on graphite is Einter = Etot(adsorbates/graphene) − Etot(isolated adsorbates in vacuum) − Etot(graphene).
Conclusions
In summary, the supramolecular self-assembly behavior of a pair of low-symmetry carboxylic acid molecules (H4OBDB and H4ADDI) and their co-assembly behaviors with a TMA bridging molecule were studied at the heptanoic acid–HOPG liquid–solid interface using an STM in combination with DFT calculations. H4OBDB and H4ADDI molecules both tend to form an O-shaped dimer via intermolecular double cyclic dimeric O–H⋯O hydrogen bonds between terminal carboxyl groups but end up forming different types of self-assembly structures. The connections between the O-shaped dimers in different self-assembly structures were investigated and discussed. We also investigated the construction of two-component co-assembly structures by mixing H4OBDB or H4ADDI molecules with bridging molecules such as TMA. The two formed co-assembly structures are similar, suggesting that the influence of different skeleton moieties on the co-assembly structure is limited. The present work demonstrates how different assembly factors are related to each other, which has some implications for the chemical structural design of low-symmetry carboxylic acid molecules and the selection of other bridging molecules in future on-surface self-assembly studies. Based on the analysis of the STM results and the DFT calculations, the mechanisms of assembly behaviors were explored.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21972031 and 22272039), the Strategic Priority Research Program of the Chinese Academy of Sciences (no. XDB36000000), the Guangdong Basic and Applied Basic Research Fund (2022A1515110876), the Guangdong Provincial Key Construction Discipline Research Capacity Enhancement Project (no. 2021ZDJS088) and the Jilin Chinese Academy of Sciences-Yanshen Technology Co., Ltd.References
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154–1196 CrossRef.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS PubMed.
- J. A. A. W. Elemans, S. Lei and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7298–7333 CrossRef CAS PubMed.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- X. M. Zhang, Q. D. Zeng and C. Wang, Sci. China: Chem., 2014, 57, 13–25 CrossRef CAS.
- L. Feng, T. Wang, Z. Tao, J. Huang, G. Li, Q. Xu, S. L. Tait and J. Zhu, ACS Nano, 2019, 13, 10603–10611 CrossRef CAS PubMed.
- S. De Feyter, P. C. Grim, J. van Esch, R. M. Kellogg, B. L. Feringa and F. C. De Schryver, J. Phys. Chem. B, 1998, 102, 8981–8987 CrossRef CAS.
- R. May, S. S. Jester and S. Höger, J. Am. Chem. Soc., 2014, 136, 16732–16735 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, B. Song, Y. Zhang, X. Jiang, M. Wang, R. Tumbleson, C. Liu, P. Wang, X.-Q. Hao, T. Rojas, A. T. Ngo, J. L. Sessler, G. R. Newkome, S.-W. Hla and X. Li, Nat. Chem., 2020, 12, 468–474 CrossRef CAS PubMed.
- S. De Feyter and F. C. De Schryver, J. Phys. Chem. B, 2005, 109, 4290–4302 CrossRef CAS PubMed.
- W. Mamdouh, H. Uji-i, J. S. Ladislaw, A. E. Dulcey, V. Percec, F. C. De Schryver and S. De Feyter, J. Am. Chem. Soc., 2006, 128, 317–325 CrossRef CAS PubMed.
- K. S. mali, J. Adisoejoso, E. Ghijsens, I. De Cat and S. De Feyter, Acc. Chem. Res., 2012, 45, 1309–1320 CrossRef CAS PubMed.
- U. Mazur and K. W. Hipps, Chem. Commun., 2015, 51, 4737–4749 RSC.
- C. Ma, J. Li, S. Zhang, W. Duan and Q. Zeng, Nanotechnology, 2021, 32, 382001 CrossRef CAS PubMed.
- S. Tan, J. Tao, W. Luo, H. Jiang, Y. Liu, H. Xu, Q. Zeng and H. Shi, Chin. Chem. Lett., 2021, 32, 1149–1152 CrossRef CAS.
- Y. Xiao, F. Cai, X. Peng, X. Kang, P. Lei, X. Li, H. Xu, X. Xunwen, T. Bin and Q. Zeng, Chin. Chem. Lett., 2021, 32, 3566–3569 CrossRef CAS.
- X. Peng, T. Meng, L. Wang, L. Cheng, W. Zhai, K. Deng, C.-Q. Ma and Q. Zeng, Chin. Chem. Lett., 2023, 34, 107568 CrossRef CAS.
- P. Lei, L. Ma, S. Zhang, J. Li, L. Gan, K. Deng, W. Duan, W. Li and Q. Zeng, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.108005.
- M. Lackinger and W. M. Heckl, Langmuir, 2009, 25, 11307–11321 CrossRef CAS PubMed.
- X. Zhang, Q. Zeng and C. Wang, RSC Adv., 2013, 3, 11351–11366 RSC.
- S. Q. Zhang, L. X. Cheng, Z. L. Gong, W. B. Duan, B. Tu, Y. W. Zhong and Q. D. Zeng, Langmuir, 2019, 35, 6571–6577 CrossRef CAS PubMed.
- J. Li, X. Zu, Y. Qian, W. Duan, X. Xiao and Q. Zeng, Chin. Chem. Lett., 2020, 31, 10–18 CrossRef CAS.
- L. Ma, P. Wang, W. Duan, B. Tu and Q. Zeng, Langmuir, 2021, 37, 11544–11551 CrossRef CAS PubMed.
- X. Li, J. Li, C. Ma, C. Chen, S. Zhang, B. Tu, W. Duan and Q. Zeng, Chin. Chem. Lett., 2021, 32, 1077–1080 CrossRef CAS.
- L. Ma, C. Ma, S. Zhang, J. Li, L. Gan, K. Deng, W. Duan, X. Li and Q. Zeng, Langmuir, 2022, 38, 4434–4441 CrossRef CAS PubMed.
- S. Zhang, J. Zhang, K. Deng, J. Xie, W. Duan and Q. Zeng, Phys. Chem. Chem. Phys., 2015, 17, 24462–24467 RSC.
- H. Dai, S. Wang, I. Hisaki, S. Nakagawa, N. Ikenaka, K. Deng, X. Xiao and Q. Zeng, Chem. – Asian J., 2017, 12, 2558–2564 CrossRef CAS PubMed.
- J. Gu, M. Wen, X. Liang, Z. Shi, M. V. Kirillova and A. M. Kirillov, Crystals, 2018, 8, 83 CrossRef.
- Q. Liang, Y. Yu, G. Feng, Y. Shen, L. Yang and S. Lei, Chem. Commun., 2020, 56, 12182–12185 RSC.
- T. Meng, P. Lei, Y. Zhang, K. Deng, X. Xiao and Q. Zeng, Chin. J. Chem., 2022, 40, 2727–2733 CrossRef CAS.
- Q. Zhou, Y. Li, Q. Li, Y. Wang, Y. Yang, Y. Fang and C. Wang, Nanoscale, 2014, 6, 8387–8391 RSC.
- D. C. Y. Nguyen, L. Smykalla, T. N. H. Nguyen, T. Rüffer and M. Hietschold, J. Phys. Chem. C, 2016, 120, 11027–11036 CrossRef CAS.
- J. Macleod, J. Phys. D: Appl. Phys., 2019, 53, 043002 CrossRef.
- M. Li, K. Deng, S. B. Lei, Y. L. Yang, T. S. Wang, Y. T. Shen, C. R. Wang, Q. D. Zeng and C. Wang, Angew. Chem., Int. Ed., 2008, 47, 6717–6721 CrossRef CAS PubMed.
- X. Peng, L. Cheng, X. Zhu, Y. Geng, F. Zhao, K. Hu, X. Guo, K. Deng and Q. Zeng, Nano Res., 2018, 11, 5823–5834 CrossRef CAS.
- C. Ma, J. Li, S. Zhang, B. Liu, W. Duan, B. Tu, F. Zhao, X. Li and Q. Zeng, J. Phys. Chem. C, 2020, 124, 23237–23242 CrossRef CAS.
- J. M. Macleod, Z. B. Chaouch, D. F. Perepichka and F. Rosei, Langmuir, 2013, 29, 7318–7324 CrossRef CAS PubMed.
- Y. Xie, C. Liu, L. Cheng, Y. Fan, H. Li, W. Liu, L. Zhu, X. Li, K. Deng, Q. Zeng and S. Han, Chin. Chem. Lett., 2022, 33, 4649–4654 CrossRef CAS.
- M. Lackinger, S. Griessl, T. Markert, F. Jamitzky and W. M. Heckl, J. Phys. Chem. B, 2004, 108, 13652–13655 CrossRef CAS.
- F. Silly, J. Phys. Chem. C, 2012, 116, 10029–10032 CrossRef CAS.
- J. Li, B. Liu, W. Duan, B. Tu and Q. Zeng, Surf. Sci., 2020, 700, 121654 CrossRef CAS.
- J. Shi, Y. Li, X. Jiang, H. Yu, J. Li, H. Zhang, D. J. Trainer, S. W. Hla, H. Wang, M. Wang and X. Li, J. Am. Chem. Soc., 2021, 143, 1224–1234 CrossRef CAS PubMed.
- J. Fang, X. Zhu, W. Luo, J. Shi, L. Wang, L. Wang, B. Tu, Q. Zeng and X. Xiao, Chin. Chem. Lett., 2022, 33, 1100–1104 CrossRef CAS.
- C. N. Morrison, S. Ahn, J. K. Schnobrich and A. J. Matzger, Langmuir, 2011, 27, 936–942 CrossRef CAS PubMed.
- J. Wang, L. M. Wang, C. Lu, H. J. Yan, S. X. Wang and D. Wang, RSC Adv., 2019, 9, 11659–11663 RSC.
- S. Zhang, C. Chen, J. Li, C. Ma, X. Li, W. Ma, M. Zhang, F. Cheng, K. Deng and Q. Zeng, Nanoscale, 2022, 14, 2419–2426 RSC.
- J. Pang, F. Jiang, M. Wu, D. Yuan, K. Zhou, J. Qian, K. Su and M. Hong, Chem. Commun., 2014, 50, 2834–2836 RSC.
- Y. Xie, H. Yang, Z. U. Wang, Y. Liu, H.-C. Zhou and J.-R. Li, Chem. Commun., 2014, 50, 563–565 RSC.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DFT-simulated single molecular models on the graphite surface and details of DFT-simulated molecular models of all assembled structures. See DOI: https://doi.org/10.1039/d2nr06740d |
| This journal is © The Royal Society of Chemistry 2023 |