
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe current status in computational exploration of Pt(IV) prodrug activation by reduction
Fortuna
Ponte†
,
Stefano
Scoditti†
 ,
Gloria
Mazzone
* and
Emilia
Sicilia
*
,
Gloria
Mazzone
* and
Emilia
Sicilia
*
Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Via P.Bucci, 87036 Rende (CS), Italy. E-mail: gloria.mazzone@unical.it; emilia.sicilia@unical.it
First published on 18th May 2023
Abstract
Octahedral PtIV complexes are considered highly promising candidates for overcoming some shortcomings of clinically approved PtII drugs. PtIV compounds, owing to their inertia, appear to be capable of resisting premature aquation and undesired binding to essential plasma proteins and have shown remarkable potential for both oral administration and for reducing side effects. Additionally, their pharmacological properties can be finely tuned by choosing appropriate axial ligands. The reduction inside the cell by biological reducing agents to the correponding active cytotoxic PtII species, accompanied by the loss of the axial ligands, is considered an essential step of their mechanism and has been extensively studied. However, a detailed understanding of the mechanism by which PtIV prodrugs are activated, which should be highly beneficial for their proper design, is lacking, and many contradictory results continue to be collected. In the hope of contributing to the advancement of knowledge in this field, this perspective focuses on the insights gained from computational studies carried out with the aim of finding answers to the many still open questions concerning the reduction of PtIV complexes in biological environments.
Introduction
Cancer is considered one of the world's most serious health problems and causes one in six deaths.1 For fighting cancer, various strategies can be practiced, including chemotherapy, radiotherapy, and surgery. Chemotherapy is a technique in which an anticancer agent with a specific mechanism of action is introduced into the organism to inhibit the growth and induce the death of tumor cells.2 The most famous chemotherapeutic agent is cisplatin, which represents one of the greatest successes in the story of medicinal inorganic chemistry. Synthesized for the first time by Michele Peyrone in 1845, cisplatin's cytotoxic activity was accidentally discovered by Barnett Rosenberg when he tried to evaluate the role of electrical current in Escherichia coli cellular division.3 After undergoing clinical trials, cisplatin was approved in 1978 by the Food and Drug Administration (FDA).4 Subsequently, various cisplatin structural analogues have been developed, including carboplatin and oxaliplatin, that have been approved for worldwide clinical use.5,6 Cisplatin and its analogues are drugs belonging to the class of DNA alkylating agents. Indeed, their mechanism of action is mostly based on the formation of DNA adducts through the binding of the PtII center to double-helix nucleobases.7 Inside the cancer cells, PtII complexes are hydrolyzed to produce the corresponding activated aqua PtII species. These active aquated species are able to form both intra- and inter-strand DNA crosslinks, mainly with nitrogen in the N7 position of guanine nucleobases, causing distortion of the DNA helix and proteins to signal for apoptosis.8–12 Both genomic and mitochondrial DNA can be damaged by the action of these drugs.13 Although widely used, the clinical utility of PtII drugs is strongly limited by severe side effects and tumor resistance. The latter is mainly due to the increased repair of damaged DNA, reduced cellular accumulation, and increased cytosolic inactivation, whereas high toxicity to healthy tissue and organs and low chemical stability cause the former.14 In spite of the enormous number of proposed and synthesized PtII alternative compounds and, in some cases, their PdII analogues,15–19 whose coordination sphere has been modified to properly tune their properties and reduce well-known shortcomings, none of them has demonstrated a superior anticancer activity than cisplatin and its approved derivatives. Among the strategies pursued to improve the therapeutic index and decrease side effects, PtIV prodrugs have received a significant amount of attention. Octahedral PtIV prodrugs are more inert to ligand substitution than their parent PtII complexes and less prone to deactivation en route before reaching the tumor cells.20 In addition, axial ligands can be properly chosen to tune both their physical and chemical properties without altering the structure of the active pharmacophore that is ultimately released.21–23 PtIV prodrugs are synthesized by oxidation of the corresponding square-planar PtII precursors, and since, in the beginning, Cl2 or H2O2 was used for carrying out the oxidation, tetraplatin and iproplatin (Scheme 1) were the first two PtIV complexes that entered clinical trials.24,25 Axial positions are occupied by two chlorido ligands in the former case and by two hydroxido ligands in the latter. The possibility of carrying out the carboxylation of the corresponding dihydroxido compounds allowed the preparation of a wide category of dicarboxylato complexes such as satraplatin26 (included in Scheme 1 and named also JM216) that, even if included in several clinical trials, has not been approved yet.27 Carboxylato ligands have been further exploited to enhance the pharmacological properties of PtIV prodrugs and add bioactive ligands to the axial positions to exert multi-action effects.23,28–32 The nature of the axial and, although to a much lesser extent, equatorial ligands is, therefore, of key importance in determining the properties of PtIV prodrugs and, in particular, their propensity to undergo two-electron reduction that provokes the breaking of the bonds between platinum and the axial ligands.33 The rate of the reduction/activation step has to be appropriately tuned to avoid occurring too rapidly before the prodrug reaches the tumor, or too slowly if the prodrug resists the reductive nature of the reducing agents.34,35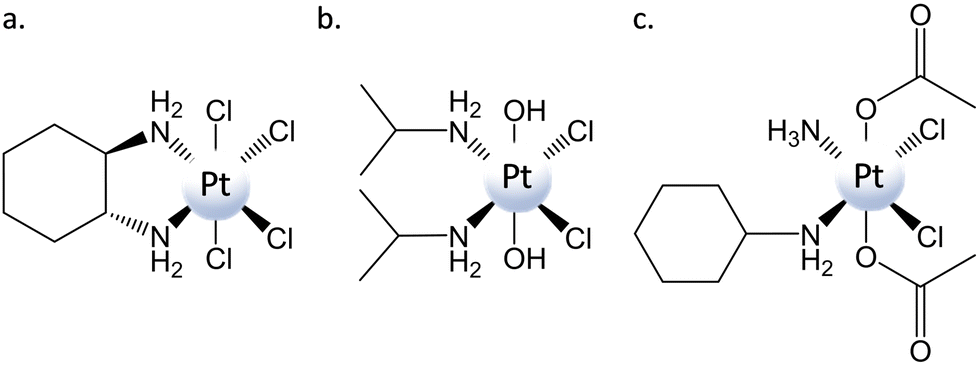 | ||
| Scheme 1 Structure of (a) tetraplatin, (b) iproplatin, and (c) satraplatin/JM216. | ||
What this comes down to is that for a successful design of PtIV prodrugs, a detailed understanding of the factors that determine the reduction rate is required. Electron transfer mechanisms for the reduction of PtIV complexes have been the subject of many studies over several decades and, on the basis of kinetic measurements, they have been classified as inner- and outer-sphere and PtII-catalysed.33,36,37 Inner-sphere involves direct contact with the reducing agent, leading to the formation of a new bond that facilitates the flow of the electrons. In the outer-sphere reduction, instead, the electrons are transferred without the establishment of any contact. The PtII catalysed mechanism is a particular kind of inner-sphere mechanism proposed by Basolo and Pearson38,39 that should involve the catalytic assistance of the produced PtII complex and, for that reason, is considered unlikely under cellular PtII complex concentrations. The rates of reduction have been commonly associated with the values of the reduction potentials, considered to be the most important parameters for determining the effectiveness of PtIV complexes to work as anticancer agents. Information concerning the ease of reduction is usually obtained by cyclic voltammetry measurements that provide the values of the potential for irreversible reduction, but not the standard redox potential. Indeed, due to the irreversibility of the reduction process, the peak potential of the irreversible cathodic response at a single scan rate is used to approximate the facility with which the metal centre of PtIV complexes is reduced40–42 and some caution has to be taken in using these values. Baik and co-workers have proposed, on the basis of the results of a series of electrochemical experiments, a decomposition scheme that separates the PtIV to PtII two-electron reduction process into electron transfer and Pt–ligand bond cleavage steps.43 DFT calculations have been employed to elucidate the mechanism, and the calculated reduction potentials have been found to be in excellent agreement with experimentally determined values that are generally more positive than measured peak potentials extracted from voltammograms. Nevertheless, numerous experiments have demonstrated that, very often, measured reduction potentials do not follow the trend in reduction rates of PtIV complexes.44,45 Other experimental methods can be used to obtain information about the PtIV reduction process, such as UV, NMR, and X-ray absorption near edge spectroscopies. All these techniques are useful to follow the disappearance of PtIV species and the formation of PtII products. However, information on the identity of the ligands bound to the metal centre is difficult to obtain, and the biological environment represents an obstacle to investigations. Very importantly, none of these methods can provide information on the mechanism by which the reduction occurs. Computational analyses are very precious alternative tools for disentangling the mechanistic details of the reduction process and have been extensively used in the course of the years to add further information about the characteristics of the reduction mechanisms generically classified as inner- and outer-sphere.46–54 Moreover, computations can be proficiently used to complement experimental outcomes.55,56 In this perspective, the main results of the computational investigations carried out on this topic will be summarized, examining both inner- and outer-sphere mechanisms viability. Although many potential biological reducing agents are present in both cells and plasma, L-ascorbic acid (AscH2) and L-glutathione are commonly believed to be the species causing the activation of PtIV prodrugs.34,37,57–59 Of the three forms of AscH2, i.e., neutral, monoanionic, and dianionic, the monodeprotonated one (AscH−) is assumed to be that with the highest concentration at physiological pH. Computations considering the presence of the other two forms will also be reported. Concerning L-glutathione, instead, L-cysteine (L-Cys) is generally used as a model of sulfur-containing bioreductants in order to reduce the required computational effort. It is worth mentioning that almost all the reported studies have been carried out exclusively by means of DFT, adopting the B3LYP exchange–correlation functional as the best performing one. The choice of the basis sets, instead, is wider. Details about the computational protocols have been reported only in a limited number of cases, while the rest refer to the original papers.
The most investigated axial ligands are chlorides, hydroxides, and carboxylates that influence the propensity of PtIV complexes to be reduced in the order Cl− > CH3COO− > OH− on the basis of what has been suggested by Hambley examining a large series of PtIV compounds.60 However, the outcomes of the investigations carried out to elucidate the role played by the identity of the axial ligands in influencing the reduction rate do not allow for pinpointing a clear trend33,35,44,57,59,61 and the examples described in the next paragraphs demonstrate that many variables have to be taken into due consideration.
Discussion
Reducing agents
A critical role in the reduction of PtIV complexes is played by the reducing agent (RA), which actively takes part in the reaction mechanism as, depending on its nature, different pathways could be in principle accessible. The RAs overly present in the physiological environment that could be implicated in the reduction of PtIV prodrugs to active PtII complexes include L-ascorbic acid or sulphur-donor biomolecules, such as glutathione, L-cysteine, and L-methionine, but even NADH, as a direct RA or indirectly involved in the reductive action of cytochrome c or haemoglobin62 (Scheme 2a). All these reductants have been taken into consideration through the exploration of PtIV prodrug reduction in recent years, though GSH and ascorbic acid remain the most used RAs. In particular, from a computational point of view, except particular examples for which other RAs such as NADH or light activated molecules have been taken into consideration,54 AscH2 and L-Cys, as the simplest models of sulphur-containing residues, are the most used for this purpose.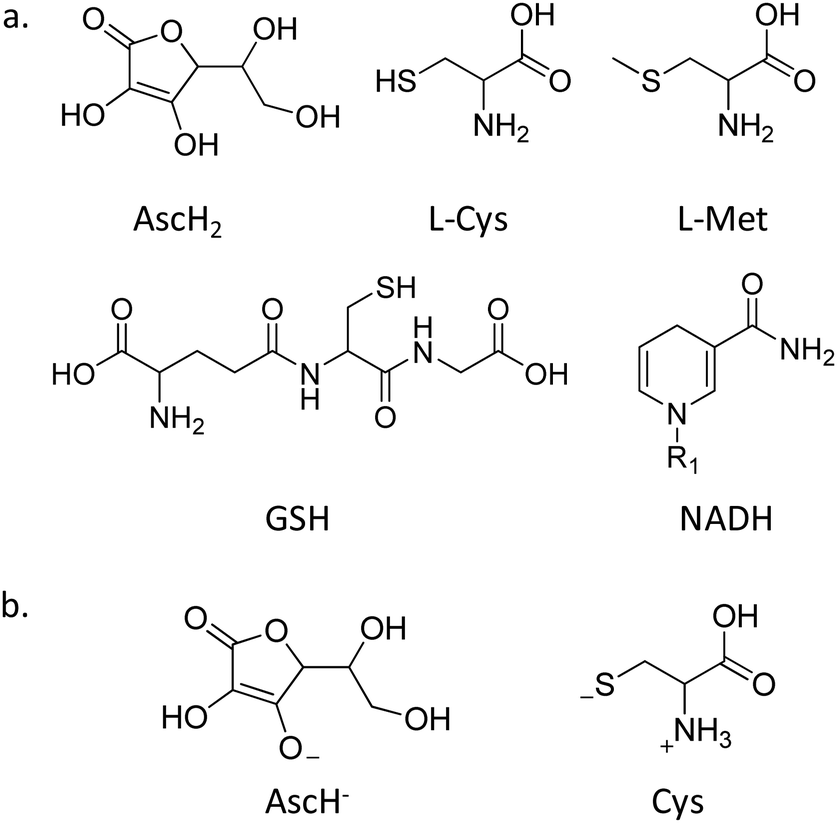 | ||
| Scheme 2 (a) Reducing agents (R1 = adenine dinucleotide); (b) the most used models for computational explorations. | ||
The active species of ascorbic acid is certainly the monoanionic ascorbate, AscH−, though the dianionic form Asc2− has been proven to be much more reactive than AscH−;45,63 however, its occurrence at physiological pH is very limited, contrary to AscH−, which is the most abundant form in such conditions. Accordingly, AscH− is widely considered the putative species for assisting the two-electron transfer for PtIV → PtII reduction. On the other hand, in physiological conditions, L-Cys exists in its zwitterionic form with intact thiol function. However, a computational study has evidenced the poor reactivity of such a form of cysteine and a low barrier in favour of the more active species formation that is reported in Scheme 2b, named Cys.47 The formation of the active species is due to a H transfer from sulphur to oxygen atoms to afford a thiolate, which is proven to be extremely more reactive than the thiol functional group.
Reaction mechanisms
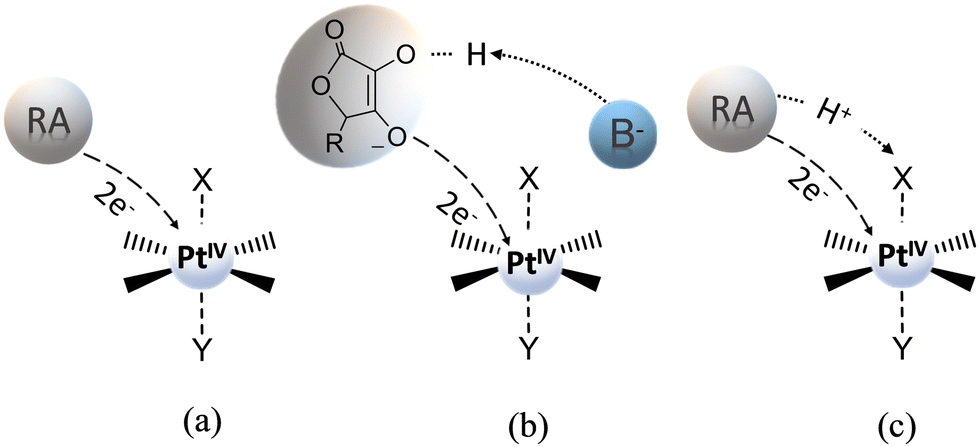
The usually invoked classification describes the reduction mechanism of PtIV complexes as inner- and outer-sphere two-electron transfer. However, this general grouping does not adequately take into consideration all the conceivable alternatives. In particular, for the inner-sphere mechanism, where the flow of the two electrons takes place through a direct interaction between the RA and the PtIV complex, several mechanistic hypotheses, that have been proposed in the course of the years and allow a more accurate description of the reduction process, are sketched in Scheme 3. The mechanism most commonly reported in the scientific literature is (a) ligand-bridge electron transfer, but three other reduction modes have been suggested and defined as (b) ligand-bridge-H transfer, (c) enolate β-carbon attack, and (d) PtII-catalysed mechanisms. In the first mechanism (a), one of the axial ligands is able to form a bridge between the PtIV centre and the reductant, facilitating the electron transfer from the RA to the metal centre, simultaneously provoking the release of the second ligand in the axial position and, then, the formation of the active square planar PtII species.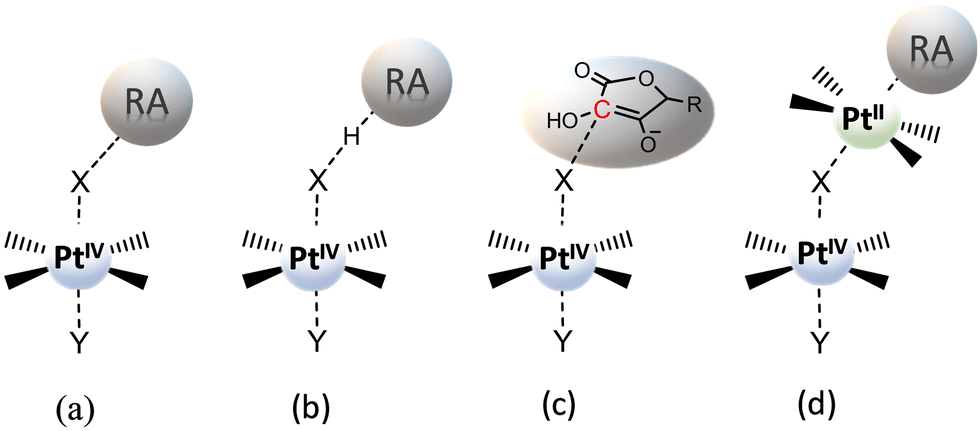 | ||
| Scheme 3 Inner-sphere mechanisms for the reduction of PtIV complexes by a generic reducing agent (RA) or a specific one (AscH−). (a) Ligand-bridge electron transfer; (b) ligand-bridge-H transfer; (c) enolate β-carbon attack; (d) Pt(II)-catalysed mechanisms. | ||
In the second mechanism (b), proposed by us for the reduction of Asplatin,53 a cisplatin-based PtIV complex, the two-electron transfer occurs in one step through the shift of a hydrido unit (H−) from the reducing agent to the bridging ligand, leading to the formation of the RA oxidized form and the concomitant loss of the trans axial ligand. The mechanism (c) was discovered in silico by Ariafard and co-workers and involves the reduction of PtIV complexes by the monodeprotonated form of ascorbic acid, AscH−. Specifically, the mechanism involves the nucleophilic attack of the enolate β-carbon of AscH− on one axial ligand (X), leading to the formation of a new C–X bond, and the simultaneous release of the second axial ligand. Finally, in the mechanism (d), proposed by Basolo and co-workers,38,39 PtIV reduction is catalysed by a PtII agent. In particular, the PtII complex is sandwiched between the RA and the PtIV complex to yield the AscH–PtII–X–PtIV–Y dimer, where X and Y are the PtIV axial ligands, and the bridging ligand mediates the two-electron transfer. During the reduction reaction, the two electrons are transferred from the RA to the PtIV metal centre, the bridging ligand passes from the PtIV complex to the PtII catalyst agent that already coordinates the RA system, and the other axial ligand is spontaneously released. A second RA molecule should help in restoring the PtII catalyst. Besides the low cellular concentration of PtII, the steric hindrance of the axial ligands of the complex undergoing the reduction could deactivate this type of catalytic mechanism.33
Concerning the outer-sphere mechanism, the propensity of PtIV complexes to be reduced is measured by their redox potential, and, experimentally, this information is usually obtained by cyclic voltammetry. As it has been underlined above, however, cyclic voltammetry measurements can provide, owing to the irreversibility of the reduction process of such complexes, only the peak potential of the irreversible cathodic response (Ep) but not the standard redox potential. Indeed, both chemical and electrochemical events are involved; two electrons are transferred, and the process can be significantly influenced by the nature of the axial ligands and the ease with which they are released. From a theoretical point of view, various computational approaches can be used for predicting reduction potentials in solvent.64 One group of such methods is based on the use of thermodynamic cycles, allowing the evaluation of condensed-phase reduction potentials from the standard-state Gibbs free energy of the corresponding reaction or half-reaction. The reaction free energy is computed as a difference in the free energies of products and reactants, with the free energy of each reactant being computed as a sum of the gas-phase free energy and the free energy of solvation. The reduction process of PtIV complexes involves the transfer of two electrons from a reducing agent so that the corresponding PtII derivative is formed and two ligands, in principle the axial ones, are released. Considering the two-electron transfer for the reaction (Scheme 4a):
| L–PtIV–L + 2e− → PtII + 2L |
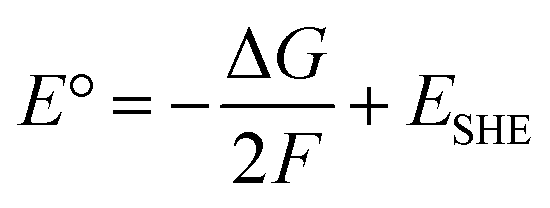 | (1) |
 | ||
| Scheme 4 Proposed outer-sphere mechanisms: (a) two-electron transfer, (b) base-assisted electron transfer, (c) proton-assisted electron transfer. | ||
ΔG represents the Gibbs free energy change that accompanies the two-electron reduction reaction reported above, F is the Faraday constant, and ESHE is the standard hydrogen electrode potential.
Adopting such an approach, Burda and coworkers have calculated the redox potential of several PtIV complexes having chlorido, hydroxido, and carboxylato ligands in axial positions.50 DFT results, obtained using numerous exchange–correlation functionals, have been compared with those of MP2 and CCSD(T) post-HF methods. In order to take into account the impact of the solvent, the implicit IEF-PCM model has been used employing scaled-UAKS radii, whereas to handle the change in the entropy contributions to Gibbs free energies in going from the gas- to the condensed-phase, the correction scheme proposed by Wertz has been used. For the ESHE, the value of −4.281 eV has been adopted. The authors have demonstrated that DFT calculations, including Wertz corrections and scaled radii, are able to reproduce experimental potentials.
Due to the unresolved controversy concerning the utility of the reduction potential obtained as the irreversible peak, Ep, in the cyclic voltammogram, Baik and co-workers have proposed experimental and theoretical approaches able to give reliable results for PtIV complexes in solution-phase redox processes.43 During the study of some PtIV cisplatin-based complex reductions, the authors have shown that the reduction occurs in two steps. Within each step, a single electron is transferred, and the process can be decomposed into electron transfer and Pt–ligand bond cleavage. In particular, in the first step, which involves a one-electron addition to the PtIV centre, the formation of a metastable six-coordinate PtIII intermediate occurs and the standard reduction potential for the PtIV/PtIII couple from the irreversible peak potential in the cyclic voltammogram, within the framework of Marcus theory, can be extracted. In the same way, the magnitude of the standard redox potential can be computationally determined as the energy change that accompanies the one-electron transfer for the reduction of the six-coordinate PtIV to the corresponding six-coordinate PtIII species with the axial ligands remaining in the metal centre coordination sphere (see Scheme 5). The reduction potential is calculated as:
| E° = −ΔGsol – ESHE | (2) |
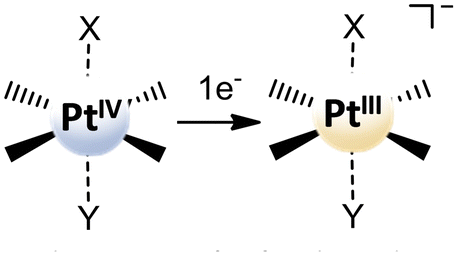 | ||
| Scheme 5 First step one-electron transfer for the reduction of six-coordinate PtIV complexes in the decomposition scheme proposed by Baik et al.43 | ||
In light of the decomposition scheme suggested by Baik et al. to be used for the estimation of the redox potential of PtIV complexes, a detailed theoretical study on the mechanism of electrochemical reduction of the prototypical PtIV anticancer complex [Pt(NH3)2Cl2(CH3COO)2] to the corresponding cisplatin active species has been performed by Re and coworkers.46 The outcomes of this investigation confirm that the initial one-electron reduction occurs through a stepwise mechanism via a metastable six-coordinate PtIII intermediate and subsequent acetate ligand detachment. Several combinations of DFT functionals and basis sets have been tested, and the results have also been used to estimate the redox potential of the complex under investigation, selecting the B3LYP/aug-cc-pVTZ level of theory as the most accurate for reproducing the experimental value.
Furthermore, an outer-sphere mechanism named “base-assisted outer-sphere” electron transfer (Scheme 4b) has been proposed and computationally tested by Ariafard and coworkers in the presence of ascorbic acid as RA.48 This mechanism has been recognized considering that, although the monodeprotonated form of ascorbic acid has the highest concentration in physiological conditions, the dianionic form, Asc2−, is several orders of magnitude more reactive. Due to this very high reactivity, PtIV complexes are immediately reduced via an outer-sphere two-electron transfer mechanism. This mechanism involves the presence of an appropriate base in the environment for accelerating the reduction reaction by deprotonating the AscH− most abundant form of the reducing agent. This deprotonation reaction requires that an energetic cost be paid as a transition state has to be overcome.
One of the first detailed computational studies on the reduction mechanism of PtIV complexes has been performed by Burda and co-workers.49 The authors have investigated the mechanism of activation by reduction of satraplatin (cis,trans,cis-[PtCl2(OAc)2(cha)(NH3)], cha =cyclohexylamine), in the presence of ascorbic acid as a RA. All three protonation forms of ascorbic acid have been considered, and a further variant of the outer-sphere mechanism, named “proton-assisted electron transfer”, has been introduced (Scheme 4c). As it can be deduced from the denomination of the mechanism, a proton shift from the RA to an axial ligand of the PtIV complex takes place, whereas two electrons pass from the reducing species to the metal center via an outer-sphere mechanism. In the examined satraplatin complex, the proton is transferred to one of the acetato ligands. As a consequence, the PtII species is released into the reaction environment, together with the formed acetic acid, the second axial ligand, and the fully oxidized form of the RA, which is dehydroascorbic acid (DHA). This mechanism is considered only for the AscH2 and AscH− forms, where the corresponding proton is present, and, when the reaction is investigated in the presence of the AscH2 agent, an additional water molecule is included in the reduction system, which acts as an acceptor of the second proton from the RA to generate DHA. It is worth mentioning that both proposed base-assisted and proton-assisted mechanisms, even if classified as outer-sphere mechanisms, depend not only on the value of the complex redox potential, but also on the presence in the environment of specific compounds able to assist the process.
Axial ligands
As underlined above, axial ligands are of pivotal importance in determining the chemical and biological properties of PtIV complexes and can be properly designed to accomplish specific aims such as modulating the reduction potential, increasing lipophilicity, interfering with cellular processes, targeting cancer cells, or targeting intracellular compartments. According to what was proposed by Gibson,33,65 axial ligands can be classified into three categories: (1) “innocent” because they are devoid of any biological activity, such as hydroxides, acetates, and chlorides; (2) targeting agents able to make the complex selective towards cancer cells; and (3)”bioactive” ligands that disturb cellular processes.In the next paragraphs, several examples of computational investigations of the reduction of PtIV complexes having ligands essentially belonging to the first class will be illustrated.
Chlorido ligands
Among the potential axial ligands, chlorides are very representative examples, as they have been included in the axial position of many PtIV complexes bearing several leaving and ancillary ligands in equatorial positions. Concerning the role played by axial ligands in influencing the propensity of PtIV complexes to be reduced, it has been reported that the presence of chlorido ligands in the axial position instead of hydroxido or carboxylato ligands should favour the reduction reaction.34 This propensity is ascribed to both the less negative reduction potential values and the possibility of forming a bridge between the platinum centre and the RA, enabling the electron transfer according to an inner-sphere mechanism (mechanism (a) in Scheme 3).Despite several studies reported in the literature for the synthesis of PtIV prodrugs bearing chlorido ligands in axial positions, only for a few of them has a detailed computational investigation been carried out to clarify the mode of release of the square-planar PtII active species.47,48,52,66,67 Among these, three examples have been selected that can be useful for elucidating the real mechanism of PtIV complex reduction. Two of them are classical PtIV-based complexes of the type [Pt(N^N)Cl4], with N^N = ethylenediamine, diaminocyclohexane (tetraplatin in Scheme 1), whose activity, once reduced, remains that of classical PtII complexes: hydrolysis and DNA platination. The third system is a PtIV–salphen complex [Pt(salphen)Cl2] able to target a structurally different DNA, the G-quadruplex.68 In the theoretical exploration, to reduce the computational effort, the complex structure has been simplified by replacing piperidine groups with hydrogen atoms and named PtIV–Sal (Scheme 6).67 The mechanisms of action of these two types of complexes will be separately addressed and discussed on the basis of the employed RA.
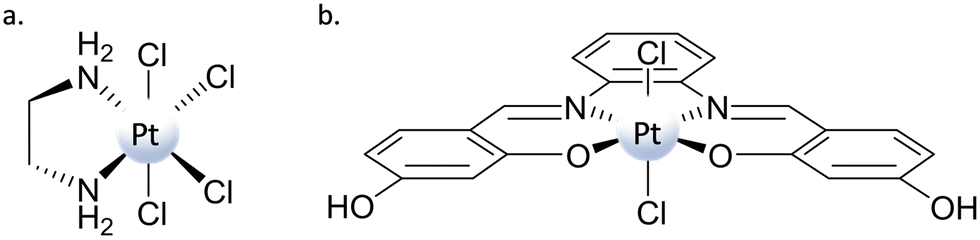 | ||
| Scheme 6 Structure of (a) [Pt(en)Cl4] and (b) PtIV–Sal complexes. | ||
 | ||
| Fig. 1 Free energy profiles describing the reduction of the tetraplatin complex by AscH−via inner sphere mechanisms (a) and (c) in water. In parentheses, electronic energies are provided. Adapted from ref. 41 with permission. Copyright 2017 by The Royal Society of Chemistry. | ||
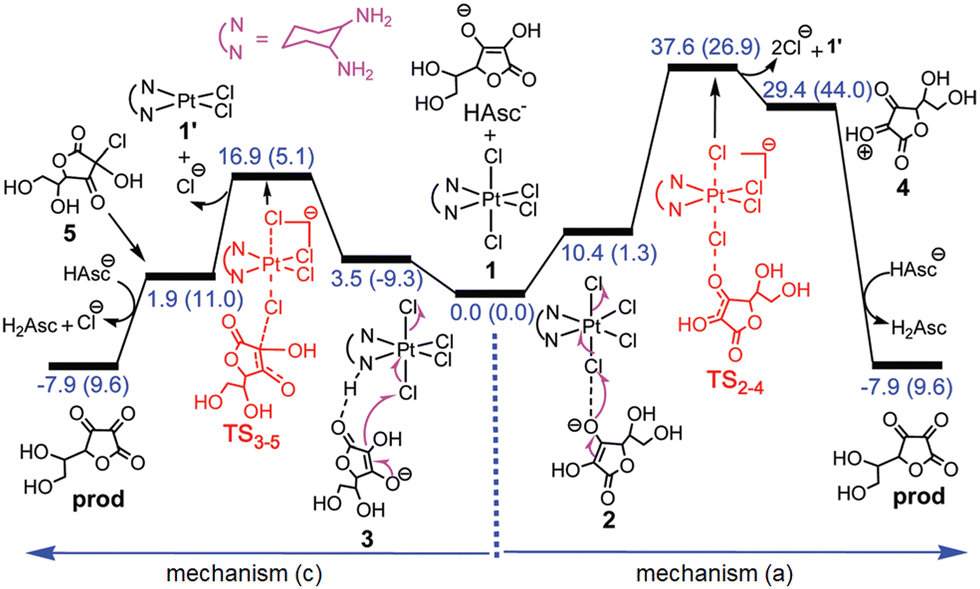
Outer-sphere mechanisms have also been explored, and on the basis of the obtained results, the novel mechanism classified as (b) base-assisted in Scheme 4 has been proposed. The assistance of both AscH− or HCO3− bases has been tested for their ability to deprotonate the ascorbate to form the very reactive Asc2− species that transfers two electrons to the PtIV complex by an outer-sphere mechanism. On the basis of the obtained results and the calculated energy barriers, the authors concluded that both inner- and outer-sphere mechanisms are viable. Nevertheless, since the enolate β-carbon attack inner-sphere reduction (Fig. 1) is calculated to be faster than the deprotonation of AscH−, the inner-sphere prevails.
Shortly after, Ariafard and co-workers added another tessera to the puzzle of PtIV complexes reduction, inspecting the reduction mechanism of [Pt(en)Cl4] (en = ethylenediamine) by sulphur-containing biological reductants, like L-Cys and L-Met.47 Such types of compounds can act according to the inner-sphere mechanism (a), with Cl− as the bridging ligand. The reduction is supposed to involve the electrophilic attack of the highly polarised chlorido ligand of the PtIV complex on the thiol.60 The reductive elimination results in the production of the RSCl species and the concomitant release of the trans ligand. The RSCl is further consumed, leading to the formation of an RSSR unit. Exploring the reaction mechanism using the most abundant zwitterionic form in the physiological environment of the RA, the authors have evidenced that the reaction can be assisted by the carboxylate group. However, the energy cost required for the Cl-bridged mechanism to occur is very high along both carboxylate-assisted and non-assisted reaction paths. Moreover, the activation energy barrier computed for the oxidation of L-Cys, mediated or not by its carboxylate group, has been computed to be considerably higher than that of L-Met, contrary to what has been experimentally observed. Since the thiol functional group is considerably more reactive when it is converted into thiolate, the authors have demonstrated that in the case of the L-Cys agent, an unusual tautomerization can occur, surpassing a low energy barrier for a transition state that permits the transfer of the proton from the S atom to one of the oxygen atoms of the carboxylato group (TS2–12). The free energy profile describing the whole mechanism that involves the active tautomer (13), formed by an H-transfer, is sketched in Fig. 2.
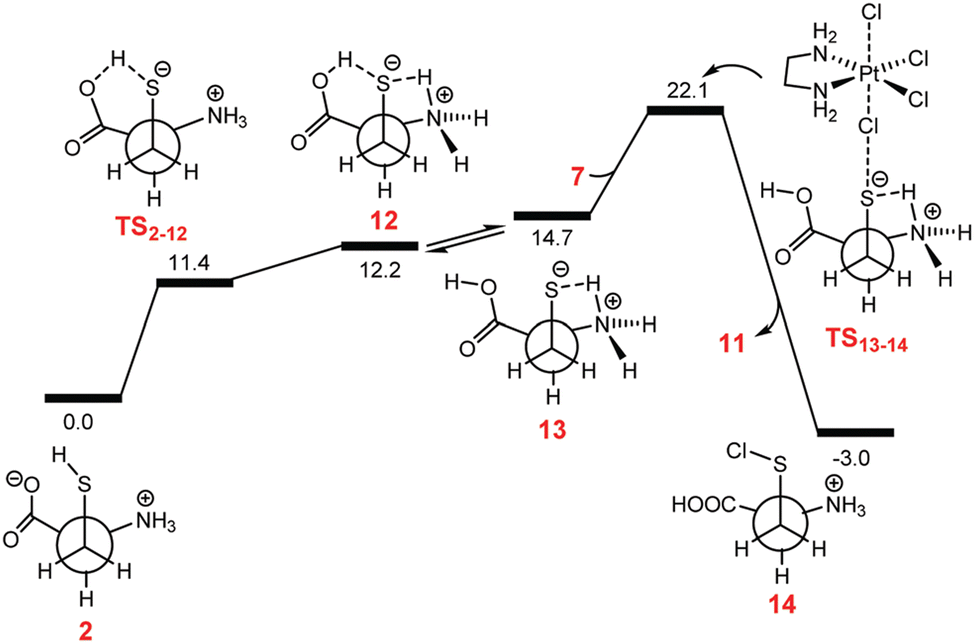 | ||
| Fig. 2 Free energy profile describing the inner-sphere ligand-bridged reduction of the [Pt(en)Cl4] complex by Cys. Reproduced from ref. 40 with permission. Copyright 2018 by The Royal Society of Chemistry. | ||
The reduction requires only 7.4 kcal mol−1 to take place, thus justifying the higher activity of L-Cys towards PtIV to PtII reduction.
Additionally, we have applied the Baik approach to both tetraplatin and [Pt(en)Cl4] complexes to estimate their redox potential. The calculated values with respect to the SHE are 0.412 eV for the former and 0.444 eV for the latter. Additionally, for these two di-chlorido PtIV complexes, highly positive values have been obtained, in line with the trend of the propensity to be reduced of PtIV complexes as a function of the identity of axial ligands.33,43 Therefore, an outer-sphere mechanism appears to be viable for both complexes.
The reduction of the PtIV–Sal complex has been explored by considering the action of two possible bioreductants: the active form of ascorbic acid, AscH−, and Cys as a model of sulphur-containing reductants. All the plausible reaction mechanisms proposed for the leaving of Cl− axial ligands, which include inner-sphere (a) ligand-bridged, (c) enolate β-carbon attack, and outer-sphere mechanisms, have been fully explored. It is worth reminding ourselves that mechanism (a) can be accessible to both the considered RAs, while mechanism (c) may only occur in the presence of ascorbate. The outcomes summarized here showed that when ascorbate is the RA, the enolate β-carbon attack is preferred over the ligand-bridged mechanism, as the corresponding activation energy barriers are 3.0 and 20.9 kcal mol−1, respectively. In addition, while the former mechanism is accompanied by an energy gain of 14 kcal mol−1, the formation of the PtII active species is endergonic by 6.0 kcal mol−1 in the latter. On the other hand, the only viable inner-sphere mechanism for the reductive action of Cys relied on the (a) ligand-bridge type, which is accompanied by S–Cl bond formation and the concomitant loss of the chloride occupying the trans position. It requires 7.2 kcal mol−1 to take place and is also favoured from a thermodynamic point of view, being exergonic by 15.5 kcal mol−1.
The outer-sphere mechanism has been explored as well, and the decomposition scheme proposed by Baik et al. (Scheme 5) has been adopted to estimate the reduction potential. Accordingly, the free energy change in solution that accompanies the first one-electron transfer leading to the formation of the six-coordinate PtIII complex has been evaluated and the reduction potential calculated employing eqn (2). The obtained value of 0.162 eV, if compared with that computed for cis-[PtCl4(NH3)2] using the same approach (0.374 eV), suggested that the PtIV–Sal complex may also be reduced following an outer-sphere mechanism.
Hydroxido ligands
The information about the ease of reduction of PtIV prodrugs gained from the measurement of the peak potential leads to the conclusion that, typically, the potentials are more negative when the axial ligands are hydroxides. However, the presence of OH− ligands, while rendering the electrochemical potential more negative and, then, the corresponding complexes less prone to reduction, offers the possibility of creating a bridge facilitating the electron transfer. As a consequence, many examples exist in the literature that evidence how the observed rates of reduction may or may not be correlated44,61 to the Ep values when PtIV complexes with hydroxido ligands are taken into consideration. Here, an example of a computational study trying to identify the correct mechanism that is operative for the reduction of PtIV complexes bearing hydroxido axial ligands is reported.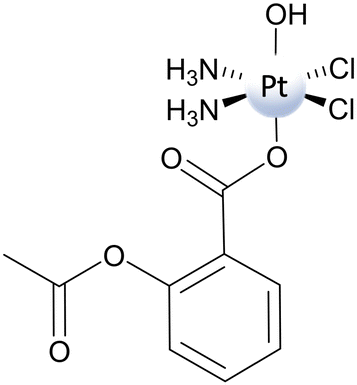 | ||
| Scheme 7 Structure of Asplatin. | ||
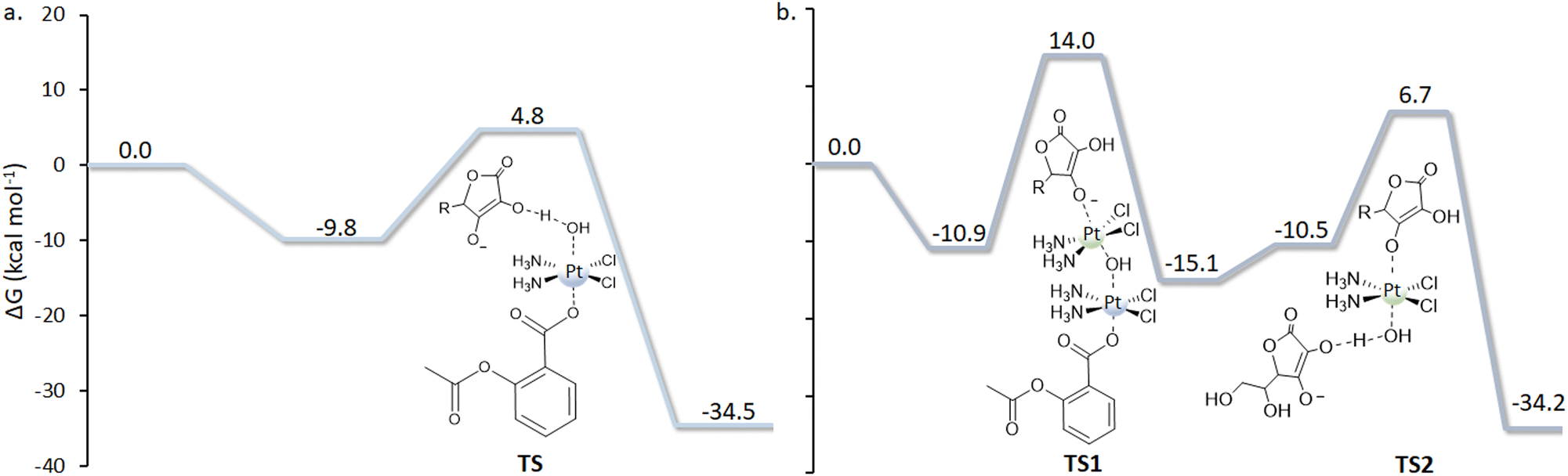 | ||
| Fig. 3 Reduction mechanism following mechanism (b), the shift of a hydride (H−) unit from the OH group of the ascorbate to the hydroxido ligand (panel a), and the PtII-catalyzed mechanism (d) (panel b). | ||
Indeed, the hydroxido ligand plays the role of a bridge, but it is the shift of a hydride (H−) unit from the OH group of the ascorbate to the hydroxido ligand that allows the two-electron transfer and the simultaneous formation of a water molecule. Trans Asp ligand release, cisplatin formation, and the oxidation of ascorbate to DHA complete the process. Therefore, the OH− ligand permits the electron transfer, but it is also directly involved in the reduction process as it generates the water molecule and leads to the formation of DHA. The reaction occurs through the TS depicted in Fig. 3a, overcoming an energy barrier of 14.6 kcal mol−1, and is calculated to be highly exergonic (−34.5 kcal mol−1). Therefore, the new type of mechanism named “ligand-bridge-H transfer”, as aforementioned, has been proposed for the first time. The investigation of mechanism (c) in Scheme 3 involves the nucleophilic attack of the enolate β-carbon of the ascorbate on the OH ligand, leading to the formation of a geminal diol product, causing the subsequent loss of the trans Asp ligand and the generation of the cisplatin active species. The height of the energy barrier that is necessary to overcome is 20.7 kcal mol−1. The subsequent step involves the elimination of water from the geminal diol product, which leads to the formation of the DHA oxidized species. In this process, an auxiliary water molecule, acting as a proton shuttle, assists the elimination, and the calculated energy barrier for the transition state is 24.4 kcal mol−1. The whole process is calculated to be exergonic by 31.8 kcal mol−1.
The (d) type reduction, instead, takes place following an autocatalytic mechanism and involves the PtII complex catalytic action that, in the reported case, is cisplatin. As described above, to carry out its catalytic action and facilitate the two-electron transfer, the PtII complex has to be sandwiched between the PtIV complex and the RA (AscH−). Indeed, in the first step, an initial dimer adduct, AscH–PtII–OH–PtIV–Asp, is formed (Fig. 3b). The hydroxido ligand, together with the deprotonated oxygen of the AscH− unit, begins to coordinate with the square planar PtII complex in the transition state TS1, and, as a final product, the new OH–PtIV–AscH complex is formed. The second ligand in the axial position of Asplatin, Asp, is released spontaneously, and cisplatin formation is achieved. Along this pathway, the two electrons are transferred through the bridge action of the OH− ligand. The process is completed when the PtIV intermediate, generated in the first step, is further reduced by a second AscH−, realized according to TS2. The second reduction takes place, overcoming an activation energy barrier of 17.2 kcal mol−1. The rate-determining step of the process involves the action of the catalyst, with the Asplatin reduction and Asc–PtIV–OH intermediate formation, and requires 24.9 kcal mol−1 to occur.
It clearly appears that, among the three explored mechanistic hypotheses, the most viable is the ligand-bridge-H transfer. This conclusion has been further confirmed by a metadynamics study of the Asplatin reduction by ascorbate in which, in order to reduce the complexity of the problem, a new approach named harmonic linear discriminant analysis (HLDA) has been applied.71 The standard redox potential of the Asplatin complex has been calculated adopting the decomposition scheme proposed by Baik and coworkers, and a value of −0.239 eV has been obtained. If this value is compared with the measured value of −0.536 eV versus the normal hydrogen electrode, it is confirmed that the peak potential is more negative than the normal potential, corresponding to a redox stability higher than the real one. Nonetheless, this negative value of the redox potential proves that the outer-sphere reduction is not viable.
The results of the investigation on the Asplatin reduction summarized here demonstrate that the inner-sphere ligand-bridge-H transfer mechanism is the preferred one, corresponding to the lowest energy barrier. The calculated redox potential, even less negative than the experimentally measured peak potential, confirms the low propensity of the complex to be reduced by an outer-sphere mechanism. Moreover, the classical inner-sphere ligand-bridge mechanism is proven to be unlikely for hydroxido ligands. The new proposed ligand-bridge-H transfer mechanism allows for the resolution of the contradiction between the measured rapidity of the hydroxido PtIV complex reduction and the impracticality of outer-sphere and classical inner-sphere ligand-bridge mechanisms.
Carboxylato ligand
The preparation of carboxylato PtIV complexes commenced later with respect to that of hydroxido and chlorido complexes that are synthesized by oxidation of the corresponding square-planar PtII precursors with Cl2 or H2O2. Only the optimization of the carboxylation process of di-hydroxido PtIV precursors has allowed the synthesis of a large number of carboxylato complexes. Among them, the diacetato satraplatin complex is the first oral deliverable platinum-containing anticancer drug that has entered a Phase III trial.72 Since no increase in overall survival has been noted, however, the drug has not obtained FDA approval. With respect to chlorido and hydroxido ligands that can act as electron bridges, carboxylato ligands are considered to be weak electron transfer bridges, and their measured peak potentials are intermediate between those of hydroxido and chlorido ligands. Furthermore, while halido and hydroxido ligands cannot be subjected to modification, carboxylato groups are the ligands mainly investigated for the synthesis of functionalized PtIV prodrugs with tuned chemical, physical, and pharmacological properties. A computational screening has been carried out by Cerón-Carrasco on a series of PtIV complexes decorating oxoplatin with five of the latest FDA-approved oncology drug sets (AOD9) coordinated through the carboxylato moiety.73Two examples of computational studies carried out on carboxylato PtIV complexes are illustrated here, aiming at highlighting that both the characteristics and the versatility of these complexes can be exploited.
One of the most recent and interesting multi-action PtIV complexes is the Ru–Pt complex proposed by Karges et al.74 This novel RuII–PtIV conjugate complex (Scheme 8) combines three biological activities: (i) the antineoplastic activity of the platinum complex; (ii) the light-mediated action of the Ru complex acting as a photosensitizer for photodynamic therapy (PDT); and (iii) the histone deacetylase (HDAC) inhibition action of 4-phenylbutyrate (PB). All these actions should be activated by the primary reduction of the conjugate to yield cisplatin, PB, and the Ru-based chromophore in the presence of a biological RA.
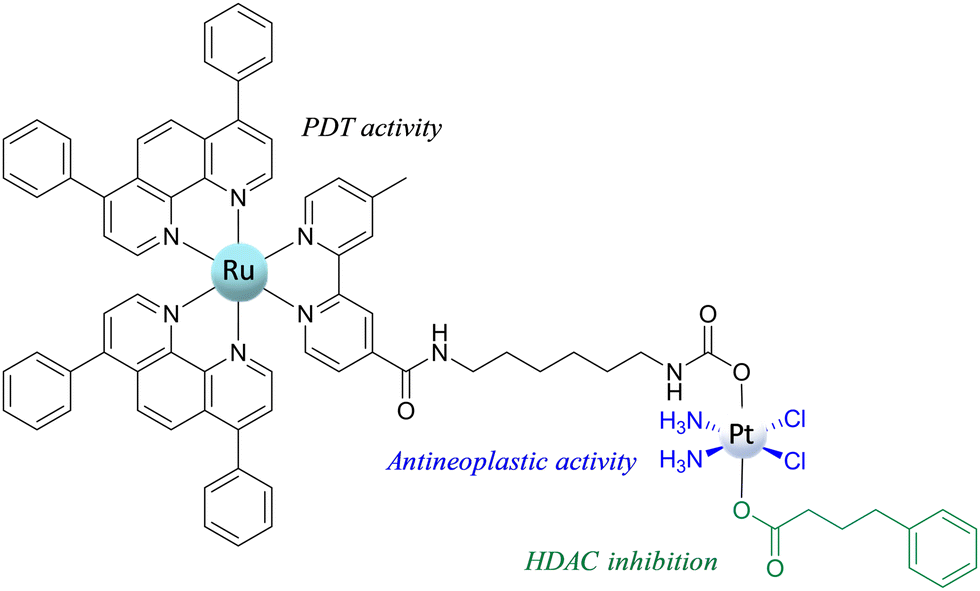 | ||
| Scheme 8 Structure of the RuII–PtIV conjugate complex. | ||
The reduction mechanism, assisted by ascorbate, of the PtIV prodrug to the PtII active species has been explored, taking into consideration all the possible modes of attack of the reductant for releasing the axial ligands and affording active cisplatin.75 Specifically, ligand-bridge-H transfer and enolate β-carbon attack in the presence of AscH− as a model of the RA have been studied by taking into account that the interaction may be established through both the axial ligands, carbamate (Ru-motif) and carboxylate (PB-motif). As in the reference paper, it is underlined that the long aliphatic chain has been used to make the RuII and PtIV portions independent, and the elucidation of the reduction mechanism has been performed considering a model of the complex reported in Scheme 9.
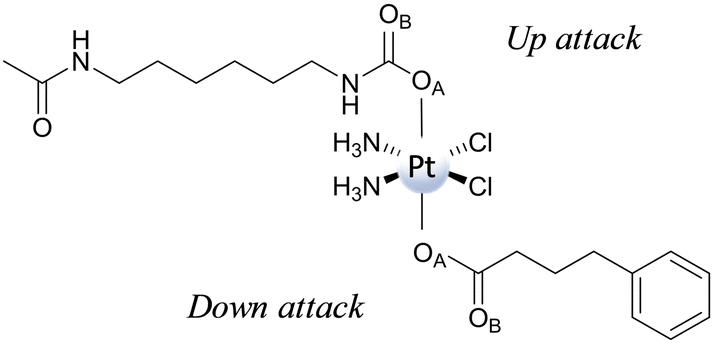 | ||
| Scheme 9 Portion of the RuII–PtIV complex used to elucidate the reduction mechanism. | ||
Then, both axial ligands have been considered to be directly involved in the reduction mechanism, distinguishing the two paths as Up and Down attacks. Furthermore, the participation of both oxygen atoms, labelled OA and OB, of the two ligands has been explored. The ligand-bridge-H transfer pathway that involves a bridge formation with the OA atom occurs, overcoming an energy barrier of 25.5 and 32.1 kcal mol−1, respectively, for Up and Down attacks. Furthermore, the reaction is exergonic by 8.6 kcal mol−1 for Up and 6.1 kcal mol−1 for Down attacks, respectively. The same reactions, but with OB playing the role of bridge, entail energy barriers of 29.4 and 30.6 kcal mol−1, respectively, for Up and Down attacks. The product is more stable than the initial adduct by 0.3 kcal mol−1 and 1.7 kcal mol−1 along the Up and Down attack pathways, respectively. From a kinetic point of view, the reduction mechanism is favoured when ascorbate approaches carbamate (Up attack). Similar behaviour has also been found for the enolate β-carbon attack, with the favoured pathway being that entailing the ascorbate interaction with the carbamate ligand from the Up side, requiring 27.1 kcal mol−1 to take place versus 35.1 kcal mol−1 needed for the Down attack. Cisplatin formation is accompanied by the generation of an ascorbate with an additional carboxylate moiety bound to the enolate β-carbon. The reaction is exergonic by 11.0 and 7.8 kcal mol−1 for Up and Down attacks, respectively. Comparing the energy profiles of the two different mechanisms, the ligand-bridge-H transfer turns out to be the favoured mechanism when functional groups such as carboxylates and carbamates are present in the axial position; the same result was found for hydroxido axial ligands. In addition, as a covalent C–O bond between ascorbate and the ligand is formed, the enolate β-carbon attack needs an additional step for the formation of final products to occur. The conclusion that the favoured reduction mechanism is the ligand-bridge-H transfer on the carbamate is consistent with the experimentally observed behaviour of a series of Pt(IV) complexes having carbamates and carboxylates as axial ligands, showing that complexes having carbamates are reduced faster.76
To evaluate whether the PtIV complex can be reduced by an outer-sphere mechanism, the standard reduction potential has been calculated using the Baik decomposition method. The computed value with respect to the SHE of −0.314 eV suggests, if compared with that of other complexes, that this complex is difficult to be reduced by this route.
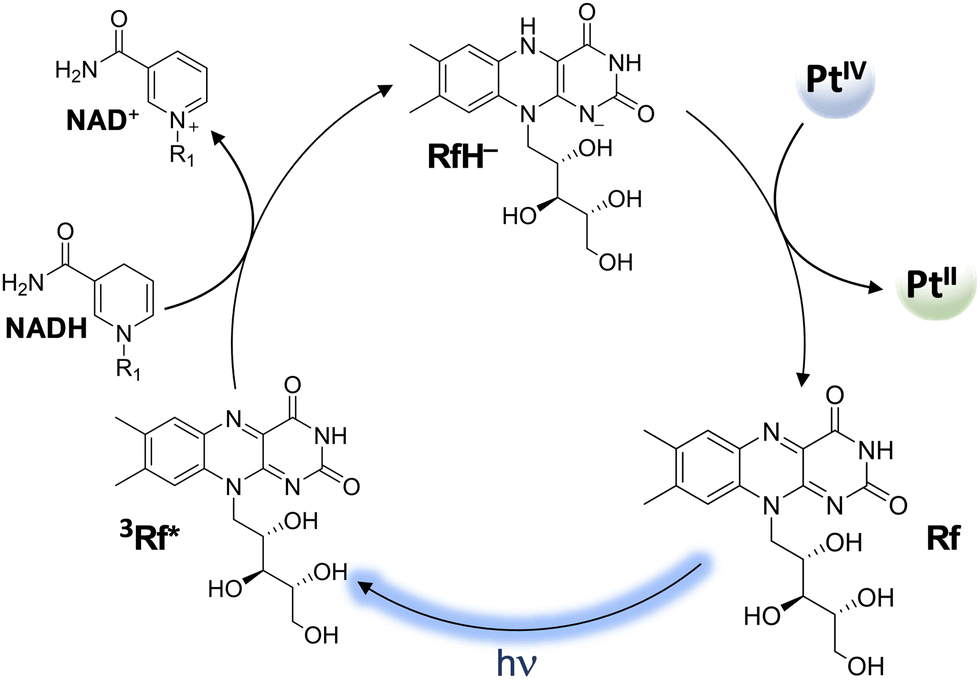 | ||
| Scheme 10 Mechanism of the photocatalytic activation of PtIV prodrugs by riboflavin. (R1 = adenine dinucleotide) | ||
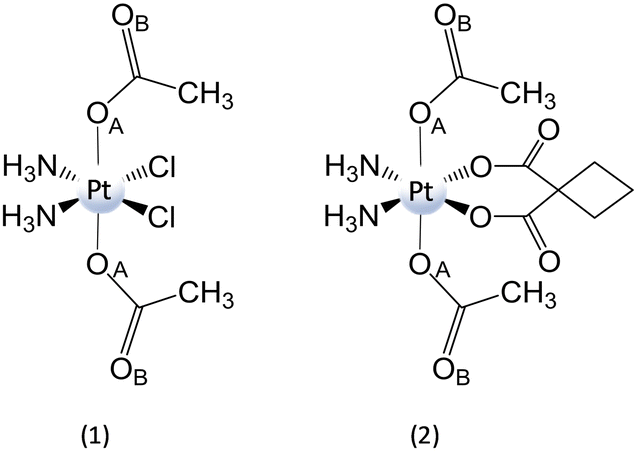 | ||
| Scheme 11 Structure of complexes (1) cis,cis,trans-[Pt(NH3)2Cl2(CH3CO2)2] and (2) cis,cis,trans-[Pt(NH3)2(CBDCA)(CH3CO2)2]. | ||
It is worth underlining that carboxylato ligands are able to form bridges with RAs by both oxygens: the one bound to the metal centre and the carbonylic one. Therefore, DFT calculations for the conversion of PtIV complexes by RfH−, MADH, and AscH− have been performed considering two different interaction modes. However, comparable trends have been calculated along both pathways, even if the pathway involving the formation of a bridge with oxygen OA is slightly preferred kinetically. Focusing only on OA, for all three donors, the hydride-donating moiety (N–H, C–H, and O–H) transfers 2e− and a proton to the OA, causing the detachment of the two axial ligands and the formation of cisplatin or carboplatin.
All the products obtained are strongly stabilized with respect to the entrance channel. The obtained free energy profiles prove that the reduction of both complexes takes place in the order RfH− > AscH− > NADH, the same trend observed experimentally.54 Moreover, RfH− results in a very efficient RA with an activation barrier lower than 10 kcal mol−1. In addition, calculations show that complex (1) having cisplatin in the square plane is reduced more readily than (2).
Moreover, the values of the reduction potential have been calculated for both complexes. The low reduction potentials of (1) (Ep = −0.324 V vs. SHE) and (2) (Ep = −0.693 V vs SHE)81 suggest that an outer-sphere mechanism is unlikely for both complexes.
Conclusions
Reduction of PtIV complexes by bioreductants appears to be the key step of their mechanism of action for eliminating the axial ligands and releasing the corresponding active PtII species that are protected from deactivation en route before reaching their biological targets. Several parameters can be varied for tuning the activity of the prodrugs, requiring that their reduction be neither too fast, in order to avoid premature activation outside the cell, nor too slow to ensure the cytotoxic PtII drug is released effectively in tumor cells. The mechanisms of reduction, classified as inner- and outer-sphere, depend essentially on the nature of the axial ligands and, when an inner-sphere mechanism is operative, also on the identity of the reducing agent. In the present paper, an overview is given of the computational studies performed to identify the correct mechanism of the decisive step for the activation of PtIV compounds. All the proposed inner- and outer-sphere mechanistic hypotheses have been taken into consideration, and several examples of computational investigations of the reduction of PtIV complexes bearing hydroxido, chloride, and carboxylato ligands have been illustrated. We hope, with the overview given here, to contribute to properly addressing all the efforts presently directed towards the design and synthesis of PtIV prodrugs with both controllable activation characteristics and enhanced properties allowing targeted treatments and mitigation of severe side effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Italian Association for Cancer Research, AIRC (ID 25578). S. S. thanks the financial support from ICSC – Centro Nazionale di Ricerca in High Performance Computing, Big Data and Quantum Computing, funded by European Union – NextGenerationEU – PNRR, Missione 4 Componente 2 Investimento 1.4. The authors gratefully acknowledge the computing time granted by the CINECA (project IsCa3_MoAMCPCT).Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, Ca-Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed.
- C. Pucci, C. Martinelli and G. Ciofani, Innovative approaches for cancer treatment: current perspectives and new challenges, Ecancermedicalscience, 2019, 13, 961 CrossRef PubMed.
- B. Rosenberg, L. Van Camp and T. Krigas, Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- E. Wiltshaw, Cisplatin in the Treatment of Cancer, Platinum Met. Rev., 1979, 23, 90–98 CAS.
- G. J. Bosl and R. J. Motzer, Testicular Germ-Cell Cancer, N. Engl. J. Med., 1997, 337, 242–254 CrossRef CAS PubMed.
- M. Morris, P. J. Eifel, J. Lu, P. W. Grigsby, C. Levenback, R. E. Stevens, M. Rotman, D. M. Gershenson and D. G. Mutch, Pelvic Radiation with Concurrent Chemotherapy Compared with Pelvic and Para-Aortic Radiation for High-Risk Cervical Cancer, N. Engl. J. Med., 1999, 340, 1137–1143 CrossRef CAS PubMed.
- A.-M. Florea and D. Büsselberg, Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects, Cancers, 2011, 3, 1351–1371 CrossRef CAS PubMed.
- B. Behmand, J.-L. Marignier, M. Mostafavi, J. R. Wagner, D. J. Hunting and L. Sanche, Radiosensitization of DNA by Cisplatin Adducts Results from an Increase in the Rate Constant for the Reaction with Hydrated Electrons and Formation of Pt I, J. Phys. Chem. B, 2015, 119, 9496–9500 CrossRef CAS PubMed.
- S. Dasari and P. Bernard Tchounwou, Cisplatin in cancer therapy: molecular mechanisms of action, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- C. F. Harrington, R. C. Le Pla, G. D. D. Jones, A. L. Thomas and P. B. Farmer, Determination of Cisplatin 1,2-Intrastrand Guanine−Guanine DNA Adducts in Human Leukocytes by High-Performance Liquid Chromatography Coupled to Inductively Coupled Plasma Mass Spectrometry, Chem. Res. Toxicol., 2010, 23, 1313–1321 Search PubMed.
- Z. H. Siddik, Cisplatin: mode of cytotoxic action and molecular basis of resistance, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- E. R. Jamieson and S. J. Lippard, Structure, Recognition, and Processing of Cisplatin−DNA Adducts, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- M. A. Fuertes, C. Alonso and J. M. Pérez, Biochemical Modulation of Cisplatin Mechanisms of Action: Enhancement of Antitumor Activity and Circumvention of Drug Resistance, Chem. Rev., 2003, 103, 645–662 CrossRef CAS PubMed.
- N. Pabla and Z. Dong, Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies, Kidney Int., 2008, 73, 994–1007 CrossRef CAS PubMed.
- T. Scattolin, V. A. Voloshkin, F. Visentin and S. P. Nolan, A critical review of palladium organometallic anticancer agents, Cell Rep. Phys. Sci., 2021, 2, 100446 CrossRef CAS.
- G. Mazzone, S. Scoditti, R. Caligiuri, L. Ricciardi, E. Sicilia, M. G. Lupo, I. Rimoldi, N. Godbert, M. La Deda, A. Ionescu, M. Ghedini, I. Aiello and G. Facchetti, Cytotoxicity of Alizarine versus Tetrabromocathecol Cyclometalated Pt(II) Theranostic Agents: A Combined Experimental and Computational Investigation, Inorg. Chem., 2022, 61, 7188–7200 CrossRef CAS PubMed.
- M. Ghedini, A. Golemme, I. Aiello, N. Godbert, R. Termine, A. Crispini, M. La Deda, F. Lelj, M. Amati and S. Belviso, Liaisons between photoconductivity and molecular frame in organometallic Pd(II) and Pt(II) complexes, J. Mater. Chem., 2011, 21, 13434 RSC.
- T. J. Carneiro, A. S. Martins, M. P. M. Marques and A. M. Gil, Metabolic Aspects of Palladium(II) Potential Anti-Cancer Drugs, Front. Oncol., 2020, 10, 2218 Search PubMed.
- M. Vojtek, M. P. M. Marques, I. M. P. L. V. O. Ferreira, H. Mota-Filipe and C. Diniz, Anticancer activity of palladium-based complexes against triple-negative breast cancer, Drug Discovery Today, 2019, 24, 1044–1058 CrossRef CAS PubMed.
- I. Ritacco, G. Mazzone, N. Russo and E. Sicilia, Investigation of the Inertness to Hydrolysis of Platinum(IV) Prodrugs, Inorg. Chem., 2016, 55, 1580–1586 CrossRef CAS PubMed.
- R. W. Gregg, J. M. Molepo, V. J. A. Monpetit, N. Z. Mikael, D. Redmond, M. Gadia and D. J. Stewart, Cisplatin neurotoxicity: the relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity, J. Clin. Oncol., 1992, 10, 795–803 CrossRef CAS PubMed.
- W. H. Ang, S. Pilet, R. Scopelliti, F. Bussy, L. Juillerat-Jeanneret and P. J. Dyson, Synthesis and Characterization of Platinum(IV) Anticancer Drugs with Functionalized Aromatic Carboxylate Ligands: Influence of the Ligands on Drug Efficacies and Uptake, J. Med. Chem., 2005, 48, 8060–8069 CrossRef CAS PubMed.
- H. Varbanov, S. M. Valiahdi, A. A. Legin, M. A. Jakupec, A. Roller, M. S. Galanski and B. K. Keppler, Synthesis and characterization of novel bis(carboxylato)dichloridobis(ethylamine)platinum(IV) complexes with higher cytotoxicity than cisplatin, Eur. J. Med. Chem., 2011, 46, 5456–5464 CrossRef CAS PubMed.
- H. Anderson, J. Wagstaff, D. Crowther, R. Swindell, M. J. Lind, J. McGregor, M. S. Timms, D. Brown and P. Palmer, Comparative toxicity of cisplatin, carboplatin (CBDCA) and iproplatin (CHIP) in combination with cyclophosphamide in patients with advanced epithelial ovarian cancer, Eur. J. Cancer Clin. Oncol., 1988, 24, 1471–1479 CrossRef CAS PubMed.
- T. J. O’Rourke, G. R. Weiss, P. New, H. A. Burris, G. Rodriguez, J. Eckhardt, J. Hardy, J. G. Kuhn, S. Fields, G. M. Clark and D. D. Von Hoff, Phase I clinical trial of ormaplatin (tetraplatin, NSC 363812), Anticancer Drugs, 1994, 5, 520–526 CrossRef PubMed.
- P. Iyengar, J. C. Hodges, R. Hughes, M. DiMaio, M. Petrone, S. Yun and H. Choy, A phase I study with Satraplatin and simultaneous chest radiation for non-small cell lung cancer, Adv. Lung Cancer, 2012, 01, 13–19 CrossRef.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Metallodrugs are unique: opportunities and challenges of discovery and development, Chem. Sci., 2020, 11, 12888–12917 RSC.
- M. S. Galanski and B. K. Keppler, Carboxylation of Dihydroxoplatinum(IV) Complexes via a New Synthetic Pathway, Inorg. Chem., 1996, 35, 1709–1711 CrossRef CAS PubMed.
- K. R. Barnes, A. Kutikov and S. J. Lippard, Synthesis, Characterization, and Cytotoxicity of a Series of Estrogen-Tethered Platinum(IV) Complexes, Chem. Biol., 2004, 11, 557–564 CrossRef CAS PubMed.
- W. H. Ang, I. Khalaila, C. S. Allardyce, L. Juillerat-Jeanneret and P. J. Dyson, Rational Design of Platinum(IV) Compounds to Overcome Glutathione-S-Transferase Mediated Drug Resistance, J. Am. Chem. Soc., 2005, 127, 1382–1383 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, M. J. McGlinchey and D. Osella, Pt(IV) antitumor prodrugs: dogmas, paradigms, and realities, Dalton. Trans., 2022, 51, 2121–2134 RSC.
- J. Sánchez-Camacho, S. Infante-Tadeo, A. C. Carrasco, S. Scoditti, Á. Martínez, F. Barroso-Bujans, E. Sicilia, A. M. Pizarro and L. Salassa, Flavin-Conjugated Pt(IV) Anticancer Agents, Inorg. Chem., 2023, 62, 5644–5651 CrossRef PubMed.
- E. Wexselblatt and D. Gibson, What do we know about the reduction of Pt(IV) pro-drugs?, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- S. Choi, C. Filotto, M. Bisanzo, S. Delaney, D. Lagasee, J. L. Whitworth, A. Jusko, C. Li, N. A. Wood, J. Willingham, A. Schwenker and K. Spaulding, Reduction and Anticancer Activity of Platinum(IV) Complexes, Inorg. Chem., 1998, 37, 2500–2504 CrossRef CAS.
- P. Gramatica, E. Papa, M. Luini, E. Monti, M. B. Gariboldi, M. Ravera, E. Gabano, L. Gaviglio and D. Osella, Antiproliferative Pt(IV) complexes: synthesis, biological activity, and quantitative structure–activity relationship modeling, JBIC, J. Biol. Inorg. Chem., 2010, 15, 1157–1169 CrossRef CAS PubMed.
- K. Lemma, D. A. House, N. Retta and L. I. Elding, Kinetics and mechanism for reduction of halo- and haloam(m)ine platinum(IV) complexes by L-ascorbate, Inorg. Chim. Acta, 2002, 331, 98–108 CrossRef CAS.
- K. Lemma, A. M. Sargeson and L. I. Elding, Kinetics and mechanism for reduction of oral anticancer platinum(IV) dicarboxylate compounds by L-ascorbate ions, J. Chem. Soc., Dalton Trans., 2000, 1167–1172 RSC.
- F. Basolo, A. F. Messing, P. H. Wilks, R. G. Wilkins and R. G. Pearson, Mechanisms of exchange and substitution reactions in platinum (IV) complexes, J. Inorg. Nucl. Chem., 1958, 8, 203–208 CrossRef CAS.
- F. Basolo and R. G. Pearson, Advances in Inorganic Chemistry and Radiochemistry, Academic Press, 1961, vol. 3, pp. 1–89 Search PubMed.
- J. J. Wilson and S. J. Lippard, Synthesis, Characterization, and Cytotoxicity of Platinum(IV) Carbamate Complexes, Inorg. Chem., 2011, 50, 3103–3115 CrossRef CAS PubMed.
- J. A. Platts, G. Ermondi, G. Caron, M. Ravera, E. Gabano, L. Gaviglio, G. Pelosi and D. Osella, Molecular and statistical modeling of reduction peak potential and lipophilicity of platinum(IV) complexes, JBIC, J. Biol. Inorg. Chem., 2011, 16, 361–372 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, I. Zanellato, I. Bonarrigo, E. Escribano, V. Moreno, M. Font-Bardia, T. Calvet and D. Osella, Synthesis, characterization and antiproliferative activity on mesothelioma cell lines of bis(carboxylato)platinum(IV) complexes based on picoplatin, Dalton Trans., 2012, 41, 3313–3320 RSC.
- M. C. McCormick, K. Keijzer, A. Polavarapu, F. A. Schultz and M. H. Baik, Understanding intrinsically irreversible, non-nernstian, two-electron redox processes: A combined experimental and computational study of the electrochemical activation of Platinum(IV) antitumor prodrugs, J. Am. Chem. Soc., 2014, 136, 8992–9000 CrossRef CAS PubMed.
- J. Z. Zhang, E. Wexselblatt, T. W. Hambley and D. Gibson, Pt(IV) analogs of oxaliplatin that do not follow the expected correlation between electrochemical reduction potential and rate of reduction by ascorbate, Chem. Commun., 2012, 48, 847–849 RSC.
- J. Dong, Y. Ren, S. Huo, S. Shen, J. Xu, H. Tian and T. Shi, Reduction of ormaplatin and cis-diamminetetrachloroplatinum(IV) by ascorbic acid and dominant thiols in human plasma: Kinetic and mechanistic analyses, Dalton Trans., 2016, 45, 11326–11337 RSC.
- I. Tolbatov, C. Coletti, A. Marrone and N. Re, Insight into the Electrochemical Reduction Mechanism of Pt(IV) Anticancer Complexes, Inorg. Chem., 2018, 57, 3411–3419 CrossRef CAS PubMed.
- A. Chipman, B. F. Yates, A. J. Canty and A. Ariafard, Reduction of a platinum(iv) prodrug model by sulfur containing biological reductants: computational mechanistic elucidation, Chem. Commun., 2018, 54, 10491–10494 RSC.
- Z. Ejehi and A. Ariafard, A computational mechanistic investigation into the reduction of Pt(IV) prodrugs with two axial chlorides by biological reductants, Chem. Commun., 2017, 53, 1413–1416 RSC.
- F. Šebesta and J. V. Burda, Interactions of Ascorbic Acid with Satraplatin and its trans Analog JM576: DFT Computational Study, Eur. J. Inorg. Chem., 2018, 1481–1491 CrossRef.
- F. Šebesta, K. Baxová and J. V. Burda, Redox Potentials for Tetraplatin, Satraplatin, Its Derivatives, and Ascorbic Acid: A Computational Study, Inorg. Chem., 2018, 57, 951–962 CrossRef PubMed.
- S. Scoditti, V. Vigna, E. Dabbish and E. Sicilia, Iodido equatorial ligands influence on the mechanism of action of Pt(IV) and Pt(II) anti-cancer complexes: A DFT computational study, J. Comput. Chem., 2021, 42, 608–619 CrossRef CAS PubMed.
- E. Dabbish, F. Ponte, N. Russo and E. Sicilia, Antitumor Platinium(IV) Prodrugs: A Systematic Computational Exploration of Their Reduction Mechanism by L-Ascorbic Acid, Inorg. Chem., 2019, 58, 3851–3860 CrossRef CAS PubMed.
- F. Ponte, N. Russo and E. Sicilia, Insights from Computations on the Mechanism of Reduction by Ascorbic Acid of PtIV Prodrugs with Asplatin and Its Chlorido and Bromido Analogues as Model Systems, Chem. – A Eur. J., 2018, 24, 9572–9580 CrossRef CAS PubMed.
- S. Scoditti, E. Dabbish, G. E. Pieslinger, E. Rezabal, X. Lopez, E. Sicilia and L. Salassa, Flavin-mediated photoactivation of Pt(IV) anticancer complexes: computational insights on the catalytic mechanism, Phys. Chem. Chem. Phys., 2022, 24, 5323–5329 RSC.
- D. Corinti, M. E. Crestoni, S. Fornarini, F. Ponte, N. Russo, E. Sicilia, E. Gabano and D. Osella, Elusive Intermediates in the Breakdown Reactivity Patterns of Prodrug Platinum(IV) Complexes, J. Am. Soc. Mass Spectrom., 2019, 30, 1881–1894 CrossRef CAS PubMed.
- D. Corinti, M. E. Crestoni, S. Fornarini, E. Dabbish, E. Sicilia, E. Gabano, E. Perin and D. Osella, A multi-methodological inquiry of the behavior of cisplatin-based Pt(IV) derivatives in the presence of bioreductants with a focus on the isolated encounter complexes, J. Biol. Inorg. Chem., 2020, 25, 655–670 CrossRef CAS PubMed.
- A. Nemirovski, I. Vinograd, K. Takrouri, A. Mijovilovich, A. Rompel and D. Gibson, New reduction pathways for ctc-[PtCl2(CH3CO2)2(NH3)(Am)] anticancer prodrugs, Chem. Commun., 2010, 46, 1842–1844 RSC.
- H. P. Varbanov, S. M. Valiahdi, C. R. Kowol, M. A. Jakupec, M. Galanski and B. K. Keppler, Novel tetracarboxylatoplatinum(iv) complexes as carboplatin prodrugs, Dalton Trans., 2012, 41, 14404–14415 RSC.
- M. Sinisi, F. P. Intini and G. Natile, Dependence of the reduction products of platinum(IV) prodrugs upon the configuration of the substrate, bulk of the carrier ligands, and nature of the reducing agent, Inorg. Chem., 2012, 51, 9694–9704 CrossRef CAS PubMed.
- M. D. Hall and T. W. Hambley, Platinum(IV) antitumour compounds: their bioinorganic chemistry, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- D. Gibson, Platinum(IV) anticancer prodrugs-hypotheses and facts, Dalton Trans., 2016, 45, 12983–12991 RSC.
- J. L. Carr, M. D. Tingle and M. J. McKeage, Satraplatin activation by haemoglobin, cytochrome C and liver microsomes in vitro, Cancer Chemother. Pharmacol., 2006, 57, 483–490 CrossRef CAS PubMed.
- K. Hindmarsh, D. A. House and R. Van Eldik, The redox kinetics of platinum(II)/(IV) complexes, Inorg. Chim. Acta, 1998, 278, 32–42 CrossRef CAS.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Computational electrochemistry: Prediction of liquid-phase reduction potentials, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- D. Gibson, Platinum(IV) anticancer agents; are we en route to the holy grail or to a dead end?, J. Inorg. Biochem., 2021, 217, 111353 CrossRef CAS PubMed.
- F. Šebesta and J. V. Burda, Reduction Process of Tetraplatin in the Presence of Deoxyguanosine Monophosphate (dGMP): A Computational DFT Study, Chem. – A Eur. J., 2016, 22, 1037–1047 CrossRef PubMed.
- V. Vigna, S. Scoditti, A. Spinello, G. Mazzone and E. Sicilia, Anticancer Activity, Reduction Mechanism and G-Quadruplex DNA Binding of a Redox-Activated Platinum(IV)–Salphen Complex, Int. J. Mol. Sci., 2022, 23, 15579 CrossRef CAS PubMed.
- S. Bandeira, J. Gonzalez-Garcia, E. Pensa, T. Albrecht and R. Vilar, A Redox-Activated G-Quadruplex DNA Binder Based on a Platinum(IV)-Salphen Complex, Angew. Chem., Int. Ed., 2018, 57, 310–313 CrossRef CAS PubMed.
- R. K. Pathak, S. Marrache, J. H. Choi, T. B. Berding and S. Dhar, The Prodrug Platin-A: Simultaneous Release of Cisplatin and Aspirin, Angew. Chem., Int. Ed., 2014, 53, 1963–1967 CrossRef CAS PubMed.
- Q. Cheng, H. Shi, H. Wang, Y. Min, J. Wang and Y. Liu, The Prodrug Platin-A: Simultaneous Release of Cisplatin and Aspirin, Chem. Commun., 2014, 50, 7427–7430 RSC.
- F. Ponte, G. M. Piccini, E. Sicilia and M. Parrinello, A metadynamics perspective on the reduction mechanism of the Pt(IV) asplatin prodrug, J. Comput. Chem., 2020, 41, 290–294 CrossRef CAS PubMed.
- C. N. Sternberg, P. Whelan, J. Hetherington, B. Paluchowska, P. H. T. J. Slee, K. Vekemans, P. Van Erps, C. Theodore, O. Koriakine, T. Oliver, D. Lebwohl, M. Debois, A. Zurlo and L. Collette, Phase III Trial of Satraplatin, an Oral Platinum plus Prednisone vs. Prednisone alone in Patients with Hormone-Refractory Prostate Cancer, Oncology, 2005, 68, 2–9 CrossRef CAS PubMed.
- J. P. Cerón-Carrasco, Theoretical Prediction of Dual-Potency Anti-Tumor Agents: Combination of Oxoplatin with Other FDA-Approved Oncology Drugs, Int. J. Mol. Sci., 2020, 21, 4741 CrossRef PubMed.
- J. Karges, T. Yempala, M. Tharaud, D. Gibson and G. Gasser, A Multi-action and Multi-target RuII–PtIV Conjugate Combining Cancer-Activated Chemotherapy and Photodynamic Therapy to Overcome Drug Resistant Cancers, Angew. Chem., Int. Ed., 2020, 59, 7069–7075 CrossRef CAS PubMed.
- S. Scoditti, G. Mazzone, N. Sanna and E. Sicilia, Computational Exploration of the Synergistic Anticancer Effect of a Multi-Action Ru(II)–Pt(IV) Conjugate, Inorg. Chem., 2022, 12903–12912 CrossRef CAS PubMed.
- S. Chen, H. Yao, Q. Zhou, M. K. Tse, Y. F. Gunawan and G. Zhu, Stability, Reduction, and Cytotoxicity of Platinum(IV) Anticancer Prodrugs Bearing Carbamate Axial Ligands: Comparison with Their Carboxylate Analogues, Inorg. Chem., 2020, 59, 11676–11687 CrossRef CAS PubMed.
- J. Gurruchaga-Pereda, V. Martínez-Martínez, E. Formoso, O. Azpitarte, E. Rezabal, X. Lopez, A. L. Cortajarena and L. Salassa, Enhancing the Photocatalytic Conversion of Pt(IV) Substrates by Flavoprotein Engineering, J. Phys. Chem. Lett., 2021, 12, 4504–4508 CrossRef CAS PubMed.
- S. Alonso-De Castro, E. Ruggiero, A. Ruiz-De-Angulo, E. Rezabal, J. C. Mareque-Rivas, X. Lopez, F. López-Gallego and L. Salassa, Riboflavin as a bioorthogonal photocatalyst for the activation of a PtIV prodrug, Chem. Sci., 2017, 8, 4619–4625 RSC.
- S. Alonso-de Castro, A. L. Cortajarena, F. López-Gallego and L. Salassa, Bioorthogonal Catalytic Activation of Platinum and Ruthenium Anticancer Complexes by FAD and Flavoproteins, Angew. Chem., Int. Ed., 2018, 57, 3143–3147 CrossRef CAS PubMed.
- X. Yang, L. Ma, H. Shao, Z. Zhou, X. Ling, M. Yao, G. Luo, S. Scoditti, E. Sicilia, G. Mazzone, M. Gao and B. Z. Tang, Riboflavin-Promoted In Situ Photoactivation of Dihydroalkaloid Prodrugs for Cancer Therapy, J. Med. Chem., 2022, 65, 15738–15748 CrossRef CAS PubMed.
- J. Gurruchaga-Pereda, V. Martínez-Martínez, E. Rezabal, X. Lopez, C. Garino, F. Mancin, A. L. Cortajarena and L. Salassa, Flavin bioorthogonal photocatalysis toward platinum substrates, ACS Catal., 2020, 10, 187–196 CrossRef CAS.
Footnote |
| † F.P. and S.S. contributed equally. |
| This journal is © the Owner Societies 2023 |