
A self-amplifying nanodrug to manipulate the Janus-faced nature of ferroptosis for tumor therapy†
Mengzhu
Zhang
,
Xiaohan
Qin
,
Zhipeng
Zhao
,
Qian
Du
,
Qian
Li
,
Yue
Jiang
and
Yuxia
Luan
 *
*
NMPA Key Laboratory for Technology Research and Evaluation of Drug Products, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China. E-mail: yuxialuan@sdu.edu.cn
First published on 21st December 2021
Abstract
Ferroptosis, an unusual non-apoptotic cell death caused by the iron-dependent accumulation of lipid peroxide, enables the flexible design of an antitumor platform. Specifically, as a positive role, ferroptosis can induce an immune response accompanied with the interferon-γ (IFN-γ)-triggered disruption of the glutathione peroxidase 4 pathway for cascade enhancement of ferroptotic cell death and ferroptosis-induced immunotherapeutic efficacy. However, as a negative role, ferroptosis also triggers inflammation-associated immunosuppression by up-regulation of the cyclooxygenase-2/prostaglandin E2 pathway and IFN-γ-associated adaptive immune resistance by up-regulation of programmed death ligand-1 (PD-L1), impeding the antitumor efficacy of multiple immune cells by immune escape. Negative and positive roles endow ferroptosis with a Janus-faced nature. It is urgent to manipulate the Janus-faced nature of ferroptosis for eliciting the maximized ferroptotic therapeutic efficacy. Herein, a self-amplifying nanodrug (RCH NPs) was designed by co-assembling hemin (ferric porphyrin), celecoxib (anti-inflammatory drug) and roscovitine (cyclin-dependent kinase 5 inhibitor) with the assistance of human serum albumin for reprograming the Janus-faced nature of ferroptosis. During hemin-triggered ferroptosis, celecoxib disrupted the inflammation-related immunosuppression while roscovitine destroyed the IFN-γ-induced up-regulation of PD-L1 via the genetic blockade effect. The RCH NPs thus demonstrated superior therapeutic effects on tumors, thanks to self-amplifying ferroptotic immunotherapy. Our work offers a conceptually innovative strategy for harnessing ferroptosis against tumors.
New conceptsFerroptosis has recently been demonstrated to be a promising approach against tumors. However, ferroptosis has an inherent Janus-faced nature in cancer therapy, compromising its therapeutic efficacy. On the one hand, ferroptosis can inherently trigger the immune response accompanied with disruption of the glutathione peroxidase 4 pathway for realizing a cascade augment of ferroptotic cell death and ferroptotic immunotherapy. On the other hand, ferroptosis also intrinsically induces immune escape, where the triggered up-regulation of cyclooxygenase-2/prostaglandin E2 and programmed death ligand-1 (PD-L1) significantly suppresses the antitumor efficacy of immune cells. It is of great interest, albeit challenging, to regulate the Janus-faced nature of ferroptosis for realizing an optimal ferroptotic therapy against tumors. Herein, a self-amplifying nanodrug was, for the first time, engineered for reshaping the Janus-faced nature of ferroptosis in cancer therapy. With the cooperation of hemin, celecoxib and roscovitine in the nanodrug, the Janus-faced nature of ferroptosis was powerfully manipulated, demonstrating a potent therapeutic efficacy against tumor progression and metastasis along with the long-lasting immunological memory response. The present nanodrug offers a conceptually innovative approach for manipulating the Janus-faced nature of ferroptosis to elicit the maximized therapeutic efficacy against tumors. |
Introduction
Ferroptosis, a newly discovered form of non-apoptotic cell death, has attracted increasing attention in tumor therapy due to its difference from apoptosis in cell morphology, genetics and biochemistry.1,2 In the mechanism, ferroptosis is caused by the iron-dependent accumulation of lipid peroxide.3 Therefore, the iron-based Fenton reaction is a potent approach to induce ferroptosis in tumor cells.4 As cancer cells have an inherent resistance to apoptosis, ferroptosis provides a more efficient strategy for suppressing tumor progress via targeting the non-apoptosis pathway.In particular, ferroptosis can induce immune response through ferroptotic tumor cells with the exposure of danger-associated molecular patterns (DAMPs) and the release of tumor-associated antigens (TAAs),5–9 where the secretion of interferon-γ (IFN-γ) by T cells is stimulated for immunotherapy. Based on the mechanism of T cell-mediated killing,10–13 the triggered IFN-γ makes tumor cells prone to ferroptosis by down-regulating the system xc− (consisting of SLC7A11 and SLC3A2) for glutathione (GSH) synthesis inhibition, wherein the detoxication of lipid peroxide against ferroptosis through the GSH-mediated glutathione peroxidase 4 (GPX4) pathway is disrupted. In a word, the ferroptosis-triggered immune response can boost the efficacy of ferroptosis, and the boosted ferroptosis augments the immune response in turn. Consequently, as a positive role, ferroptosis itself possesses a cascade enhancement of ferroptotic cell death and ferroptosis-induced immunotherapy once ferroptosis is triggered, thanks to the interplay of ferroptosis with the inherent induced IFN-γ.
Despite the positive role of ferroptosis in tumor therapy, the intrinsically boosted immunotherapeutic efficacy is far from satisfactory as there also exits an inherent negative role in the ferroptosis-triggered immunotherapy. On the one hand, ferroptosis specifically induces inflammation-associated immunosuppression by up-regulating cyclooxygenase-2 (COX-2) along with the COX-2-derived up-regulation of prostaglandin E2 (PGE2, a proinflammatory factor) in tumor cells.14–18 As a result, ferroptosis inherently stimulates PGE2-based inflammation-associated immunosuppression, upon which ferroptotic tumor cells gain the privilege of immune escape and resistance to immunotherapy, impeding the immunotherapeutic efficacy. On the other hand, although the ferroptosis-induced T cell-secreted IFN-γ could kill the tumor cells, it is also an accomplice of adaptive immune resistance, where IFN-γ promotes T cell dysfunction through the IFN-γ-induced up-regulation of tumoral programmed death ligand-1 (PD-L1) to rescue tumor cells from immune elimination.19–21 Hence, as a negative role, ferroptosis also inherently elicits immune escape involving inflammation-associated immunosuppression by up-regulation of the COX-2/PGE2 pathway and IFN-γ-associated adaptive immune resistance by up-regulation of PD-L1, resulting in the major obstacle for ferroptotic immunotherapy.
Taken altogether, negative and positive roles endow ferroptosis with a Janus-faced nature, where ferroptosis-driven immune escape is regarded as “a limiting factor”, severely limiting the effectiveness of ferroptosis. Therefore, the design of a ferroptosis-based nanoplatform to manipulate the Janus-faced nature of ferroptosis for eliciting the maximized ferroptotic therapy is in urgent demand.
Herein, for the first time, we rationally designed a self-amplifying nanodrug to reprogram the Janus-faced nature of ferroptosis, which could harness the positive role of ferroptosis along with reversing its negative role to achieve the optimal ferroptotic tumor therapy. Our nanodrug (RCH NPs) is achieved by co-assembling hemin (Hemin, ferric porphyrin), celecoxib (Celeco, a non-steroidal anti-inflammatory drug), and roscovitine (Rosco, a cyclin-dependent kinase 5 (Cdk5) inhibitor) with human serum albumin (HSA). In RCH NPs, hemin acts as a ferroptosis inducer by converting endogenous hydrogen peroxide (H2O2) into cytotoxic hydroxyl radicals (˙OH) for lipid peroxidation through the Fenton reaction. Celecoxib selectively inhibits the inflammatory COX-2/PGE2 pathway that is up-regulated by ferroptosis to relieve inflammation-associated immunosuppression while roscovitine inhibits the Cdk5 pathway to suppress IFN-γ-dependent PD-L1 gene transcription and abolish adaptive immune resistance through the genetic blockade effect (Scheme 1). Moreover, HSA not only facilitates the binding of hydrophobic drug molecules via hydrophobic pockets for efficacious drug delivery but also promotes accumulation at tumor tissues by albumin-binding proteins overexpressed in the tumor. Our RCH NPs demonstrated excellent therapeutic efficacy against tumor progression and metastasis, along with a long-lasting immunological memory response, thanks to the potent manipulation of the Janus-faced nature of ferroptosis. Our work thus offers a conceptually innovative approach for eliciting the maximized therapeutic efficacy of ferroptosis against tumors.
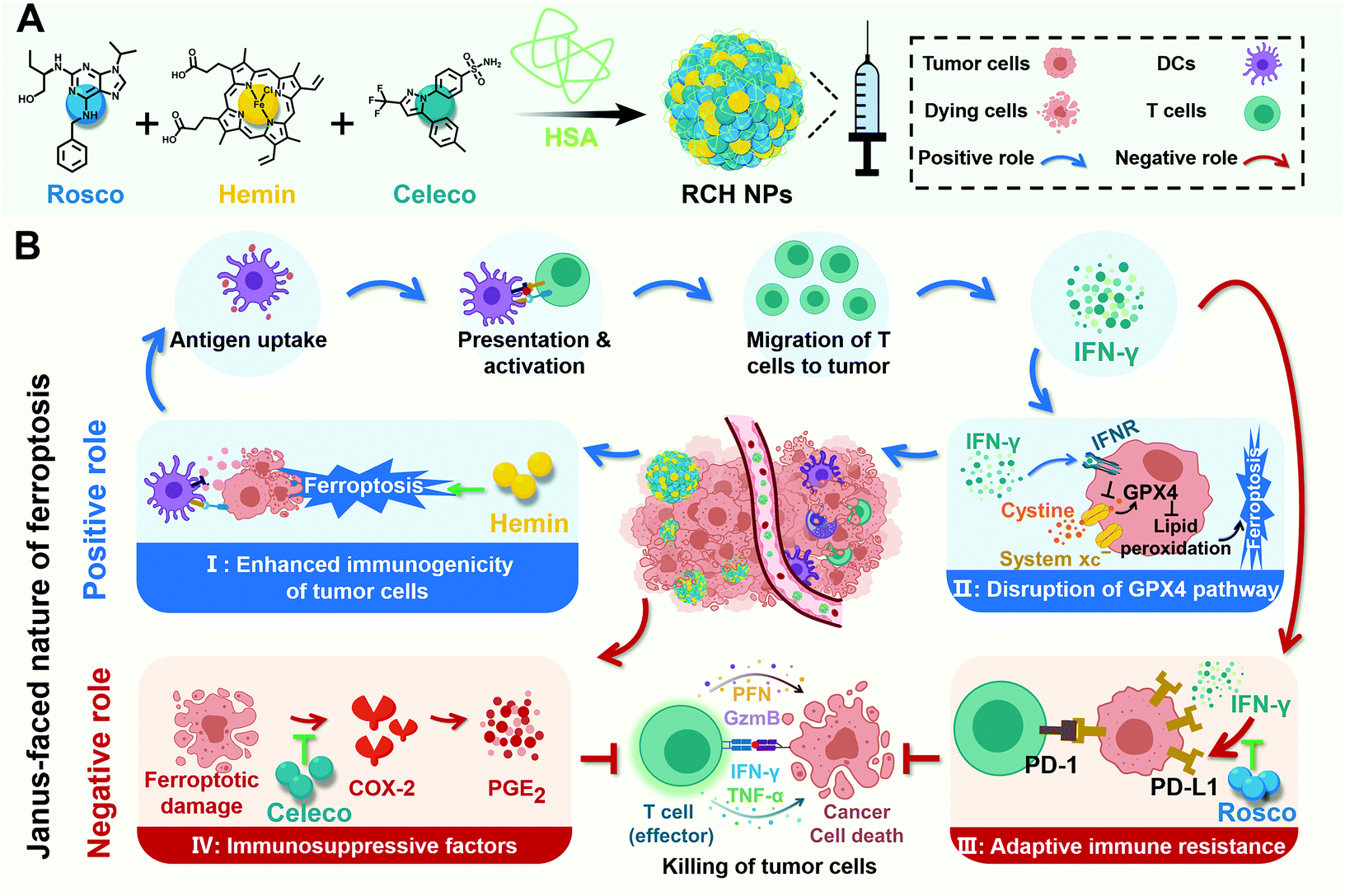 | ||
| Scheme 1 (A) Illustration of the fabrication of RCH NPs and (B) schematic diagram of ferroptotic immunotherapy via RCH NPs reprograming the Janus-faced nature of ferroptosis to abolish inherent ferroptosis-driven immune escape. | ||
Results and discussion
Preparation and characterization of RCH NPs
The RCH NPs nanodrug was achieved through the co-assembly of hemin, celecoxib and roscovitine with the help of HSA. The brown-colored solution of as-prepared RCH NPs displayed a remarkable Tyndall effect upon laser irradiation (the inset in Fig. 1A). As shown in Fig. 1A, the typical UV-Vis absorbance peaks demonstrated the successful co-assembly of hemin, celecoxib and roscovitine for achieving the RCH NPs. The encapsulation efficiency of hemin, celecoxib and roscovitine in RCH NPs was measured to be 71.20%, 56.96%, 33.47%, respectively. The transmission electron microscopy (TEM) image demonstrated that RCH NPs showed a spherical morphology (the inset in Fig. 1B). Dynamic light scattering (DLS; Fig. 1B) indicated that the size of the RCH NPs was 143.6 ± 1.4 nm (PDI: 0.154 ± 0.007). As illustrated in Fig. S1 (ESI†), the particle-size results for RCH NPs at different times in water or phosphate buffered saline (PBS, pH 7.4) verified the excellent stability of the RCH NPs. The RCH NPs possessed a negative surface charge with a zeta potential of −19.03 ± 0.47 mV (Fig. S2, ESI†).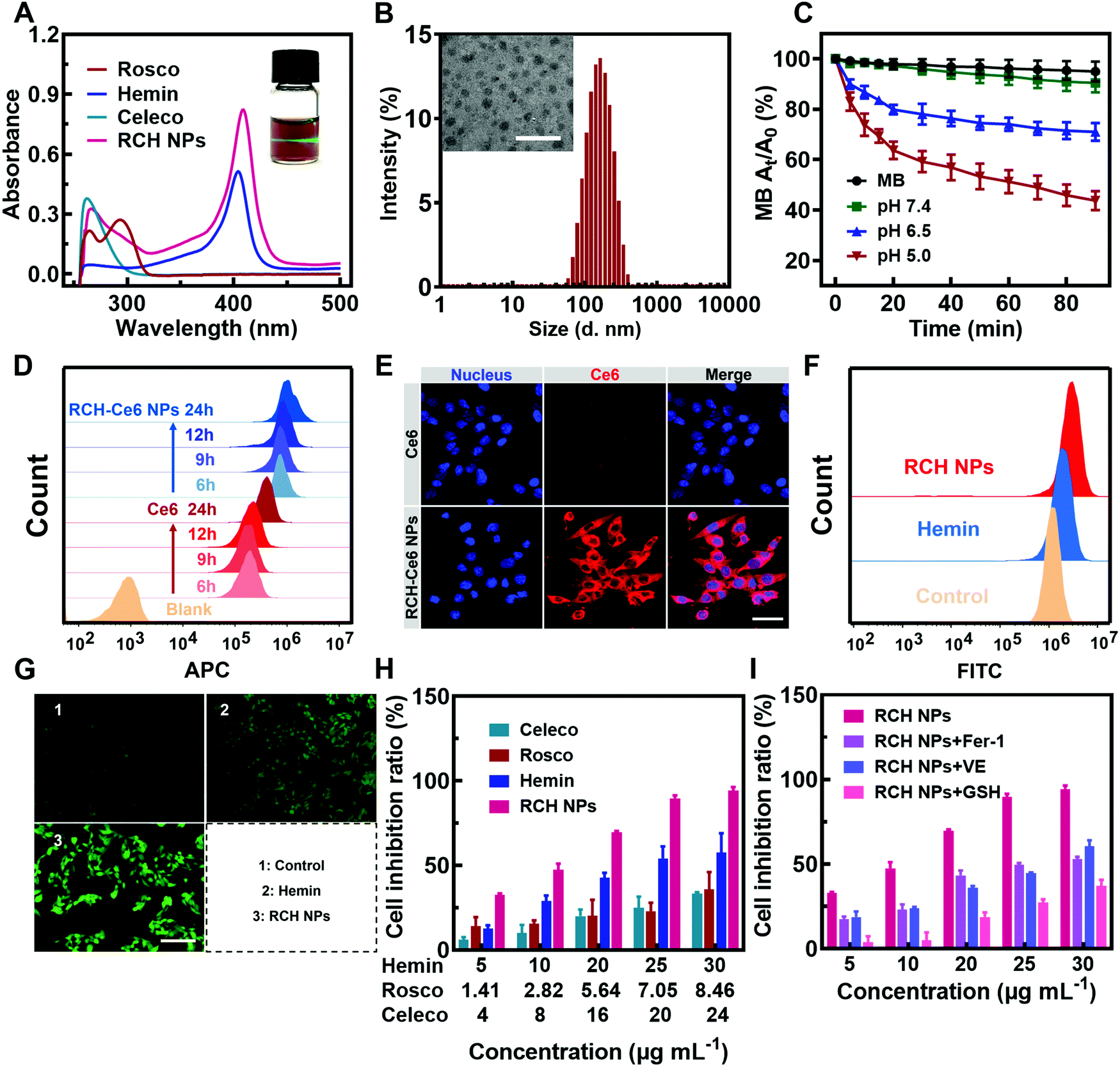 | ||
| Fig. 1 (A) UV-Vis spectra of roscovitine, hemin, celecoxib and RCH NPs as well as the image displaying the Tyndall effect of RCH NPs (inset). (B) Size distribution and the TEM image (inset) of RCH NPs. Scale bar: 400 nm. (C) MB degradation induced by the Fenton reaction of RCH NPs at different pH (7.4, 6.5 and 5.0). (D) Flow cytometry results and (E) fluorescent images of cell uptake for the free Ce6 and RCH–Ce6 NPs. Scale bar: 40 μm. (F) Flow cytometry results and (G) fluorescent images of intracellular ˙OH generation in PBS, hemin and RCH NPs. Scale bar: 200 μm. (H) Cytotoxicity test of celecoxib, roscovitine, hemin and RCH NPs on B16F10 cells after 24 h of incubation. (I) Ferroptosis in RCH NPs-mediated cytotoxicity: cellular inhibition ratios under the treatment of RCH NPs co-incubating with various ferroptosis inhibitors such as Fer-1, VE, and GSH. | ||
In vitro release behavior of RCH NPs
The release property of roscovitine at different pH values (7.4, 6.5 and 5.0) was evaluated. As displayed in Fig. S3 (ESI†), nearly 72% of roscovitine was released in the acidic pH (5.0) of the lysosome, while only 35% was released at physiological pH (7.4) within 36 h. Roscovitine exhibited pH-responsive release properties. This result suggested that the acidic environment in tumor cells could effectively trigger drug release after the RCH NPs were internalized. Furthermore, after incubation for 2, 4, 6 and 9 h, the medium was collected for characterization using DLS. The DLS result exhibited an evolutional disassembly behavior of RCH NPs along with time (Fig. S4, ESI†). In contrast to the initial nanoparticles, the particle size of the RCH NPs decreased significantly after 2 h of incubation; then the nanoparticles progressively dissociated into smaller particles after incubation for another 2, 4, and 7 h, identifying the disassembly of RCH NPs over time. Changes in the particle size of the RCH NPs further confirmed the release behavior of drugs from the nanodrug in the acidic tumor environment.Evaluation of RCH NPs-mediated antitumor effect in vitro
The Fenton activity of RCH NPs was evaluated using ultraviolet spectrophotometry based on the ˙OH detection probe methylene blue (MB).22 The decrease in the typical UV-Vis absorbance peak of MB at 660 nm reflected the performance of producing ˙OH. As shown in Fig. 1C, determination of the Fenton activity of RCH NPs at various pH values was conducted. The RCH NPs showed negligible Fenton activity at pH 7.4. By contrast, the Fenton activity of RCH NPs increased immensely as the pH was reduced to 5.0. Benefiting from this unique feature, the Fenton reaction induced by the RCH NPs enabled the response to the acidic tumor microenvironment and avoided damage to normal tissues at the same time. To further prove the Fenton activity of RCH NPs, the typical oxidation reaction of TMB (3,3′,5,5′-tetramethylbenzidine) was utilized to detect the generation of ˙OH in pH 5.0 media. Compared with the mixture of TMB and H2O2, there was an obvious absorbance peak at 655 nm after adding the RCH NPs (Fig. S5, ESI†), which is the typical UV-Vis absorbance peak of oxidized TMB (oxTMB). As shown in Fig. S6 (ESI†), the oxidation reaction of TMB became stronger along with time. The results confirmed that the RCH NPs could act as a Fenton catalyst to generate ˙OH for ferroptosis.The internalization of the drug into cells is the prerequisite for RCH NPs to exert their therapeutic effect. The uptake behavior of RCH NPs in B16F10 cells was investigated using flow cytometry and laser scanning confocal microscopy (LSCM). Chlorin e6 (Ce6) was chosen as a fluorescent indicator because RCH NPs had no effective fluorescent property. In contrast to tumor cells treated with free Ce6, cells of the RCH–Ce6 NPs group exhibited a significantly stronger red fluorescence intensity (Fig. 1D and E), indicating the higher uptake efficiency of RCH NPs, which was ascribed to the HSA-governed binding effect. The better internalization of the HSA-based nanodrug results from the albumin-binding protein that is overexpressed by B16F10 cells.23–25 In addition, the cellular fluorescence signal, which represents the uptake efficiency, became stronger with time, exhibiting a time-dependent manner.
Using 2′,7′-dichlorofluorescein diacetate (DCFH–DA) as the probe, the in vitro Fenton-reaction-triggered production of ˙OH was investigated using flow cytometry and fluorescence microscopy. As illustrated in Fig. 1F, the RCH NPs group exhibited a much stronger fluorescence than the other groups, revealing its excellent capability for ˙OH generation. Qualitative analysis via fluorescence microscopy indicated results that were consistent with flow cytometry (Fig. 1G), demonstrating that our designed RCH NPs were an efficient iron-based nanoplatform for ferroptosis due to their outstanding ˙OH generation.
The in vitro cytotoxicity of RCH NPs was investigated using the MTT assay. As can be seen from Fig. 1H, the RCH NPs markedly induced a higher level of cell inhibition against B16F10 cells in comparison with free celecoxib, roscovitine and hemin. The contribution of ferroptosis to the RCH NPs-induced cytotoxicity in B16F10 cells was further investigated. Compared with the group of RCH NPs, the addition of ferroptosis inhibitors such as ferrostatin-1 (Fer-1), vitamin E (VE), and GSH, which have properties of radical-trapping or antioxidation in RCH NPs groups, saved cells from ferroptosis, confirming that ferroptosis played an essential role in RCH NPs-mediated cytotoxicity (Fig. 1I). These results demonstrated that RCH NPs could kill tumor cells effectively and were adopted as a brilliant ferroptosis inducer.
In vitro antitumor immune response of RCH NPs
The immunogenic cell death (ICD)-inducing ability of RCH NPs based on ferroptosis was examined via release of the high mobility group box 1 (HMGB1) from the nucleus, the exposure of calreticulin (CRT) on the cell surface and the secretion of adenosine triphosphate (ATP). Compared with other groups, the cells of the RCH NPs group were observed to enhance HMGB1 release from the nucleus (Fig. 2A) and a larger amount of CRT exposure on the surface (Fig. 2B). The ATP content secreted by tumor cells was further examined using a microplate reader. As displayed in Fig. S7 (ESI†), the highest ATP secretion was observed in the cells treated with RCH NPs, which is 2.6-fold higher than that of the control group. The above results thus confirmed that RCH NPs could generate potent ICD for eliciting antitumor immunotherapy.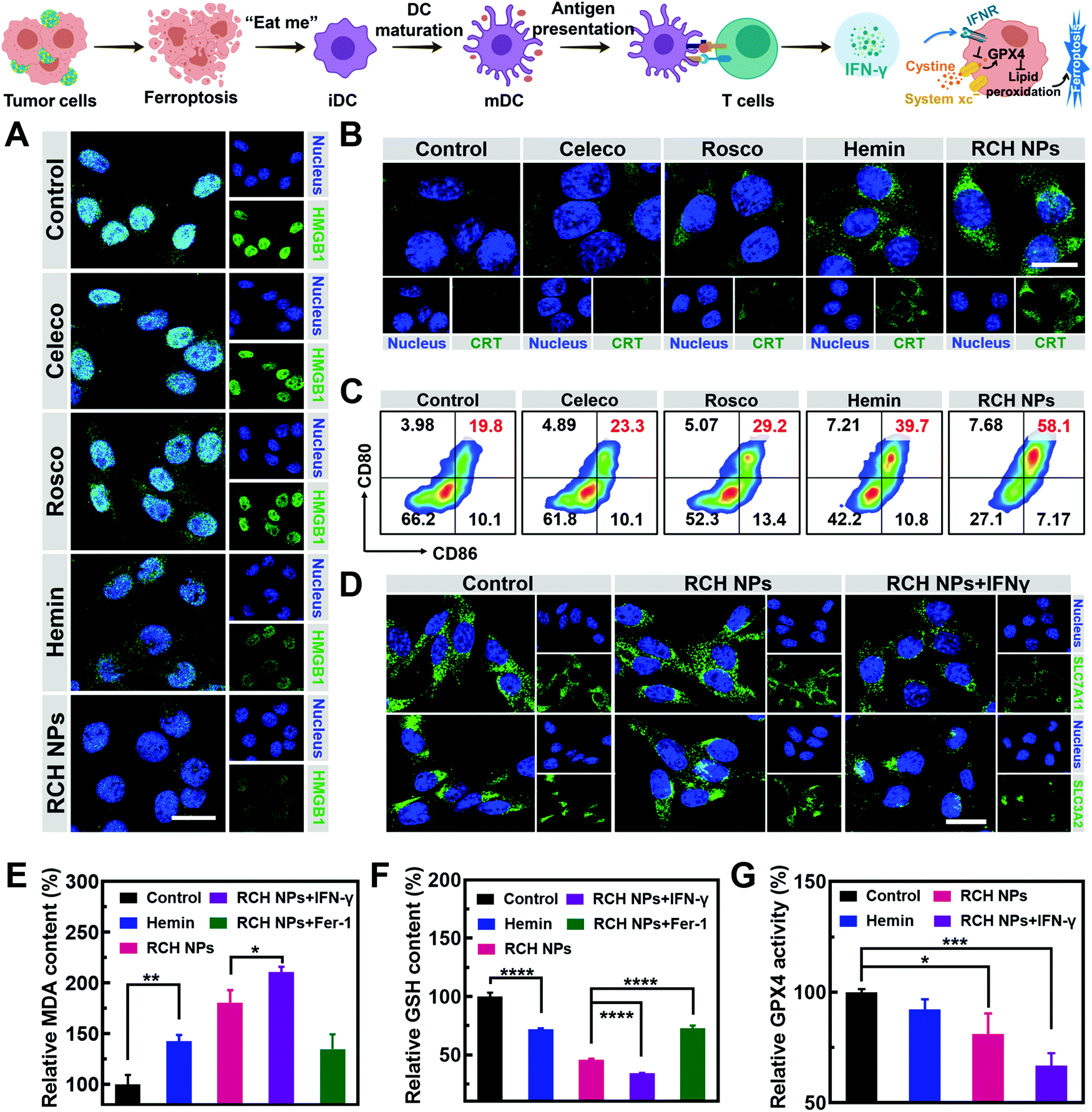 | ||
| Fig. 2 Fluorescent images of (A) HMGB1 release from the cell nucleus and (B) CRT exposure on the B16F10 cell membrane after different treatments. Scale bar: 20 μm. (C) Flow cytometry results of DC maturation of different groups. (D) Fluorescent images of SLC7A11 and SLC3A2 expression under different treatments. Scale bar: 20 μm. (E) MDA content, (F) GSH content, and (G) activity of the GPX4 in B16F10 cells during incubation with different formulations. | ||
As the immunogenicity triggered by RCH NPs was expected to arouse a significant antitumor immune response, dendritic cell (DC) activation and maturation triggered by ICD were assessed to understand the immune response, which was regarded as further evidence of the positive role of ferroptosis in antitumor immunity as detailed above. Bone-marrow-derived dendritic cells (BMDCs) from C57BL/6 mice were incubated with different groups of tumor cells. Flow cytometry was utilized to analyze the proportion of the matured DCs (CD11c+CD80+CD86+). The RCH NPs group induced considerable DC maturation, demonstrating 2.93-fold greater efficiency than that of the control group (Fig. 2C). These results indicated that RCH NPs could successfully activate the immune response for efficient immunotherapy wherein the enhanced immunogenicity of tumor cells and DC maturation were demonstrated.
As an aspect of the Janus-faced nature of ferroptosis, the positive role of ferroptosis in antitumor therapy was considered to participate in T cell-mediated immunotherapy by IFN-γ. IFN-γ derived from cytotoxic T cells can be significantly up-regulated by the ferroptosis-activated immune response. As a killing mechanism of T cell-mediated immunotherapy, IFN-γ could in turn sensitize tumor cells to ferroptosis by reducing the expression of SLC7A11 and SLC3A2 to enhance ferroptosis-specific lipid peroxidation, as reported.10,11 From in vitro cellular experiments, IFN-γ was added directly to incubate tumor cells. As shown in Fig. 2D, LSCM was utilized to assess the expression level of SLC7A11 and SLC3A2. The fluorescence of the two proteins of tumor cells showed negligible changes after treatment with RCH NPs. By contrast, the cell fluorescence decreased significantly after co-incubation with IFN-γ. The above results revealed that IFN-γ derived from the activated immune system could down-regulate SLC7A11 and SLC3A2, resulting in a cascade enhancement of ferroptotic cell death and ferroptosis-induced immunotherapy.
As the vital biomarker of ferroptosis, the accumulation of lipid peroxide inside tumor cells determines the lethality of ferroptosis, wherein malondialdehyde (MDA), working as a lipid oxidation end-product, shows increased levels during ferroptosis.26,27 By contrast, GSH, a major nutrient involved in multiple aspects of oxidation–reduction in tumor cells, would be consumed by cytotoxic ˙OH generated from the Fenton reaction in ferroptosis.28 With both as biomarkers of ferroptosis, the extent of MDA production (Fig. 2E) and GSH depletion (Fig. 2F) induced by RCH NPs was further investigated in B16F10 cells to confirm the intrinsic characteristics of ferroptosis. RCH NPs exhibited more efficient MDA production and GSH depletion than free hemin, thanks to the enriched accumulation of RCH NPs through nanosize-amplified cellular uptake. The more efficient MDA production and GSH depletion demonstrated the more effective intracellular lipid peroxidation for ferroptosis. As a control, the RCH NPs group with ferroptosis inhibitor Fer-1 exhibited less efficient MDA production and GSH depletion. In addition, an enhanced extent of MDA production and GSH depletion by RCH NPs with IFN-γ co-incubation was also observed, revealing that the immune response could augment ferroptosis-specific lipid peroxidation by IFN-γ secretion for facilitating ferroptosis. Taken altogether, the ferroptosis-induced immune response could in turn enhance ferroptosis via the secreted IFN-γ, realizing self-amplification between ferroptosis and immunotherapy in a cascade manner.
Ferroptosis can be promoted via the deactivation of GPX4.29 GPX4-catalyzed detoxication of the lipid peroxide to non-toxic lipid alcohols needs GSH to act as the cofactor. Inspired by the excellent results in the GSH depletion assay with RCH NPs, the activity of GPX4 was further examined. According to Fig. 2G, hemin displayed a slight inhibition of GPX4 activity in contrast to the control group. However, the GPX4 activity decreased markedly in tumor cells of the RCH NPs group, and this trend became more obvious with the addition of IFN-γ, further indicating that IFN-γ was helpful in making the tumor cells more efficient to ferroptosis. This could be ascribed to the blockage of GSH synthesis via the inhibition of SLC7A11 and SLC3A2 governed by IFN-γ, as mentioned above.
Disruption of inherent ferroptosis-driven immune escape by RCH NPs
Ferroptotic tumor cells are biologically characterized by the up-regulated COX-2 along with the COX-2-derived up-regulation of PGE2.14–16 However, PGE2 is a major inflammation-associated immunosuppressive factor that endows ferroptotic tumor cells with immune escape for suppressing the antitumor immunity.17,18 Thus, as a negative role, ferroptosis inherently stimulates inflammation-associated immunosuppression (Fig. 3A). Our RCH NPs are composed of celecoxib, a non-steroidal anti-inflammatory drug, which enables the tailoring of the COX-2 activity. To uncover the regulation of COX-2 by our RCH NPs during ferroptosis, the in vitro and in vivo COX-2 expression levels were investigated with the control experiments. Considering the uptake efficiency and water solubility between the free hydrophobic hemin drug and hemin-based nanoparticles, the assembly of hemin and HSA (defined as H@H NPs) was utilized as a control for comparison with the reprogramming role of the RCH NPs. For western blot analysis at the cellular level (Fig. 3B and C), a higher level of COX-2 for the H@H NPs group was observed over that of the NS (normal saline) group, indicating that the up-regulated COX-2 level was triggered by hemin-mediated ferroptosis. Notably, the level of COX-2 for the RCH NPs group significantly decreased compared with that of the H@H NPs group, thanks to the inhibition of COX-2 by celecoxib within the RCH NPs. The results of cellular fluorescence images were in agreement with that of western blot analysis (Fig. 3D). Moreover, for the immunofluorescence-stained images of tumor sections (Fig. 3E), the stronger COX-2 expression of the H@H NPs group over the NS group and the weaker COX-2 expression of RCH NPs compared with the H@H NPs group were observed, consistent with the results at the cellular level. The above results thus demonstrated that the RCH NPs could powerfully suppress the COX-2 activity. The secretion of PGE2 was further assessed using an ELISA kit to verify the proinflammatory role triggered by ferroptosis and the anti-inflammatory role mediated by the RCH NPs. As shown in Fig. 3F, the H@H NPs promoted the secretion of PGE2 in tumor cells compared with the control group, indicating that ferroptosis inherently triggered PGE2-based inflammation-associated immunosuppression. However, the PGE2 content of the RCH NPs group was significantly lower than that of the H@H NPs group due to the unique role of celecoxib, further demonstrating that RCH NPs could potently inhibit the inflammatory COX-2/PGE2 pathway up-regulated by ferroptosis to relieve inflammation-associated immunosuppression.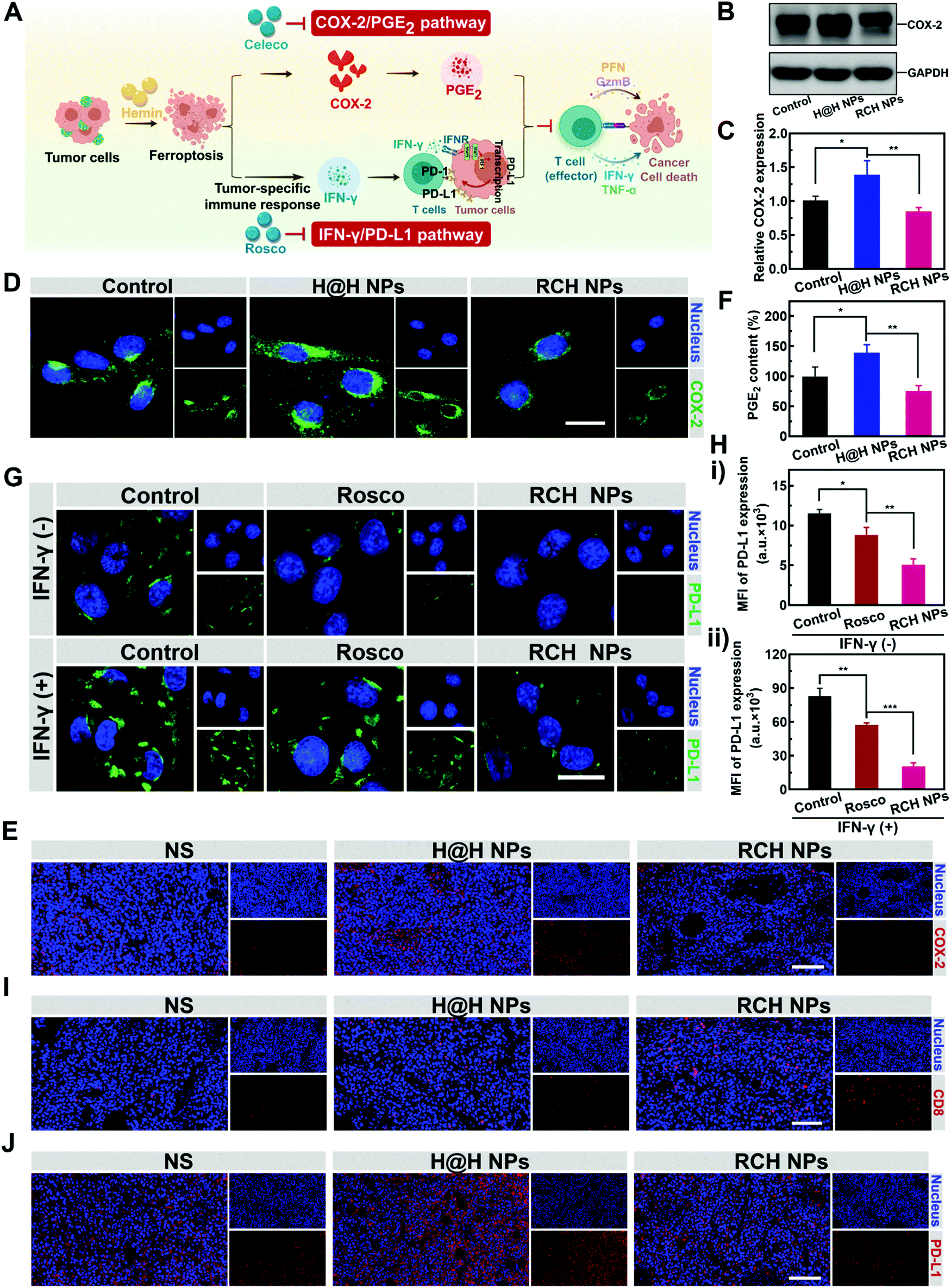 | ||
| Fig. 3 (A) Illustration showing that the RCH NPs abrogate ferroptosis-driven immune escape. (B) Western blot results, (C) corresponding quantitative determination and (D) fluorescent images of the COX-2 expression of different groups (scale bar: 20 μm). (E) Immunofluorescence-stained images of tumor sections for COX-2 expression analysis. Scale bar: 200 μm. (F) ELISA results of PGE2 secretion with different treatments. (G) Fluorescent images (scale bar: 20 μm) and (H) flow cytometry results of PD-L1 expression on B16F10 cells after different treatments. (I) Immunofluorescence-stained images of tumor sections for CD8+ T cell infiltration analysis. Scale bar: 200 μm. (J) Immunofluorescence-stained images of the tumor sections for PD-L1 expression analysis. Scale bar: 200 μm. | ||
The effective immune response elicited by ferroptosis could enhance the production of IFN-γ via the recruitment of cytotoxic T cells. However, as another negative role, ferroptosis-stimulated IFN-γ production could act as an accomplice of adaptive immune resistance to promote the expression of PD-L1 on tumor cells by inducing interferon regulatory factors 1 (IRF1), thus impairing the antitumor immunotherapy effect (Fig. 3A). Recent literature reports have identified that Cdk5 inhibition could lead to the persistent expression of PD-L1 transcriptional repressors, interferon regulatory factors 2 (IRF2), and interferon regulatory factor 2 binding protein 2 (IRF2BP2), which not only directly down-regulated the tumoral PD-L1 expression by virtue of genetic blockage, but also blunted the cascade reaction between PD-L1 up-regulation and IFN-γ stimulation via the competition between IRF1 and IRF2.30–32 Thus the roscovitine-based nanodrug can reverse the adaptive immune resistance by disrupting the IFN-γ-governed PD-L1 up-regulation. To understand the efficacy of roscovitine in RCH NPs for regulation of the expression of PD-L1 in cellular experiments, we assessed the PD-L1 expression level on the surface of B16F10 cells after treatment via IFN-γ alone (the control group), free roscovitine + IFN-γ and RCH NPs + IFN-γ for 24 h. As elucidated in Fig. 3G and H, for the group of IFN-γ alone, IFN-γ stimulation significantly up-regulated the PD-L1 expression level on the B16F10 cells, indicating severe adaptive immune resistance induced by the IFN-γ. However, the groups of both free roscovitine + IFN-γ and RCH NPs + IFN-γ exhibited the effective inhibition of PD-L1 expression, where RCH NPs + IFN-γ demonstrated a greater efficiency than free roscovitine + IFN-γ. Therefore, even under IFN-γ stimulation, the RCH NPs possessed an excellent ability for the persistent suppression of PD-L1 expression, indicating that the RCH NPs-induced attenuation of PD-L1 expression was irreversible for subsequent IFN-γ stimulation, thanks to the roscovitine in the RCH NPs. As a result, the RCH NPs could precisely reverse adaptive immune resistance in ferroptosis. To further understand the efficacy of the RCH NPs in regulating the IFN-γ-induced PD-L1 expression in ferroptosis, we conducted an in vivo study within tumors. We investigated the quantity of intratumoral cytotoxic T cells (CD8+ T cells) after different treatments. As depicted in Fig. 3I, the effective immune response elicited by ferroptosis significantly promoted the recruitment of cytotoxic T cells in the RCH NPs group, which in turn enabled the stimulation of IFN-γ secretion. For the immunofluorescence-stained images of tumor sections in determining the PD-L1 expression (Fig. 3J), the stronger PD-L1 expression of the H@H NPs group over the NS group and the weaker PD-L1 expression of the RCH NPs group compared with the H@H NPs group were observed, which was consistent with the above results at the cellular level. From the results either at the cellular level or in vivo, our RCH NPs could potently blunt the cascade reaction between IFN-γ stimulation and PD-L1 up-regulation for reversing the adaptive immune resistance induced by IFN-γ in ferroptosis, enabling efficient ferroptotic immunotherapy.
Taken together, the inherent negative roles of ferroptosis in immunotherapy can be potently solved by our RCH NPs, either via eliminating inflammation-associated immunosuppression or via reversing the adaptive immune resistance for maximizing the therapeutic efficacy of ferroptosis (Fig. 3A).
In vivo antitumor performance of RCH NPs
The feasibility of the RCH NPs as an intravenous injection preparation was evaluated. According to Fig. S8 (ESI†), the hemolysis ratio of the RCH NPs was below 5% at each concentration, indicating that intravenous administration was feasible for our biocompatible RCH NPs. Due to the lack of effective fluorescent properties for the RCH NPs, RCH–Ce6 NPs were utilized as a substitute for the RCH NPs in the fluorescence imaging study, where the RCH–Ce6 NPs were prepared in a similar way to that of the RCH NPs. The fluorescence imaging results, to understand the in vivo distribution and intratumoral accumulation of Ce6-loaded RCH NPs after intravenous administration, are shown in Fig. 4A. The total fluorescence intensity (TFI) of the RCH–Ce6 NPs group in the tumor tissue was markedly stronger than that of the free Ce6 group, demonstrating the superior tumor accumulation, thanks to the enhanced permeability and retention (EPR) effect along with the HSA-governed interaction with albumin-binding proteins overexpressed in tumors.33–35 In addition, the fluorescence signal results of normal organs and tumors (Fig. 4A and Fig. S9, ESI†) revealed the tissue-distribution characteristics of the nanodrug. The strong fluorescence signal of tumors in ex vivo imaging further confirmed the superior accumulation of RCH–Ce6 NPs at the tumor site. Notably, a weak fluorescence intensity was detected in the spleen, heart and lungs of both groups, indicating the low accumulation and distribution in all three of these organs, which could result in modest toxic effects. The strong fluorescence intensity of the RCH–Ce6 NPs in the liver and kidneys might be attributed to its metabolic and excretory pathways.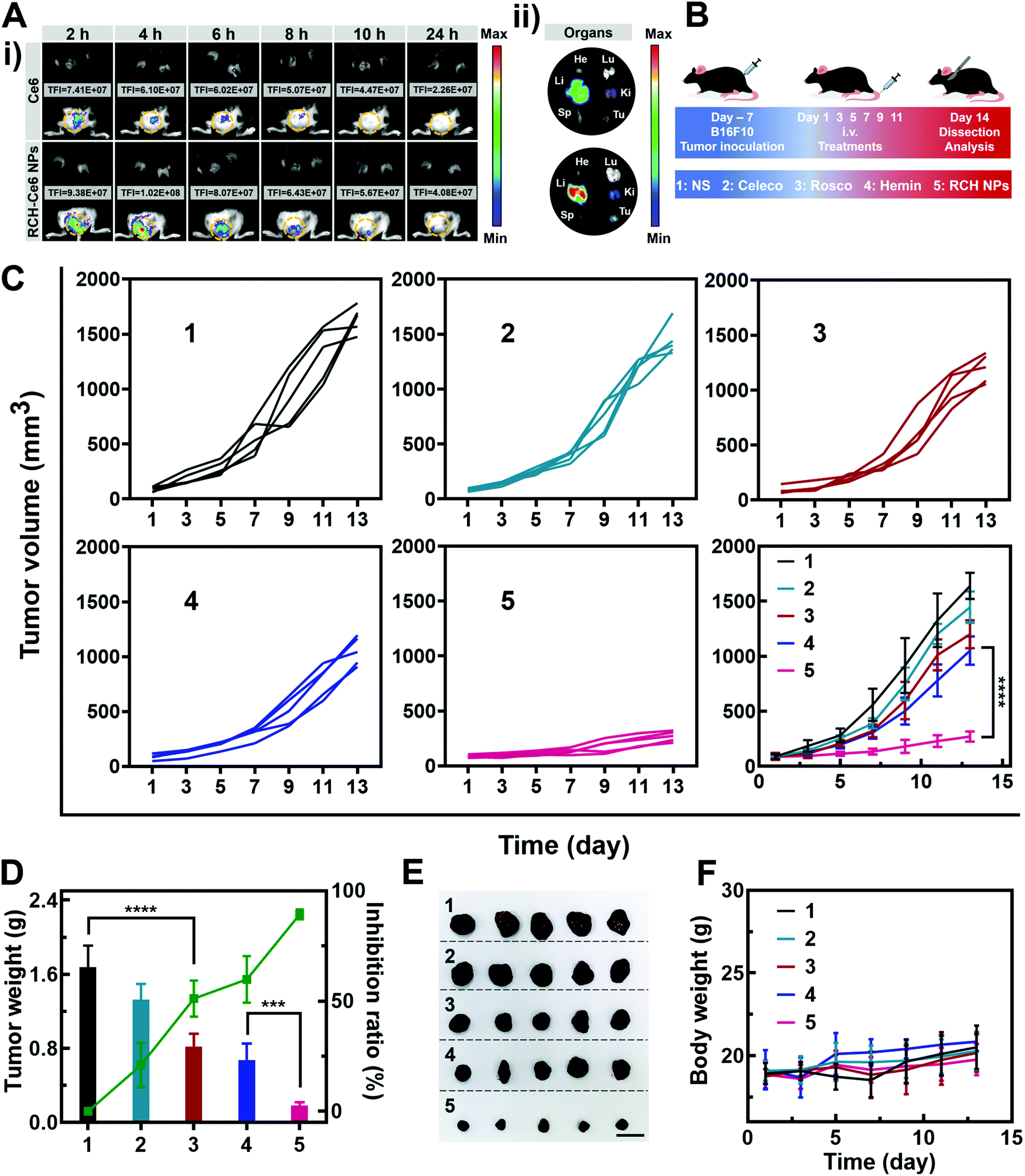 | ||
| Fig. 4 (A) Fluorescence photographs of (i) the mice after injection of free Ce6 and RCH–Ce6 NPs and (ii) the main organs and tumors (He, heart; Sp, spleen; Ki, kidney; Li, liver; Lu, lung; and Tu, tumor) excised 24 h after injection. (B) Schematic illustration for the experiment with RCH NPs to inhibit tumor growth. (C) Average tumor-growth curves and individual tumor-growth kinetics of different treated mice. (D) Change in tumor weight, and tumor inhibition ratio of different groups (n = 5). (E) Photographs of excised tumors of different groups (n = 5). Scale bar: 2 cm. (F) Body weight of different groups (n = 5). The groups include 1, NS; 2, Celeco; 3, Rosco; 4, Hemin; and 5, RCH NPs. | ||
The antitumor performance was investigated in B16F10 tumor-bearing mice, which were treated with NS, free celecoxib, roscovitine, hemin, and RCH NPs via intravenous administration every 2 days (Fig. 4B). The tumor volume was monitored during the treatment period (Fig. 4C). The celecoxib group showed the smallest inhibition of tumor growth, while the groups of roscovitine and hemin showed moderate tumor-growth suppression. However, the RCH NPs displayed the highest inhibition efficacy for tumors, where the tumor growth was significantly suppressed during the course of treatment. Moreover, the tumor weight and inhibition ratio further confirmed the superiority of the RCH NPs in tumor therapy (Fig. 4D). Photographs of the excised tumors after different treatments were shown in Fig. 4E. The results of hematoxylin–eosin (H&E) staining revealed there was no visible pathological damage in the main organs along with massive cell death and tissue damage in the tumor sites (Fig. S10, ESI†), demonstrating the good biocompatibility and outstanding antitumor effect of the RCH NPs. Moreover, the body-weight differences of the mice in all groups were negligible during the treatment (Fig. 4F). As two common blood biochemical markers, alanine amino transferase (ALT) and aspartate aminotransferase (AST) were detected to evaluate the safety issues. As displayed in Fig. S11 and S12 (ESI†), compared with the NS group, the content of ALT and AST in the RCH NPs group showed negligible changes. The results of blood chemistry further revealed that the RCH NPs had good biological safety. These results thus confirmed that our RCH NPs exhibited a powerful antitumor therapeutic performance with good biocompatibility.
In vivo immune response of RCH NPs
The efficient antitumor performance of the RCH NPs benefited not only from the ferroptosis-triggered direct killing effect but also the antitumor immunotherapeutic capability activated by reprogramming the Janus-faced nature of ferroptosis. To understand the immune response caused by our RCH NPs, flow cytometry analysis was employed to examine the ferroptosis-triggered immune response. As the typical sentinels of the immune system, DCs in tumor draining lymph nodes (TDLNs) were assayed via staining with the markers of CD80 and CD86 to quantitatively determine the proportion of matured DCs (CD11c+CD86+CD80+). Compared with the NS group, the RCH NPs group demonstrated a 2.9-fold higher ratio of the matured DCs (Fig. 5A and B), indicating its powerful capability of RCH NPs in promoting DC maturation with effective initiation of the immune response, thanks to the enhanced immunogenicity by ferroptosis.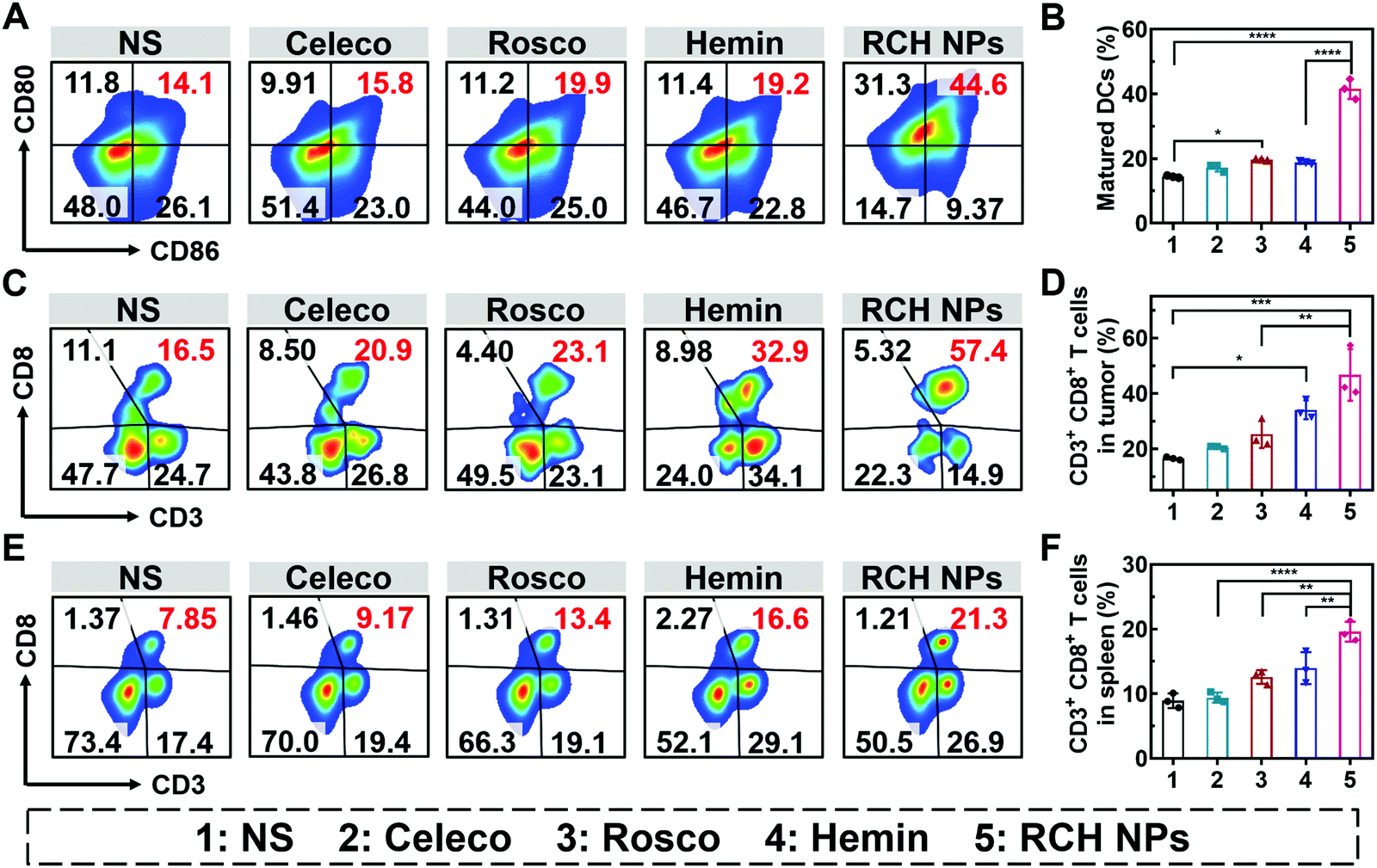 | ||
| Fig. 5 (A) Flow analysis and (B) quantitative result of the percentages of maturated DCs in the TDLNs under different treatments (n = 3). (C) Flow analysis and (D) quantitative result of the percentages of tumor infiltrating CD8+ T lymphocytes of various treated mice (n = 3). (E) Flow analysis and (F) quantitative result of CD8+ T lymphocytes in splenic lymphocytes of various treated mice (n = 3). The groups include 1, NS; 2, Celeco; 3, Rosco; 4, Hemin; and 5, RCH NPs. | ||
The antitumor immune effect is primarily governed by CD8+ cytotoxic T lymphocytes (CTLs, CD3+CD8+), which release cytotoxins to kill tumor cells directly.36,37 As shown in Fig. 5C and D, the tumor infiltration of CD8+ T cells in the hemin group was significantly increased to 2.1-fold compared with the NS group, indicating that ferroptosis could elicit an effective local immune response for immunotherapy. In particular, the tumor infiltration of CD8+ T cells of the RCH NPs-treated group was further elevated up to 2.8-fold compared with the NS group. These results clearly indicated that the RCH NPs could induce a more potent local immune response at the tumor tissue site via reprograming the Janus-faced nature of ferroptosis.
Furthermore, T lymphocytes in the spleen were analyzed to validate the systemic immune response triggered by the RCH NPs. The proportion of splenic CD8+ T lymphocytes was greatly elevated in the RCH NPs group, revealing that systemic immunity had been induced (Fig. 5E and F). The secretion of cytokines including interleukin-2 (IL-2), tumor necrosis factor-α (TNF-α) and IFN-γ were further analyzed via ELISA. As presented in Fig. S14 (ESI†), the RCH NPs efficiently up-regulated the levels of these three T-helper 1 (Th1)-type cytokines, further revealing that the remarkable systemic immune response had been triggered by treatment with the RCH NPs. As a result, apart from the direct killing effect caused by ferroptosis, the excellent antitumor effect of our RCH NPs was also attributed to their ability to facilitate the maturation of DCs, activating the immune effector cells, and elevating the levels of crucial immune cytokines, which accordingly led to a powerful antitumor immunotherapeutic efficacy.
The abscopal effect of RCH NPs for bilateral model
Inspired by the excellent results of the in vivo immune response triggered by the RCH NPs on B16F10 tumor-bearing mice, the abscopal effect of RCH NPs was further investigated in bilateral tumor-bearing mice, which were treated with NS and RCH NPs via intratumoral (primary tumor) administration every 4 days for a total of 3 times. The volume of the primary and abscopal tumors was monitored during the treatment period. Compared with the NS group, the primary tumor growth of the RCH NPs-treated mice was significantly inhibited, as shown in Fig. S15 (ESI†). For the abscopal tumors, the RCH NPs indicated a potent inhibition effect on the tumor growth, superior to the NS group (Fig. S16, ESI†). The results of the tumor weight and inhibition ratio further confirmed the excellent abscopal effect of the RCH NPs in antitumor therapy (Fig. S17 and S18, ESI†), which was mainly attributed to the triggered immunotherapy in untreated abscopal tumors. In addition, the body-weight differences of the mice in two groups were negligible during the treatment (Fig. S19, ESI†). Moreover, CTLs in abscopal tumors were analyzed using flow cytometry to systematically reveal the immunotherapeutic effect of RCH NPs. After treatment with RCH NPs, the tumor infiltration of CD8+ T cells in abscopal tumors was improved, and was 2.3-fold higher than that of the NS group (Fig. S20, ESI†). These results suggested that our RCH NPs are an effective antitumor nanodrug to fight against both primary tumors and abscopal tumors via an enhanced immune response.In vivo anti-metastasis evaluation of RCH NPs
Tumor metastasis is the main factor dominating the survival of cancer patients.38 The anti-metastasis capability of our designed RCH NPs was further examined using an established lung metastatic model based on the intravenous injection of B16F10 cells into tumor-bearing mice (Fig. 6A). After treatment with various formulations, the lungs of the mice were recovered to examine the metastatic lesions. As presented in Fig. 6B, metastatic nodules of the NS group appeared on the lung surface in large quantities, the number of which was approximately 60. However, the number of lung metastasis nodules in the other groups were decreased by different degrees. In particular, the number of lung metastasis nodules in the RCH NPs group decreased by 87.2% compared with the NS group. H&E images of the lungs further confirmed that the RCH NPs group displayed the minimal extent of cancerization (Fig. 6C). The potent ability of inhibiting lung metastasis by the RCH NPs benefited from the powerful self-activated systemic immune response. Therefore, our RCH NPs could act as a pre-eminent nanodrug for suppressing tumor metastasis.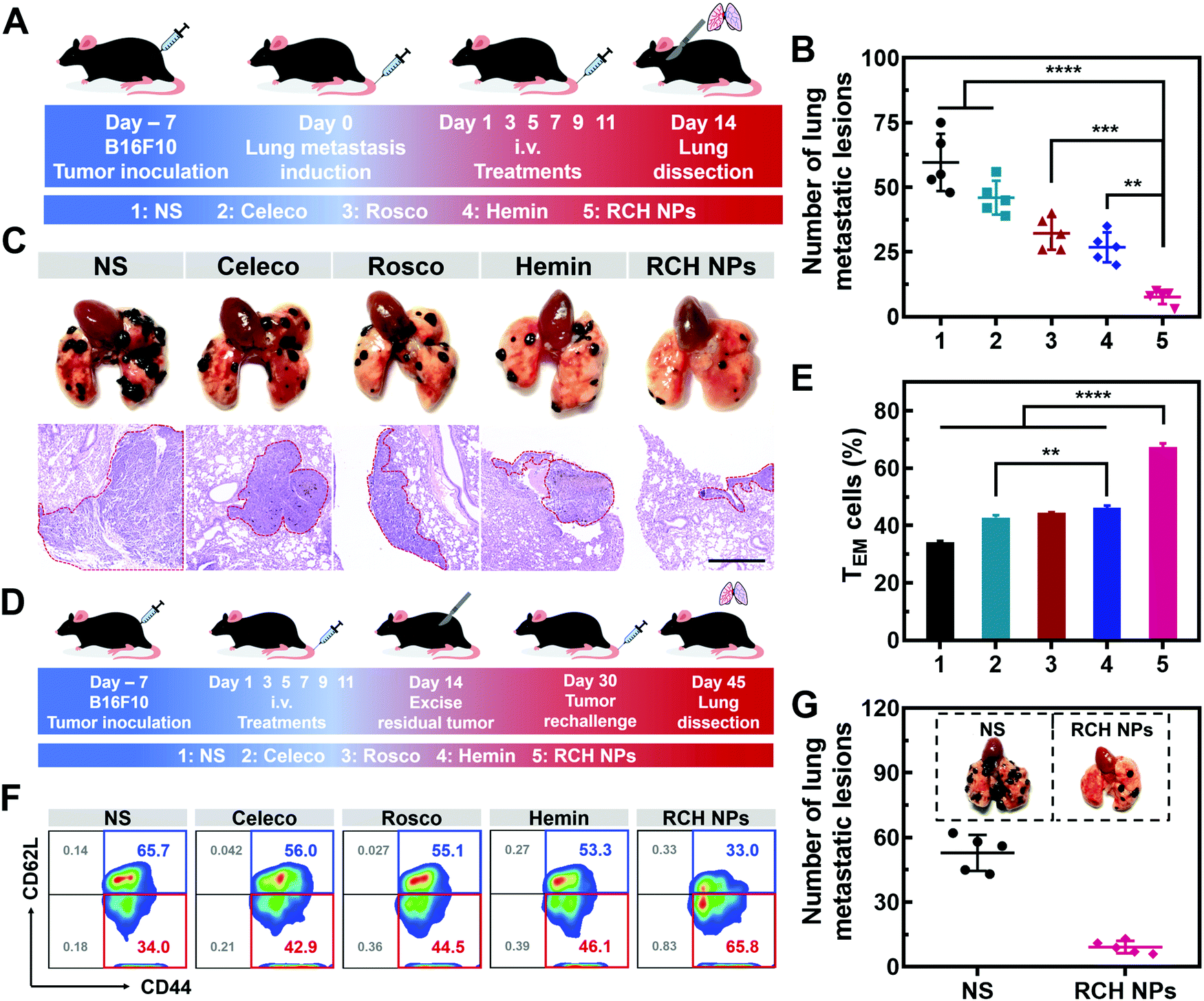 | ||
| Fig. 6 (A) Schematic illustration of the experimental approach for RCH NPs inhibiting lung metastasis. (B) Numbers of the lung metastatic lesions in different treated mice (n = 5). (C) Camera images (above) and H&E staining images (below) of lung metastasis in B16F10 tumor-bearing mice. Scale bar: 500 μm. (D) Schematic illustration of the immunological memory evaluation. (E) The proportion of effector memory T cells in the spleens from different treated mice (n = 3). (F) Flow cytometry results of the ratio of memory T cells in spleens. (G) Camera images of lung metastasis (n = 5). The groups include 1, NS; 2, Celeco; 3, Rosco; 4, Hemin; and 5, RCH NPs. | ||
In vivo long-term immune response evaluation of RCH NPs
Memory T cells play a crucial role in the long-term antitumor effect with the hallmarks of long-lasting self-renewal, replication and survival in body.39,40 Hence, inspired by the results of the excellent systemic immunological response, the percentages of memory T cells were further tested to explore the capability of the immune memory caused by the RCH NPs. As illustrated in Fig. 6D for 16 days after the surgical resection, the spleens were separated from the mice of all groups to quantitatively analyze the percentages of central memory T cells (TCM, CD3+CD8+CD62L+CD44+) and effector memory T cells (TEM, CD3+CD8+CD62L–CD44+). Compared with the NS group, the percentage of TEM in the RCH NPs group of mice was clearly elevated from 34.3 ± 0.4% to 67.3 ± 1.3% (Fig. 6E and F), while the percentage of TCM was reduced from 65.3 ± 0.4% to 31.9 ± 1.0% (Fig. S21, ESI†). These results confirmed that the powerful immunological memory had been elicited by the RCH NPs. We further explored the performance of RCH NPs for long-term tumor inhibition via rechallenging the mice with B16F10 cells at 16 days post-tumor excision. From Fig. 6G, the lungs of the mice treated with NS exhibited many metastatic lesions, while the lung metastasis of the mice treated with the RCH NPs was significantly reduced. As a result, our RCH NPs enabled a potent immunological memory with the capacity for long-term tumor-growth suppression.Conclusion
In summary, a self-amplifying nanodrug was precisely designed to reprogram the Janus-faced nature of ferroptosis via harnessing the positive role of ferroptosis along with reversing its inherent negative role to achieve optimal ferroptotic tumor therapy. As a positive role, the RCH NPs demonstrated powerful ferroptotic damage along with inducing an immune response, where the generated IFN-γ triggered disruption of the GPX4 pathway for cascade enhancement of ferroptotic cell death and ferroptosis-induced immunotherapeutic efficacy. In particular, the inherent negative roles of ferroptosis in immunotherapy were potently solved by our RCH NPs via both eliminating inflammation-associated immunosuppression and reversing IFN-γ-related adaptive immune resistance for maximizing the immunotherapeutic efficacy, thanks to the cooperation of celecoxib and roscovitine in the nanodrug. Our designed nanodrug (RCH NPs), which consists of hemin, celecoxib and roscovitine, is a promising multifunctional drug-delivery system for biomedical applications, and demonstrates powerful therapeutic efficacy against tumor progression and metastasis along with a long-lasting immunological memory response. Compared with traditional ferroptosis-based inorganic nanoplatforms (such as iron-based metal–organic frameworks), the RCH NPs demonstrate excellent biological safety and a more efficient immunotherapy effect, with great potential for further clinical development. The present work offers a conceptually innovative approach for manipulating the Janus-faced nature of ferroptosis to elicit maximized therapeutic efficacy against tumors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC, No. 82061148009, No. 21872083, and No. 81903558). The authors acknowledge the Pharmaceutical Biology Sharing Platform and the Advanced Medical Research Institute of Shandong University.References
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison, 3rd and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS PubMed.
- B. Hassannia, P. Vandenabeele and T. Vanden Berghe, Cancer Cell, 2019, 35, 830–849 CrossRef CAS PubMed.
- C. Liang, X. Zhang, M. Yang and X. Dong, Adv. Mater., 2019, 31, e1904197 CrossRef PubMed.
- F. Zhang, F. Li, G. H. Lu, W. Nie, L. Zhang, Y. Lv, W. Bao, X. Gao, W. Wei, K. Pu and H. Y. Xie, ACS Nano, 2019, 13, 5662–5673 CrossRef CAS PubMed.
- Q. Wen, J. Liu, R. Kang, B. Zhou and D. Tang, Biochem. Biophys. Res. Commun., 2019, 510, 278–283 CrossRef CAS PubMed.
- B. Ding, P. Zheng, F. Jiang, Y. Zhao, M. Wang, M. Chang, P. Ma and J. Lin, Angew. Chem., Int. Ed., 2020, 59, 16381–16384 CrossRef CAS PubMed.
- Q. Jiang, K. Wang, X. Zhang, B. Ouyang, H. Liu, Z. Pang and W. Yang, Small, 2020, 16, e2001704 CrossRef PubMed.
- D. Tang, R. Kang, T. V. Berghe, P. Vandenabeele and G. Kroemer, Cell Res., 2019, 29, 347–364 CrossRef CAS PubMed.
- W. Wang, M. Green, J. E. Choi, M. Gijón, P. D. Kennedy, J. K. Johnson, P. Liao, X. Lang, I. Kryczek, A. Sell, H. Xia, J. Zhou, G. Li, J. Li, W. Li, S. Wei, L. Vatan, H. Zhang, W. Szeliga, W. Gu, R. Liu, T. S. Lawrence, C. Lamb, Y. Tanno, M. Cieslik, E. Stone, G. Georgiou, T. A. Chan, A. Chinnaiyan and W. Zou, Nature, 2019, 569, 270–274 CrossRef CAS PubMed.
- X. Lang, M. D. Green, W. Wang, J. Yu, J. E. Choi, L. Jiang, P. Liao, J. Zhou, Q. Zhang, A. Dow, A. L. Saripalli, I. Kryczek, S. Wei, W. Szeliga, L. Vatan, E. M. Stone, G. Georgiou, M. Cieslik, D. R. Wahl, M. A. Morgan, A. M. Chinnaiyan, T. S. Lawrence and W. Zou, Cancer Discovery, 2019, 9, 1673–1685 CrossRef CAS PubMed.
- Y. Su, B. Zhao, L. Zhou, Z. Zhang, Y. Shen, H. Lv, L. H. H. AlQudsy and P. Shang, Cancer Lett., 2020, 483, 127–136 CrossRef CAS PubMed.
- M. A. Badgley, D. M. Kremer, H. C. Maurer, K. E. Delgiorno, H. J. Lee, V. Purohit, I. R. Sagalovskiy, A. Ma, J. Kapilian, C. E. M. Firl, A. R. Decker, S. A. Sastra, C. F. Palermo, L. R. Andrade, P. Sajjakulnukit, L. Zhang, Z. P. Tolstyka, T. Hirschhorn, C. Lamb, T. Liu, W. Gu, E. S. Seeley, E. Stone, G. Georgiou, U. Manor, A. Iuga, G. M. Wahl, B. R. Stockwell, C. A. Lyssiotis and K. P. Olive, Science, 2020, 368, 85–89 CrossRef CAS PubMed.
- W. S. Yang, R. SriRamaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji, C. B. Clish, L. M. Brown, A. W. Girotti, V. W. Cornish, S. L. Schreiber and B. R. Stockwell, Cell, 2014, 156, 317–331 CrossRef CAS PubMed.
- D. Tang, X. Chen, R. Kang and G. Kroemer, Cell Res., 2021, 31, 107–125 CrossRef CAS PubMed.
- D. Li and Y. Li, Signal Transduction Targeted Ther., 2020, 5, 108 CrossRef PubMed.
- P. Kalinski, J. Immunol., 2012, 188, 21–28 CrossRef CAS PubMed.
- D. Wang and R. N. DuBois, Carcinogenesis, 2015, 36, 1085–1093 CrossRef CAS PubMed.
- J. L. Benci, L. R. Johnson, R. Choa, Y. Xu, J. Qiu, Z. Zhou, B. Xu, D. Ye, K. L. Nathanson, C. H. June, E. J. Wherry, N. R. Zhang, H. Ishwaran, M. D. Hellmann, J. D. Wolchok, T. Kambayashi and A. J. Minn, Cell, 2019, 178, 933–948.e914 CrossRef CAS PubMed.
- S. Spranger, R. M. Spaapen, Y. Zha, J. Williams, Y. Meng, T. T. Ha and T. F. Gajewski, Sci. Transl. Med., 2013, 5, 200ra116 Search PubMed.
- P. Sharma and J. P. Allison, Science, 2015, 348, 56–61 CrossRef CAS PubMed.
- H. Tian, M. Zhang, G. Jin, Y. Jiang and Y. Luan, J. Colloid Interface Sci., 2021, 587, 358–366 CrossRef CAS PubMed.
- J. Park, B. Sun and Y. Yeo, J. Controlled Release, 2017, 263, 90–101 CrossRef CAS PubMed.
- B. Hoang, M. J. Ernsting, A. Roy, M. Murakami, E. Undzys and S. D. Li, Biomaterials, 2015, 59, 66–76 CrossRef CAS PubMed.
- C. Neuzillet, A. Tijeras-Raballand, J. Cros, S. Faivre, P. Hammel and E. Raymond, Cancer Metastasis Rev., 2013, 32, 585–602 CrossRef CAS PubMed.
- T. Liu, W. Liu, M. Zhang, W. Yu, F. Gao, C. Li, S. B. Wang, J. Feng and X. Z. Zhang, ACS Nano, 2018, 12, 12181–12192 CrossRef CAS PubMed.
- B. Yu, B. Choi, W. Li and D. H. Kim, Nat. Commun., 2020, 11, 3637 CrossRef CAS PubMed.
- M. Gao, J. Deng, F. Liu, A. Fan, Y. Wang, H. Wu, D. Ding, D. Kong, Z. Wang, D. Peer and Y. Zhao, Biomaterials, 2019, 223, 119486 CrossRef CAS PubMed.
- L. Chen, Z. Lin, L. Liu, X. Zhang, W. Shi, D. Ge and Y. Sun, ACS Biomater. Sci. Eng., 2019, 5, 4861–4869 CrossRef CAS PubMed.
- R. D. Dorand, J. Nthale, J. T. Myers, D. S. Barkauskas, S. Avril, S. M. Chirieleison, T. K. Pareek, D. W. Abbott, D. S. Stearns, J. J. Letterio, A. Y. Huang and A. Petrosiute, Science, 2016, 353, 399–403 CrossRef CAS PubMed.
- Q. Li, Z. P. Zhao, X. H. Qin, M. Z. Zhang, Q. Du, Z. H. Li and Y. X. Luan, Adv. Funct. Mater., 2021, 31, 2104630 CrossRef CAS.
- Q. Li, Y. Zhou, W. He, X. Ren, M. Zhang, Y. Jiang, Z. Zhou and Y. Luan, J. Controlled Release, 2021, 338, 33–45 CrossRef CAS PubMed.
- C. Zhou, X. Song, C. Guo, Y. Tan, J. Zhao, Q. Yang, D. Chen, T. Tan, X. Sun, T. Gong and Z. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 42534–42548 CrossRef CAS PubMed.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2013, 4, 81–89 CrossRef PubMed.
- M. Zhang, T. W. Herion, C. Timke, N. Han, K. Hauser, K. J. Weber, P. Peschke, U. Wirkner, M. Lahn and P. E. Huber, Neoplasia, 2011, 13, 537–549 CrossRef CAS PubMed.
- Q. Chen, L. Xu, C. Liang, C. Wang, R. Peng and Z. Liu, Nat. Commun., 2016, 7, 13193 CrossRef CAS PubMed.
- B. Farhood, M. Najafi and K. Mortezaee, J. Cell. Physiol., 2019, 234, 8509–8521 CrossRef CAS PubMed.
- S. Zhou, Q. Shang, N. Wang, Q. Li, A. Song and Y. Luan, J. Controlled Release, 2020, 328, 617–630 CrossRef CAS PubMed.
- T. Walzer, C. Arpin, L. Beloeil and J. Marvel, J. Immunol., 2002, 168, 2704–2711 CrossRef CAS PubMed.
- M. Yu, X. Duan, Y. Cai, F. Zhang, S. Jiang, S. Han, J. Shen and X. Shuai, Adv. Sci., 2019, 6, 1900037 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00506e |
| This journal is © The Royal Society of Chemistry 2022 |