
Single atomic Fe–N4 active sites and neighboring graphitic nitrogen for efficient and stable electrochemical CO2 reduction†
Leta
Takele Menisa‡
ab,
Ping
Cheng‡
 cd,
Xueying
Qiu
a,
Yonglong
Zheng
ae,
Xuewei
Huang
af,
Yan
Gao
*a and
Zhiyong
Tang
*a
cd,
Xueying
Qiu
a,
Yonglong
Zheng
ae,
Xuewei
Huang
af,
Yan
Gao
*a and
Zhiyong
Tang
*a
aCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing, 100190, P. R. China. E-mail: gaoyan@nanoctr.cn
bCollege of Science, University of Shanghai for Science and Technology, Shanghai, 200093, China
cCollege of Natural and Computational Sciences, Department of Chemistry, Haramaya University, P.O. Box 138, Dire Dawa, Ethiopia
dSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin, 150080, China
eInstitute of Advanced Synthesis (IAS), and School of Chemistry and Molecular Engineering, Jiangsu National Syner-getic Innovation Centre for Advanced Materials, Nanjing Tech University, 211816, Nanjing, China
fGreen Catalysis Center, and College of Chemistry, Zhengzhou University, Zhengzhou, Henan 450001, P. R. China
First published on 9th June 2022
Abstract
Single atomic Fe–Nx moieties have shown great performance in CO2-to-CO conversion. However, understanding the structural descriptors that determine the activity of Fe–Nx remains vague, and promising strategies to enhance their catalytic activity are still not clear. Herein, we used a high-temperature pyrolysis strategy and post-synthesis acid treatment for the direct growth of a single Fe–Nx site adjacent to graphitic nitrogen for the electrochemical CO2 reduction reaction. This strategy could significantly reduce the amount of pyridinic and pyrrolic N atoms, while graphitic N surrounding the Fe–Nx site predominantly increases. An experimental study combined with density functional theory revealed that the increase in the neighboring graphitic N decreases the number of electrons transferred between CO and the catalyst for FeN4-2N-3 and FeN4-4N-3, which results in the decrease of the adsorption strength of CO and the energy barrier for desorbing CO*. The as-synthesized Fe–Nx neighbored by graphitic nitrogen exhibited maximum faradaic efficiency of 91% at a lower overpotential of 390 mV. Due to the increase in the graphitic N, the catalysts perform efficiently for 35 h without any drop in current density.
New conceptsElectrochemical CO2 reduction reaction is a promising strategy for CO2 conversion to carbonaceous products. In specific, supported single metal atoms have been widely studied for efficient CO2 reduction. In such a system, the contribution of the metal center is well studied and reported. However, as supported single metal atoms require binding sites like nitrogen to anchor the metal atom, the participation and atomic concentration of N moieties are not well known. Herein, we used experimental analysis combined with density functional theory to pinpoint the participation of graphitic N neighboring Fe–N4 during electrochemical CO2 reduction reaction. Experimental results combined with the DFT study revealed that the increase in neighboring graphitic N decreases the number of electrons transferred between CO and Fe–N4 sites. The decrease in electron transfer results in a decrease in the adsorption strength of CO, facilitating the detachment of CO from the catalyst surface easily. |
1. Introduction
Electrochemical conversion of CO2 to value-added chemicals using renewable electricity provides a promising approach to mitigate both global warming and the energy crisis.1–3 However, due to the chemical inertness of CO2 molecules and multi-electron transfer nature of the reaction, this technique is still challenged by high reaction barriers and the competing proton reduction reaction.4,5 In the past decade, heterogeneous electrocatalysts based on noble metals Pd, Ag, Au, and their alloys have shown to be active for carbon dioxide reduction reaction (CO2RR) in aqueous solutions at low overpotentials; however, high cost limits their applications.6–12 Thus, it is desirable to develop efficient and selective heterogeneous electrocatalysts for CO2RR based on inexpensive and earth-abundant metals.Recently, supported metal nanoparticles gained considerable attention, particularly when the metal nanoparticle size is reduced to single atom catalysts (SACs) as the rising number of low-coordinated metal atoms functions as the catalytically active sites.13,14 Single metal anchored on N-doped carbon has been widely employed for electrocatalysis reactions such as CO2,15,16 ORR,17–21 and HER.22,23 In particular, numerous single metals (M = Fe, Co, Ni, Cu, Zn, Mn)-nitrogen (N)-doped carbon (C) electrocatalysts for CO2RR have been reported with M–Nx as the active sites.13,24–29 These catalysts have distinct chemical properties and activities from their respective metal nanoparticles and molecular catalysts. Pyrolyzed iron/nitrogen-doped carbons (Fe–N–C) have been regarded as one of the most promising SACs for CO2RR.30–32 Nevertheless, most studies have focused on tuning the coordination environment of Fe–Nx active sites, while the key atomic/electronic structures that determine the intrinsic activity of the Fe center still remain unclear. Previous studies on Fe-based catalysts revealed that unique electronic structures such as FeN428,30 and FeN54 are regarded as active sites for electrochemical CO2 reduction reactions.30 Despite this, numerous studies has been carried out in the past focusing only on the metal active centers. Moreover, metal-free N-doped carbon catalysts are also reported as potential candidates for the CO2RR, wherein it is unreasonable to ignore the impact of different configurations of nitrogen in the Fe–N–C matrix. Considering different N moieties exist in Fe–N–C catalysts, the impact of the different configurations and atomic concentrations of N-moieties on CO2RR activities are not yet deeply understood.33–35 Recently, Liu et al. identified the importance of graphitic N in a FeN4/Graphitic N complex. However, the atomic concentration of graphitic N is still not well determined.36 Further, Zhang et al. constructed atomic iron dispersed on nitrogen-doped graphene. In their DFT calculation, they considered three free energy diagrams with different graphitic nitrogen doping configurations. They reported that the nitrogen substitution on graphene improved the catalytic activity of the Fe–N4 moieties by lowering the energy barrier of COOH* formation and facilitating the CO* desorption step. However, the atomic concentration of graphitic N was not further investigated.28 Various other studies on metal-free nitrogen-doped carbon also revealed that catalysts dominated by graphitic N are superior to other N types to drive highly selective CO2RR to CO against the hydrogen evolution reaction.37–39 Additionally, the degree of graphitization can be affected by different carbon supports. For example, ZIF35,40,41 and carbon nanotubes42–44 feature an intrinsic drawback of low graphitization degree, leading to relatively low electron conductivity and poor catalyst stability. In this regard, anchoring Fe–Nx active sites on a high-graphitization-degree carbon helps in the easy determination of the atomic concentration of different N moieties.45 Activated carbon black offers an excellent support for atomically dispersed single atom catalysts.25,46–50 In addition, the co-doping of N and metal facilitates the degree of graphitization under high temperature pyrolysis on activated carbon black.
Herein, we synthesized a single-atom catalyst (SACs) consisting of atomically dispersed FeNx sites supported on N-doped carbon through a facile pyrolysis strategy to study the effect of neighboring graphitic N surrounding Fe–Nx site in the electrochemical CO2 reduction reaction. For comparison, we have also synthesized metal-free N–C and N-free Fe–C catalysts. The Fe-SAC was synthesized through facile thermal pyrolysis of melamine absorbed on carbon black. The presence of carbon black not only increases the electrical conductivity of the as-synthesized catalyst but also provides a higher surface area. On the other hand, melamine provides a rich nitrogen source to introduce the N-dopant into carbon and to anchor an individual iron atom for the formation of a Fe–Nx active site. The degree of graphitization was further studied by pyrolyzing Fe-SACs at different temperatures and post-synthesis acid treatment. The synthesized Fe-SACs exhibited high CO faradaic efficiency reaching 91% at a low overpotential of 0.39 V vs. RHE. Our experimental analysis combined with DFT calculation concludes the enhanced performance was attributed to the synergistic effect of Fe–N4 and neighboring graphitic N in Fe-SACs after acid treatment. Specifically, different graphitic N configurations neighboring Fe–N4 moieties can effectively induce a higher filing degree of d-orbitals by transferring electrons, which can optimize the binding energy of key reaction intermediates.
2. Results and discussion
2.1. Synthesis and characterization of Fe-SACs
The optimal catalyst denoted as Fe-SAC was obtained by pyrolyzing a mixture of iron nitrate as the metal precursor, melamine as N ligand, and carbon black as support at 800 °C in an argon atmosphere. The as-synthesized catalysts were treated with acid before further use (Fig. 1a). The results obtained from elemental analysis (EA, for nitrogen) and inductively coupled plasma mass spectrometry (ICP-MS, for Fe) indicated the final content of Fe and N in Fe-SACs were 0.18 wt% and 1.16 wt% (Table S1, ESI†), respectively. For comparison, Fe-free N–C, an N-free Fe–C, and pure pristine C were synthesized with the same protocol as that of Fe-SACs with minor modifications (ESI†). The distribution of Fe on the N-doped carbon was analyzed by HR-TEM and HAADF-STEM. The HR-TEM (Fig. 1b) analysis showed pristine carbon without aggregation of Fe atom as either clusters or bigger nanoparticles. Due to the co-doping of Fe and N in the graphene planes of carbon, the (002) interplanar distance has become larger compared to that of intact graphite (0.35 nm vs. 0.33 nm) (Fig. 1d). Further, aberration-corrected HAADF-STEM (Fig. 1e) in sub-angstrom size depicts the formation of isolated and uniformly dispersed Fe atoms as bright spots on the defective carbon support, confirming the atomic distribution of metal centers on the surface of the carbon. Furthermore, the homogeneous distribution of both N and Fe on carbon was confirmed by the energy-dispersive X-ray spectroscopy (EDX) (Fig. 1c). X-Ray diffraction (XRD) was employed to characterize the phase composition of the as-prepared catalysts (Fig. 1g). In contrast with the amorphous carbon structure of the N–C catalyst prepared, new sharp peaks assigned to the (002) basal planes of carbon were observed in Fe-SACs. The relatively sharp and intense peaks at 25.6° and 44.0° in Fe-SACs indicate that the additional pyrolysis and Fe doping have resulted in a higher degree of crystallization and graphitization. Thus, some new carbon is formed after doping of Fe and the high-temperature pyrolysis process.51 Moreover, XRD also confirmed the absence of peaks associated with Fe2O3 or Fe3O4, indicating that the metal is not aggregated to the nanoparticle.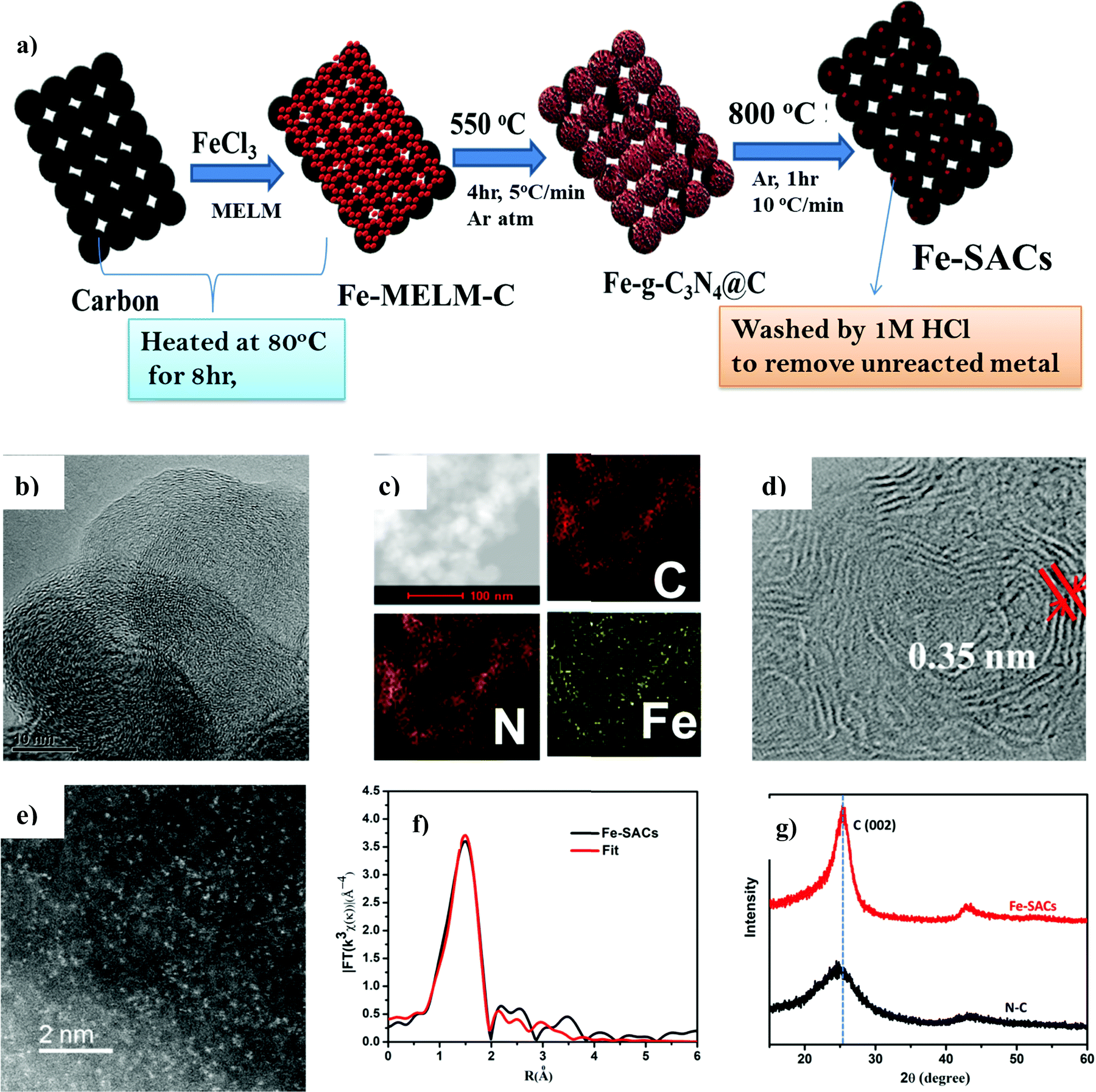 | ||
| Fig. 1 Synthesis and Characterization of Fe-SACs (a) Synthesis strategy of Fe-SACs, (b) HRTEM image of Fe-SACs (c) elemental composition of Fe-SACs, (d) aberration corrected high resolution-TEM image of Fe-SACs, (e) aberration corrected HAADF-STEM image of Fe-SACs, (f) EXAFS fitting curve, and (g) XRD diffraction pattern of N–C and Fe-SACs. | ||
Additionally, Fe K-edge was used to analyze quantitatively the structural parameters for the atoms surroundings the central atoms. It can be seen that Fe–N4 produces a satisfactory fit, indicating that Fe–N4 is the main active form of the Fe-SAC catalyst (Fig. 1f and Table S2, ESI†).
2.2. Evaluation of the electrocatalytic activity of Fe-SACs
The catalytic activity of Fe-SACs for the electrochemical CO2 reduction reaction was evaluated by linear sweep voltammetry (LSV) in Argon saturated 0.1 M KHCO3 (pH = 7.2) and CO2-saturated 0.1 M KHCO3 solutions (pH = 6.8) (Fig. 2a). The onset potential of Fe-SACs was more positive in the CO2 saturated 0.1 M KHCO3 solution compared with that in the Argon saturated 0.1 M KHCO3 solution. The cathodic current in the CO2 saturated 0.1 M KHCO3 solution was also higher than that in the Argon saturated 0.1 M KHCO3 solution, suggesting the efficient reduction of CO2 over the Fe-SAC electrode (Fig. S4a, ESI†). The resulting current density increased abruptly below −0.40 V because of two competing reactions, CO2RR and HER. It was found that Fe-SACs perform far superior to N doped carbon. Based on this fact, it can be seen that the large response of Fe-SACs to CO2RR could be mainly attributed to the synergistic effect between doped N and Fe atoms, similar to that observed for oxygen reduction reactions (ORR). The synergistic effect can be further confirmed by measuring the electrochemical surface area. The dramatic increase in the electrochemical surface area (ECSA) after Fe and N co-doping on carbon suggest a synergistic effect between the Fe site and N during CO2RR (Fig. S5, ESI†). In addition to the Fe–Nx active site, the increased degree of graphitization has also facilitated the performance of Fe-SACs, as demonstrated by the increased current density when compared to Fe-SACs prepared at a low pyrolysis temperature after acid leaching (Fig. S4b, ESI†).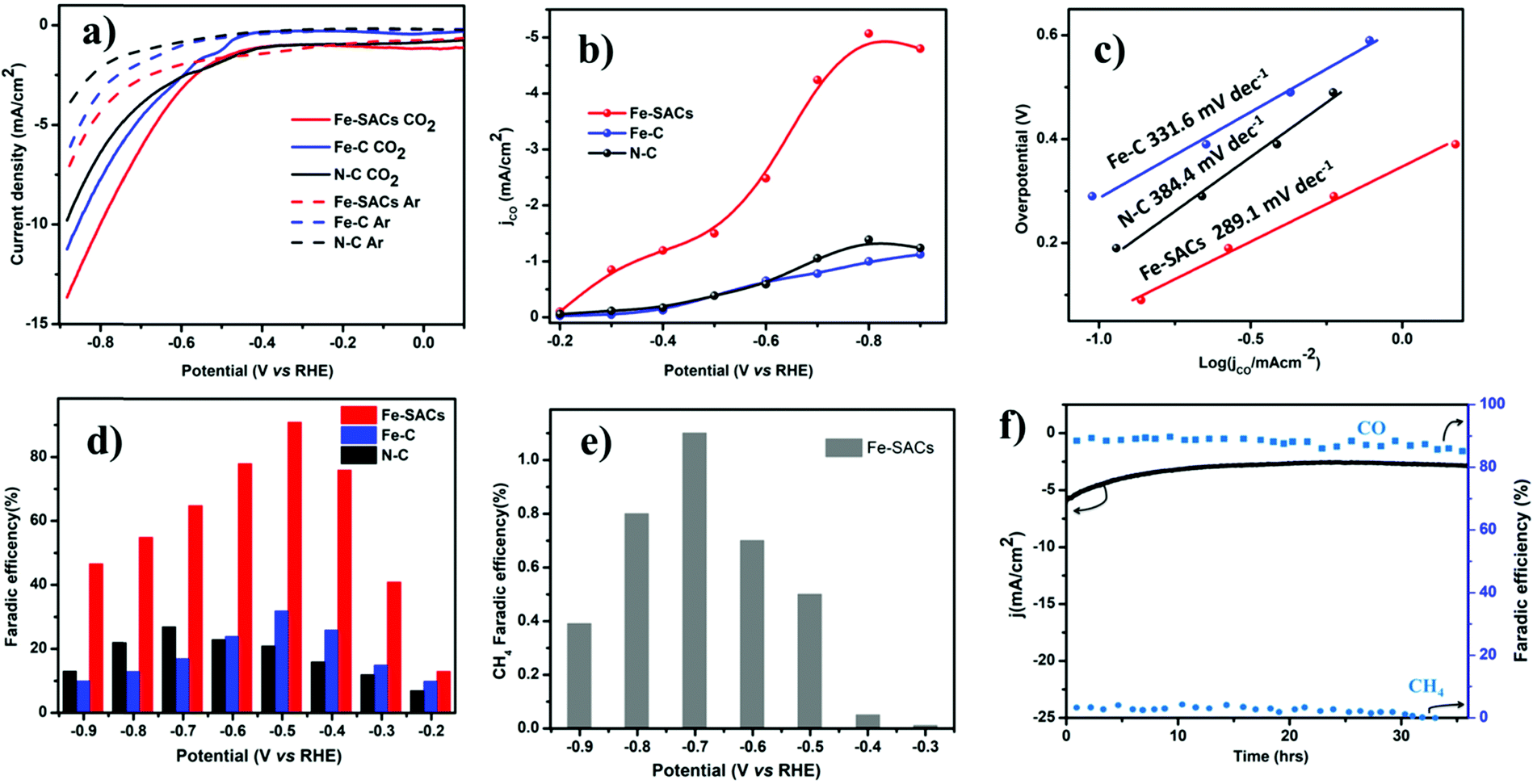 | ||
| Fig. 2 CO2RR catalytic performance of the as-synthesized catalysts. (a) LSV scans (5 mV s−1) for the Fe-SACs, Fe–C, and N–C catalysts in Ar- and CO2-saturated KHCO3 solutions (0.1 M). (b) Partial current density of Fe-SACs. (c) Tafel plots of the partial CO current density for relevant catalysts at different applied potentials. (d) Faradaic efficiencies (FEs) of CO and H2 at various applied potentials on different catalysts. (e) FEs of CH4 at different applied potentials for FE-SACs and (f) long-term stability of Fe-SACs at a potential load of −0.5 V vs. RHE and the corresponding FEs of CO and CH4. | ||
We performed potential controlled electrolysis to investigate CO2 reduction on Fe-SACs. CO is the major gas detected by online gas chromatography (Fig. 2d) and no other liquid product was detected by 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S7, ESI†). It was observed that the Fe-SAC catalyst exhibited the highest selectivity towards CO production with a maximum FE of 91.0% at −0.5 V (vs. RHE) corresponding to an overpotential of 0.39 V, which is among the best efficiency reported for carbon-based materials. Compared to pristine N-doped carbon that shows 28% FE all over investigated potentials (Fig. 2d), the performance exhibited by Fe-SACs was far superior, suggesting that the nitrogen dopant alone is not a major contributor to the enhanced electrocatalytic performance of CO2RR.52 Further, a small fraction of hydrocarbon methane (CH4) was detected above −0.3 V vs. RHE potential. A maximum FE of 1.1% at about −0.7 V vs. RHE was recorded for methane (Fig. 2e). Moreover, Fe–C and Fe-nanoparticles are synthesized with the same protocol as that of Fe-SACs in absence of the N ligand for Fe–C, and tripling the Fe precursor in the latter case. Both catalysts exhibited low performance (Fe–C, 31% FE and Fe–NP, 43% FE, Fig. S5, ESI†) toward CO2 reduction, indicating that the presence of Fe alone is also not enough for CO2 reduction. Thus, based on the performance results above, the synergistic effect between N and atomic Fe species on carbon contributes to the overall electrochemical CO2 reduction activity of Fe-SACs, unlike N-doped carbon or Fe-based nanoparticles. Moreover, the performance exhibited by Fe-SACs is better when compared to different pyrolyzed Fe-SACs, indicating the impact of the increased degree of graphitization on the CO2RR performance (Fig. S4b, ESI†). The much higher graphitization degree of the Fe-SACs not only improved the CO selectivity but also enabled improved CO2RR electrocatalytic stability. To corroborate, a prolonged CO2 reduction was carried out to analyze the stability of the Fe-SACs and it was found that Fe-SACs reduce CO2 without drops in activity and current density for about 35 h (Fig. 2f). The catalyst shows above 85% FE to CO during the entire long-term electrolysis at −0.5 V vs. RHE, confirming the remarkable durability of Fe-SACs for CO2RR.
The kinetics of CO2 reduction was further studied using Tafel analysis (Fig. 2c). Compared to N-doped carbon with a Tafel slope of 384.4 mV dec−1, the Tafel slope for Fe-SACs (289.1 mV dec−1) was small. This clearly indicates faster kinetics for electron transfer from Fe-SACs to CO2 than that in N–C, indicating the synergistic effect between doped N and Fe in the form of Fe–N4. The decreased Tafel slope of the Fe-SACs illustrates that the initial electron transfer to a CO2 molecule is largely accelerated to form the *CO2 intermediate.
2.3. Studying the origin of enhanced performance
To better understand the origin of enhanced CO2RR over Fe-SACs, we carried out further characterizations of the catalysts. Nitrogen physisorption isotherms (Fig. S2 and Table S2, ESI†) showed an increase in the Brunauer–Emmett–Teller (BET) surface area after co-doping both Fe and N (217 m2 g−1 for Fe-SACs and 167 m2 g−1 for N–C) and resulted in the formation of more micropores and mesopores on the carbon. The doping of Fe and N could also increase defects in carbon (Fig. S3, ESI†). This was verified by the increase in ID/IG ratio in the Raman spectrum relative to pristine N–C (Table S1, ESI†). Raman spectra of Fe-SACs exhibited graphitic D and G bands at ≈1342 and 1587 cm−1, respectively, and a 2D band appears at ≈2692 cm−1, confirming the local carbon structures contain both graphitic and disordered carbon atoms. The increased degree of graphitization of the Fe-SACs relative to the other catalysts prepared at different pyrolysis temperatures was also evidenced by the lowered D band to G band intensity ratio and sharpened peaks in the Raman spectra (Fig. S3, ESI†).The structure-performance relationship was further revealed by X-ray photon spectroscopy (XPS) (Fig. 3c and d) by analyzing the chemical components of the Fe site and doped N (Table S1, ESI†). Deconvoluted high-resolution N 1s spectra (Fig. 3d) for Fe-SACs showed that the N 1s spectrum was deconvoluted into pyridinic N (∼398.5 eV), Nx–Fe (∼399.4 eV), pyrrolic N (∼400.3 eV), graphitic N (∼401.2 eV), and N–Ox (∼403.5 eV).53,54 On the other hand, N 1s spectra of N–C showed four peaks corresponding to pyridinic, pyrrolic, graphitic, and oxidized N (Fig. 3d and Fig. S9, ESI†). From the quantitative analysis (Table S1, ESI†) based on the raw XPS fine spectral data, it was found that Fe-SACs contains a lower content of atomic Fe–Nx sites than the Fe-SACs prepared by treating at a lower pyrolysis temperature. The decrease in the Fe species content can be attributed to the volatilization of metal species during high-temperature pyrolysis.55 Additionally, the content of graphitic N increases with pyrolysis temperature, while the contents of pyridinic and pyrrolic N decrease significantly. However, this can be related to the increment of graphitization degree caused by the high-temperature annealing and post-synthesis acid treatment, during which both pyrrolic and pyridinic N were converted to thermodynamically more stable graphitic N. Further, the increased intensity of π–π* in XPS of C 1s peak confirms the increase in the degree of graphitization (Fig. S10, ESI†). The increased graphitic N surrounding the Fe–Nx sites could dramatically impact the spin state of the Fe center and its catalytic properties, as can be observed both experimentally and theoretically. The oxidation state of Fe was also analyzed by XPS based on the Fe 2p photoelectrons signals (Fig. 3d and Fig. S9, ESI†), in which two pairs of peaks could be assigned to the Fe2+ (711.2 and 725.1 eV) and Fe3+ (712.9 and 726.7 eV), with a satellite peak at 718.8 eV. The binding energy values and associated shakeup structure point to Fe3+ species thus; majority of Fe2+ was oxidized to Fe3+ in the presence of organic compound and higher temperature pyrolysis similar to previous report.56,57
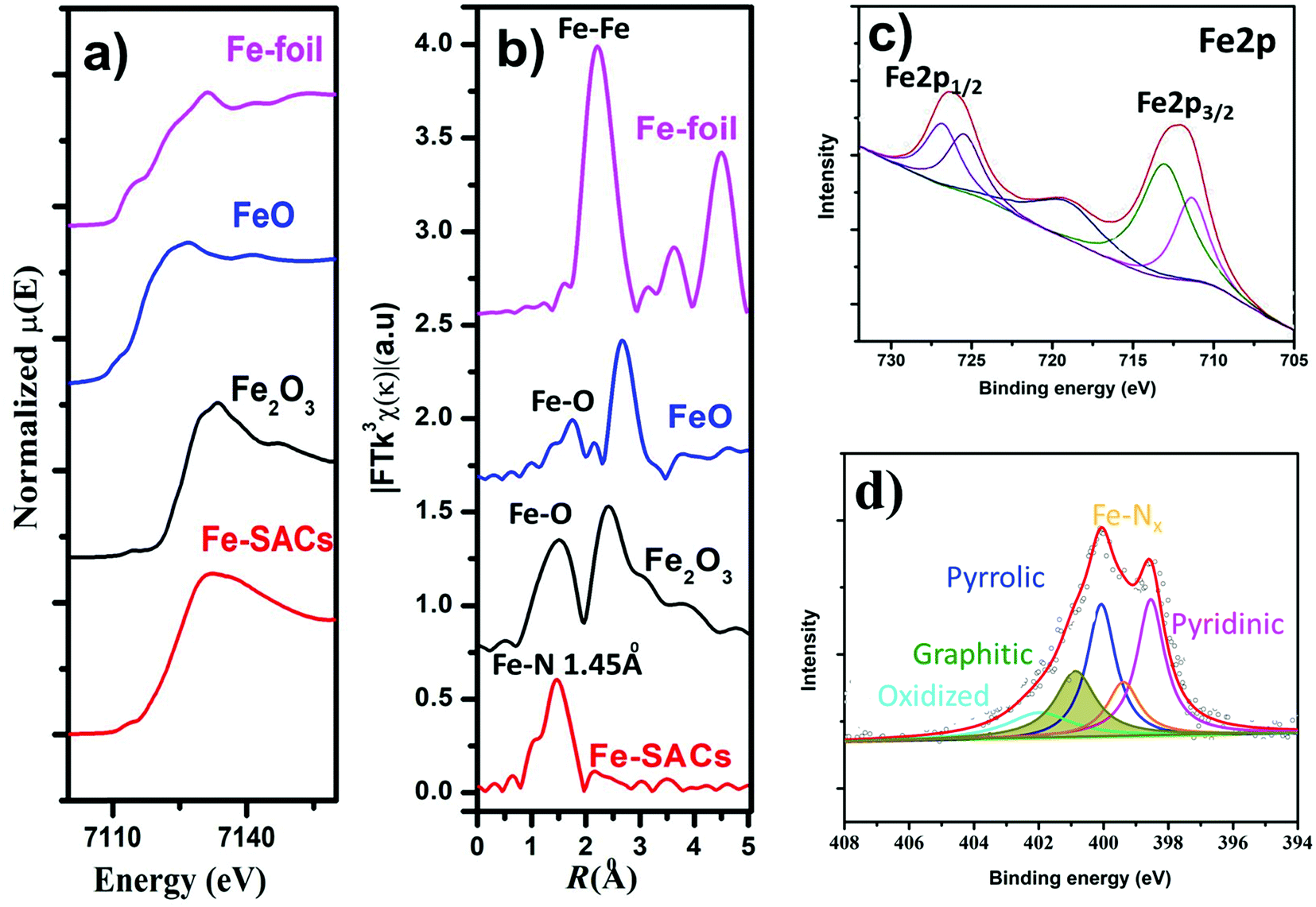 | ||
| Fig. 3 Fe K-edge XANES, EXAFS, and XPS spectra of Fe-SACs samples. (a) Fe K-edge XANES spectra of synthesized catalysts compared with different Fe samples. (b) Fourier transformed (FT) extended X-ray absorption fine structure (EXAFS) of the samples. (c) High-resolution Fe 2p XPS spectra of the Fe-SACs and d) High-resolution N 1s XPS spectra of the Fe-SACs. | ||
The local structural and electronic states of the Fe atom were analyzed by X-ray absorption near edge structure (XANES) measurements of the as-synthesized catalysts. The edge of XANES spectra of both Fe-SAC catalysts shifted towards a higher binding energy compared to that of the Fe foil (Fig. 3a), suggesting a positive charge state of Fe atoms in the as-synthesized Fe-SACs.4,20,58,59 Further, additional structural information was also inferred from extended X-ray absorption fine structure (EXAFS) spectra at the Fe K-edge (Fig. 3b). The Fourier transformed (FT) k3χ(κ) spectrum exhibited a dominant peak centered at the R space of 1.45, corresponding to the Fe–N bond, similar to the previously reported Fe–N peak in FePC,53 corroborating the existence of the Fe–Nx coordination and the Fe–O interaction of FeO sites around 1.62 Å. The absence of the peak at approximately 2.18 Å (corresponding to the Fe–Fe bond) further confirms the atomic dispersion of the isolated Fe atoms in Fe-SACs.60 Considering the enhanced electrochemical performance of Fe-SACs, the results suggest that the CO2 reduction activity in Fe-SAC samples is predominantly derived from the active sites of Fe–N coordinated together with neighboring graphitic N. Further, to understand the observed durability of the catalysts, we obtained the XPS spectra and HRTEM for Fe-SACs after prolonged CO2RR at −0.5 V vs. RHE (Fig. S11 and Table S1, ESI†). The content of M–Nx shows negligible variation, which is consistent with the observed stable FECO during the prolonged stability test (Fig. 2f). Moreover, HRTEM shows the absence of nanoparticles or clusters on carbon after 35 h of reaction. All these results indicate that the observed remarkable durability of Fe-SACs for CO2RR (Fig. 2f) could be due to the active component of M–Nx on the catalyst.
2.4. The effect of increased neighboring graphitic N
To obtain atomic insight into the impact of neighboring graphitic N on the FeN4 sites, we conducted spin-polarized density functional theory (DFT) calculations to investigate the effect of doping different amounts of graphitic N in the neighboring carbon of the FeN4 site on the electronic structure and CO2RR activity. A total of 27 different substituting structures of graphitic N were constructed (Fig. 4a and Fig. S12, ESI†) and marked as xN–y (x = 1, 2, 3, 4), where x denotes the number of doped graphitic N atoms and y stands for different configurations of the doped graphitic N. Their activities toward CO2RR were evaluated (Fig. 4b and Fig. S13, ESI†). CO2RR on FeN4 without any surrounding graphitic N has to overcome a relatively high barrier of 0.702 eV to form *COOH. Additionally, the desorption of *CO is also not easy in absence of graphitic N with the barrier of 0.500 eV, as shown in Fig. 4b. The presence of neighboring graphitic N can significantly decrease the barrier of forming COOH* and desorption of CO*, though different atomic configurations of the graphitic N also play a role. In particular, FeN4 with two neighboring graphitic N (FeN4-2N-3) has the best optimum value with the barrier of its rate-determining step (COOH* forming) of 0.36 eV. This result is comparable with the obtained lowest overpotential of 0.34 V in our experimental analysis.45 Furthermore, we investigate the Bader charge of *COOH and *CO on the five catalysts (FeN4, FeN4-1N-3, FeN4-2N-3, FeN4-3N-6, and FeN4-4N-3) (Table S3, ESI†). The result demonstrates that the presence of neighboring graphitic N increases the number of electrons transferred between COOH and the catalyst, which results in an increase in the COOH adsorption strength. The strong adsorption strength of COOH* contributes to the decrease of the COOH* forming a barrier. Similarly, the presence of neighboring graphitic N decreases the number of electrons transferred between CO and the catalyst for FeN4-2N-3 and FeN4-4N-3, which results in the decrease of the CO adsorption strength, decreasing the energy barrier for desorbing CO*. For FeN4-1N-3 and FeN4-3N-6, the number of electrons transferred between CO and the catalyst was not significantly decreased compared with that of FeN4 because of their slightly distorted structures. These results indicate a potential promotional effect of graphitic nitrogen on Fe–N4 for electrochemical CO2 reduction. Therefore, the highly efficient activity of CO2 reduction to CO could be attributed to the synergetic effect of the Fe–N4 moieties and neighboring graphitic nitrogen on the carbon surface.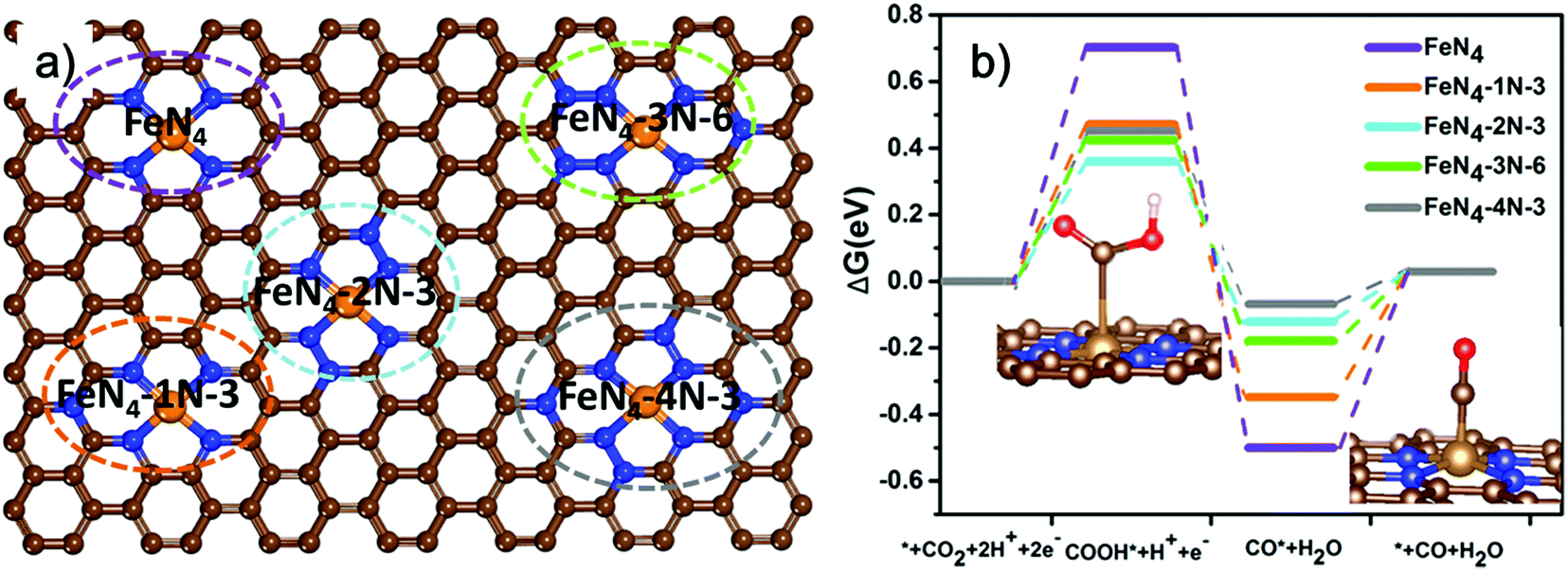 | ||
| Fig. 4 DFT study of Fe-SACs with different graphitic N configurations: (a) representative configurations of FeN4 with different surrounding graphitic N atoms and (b) free energy diagram for electrochemical CO2 reduction to CO on Fe–N4 moieties embedded on carbon sheets inset: optimized geometries of reaction intermediates (gray (C), Orange (Fe) Blue (N) Red (O) and light white (H). | ||
3. Conclusion
In conclusion, atomically dispersed Fe-SACs were successfully synthesized on N-doped carbon through a facile thermal pyrolysis method. Subsequent post-treatments including acid leaching and thermal annealing resulted in significantly elevated content of graphitic nitrogen neighboring the atomic Fe–Nx sites, leading to enhanced CO2RR activity. The as-synthesized Fe-SAC catalysts exhibited enhanced CO2 reduction activity with the maximum faradaic efficiency of 91% at −0.5 V vs. RHE, which is equal to an overpotential of 390 mV. The increased activity of Fe-SACs originates from the synergistic effect between the Fe center and neighboring graphitic N. Moreover, DFT predicted calculations also revealed that increasing the contents of neighboring graphitic N can significantly decrease the value of ΔG*CO. The different atomic configurations of the graphitic N also is a contributing factor to observed selectivity. In particular, FeN4 with two neighboring graphitic N (FeN4-2N-3) has the best optimum value of ΔG*CO and therefore the lowest overpotential of 0.34 V was achieved.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors acknowledge financial support from the Strategic Priority Research Program of Chinese Academy of Sciences (XDB36000000, Z. Y. T.), National Key R&D Program of China (2021YFA1200302, Z. Y. T.), and National Natural Science Foundation of China (51772057, Y. G.,92056204, 21890381 and 21721002, Z. Y. T.).References
- H. Shen, E. Gracia-Espino, J. Ma, H. Tang, X. Mamat, T. Wagberg, G. Hu and S. Guo, Nano Energy, 2017, 35, 9–16 CrossRef CAS.
- S.-T. Gao, S.-Q. Xiang, J.-L. Shi, W. Zhang and L.-B. Zhao, Phys. Chem. Chem. Phys., 2020, 22, 9607–9615 RSC.
- S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang and J. Lu, Angew. Chem., 2019, 131, 15013–15018 CrossRef.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan and C. Cao, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- C. Rogers, W. S. Perkins, G. Veber, T. E. Williams, R. R. Cloke and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 4052–4061 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., 2016, 128, 9900–9904 CrossRef.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm and R. De Marco, Adv. Mater., 2018, 30, 1706287 CrossRef PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS PubMed.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Chemistry, 2017, 3, 812–821 CrossRef.
- H. Shen, E. Gracia-Espino, J. Ma, K. Zang, J. Luo, L. Wang, S. Gao, X. Mamat, G. Hu and T. Wagberg, Angew. Chem., Int. Ed., 2017, 56, 13800–13804 CrossRef CAS PubMed.
- J.-D. Yi, R. Xu, Q. Wu, T. Zhang, K.-T. Zang, J. Luo, Y.-L. Liang, Y.-B. Huang and R. Cao, ACS Energy Lett., 2018, 3, 883–889 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang and D. Wang, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian and W. C. Cheong, Angew. Chem., Int. Ed., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- W. Zhou, J. Jia, J. Lu, L. Yang, D. Hou, G. Li and S. Chen, Nano Energy, 2016, 28, 29–43 CrossRef CAS.
- P. Su, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, Small, 2016, 12, 6083–6089 CrossRef CAS PubMed.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS PubMed.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou and D. Du, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan and X. Cheng, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie and A. S. Jalilov, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- L. T. Menisa, P. Cheng, C. Long, X. Qiu, Y. Zheng, J. Han, Y. Zhang, Y. Gao and Z. Tang, Nanoscale, 2020, 12, 16617–16626 RSC.
- T. N. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS.
- W. Ju, A. Bagger, X. Wang, Y. Tsai, F. Luo, T. Möller, H. Wang, J. Rossmeisl, A. S. Varela and P. Strasser, ACS Energy Lett., 2019, 4, 1663–1671 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal., B, 2019, 241, 113–119 CrossRef CAS.
- C. Liu, Y. Wu, K. Sun, J. Fang, A. Huang, Y. Pan, W.-C. Cheong, Z. Zhuang, Z. Zhuang and Q. Yuan, Chemistry, 2021, 7, 1297–1307 CrossRef CAS.
- Z. Zhang, L. Yu, Y. Tu, R. Chen, L. Wu, J. Zhu and D. Deng, Cell Rep. Phys. Sci., 2020, 1, 100145 CrossRef CAS.
- J. Li, W.-Y. Zan, H. Kang, Z. Dong, X. Zhang, Y. Lin, Y.-W. Mu, F. Zhang, X.-M. Zhang and J. Gu, Appl. Catal., B, 2021, 298, 120510 CrossRef CAS.
- Z. Shu, G. Ye, J. Wang, S. Liu, Z. He, W. Zhu, B. Liu and M. Liu, Ionics, 2021, 27, 3089–3098 CrossRef CAS.
- Y. Wang, P. Hou, Z. Wang and P. Kang, ChemPhysChem, 2017, 18, 3142–3147 CrossRef CAS PubMed.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X. D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang and Y. Liang, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li and C. Ma, Angew. Chem., Int. Ed., 2017, 56, 7847–7852 CrossRef CAS PubMed.
- D. Xia, X. Yang, L. Xie, Y. Wei, W. Jiang, M. Dou, X. Li, J. Li, L. Gan and F. Kang, Adv. Funct. Mater., 2019, 29, 1906174 CrossRef CAS.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. D. Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin and J. Bao, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang and J. Liu, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- W.-J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L.-J. Zhang, J.-Q. Wang, J.-S. Hu, Z. Wei and L.-J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang and D. Su, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- Q. Hao, D.-X. Liu, R. Deng and H.-X. Zhong, Front. Chem., 2022, 9, 837580 CrossRef PubMed.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu and C. Wu, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng and A. I. Rykov, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nh00143h |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |