
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect CO2 capture and conversion to fuels on magnesium nanoparticles under ambient conditions simply using water†
Sushma A.
Rawool
a,
Rajesh
Belgamwar
a,
Rajkumar
Jana
b,
Ayan
Maity
a,
Ankit
Bhumla
a,
Nevzat
Yigit
c,
Ayan
Datta
 b,
Günther
Rupprechter
c and
Vivek
Polshettiwar
*a
b,
Günther
Rupprechter
c and
Vivek
Polshettiwar
*a
aDepartment of Chemical Sciences, Tata Institute of Fundamental Research (TIFR), Mumbai, India. E-mail: vivekpol@tifr.res.in; Tel: +91 8452886556
bSchool of Chemical Sciences, Indian Association for the Cultivation of Science, Kolkata, India
cInstitute of Materials Chemistry, Technische Universität Wien, Getreidemarkt 9/BC/165, 1060, Vienna, Austria
First published on 31st March 2021
Abstract
Converting CO2 directly from the air to fuel under ambient conditions is a huge challenge. Thus, there is an urgent need for CO2 conversion protocols working at room temperature and atmospheric pressure, preferentially without any external energy input. Herein, we employ magnesium (nanoparticles and bulk), an inexpensive and the eighth-most abundant element, to convert CO2 to methane, methanol and formic acid, using water as the sole hydrogen source. The conversion of CO2 (pure, as well as directly from the air) took place within a few minutes at 300 K and 1 bar, and no external (thermal, photo, or electric) energy was required. Hydrogen was, however, the predominant product as the reaction of water with magnesium was favored over the reaction of CO2 and water with magnesium. A unique cooperative action of Mg, basic magnesium carbonate, CO2, and water enabled this CO2 transformation. If any of the four components was missing, no CO2 conversion took place. The reaction intermediates and the reaction pathway were identified by 13CO2 isotopic labeling, powder X-ray diffraction (PXRD), nuclear magnetic resonance (NMR) and in situ attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR), and rationalized by density-functional theory (DFT) calculations. During CO2 conversion, Mg was converted to magnesium hydroxide and carbonate, which may be regenerated. Our low-temperature experiments also indicate the future prospect of using this CO2-to-fuel conversion process on the surface of Mars, where CO2, water (ice), and magnesium are abundant. Thus, even though the overall process is non-catalytic, it could serve as a step towards a sustainable CO2 utilization strategy as well as potentially being a first step towards a magnesium-driven civilization on Mars.
Introduction
Excessive CO2 emissions are a major cause of climate change, and hence reducing the CO2 levels in the Earth's atmosphere is key to limit adverse environmental effects. Rather than just capturing and storing CO2, it would be desirable to use it as carbon feedstock for fuel production, to achieve the target of “net-zero-emissions energy systems”.1–3 Some efficient protocols have been reported for CO2 conversion,2–8 but they required external energy by photo-, electro- or thermal activation (which also requires gaseous hydrogen). Thus, any process that could use water as a hydrogen source at room temperature and atmospheric pressure would be highly sustainable, but no such processes have been reported in the literature to date.Herein, based on our work in heterogeneous catalysis,9–13 we report the use of Mg nanoparticles (and bulk) to directly react CO2 with water at room temperature and atmospheric pressure, forming methane, methanol, and formic acid without requiring external energy sources. Magnesium is the 8th most abundant element in the Earth's crust and 4th most common element in the Earth (after iron, oxygen and silicon).
Both pure CO2 and CO2 in the air were used, while water acted as a hydrogen source. Nanoparticles of Mg are air-stable and can be handled in an open environment. Mg-based catalysts/materials are widely used for organic transformations,14–20 hydrogen generation, and thermal and electric energy production,21–23 and there have been few studies of CO2 and Mg metal/metal oxide interactions.24–32 However, so far, there are no reports employing Mg for CO2 conversion to methane, methanol, and formic acid. Herein, we present the first report of how Mg can convert CO2 to fuel [CO2 + H2O + Mg → CH4 + CH3OH + HCOOH + H2 + Mg5(CO3)4(OH)2·4H2O], which proceeds at room temperature without any external energy. The mechanism of this process is determined by several independent (also in situ) techniques, backed-up by theoretical calculations. This study demonstrates the first step towards sustainable CO2 utilization to produce fuel and chemicals.
Results and discussion
Transmission electron microscopy (TEM) indicated that the Mg nanoparticles (NPs) were aggregates with sizes ranging from 200 to 500 nm (Fig. 1a, inset), composed of nanocrystals. Powder X-ray diffraction (PXRD, Fig. 1a) analysis confirmed the crystallinity of the Mg nanoparticles, with N2 sorption indicating a specific surface area of 13 m2 g−1 (Fig. S1†). Elemental analysis by EDX showed the presence of Mg and oxygen (Table S1†), indicating the presence of the MgO layer on the Mg NP surface. The average crystallite size calculated using Scherrer's equation was found to be 32 nm. The CO2 reduction reaction on Mg NPs in water was evaluated in a batch reactor at room temperature and atmospheric pressure. The products were identified and quantified by online gas chromatography (micro GC) with a thermal conductivity detector (TCD). Conversion of CO2 in air yielded 3.5 μmol methane per gram Mg in just 10 minutes (Fig. 1b). For pure CO2 gas, ∼100 μmol methane g−1 was obtained in 15 minutes (Fig. 1c).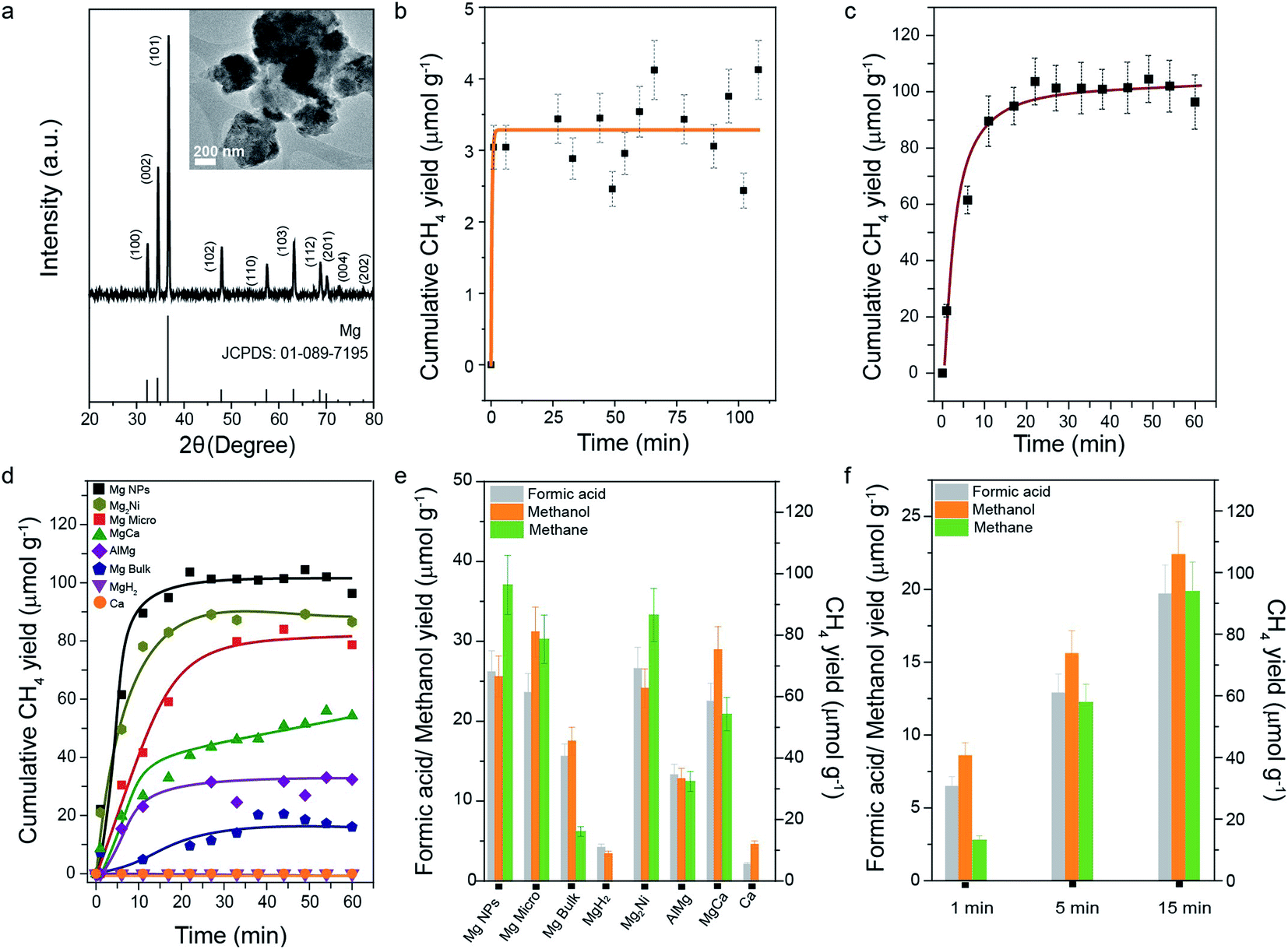 | ||
| Fig. 1 (a) PXRD patterns (TEM image in the inset) of Mg NPs along with the standard XRD pattern of Mg (JCPDS no. 01-089-7195); cumulative methane yield with reaction time for CO2 reduction by Mg NPs using (b) CO2 directly from the air and (c) 99.99% CO2; (d) comparison of yields of various metals and Mg–metal alloys (99.99% CO2); (e) methanol, formic acid, and methane yields after1 h, for CO2 (99.99%) reduction on Mg NPs, micro powder and bulk Mg, Mg–metal alloys, MgH2 and Ca metal; (f) methane, methanol, and formic acid yield quantified after 1, 5, and 15 min, for reduction of CO2 using Mg NPs. All reactions were carried out in water at room temperature and atmospheric pressure. Error bars are omitted in (d) for clarity and were less than 10%. The selectivity for carbon-containing products such as methane, methanol and formic acid is given in Fig. S2.† | ||
Next, we studied the performance of bulk Mg powders and Mg-alloys with Al, Ni, and Ca. Even bulk (granules) and micron-sized (powder) Mg converted CO2 to methane with good yield (Fig. 1d and Table S2†), but at a slower rate than nanoparticles. This indicated that the nano-size of the Mg NPs, leading to high specific surface area (SSA), facilitated efficient chemisorption of CO2 and water on the Mg surface, accelerating methane formation. MgH2 and pure calcium did not produce any methane (Fig. 1d), suggesting that the bare Mg0 surface was necessary. Mg–Al, Mg–Ni, and Mg–Ca alloys showed less methane formation than Mg NPs (Fig. 1d and Table S2†), indicating a unique behavior of pure Mg for CO2 conversion. The liquid reaction mixtures were analyzed by 1H nuclear magnetic resonance (NMR) spectroscopy and the formation of methanol and formic acid was also observed (Fig. 1e and f). Mg NPs yielded 25.6 μmol g−1 of methanol and 26.2 μmol g−1 of formic acid. Bulk Mg, as well as its alloys, also yielded methanol and formic acid, but a lower amount than yielded by Mg NPs. MgH2 and Ca produced only minute amounts of methanol and formic acid, confirming their lower ability to adsorb and activate CO2. The Mg nanoparticles showed selectivity of 65.8, 16.7 and 17.2% for methane, methanol and formic acid production, respectively (Fig. S2†).
CO2 conversion process efficiency comparison
Table 1 summarizes some of the best (to our knowledge) CO2 (from the air or pure) conversion processes under ambient conditions.11,33–36 Our Mg process produced 314 μmol g−1 h−1 total products when pure CO2 was used, which was significantly higher than those produced by the best-known catalysts, Pd7Cu1/TiO2 (19.6 μmol g−1 h−1),35 or Rb0.33WO3 (15 μmol g−1 h−1),33 both using high-power UV light (Table 1, S. N. 3 and 4). The Mg-assisted CO2 conversion efficiency was also better than that of Au-catalysts (0.063–0.13 μmol g−1 h−1, Table 1, S. N. 6–8).11,34,36 Even for CO2 directly from the air, Mg showed a productivity of 59 μmol products g−1 h−1, while the highest reported value so far was 2.25 μmol products g−1 h−1 using Rb0.33WO3, which requires high power UV light (Table 1, S. N. 5). Thus, Mg NPs are a very good alternative to photocatalysts for sustainable CO2 to fuel conversion under ambient conditions.| S. N. | Material | CO2 source | Energy source | Reaction time (h) | Methane yield (μmol g−1) | Methanol yield (μmol g−1) | Product (methane + methanol) formation rate (μmol g−1 h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a No external energy means energy available in the reaction vessel at room temperature is sufficient for this reaction to proceed. | ||||||||
| 1 | Mg NPs | Pure CO2 | No external energya | 0.4 | 100 ± 10% | 25.6 ± 10% | 314 | This work |
| 2 | Mg NPs | CO2 from air | No external energya | 0.1 | 3.5 ± 10% | 2.4 ± 12% | 59 | This work |
| 3 | Pd7Cu1/TiO2 | Pure CO2 | UV light (2 mW cm−2) | 1 | 19.6 | 0 | 19.6 | 35 |
| 4 | Rb0·33WO3 | Pure CO2 | UV to NIR (300 W Xe lamp) | 4 | 0 | 60 | 15 | 33 |
| 5 | Rb0·33WO3 | CO2 from air | UV to NIR (300 W Xe lamp) | 4 | 0 | 9 | 2.25 | 33 |
| 6 | Au–Co | Pure CO2 | VIS light (100 mW cm−2) | 1 | 0.13 | 0 | 0.13 | 36 |
| 7 | Black gold | Pure CO2 | VIS to NIR light (1 W cm−2) | 12 | 1.5 | 0 | 0.125 | 11 |
| 8 | Au NPs | Pure CO2 | VIS light (300 mW cm−2) | 10 | 0.63 | 0 | 0.063 | 34 |
The comparison of a non-catalytic process with catalytic ones seems inappropriate. However, this is intentionally made to demonstrate that this Mg process, even being not catalytic, is still efficient and more sustainable than reported room temperature catalytic processes. For example, the Mg protocol yields 100 μmol g−1 methane in 0.4 h and to achieve the same methane yield, an Au catalyst needed 1587 h, i.e. 66 days (and solar energy for 66 days).34 None of the reported room temperature nano-catalysts showed stability for 1587 h and were often deactivated in just 12 to 24 h (Table 1). Thus, catalysts are not-consumed by definition, but in reality, most of them get deactivated in a few hours (“indirectly consumed”). The deactivated catalysts can be ideally regenerated/activated, but this has a substantial energy penalty and hydrogen gas consumption (and in the case of deactivation by nanoparticle sintering, no regeneration is possible). In contrast, Mg regeneration can be carried out by using a solar energy-driven laser without any hydrogen gas and for only 1 USD per kg.21
To prove that CO2 was indeed the carbon source of products, a control experiment was conducted in argon gas (without CO2), in which the formation of methane, methanol and formic acid was not observed. For further confirmation, we used isotopically labeled 13CO2 and analyzed the products by GC (Fig. S2†) and 13C NMR spectroscopy (Fig. 2a). The 13C NMR spectrum (Fig. 2a) indicated methanol and formic acid. As a further control experiment, unlabeled CO2 was used. The formation of methanol and formic acid was evident from 1H NMR, whereas no 13C signal was obtained with the same NMR pulse sequence and the total number of scans (Fig. S1b†). This was due to the low natural abundance (1%) of 13C in unlabeled CO2 (Fig. S1b†). The reaction products were monitored after 0, 1, 5, 15, 30 and 60 min reaction time, using a GC with TCD, flame ionization detector (FID) and mass spectrometer (MS). The reduction of 13CO2 produced isotopic 13CH4 (m/z = 17, Fig. S1g†), which confirmed CO2 as a carbon source for methane, methanol, and formic acid.
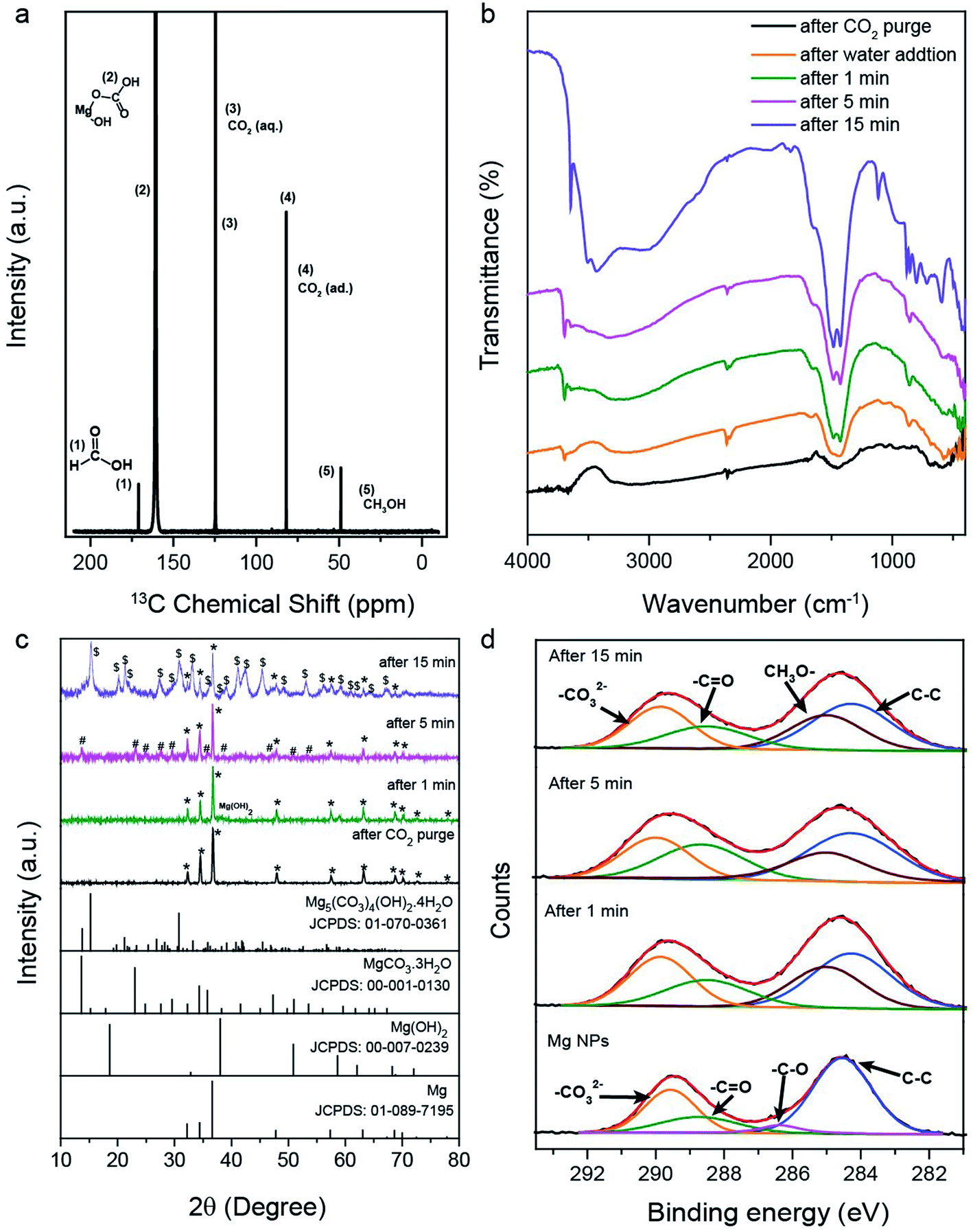 | ||
Fig. 2 (a) 13C NMR spectrum of the reaction mixture (using labeled 13CO2) after 1 h of reaction on Mg NPs; (b) FTIR spectra, (c) PXRD patterns and (d) XPS C 1s spectra of solid products recovered after 1, 5, and 15 min of the reaction of CO2 with Mg NPs and water, at room temperature and atmospheric pressure. (*Mg metal, #MgCO3·3H2O and $Mg5(CO3)4(OH)2·4H2O). Standard XRD patterns of Mg (JCPDS no.: 01-089-7195), Mg(OH)2 (JCPDS no.: 00-007-0239), MgCO3·3H2O (JCPDS no.: 00-001-0130) and Mg5(CO3)4(OH)2·4H2O (JCPDS no.: 01-070-0361) are included in (c). In (d), orange –CO32−, green –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, violet –C–O, blue –C–C, and purple –CH3–O. O, violet –C–O, blue –C–C, and purple –CH3–O. | ||
Reaction mechanism of Mg mediated CO2 conversion
To examine the overall reaction pathway, we characterized surface species and (intermediate) products after CO2 and water addition, and after 1, 5 and 15 min reaction time. Increasing levels of methane, methanol and formic acid yields were observed upon increase in reaction time (Fig. 1f). The solid reaction mixture was then isolated, dried and analyzed by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and PXRD (Fig. 2b and c). The IR spectrum of the Mg NPs after exposure to only CO2 showed a broad absorption at 1259–1621 cm−1, indicating adsorbed carbonate species.37 The FTIR spectrum of the sample after 15 min reaction time contained bands corresponding to carbonate species of basic magnesium carbonate [Mg5(CO3)4(OH)2·4H2O].38,39 IR analysis thus indicated the formation of magnesium carbonate within 1 and 5 min, and basic magnesium carbonate after 15 min of reaction. Corresponding PXRD patterns (Fig. 2c) confirmed the formation of magnesium carbonate at 5 min and basic carbonate after 15 min. A small amount of Mg was still present at 15 min of reaction time, although product formation was nearly stopped at this stage. However, when we recorded the PXRD patterns of the products recovered after 1 h of reaction (Fig. S4†), the Mg signal was not observed. This indicates that the small amount of Mg present at 15 min is catalytically active; however, its contribution was so small that the increase in product yield was not detected in the GC, although those traces of Mg reacted and vanished during 1 h reaction time.The reaction mechanism was also investigated by ex situ X-ray photoelectron spectroscopy (XPS), with Mg 2p, C 1s, and O 1s spectra of the solid materials acquired after 1, 5, and 15 minutes of reaction (Fig. 2d and S3†). For Mg NPs (before reaction), C 1s indicated adsorbed carbonates (289.4 eV; orange), carbonyl (288.7 eV; green), C–O (286.4 eV; magenta) and carbon (284.5 eV; blue) (Fig. 2d).40,41 The C 1s spectra of samples recovered after different reaction times show peaks at 285.0, 288.6, 289.9 and 284.2 eV, corresponding to –OCH3,42 the carbonyl group of formate, carbonate, and support carbon, respectively. The contribution of carbonyl, carbonate and –OCH3 species increased with reaction time, indicating that they are the intermediates formed during the reaction (Table S3†).
The XPS data (Fig. S3a†) are in accordance with ex situ FTIR (Fig. 2b) and PXRD (Fig. 2c) analysis. The O 1s XPS peaks at 529.39 (blue), 531.29 (green) and 532.78 eV (orange) correspond to MgO,43 Mg(OH)2 and magnesium carbonate species, respectively (Fig. S3a†).40 In O 1s, magnesium methoxide and hydroxide appear at a similar binding energy of 531.29 eV.42 As the reaction proceeded, the contributions of carbonate, hydroxide and methoxide were found to increase. The Mg 2p of the solid recovered at different time intervals showed four different peaks at 48.6, 49.3, 50.3 and 51.07 eV, again attributed to Mg,44 MgO, Mg(OH)2/Mg–OCH3 and magnesium carbonate, respectively (Fig. S3b†);40,42,44,45 the latter three contributions increased as the reaction proceeded, indicating the CO2 conversion to products via these intermediates.
To corroborate that (basic) carbonate species were reaction intermediates, the reaction progress of the purged reactor was then further monitored with time (Fig. S4†) and, the formation of methane was still observed, confirming that basic magnesium carbonates were reaction intermediates, which were then converted to methane. These studies suggest that water, CO2, and surface atoms of the Mg NPs react to produce intermediate basic magnesium carbonate, while Mg atoms in the sub-surface layers acted as an electron donor for the reduction step. The effect of the MgO shell (surface layer) was studied by Mg oxidation in air at 80 °C for 1, 3, and 11 days (Fig. S5a†). There was no significant effect on product yield (Fig. S5b†), indicating no role of MgO, and even pure MgO showed no methane, methanol and formic acid formation.
In situ attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) was employed to examine the mechanistic pathways under reaction conditions of CO2 conversion on Mg NPs by using water (Fig. 3). When CO2 gas was passed over the (pretreated, Fig. S6†) Mg surface at room temperature, a doublet appeared from the asymmetric stretching mode of CO2 gas, which after switching to He transformed into a singlet at 2341 cm−1, characteristic of the stretching mode of CO2 chemisorbed on the Mg surface (Fig. 3a). The presence of additional weak absorption bands in the region of 2800–3000 cm−1, corresponding to the C–H stretching modes (inset of Fig. 3b), indicates that the conversion of CO2 into hydrocarbons was initiated in the presence of H2O traces. Furthermore, when the Mg NPs were first exposed to water, He, and then CO2 gas, signals for unidentate carbonate (1511, 1392 and 1052 cm−1), bidentate carbonate (1666, 1346 and 1007 cm−1), bicarbonates (1626, 1409 and 1273 cm−1), MgCO3·3H2O (1488, 1433 and 1063 cm−1), and Mg5(CO3)4(OH)2·4H2O (1480, 1426 and 1143 cm−1) were observed (Fig. S7 and S8†).37–39,46 Also, peaks at 1530, 1329 and 1356 cm−1, for asymmetric stretching, νas(OCO), symmetric stretching, νs(OCO) and bending of C–H of formate adsorbed on the Mg surface were observed, respectively.47
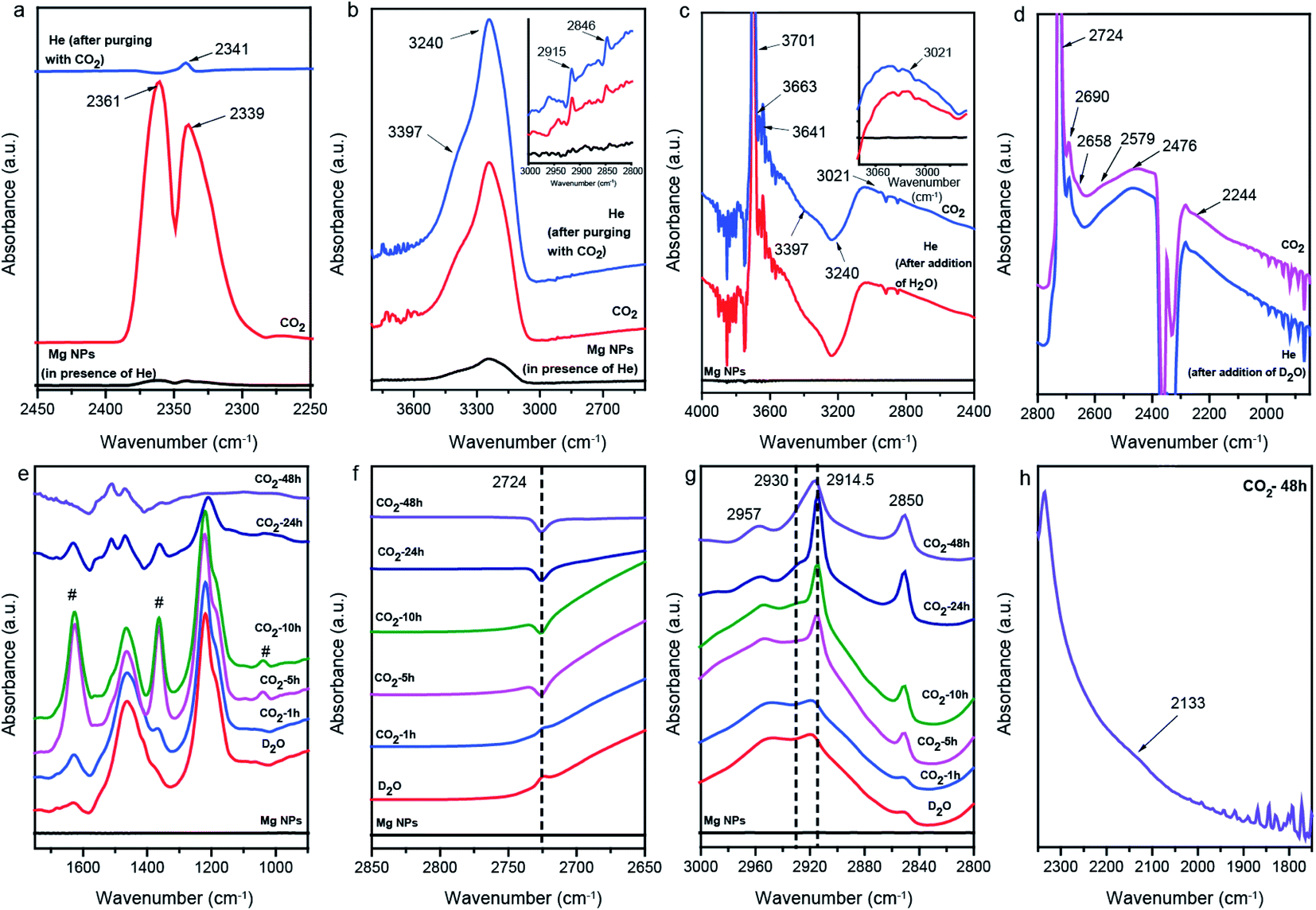 | ||
| Fig. 3 In situ ATR-FTIR spectra: (a and b) in He, CO2, and again He, (c) after adding H2O to the Mg NPs and subsequently purging with He and CO2 gas, (d) after adding D2O to the Mg NPs and subsequently purging with He and CO2 gas, and (e–h) after adding D2O to the Mg NPs and then purging with CO2 for 1, 5, 10, 24 and 48 h. | ||
The absorption band at 3701, 3663 and 3641 cm−1 in H2O shifted to 2724, 2690 and 2658 cm−1 in D2O, characteristic of isolated –OD groups, surface –OD of Mg–OCD2OD and –OD of bicarbonates, respectively (Fig. 3c and d).37,48 The νC–H stretching band observed at 3021 cm−1 (inset of Fig. 3c) shifted to a lower frequency of 2244 cm−1 by a factor of 1.35, and was assigned to the stretching of adsorbed CD4 groups.49
To trap the species formed during the reaction, CO2 gas was directly passed into the D2O (liquid) and ATR spectra were recorded at different time intervals. The spectra recorded after 1 h CO2 purging showed characteristic absorption bands at 1626 and 1363 cm−1, corresponding to DO–Mg–OC(O)OD species (marked as # in Fig. 3e).50 The decrease in the intensity of the 2724 cm−1 O–D stretching of isolated –OD groups with time clearly indicates that CO2 interacted with these –OD groups, leading to the formation of bicarbonate species (Fig. 3f). After longer purging with CO2 (10, 24 and 48 h), it was observed that the intensity of the bands attributed to the DO–Mg–OC(O)OD species decreased and new peaks at 1511 and 1468 cm−1 appeared, corresponding to the asymmetric and symmetric stretching of νC–O of the magnesium carbonate. From this observation, it can be inferred that the DO–Mg–OC(O)OD species were converted to hydrocarbons (Fig. 3g).51 A new weak band at 2133 cm−1, attributed to the C–D stretching of the CD3 group adsorbed on the Mg surface, and assigned based on the CD3 stretching mode at 2121 cm−1 over Ge, was reported by Knapp et al.52 (Fig. 3h).
Proposed reaction mechanism
Based on the ex situ and in situ results, a mechanism of Mg-assisted CO2 conversion can be deduced (Fig. 4). First, the CO2 molecule physisorbs on the Mg surface, followed by the formation of a chemisorbed bent anionic CO2− surface species (bonded to an Mg atom via an oxygen atom). As such a CO2− species is unstable;19 it reacts with water to form basic magnesium carbonate (I). Next, a second Mg–O bond forms, creating a four-membered ring (II).19 An electron from Mg and a proton from water convert this cyclic species (II) to a carboxyl radical (III). The free hydrogen atom (radical), that originated from the reduction of a proton by an electron (from Mg), then converts the carboxyl radical to a formic acid surface intermediate (IV), which upon hydrolysis yields formic acid. The formic acid surface species (IV) then accepts further H˙ to form a dihydroxymethyl radical intermediate (V), which upon addition of another H˙ leads to a methanol surface intermediate (VI), yielding methanol. Species (VI) on further reduction forms a methyl radical intermediate (VII), which is then reduced by H˙ to yield methane. The formed magnesium hydroxide (IX) reacts with CO2 to give basic carbonate (I). Ideally, the reaction cycle should continue, but the product concentration saturated after approximately 15 min and no further reaction was observed due to the nearly complete conversion of Mg to basic carbonate, leaving no unreacted Mg to act as an electron donor. Mg can then be regenerated, e.g. by using a solar-energy pumped laser.53,54 | ||
| Fig. 4 Plausible reaction mechanism of CO2 conversion to methane, methanol, and formic acid by Mg using water. Mg in the scheme represents the Mg surface of nanoparticles. The upward arrow after H2 (in step I) indicates hydrogen gas release. | ||
To confirm that a water proton was reduced to a H˙ radical by an electron from Mg, CO2 conversion was carried out using gaseous hydrogen (with no water present). When H2 was used, no methane, methanol and formic acid formation occurred, supporting the proposed role of water in the mechanism.
Another aspect to consider is that if Mg(0) transformed into Mg(II) upon reaction with water, it would no longer be able to provide electrons for CO2 reduction. Once Mg metal is exposed to water, a layer (or shell) of poorly soluble magnesium hydroxide forms on the Mg NP surface. The presence of this layer on the surface restricts the contact of H2O with sub-surface Mg metal present underneath. However, in the presence of CO2, this magnesium hydroxide layer interacts with CO2 to form basic magnesium carbonate, which has a higher water solubility than magnesium hydroxide. The presence of metallic Mg during the reaction was observed in the PXRD pattern (Fig. 2c), while its interactions with CO2 were detected in FTIR (Fig. 2b and 3) and NMR (Fig. 2a) spectra. The Mg atoms in the sub-surface layers transfer electrons to basic carbonate on the surface layers, converting it to methane, methanol and formic acid. As the basic magnesium carbonate is soluble in water, it does not restrict the access of fresh reactant molecules (H2O and CO2) to the Mg atoms present underneath the layer. This process continues till Mg metal is consumed. Thus, various ex situ and in situ studies fully support the reaction mechanism proposed in Fig. 4.
Density functional theory (DFT) study of the CO2 conversion process
In order to confirm and refine the proposed mechanistic pathways, we have performed a density functional theory (DFT) study, based on first-principles calculations using the Vienna Ab Initio Simulation Package (VASP).55 CO2 activation is the key step in the CO2 reduction reaction.56–58 Upon adsorption on the Mg surface, the linear CO2 molecule gets bent, with one of the oxygen atoms strongly adsorbing on Mg while the carbon atom weakly interacts with Mg (Fig. 5a, S9 and Table S4†). Based on the experimental results, the activation of CO2 may proceed through three different probable pathways (Fig. 5a and S9†) towards MgCO3 formation. As depicted in Fig. 5a, S9 and S10† both pathways I & II (CO2 adsorption on pristine and hydrated Mg (Mg–OH(ad) + H(ad)) surfaces, respectively) are thermodynamically favorable as all the steps are downhill of the potential energy surface (PES). In contrast, pathway-III (where CO2 activation occurs on the Mg(OH)2 surface) is thermodynamically unfavorable, due to the uphill CO2 adsorption step in the PES (Fig. S11, S9e and f†). Furthermore, CO2 is activated more effectively in pathways I & II than in pathway-III (Fig. S9 and Table S4†), following an activation order: pathway-I ≥ pathway-II ≫ pathway-III. The CO2 adsorption energy of the most feasible pathway (pathway-I) is −0.31 eV, paralleled by lengthening of the CO bond from 1.16 Å (equilibrium bond length) to 1.22 and 1.19 Å, and a change of the OCO bond angle from 180 to 141°. Both clearly suggest a more efficient CO2 activation.56–58 In addition, Bader charge analysis59,60 indicates a charge transfer of 0.17e from Mg to CO2 and a charge density difference (CDD) plot further supports CO2 activation through negative charge localization on the carbon atom (Fig. S12†).
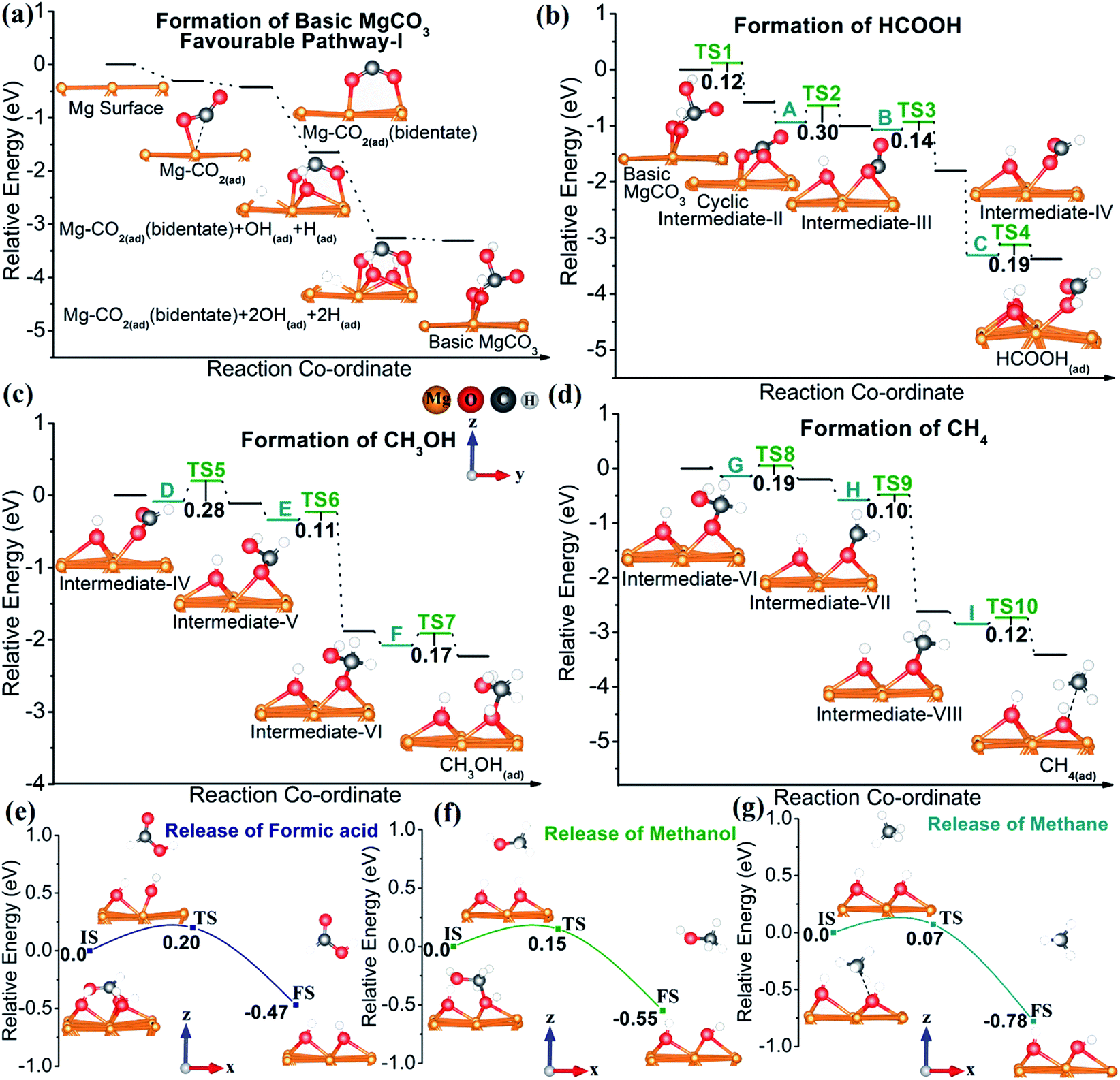 | ||
| Fig. 5 Potential energy surface (PES) for CO2 reduction: (a) formation of basic MgCO3via pathway-I (for the PES of pathways-II &III see Fig. S10 and S11 in the ESI†), (b) formation of formic acid, (c) formation of methanol, and (d) formation of methane; release steps of different products from the Mg NP surface during CO2 reduction to (e) formic acid, (f) methanol, and (g) methane. z,y coordinates shown in (c) are the same for (a, b and d). | ||
Fig. 5b shows the relative energy diagram of all elementary steps involved in formic acid formation from basic MgCO3. The downhill pathway from basic MgCO3 to surface-adsorbed HCOOH proves the thermodynamic feasibility of HCOOH formation. As evident from Fig. 5b and S13a–c,† the low activation barriers (i.e. ≤0.3 eV) also suggest kinetic viability of the proposed pathway at room temperature and ambient pressure. The high stabilization energy of C (i.e. hydrated intermediate-IV) accounts for the formation of formic acid. The formation of methanol also shows a downhill pathway for each elementary step (Fig. 5c and S13d–f). Apart from this, the methanol pathway is kinetically feasible with activation barriers of the elementary steps less than 0.3 eV (Fig. 5c). Interestingly, the route to CH4 formation is not only thermodynamically highly feasible, with each of the elementary steps downhill, but also kinetically accessible under the experimental conditions. A close look at the relative energy diagram (Fig. 5d) demonstrates that the formation of surface-adsorbed CH4 (CH4(ad)) on Mg occurs from intermediate-VIII, via I abstracting two protons from adjacent sites (Fig. S13i†). The reaction pathway for CH4 formation reveals that the breakage of the C–O bond (carbon atom from CH4(ad) and the O atom adsorbed on Mg) is essential for the formation of gaseous CH4 (Fig. 5e and S14c†). A detailed analysis of CH4(ad) discloses that the C–O bond distance for CH4(ad) is 2.6 Å while the equilibrium C–O bond length is 1.43 Å (Fig. S9 and Table S4†). This pronounced lengthening of the C–O bond in the CH4(ad) state clearly indicates the high feasibility of CH4 formation, both thermodynamically and kinetically. Accordingly, a close analysis of HCOOH(ad) and CH3OH(ad) reveal the necessity of Mg–O (bond between the Mg surface and O atom of HCOOH(ad)) and C–O (C atom of CH3OH(ad) and the adsorbed O atom on Mg) bond breaking, respectively. As expected, these bonds are elongated with respect to their equilibrium bond lengths, indicating a weak interaction between the Mg surface and these adsorbed species (Fig. 5f–g, S14a, b and Table S5†). Most importantly, the extent of bond elongation follows the order HCOOH(ad) ≤ CH3OH(ad) < CH4(ad), indicating that the formation of molecular CH4 is most feasible. Therefore, a relatively low kinetic barrier and high reaction energy (Er) make methane formation more feasible than that of HCOOH and CH3OH (Er for CH4 is −0.68 eV, whereas for HCOOH and CH3OH it is −0.26 and −0.32 eV, respectively).57
The release of the product species also affects the reaction rate,10,57 with HCOOH and CH3OH having low desorption activation barriers from Mg of 0.2 and 0.15 eV, respectively (Fig. 5e and f). However, methane desorption from the Mg surface is remarkably facile with a low activation barrier of 0.07 eV for activation compared to those of HCOOH (c.f. activation barrier = 0.20 eV) and CH3OH (c.f. activation barrier = 0.15 eV) (Fig. 5g). Hence, based on both experimental findings and theoretical calculations, the Mg NP surface can indeed convert CO2 to HCOOH, CH3OH, and CH4,, with the latter dominating at room temperature and ambient pressure.
There are a few “uphill” increments in energy barriers during various elementary steps, as well as during desorption (to release product species from the Mg surface), which need to be overcome before the following step(s) can take place. As evident from the relative energy diagram (Fig. 5), the maximum activation energy barrier for the transition state is 0.3 eV, i.e. 29 kJ mol−1, which is in the range of room temperature activation energy barriers for elementary reactions. Along with that, the desorption activation barriers for formic acid, methanol and methane are low (0.2, 0.15 and 0.07 eV respectively). Therefore, the activation energy barriers for the transition states, as well as the desorption steps can be easily overcome at room temperature.
Furthermore, magnesium has a standard electrode potential of −2.69 V vs. the standard hydrogen electrode. As the standard reduction potential of Mg is more negative than the reduction of water, Mg readily reacts with water to form hydrogen.23 The reaction of magnesium with water (Mg + H2O → Mg(OH)2 + H2) is exothermic (ΔH = −354 kJ mol−1),22 and this released energy can also be used to overcome the energy barriers. Thus, under reaction conditions, it is basically the surrounding environment that provides the small energy to overcome the activation barriers. Note that this is a “no external energy” process and not a “no energy” process.
Process sustainability
The crucial magnesium electrolysis step to produce magnesium from Mg2+ requires low energy,61 and Mg is one of the metals with the lowest energy demand for production (Fig. S15†).62,63 CO2 produced by other metals, as compared to Mg, is summarised in Fig. S16,† indicating that Mg is one of the metals producing the lowest amount of CO2 during production. If one compares the price of methane produced by our Mg protocol with the price of methane produced by other reported protocols (Table S6†), one can see nearly a million times difference in cost. This is because Mg is 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 times cheaper than all other well-known metal catalysts for CO2 reduction, and the Mg process has far better CO2 conversion efficiency/productivity than the reported catalysts.
000–50000 times cheaper than all other well-known metal catalysts for CO2 reduction, and the Mg process has far better CO2 conversion efficiency/productivity than the reported catalysts.
Hydrogen yield with reaction time during the reduction of CO2 at room temperature and ambient pressure is given in Fig. S17.† Notably, in the absence of CO2, Mg does not react efficiently with water and hydrogen yield was extremely low, 100 μmol g−1 as compared to 42000 μmol g−1 in the presence of CO2. This was due to poor solubility of magnesium hydroxide formed by the reaction of Mg with water, restricting the internal Mg surface to react further with water. However, in the presence of CO2, magnesium hydroxide gets converted to carbonates and basic carbonates, which are more soluble in water than magnesium hydroxide and get peeled off from Mg, exposing a fresh Mg surface to react with water. Thus, this protocol can even be used for hydrogen production (940 mL g−1), which is nearly 420 times more than the hydrogen produced by the reaction of Mg with water alone (2.24 mL g−1).
Using this protocol, 1 kg of magnesium via simple reaction with water and CO2 produces 2.43 liters of methane, 940 liters of hydrogen (Fig. S17†) and 3.85 kg of basic magnesium carbonate (green cement), and also small amounts of methanol, and formic acid. Thus, it is important to note that hydrogen was the predominant product, as the reaction of water with magnesium was preferred over the reaction of CO2 and water with magnesium.
Notably, this entire production happens in just 15 minutes, at room temperature and atmospheric pressure, in this exceptionally simple and safe protocol. Unlike other metal nanoparticles, the nano-Mg powder is extremely stable and can be handled in air without any loss in activity. If the use of fossil fuels and natural methane gets banned or restricted, there will be a need for a CO2 neutral process for various chemicals and fuels (methane, methanol, formic acid and hydrogen). This Mg protocol will then be the cheapest of any other reported CO2 conversion protocols in the literature to date. Further considerations with respect to the Mg process sustainability (energy demand, CO2 production and global warming impact) are discussed in ESI, Fig. S15 and S16.†
Regeneration of the consumed Mg NPs is required to make the whole CO2 conversion process feasible and this can be achieved via a commercially available process.21,53 It is thus essential to note that although no external energy is required during the CO2 conversion process, external energy is required to regenerate Mg. Recently, Mg was regenerated for ∼1 USD per kg of Mg using a solar-energy pumped laser.53,54 The product magnesium hydroxide carbonate can also be used as green cement.64 Accordingly, the Mg-assisted CO2 conversion process is economical, even though it is non-catalytic. Also, the Mg nanopowder is stable due to the presence of a thin MgO passivation surface layer and can be stored in air without loss of activity (Fig. S5†). There was no need to remove the thin MgO layer before the reaction, as it reacted with water to form magnesium hydroxide. This makes the overall process safe.
Magnesium assisted CO2 conversion to fuel on the Mars’ surface
Planet Mars’ environment has 95.32% of CO2, while its surface has water in the form of ice. Recently, the presence of magnesium on Mars in abundant amounts was also reported.65–67 Therefore, to explore the possibility of the use of this Mg-assisted CO2 conversion process on Mars, we carried out this Mg-assisted CO2 conversion at −1 to +1 °C temperature. Notably, methane was produced in a reasonable amount (35 μmol g−1) in just 20 minutes, although at a slower rate as compared to the room temperature reaction (Fig. S18†), due to the low reaction temperature. A good amount of methanol (28 μmol g−1) and formic acid (37 μmol g−1) formation was also observed. These results indicate the potential of this Mg-process to be used in the Mars’ environment, a step towards magnesium utilization on Mars, although more detailed studies are needed.Conclusions
Magnesium converted CO2 (directly from the air or pure) at room temperature and atmospheric pressure to methane, methanol, and formic acid, using water as the hydrogen source. No external energy (such as light, heat, or electricity) was required. The reaction was very fast, with a maximum product yield after 15 min. Hydrogen was predominant as compared to carbon-based products, as the reaction of water with magnesium was favored over the reaction of CO2 and water with magnesium. Although Mg was converted to basic magnesium carbonate, its regeneration to Mg NPs is possible using a solar-energy pumped laser process.We observed a unique ability of Mg not only to chemisorb and activate CO2 but also to transfer electrons to facilitate CO2 reduction. We observed a distinctive cooperative action of magnesium, basic magnesium carbonate, CO2, and water to convert CO2 to methane, methanol, and formic acid. 13CO2 isotopic labeling experiments, ex situ PXRD, NMR, FTIR, XPS and in situ ATR studies identified the relevant reaction intermediates and reaction mechanism, which was supported by density functional theory (DFT).
Thus, it was demonstrated for the first time that magnesium (nanoparticles and bulk), an inexpensive and the eighth-most abundant element, converted pure CO2 (as well as CO2 from the air) to fuel at room temperature using only water as the hydrogen source. This Mg-process has the potential to be used in the Mars’ environment for fuel production.
Experimental
Characterization
Electron microscopy imaging was performed on an FEI Tecnai transmission electron microscope. Samples were prepared by drop-casting diluted ethanolic suspensions of the relevant powder onto a copper grid (for TEM). PXRD patterns were obtained using a Panalytical X'Pert Pro powder X-ray diffractometer with Cu-Kα radiation. FTIR spectra were recorded using a JASCO FT/IR-4700. N2 adsorption measurements were performed using a Micromeritics 3-Flex Surface Analyzer. The samples were degassed at 110 °C overnight before analysis. The NMR study was carried out using a Bruker AVANCE II spectrometer B0 = 9.4T (V0 = 800 MHz for 1H). Methanol and formic acid were identified by 1H NMR spectroscopy and quantified by using the external calibration method. 13C NMR spectroscopy was used to identify the liquid products of the labeled 13CO2 experiment, and gaseous products of the labeled 13CO2 experiment were analyzed by using a gas chromatograph equipped with a TCD, FID and mass spectrometer (Agilent 7890B, Agilent Technologies), and an Agilent hybrid column CP7430 and helium as a carrier gas. XPS analysis was carried out using a Thermo Kα+ spectrometer with micro-focused and monochromated Al-Kα radiation (1486.6 eV) as the X-ray source. A pellet of the magnesium powder was made and fixed on carbon tape. The carbon signal at 284.6 eV was used as an internal reference. Peak fitting of the spectra was performed using an XPSPEAK 4.1 by using a Shirley background.CO2 reduction experiments
CO2 conversion was carried out by taking 50 mg of Mg (or other metals/alloys) in a 350 mL batch reactor. The Mg nanopowder, its alloy and other metals were purchased from Nanoshel Inc. with purity > 99.9%. All the samples were used without further purification.Initially, 50 mg of magnesium was taken in a round bottom (RB) flask with a volume of 350 mL and then sealed with a rubber septum. With the help of a needle, CO2 gas was purged into the RB flask for 30 min (optimized) for better contact with Mg surfaces. The flow of the gas was controlled using a mass flow controller (MFC). After purging, the magnesium surface was saturated with CO2 and the RB flask was also filled with CO2, and then DI water was injected into the RB flask quickly in ∼20 s while the reaction mixture was stirred using magnetic bid. This allowed all three reaction components (Mg, CO2 and water) to be in good contact with each other. Note that the reaction was initiated as soon as water was added. The progress of this fast reaction was monitored with a Micro GC connected online to the RB reactor, as shown in ESI-experimental section.†
Methane and hydrogen were quantified using an Agilent Technologies 490 Micro GC with a molecular-sieve column and TCD detector. Liquid products such as methanol and formic acid were identified and quantified by 1H NMR spectroscopy. The detailed procedure for the detection of the liquid products by 1H NMR spectroscopy is given in the ESI.†
Instead of pure CO2, air was purged through Mg NPs taken in a batch reactor for 4 h with the flow rate of 200 mL min−1 in order to carry out the reaction using CO2 present in the air. Isotopically labeled experiments were carried out by using 13CO2 and gaseous products were analyzed by using a GC coupled with a TCD, FID and mass spectrometer (Agilent 7890B, Agilent Technologies). For GC-MS analysis, helium was used as the carrier gas and an Agilent hybrid column CP7430 as the stationary phase.
In situ attenuated total reflection (ATR) infrared spectroscopy
In situ ATR-FTIR studies were carried out to examine the mechanistic pathways.68 Before in situ ATR-FTIR studies, the temperature-programmed desorption (TPD) experiment was carried out under a He flow using 20 mg catalysts with a heating rate of 10 °C min−1 up to 350 °C. Analysis of desorbed species was carried out by using a differentially-pumped quadrupole mass spectrometer (Prisma Plus QMG 220, Pfeiffer Vacuum).In situ ATR-FTIR experiments were performed with a Vertex 70 (Bruker Optics) spectrometer equipped with a liquid-nitrogen-cooled mercury cadmium telluride (MCT) detector and a commercial mirror unit. The ZnSe crystal (52 mm × 20 mm × 2 mm) was coated with a film of the catalyst and was placed into the vertical commercial reactor-cell (Specac). The background was recorded with the catalyst under a He flow at room temperature, but in the absence of the liquid phase. Thereafter, the reactants or He were flown through the system. A series of consecutive (in situ) spectra (256–512 scans per spectrum; resolution 4 cm−1) were then collected.
Computational details
All the spin-polarized calculations were performed within the framework of density functional theory (DFT), using the plane–wave technique as implemented in the Vienna Ab Initio Simulation Package (VASP).55 The exchange-correlation energy was accounted within the generalized gradient approximation method (GGA) parameterized by Perdew–Burke–Ernzerhof (PBE).69 We used the projector augmented wave potential (PAW) to treat the ion–electron interactions. The DFT-D2 empirical correction method proposed by Grimme was applied for describing the effect of van der Waals interactions.70 In all computations, the kinetic energy cut off is set to be 500 eV in the plane-wave expansion. All the structures were fully relaxed (lattice constant and atomic position) using the conjugated gradient method and the convergence threshold was set to be 10−4 eV in energy and 0.01 eV Å−1 in force. For geometry optimization, the Brillouin zone was sampled using a 5 × 5 × 1 Monkhorst–Pack k-point mesh. In order to calculate the charge transfer between the surface adsorbed CO2 and Mg NP surface slab, we chose the Bader charge-population analysis method.59,60 For structure optimization of the CO2 molecule in the gas phase, we used the B3LYP/6-31+G(d,p) method as implemented in Gaussian 09.71 The climbing image nudge elastic band method (CI-NEB) was used to calculate the activation barriers and six intermediate images were considered along the minimum energy path (MEP) to search for transition states for the intermediate states toward CO2 reduction.72 For all the pathways, the activation barriers (ΔE‡) were calculated using the following equations: ΔE‡ = ETS − EIS and ΔE = EFS − EIS; where IS, TS and FS correspond to the initial state, transition state and final state, respectively. The adsorption energy (Ead) of adsorbates (i.e. intermediate molecules and/or atoms on the Mg surface) was defined as:57Ead = EA/S − EA − ES where EA/S, EA and ES represent total energies of the Mg surface slab with adsorbates, only adsorbates and the Mg surface respectively. The reaction energy (Er) was calculated using the following equation67Er = EFS − EIS. The Mg NP surface was modelled considering a four-layer periodic surface slab consisting of a 3 × 3 surface cell (9.15 × 9.15 Å2) with 36 Mg atoms. To avoid the spurious interactions between the neighboring slabs, a vacuum layer of 20 Å is used in the direction perpendicular to the surface (along the Z-direction) and the nearest distance between the two adsorbed molecules in the adjacent supercell is ∼6 Å.Author contributions
V. P. proposed the research direction and designed and planned the experiments. V. P., S. R., and R. B. performed various CO2 conversion experiments and characterization, assisted by A. M. and A. B. NMR and FT-IR studies were performed by S. R. ATR and TPD experiments were conducted by N. Y. and G. R. DFT calculations were carried out by R. J. and A. D. All the data were analyzed by V. P. and S. R. V. P. wrote the manuscript with inputs from S. R., which was commented on by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Atomic Energy (DAE), Government of India (under project no. R&D-TIFR-RTI4003). We acknowledge the EM, XRD, and NMR Facility of TIFR, Mumbai. We thank Ms. Mamata Joshi for her help in setting up various NMR experiments. G. R. acknowledges the Austrian Science Fund (FWF) (DK+Solids4Fun W1243) and TU Wien (DK CO2Refinery). A. D. thanks DIA-SERB for funding computational resources.References
- S. J. Davis, et al., Net-zero emissions energy systems, Science, 2018, 360, eaas9793 CrossRef PubMed.
- C. Breyer, M. Mahdi Fasihi, C. Bajamundi and F. Creutzig, Direct air capture of CO2: a key technology for ambitious climate change mitigation, Joule, 2019, 3, 2053–2065 CrossRef.
- O. S. Bushuyev, P. D. Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, What should we make with CO2 and how can we make it?, Joule, 2018, 2, 825–832 CrossRef CAS.
- K. S. Lackner, S. Brennan, J. M. Matter, A.-H. Alissa Park, A. Wright and B. van der Zwaan, The urgency of the development of CO2 capture from ambient air, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 13156–13162 CrossRef CAS.
- P. G. O'Brien, A. Sandhel, T. E. Wood, A. A. Jelle, L. B. Hoch, D. D. Perovic, C. A. Mims and G. A. Ozin, Photomethanation of gaseous CO2 over Ru/silicon nanowire catalysts with visible and near-infrared photons, Adv. Sci., 2014, 1, 1400001 CrossRef.
- D. W. Keith, G. Holmes, D. St. Angelo and K. Heide, A process for capturing CO2 from the atmosphere, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. García, Complete photocatalytic reduction of CO2 to methane by H2 under solar light irradiation, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- T. N. Huan, D. A. D. Corte, S. Lamaison, D. Karapinar, L. Lutz, N. Menguy, M. Foldyna, S. –H. Turren-Cruz, A. Hagfeldt, F. Bella, M. Fontecave and V. Mougel, Low-cost high-efficiency system for solar-driven conversion of CO2 to hydrocarbons, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 9735–9740 CrossRef CAS PubMed.
- R. Belgamwar, A. Maity, T. Das, S. Chakraborty, C. P. Vinod and V. Polshettiwar, Lithium silicate nanosheets with excellent capture capacity and kinetics with unprecedented stability for high-temperature CO2 capture, Chem. Sci., 2021, 12 10.1039/d0sc06843h.
- A. K. Mishra, R. Belgamwar, R. Jana, A. Datta and V. Polshettiwar, Defects in nanosilica catalytically convert CO2 to methane without any metal and ligand, Proc. Natl. Acad. Sci. U.S.A, 2020, 117, 6383–6390 CrossRef CAS PubMed.
- M. Dhiman, A. Maity, A. Das, R. Belgamwar, B. Chalke, Y. Lee, K. Sim, J.-W. Jwa-Min Nam and V. Polshettiwar, Plasmonic colloidosomes of black gold for solar energy harvesting and hotspots directed catalysis for CO2 to fuel conversion, Chem. Sci., 2019, 10, 6594–6603 RSC.
- A. Maity, R. Belgamwar and V. Polshettiwar, Facile synthesis protocol to tune size, textural properties & fiber density of dendritic fibrous nanosilica (DFNS): Applications in catalysis and CO2 capture, Nat. Prot., 2019, 14, 2177–2204 CrossRef CAS.
- A. Maity, S. Chaudhari, J. J. Titman and V. Polshettiwar, Nanosponges of acidic amorphous aluminosilicate for catalysis, plastic degradation and CO2 to fuel conversion, Nat. Commun., 2020, 11, 3828 CrossRef.
- P. Knochel, A flash of magnesium, Nature Chem, 2009, 1, 740 CrossRef CAS PubMed.
- E. C. Ashby, A. B. Goel and R. N. DePriest, Evidence for an electron transfer mechanism in the reduction of ketones by main group metal hydrides, J. Am. Chem. Soc., 1980, 102, 7779–7780 CrossRef CAS.
- Y. Yang, M. D. Anker, J. Fang, M. F. Mahon, L. Maron, C. Weetman and M. S. Hill, Hydrodeoxygenation of isocyanates: snapshots of a magnesium-mediated C=O bond cleavage, Chem. Sci., 2017, 8, 3529–3537 RSC.
- C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot and M. F. Mahon, Magnesium-catalysed nitrile hydroboration, Chem. Sci., 2016, 7, 628–641 RSC.
- N. L. Lampland, M. Hovey, D. Mukherjee and A. D. Sadow, Magnesium-catalyzed mild reduction of tertiary and secondary amides to amines, ACS Catal., 2015, 5, 4219–4226 CrossRef CAS.
- D. Yang, L. Wang, F. Han, D. Li, D. Zhao and R. Wang, Intermolecular enantioselective dearomatization reaction of β-naphthol using meso-aziridine: a bifunctional in situ generated magnesium catalyst, Angew. Chem., Int. Ed., 2015, 54, 2185–2189 CrossRef CAS PubMed.
- R. Lalrempuia, A. Stasch and C. Jones, The reductive disproportionation of CO2 using a magnesium(I) complex: analogies with low valent f-block chemistry, Chem. Sci., 2013, 4, 4383–4388 RSC.
- R. K. Kostanyan, Use of magnesium as renewable energy source, Int. J. Chem. Mol. Eng., 2017, 11, 416–421 Search PubMed.
- M. Huang, L. Ouyang, J. Ye, J. Liu, X. Yao, H. Wang, H. Shao and M. Zhu, Hydrogen generation via hydrolysis of magnesium with seawater using Mo, MoO2, MoO3 and MoS2 as catalysts, J. Mater. Chem. A, 2017, 5, 8566–8575 RSC.
- M. Uda, H. Okuyama, T. S. Suzuki and Y. Sakka, Hydrogen generation from water using Mg nanopowder produced by arc plasma method, Sci. Technol. Adv. Mater., 2012, 13, 025009 CrossRef PubMed.
- R. G. Copperthwaite, P. R. Davies, M. A. Morris, M. W. Roberts and R. A. Ryder, The reactive chemisorption of carbon dioxide at magnesium and copper surfaces at low temperature, Catal. Lett., 1988, 1, 11–19 CrossRef CAS.
- S. Campbell, P. Hollins, E. McCash and M. W. Roberts, Reaction of carbon dioxide with the magnesium(0001) surface, J. Electron Spectrosc. Relat. Phenom., 1986, 39, 145–153 CrossRef CAS.
- D. Ochs, M. Brause, B. Braun, W. Maus-Friedrichs and V. Kempter, CO2 chemisorption at Mg and MgO surfaces: a study with MIES and UPS (He I), Surface Sci, 1998, 397, 101–107 CrossRef CAS.
- A. F. Carley, G. Hawkins, S. Read and M. W. Roberts, Reactions of co-adsorbed carbon dioxide and dioxygen at the Mg(0001) surface at low temperatures, Top. Catal., 1999, 8, 243–248 CrossRef CAS.
- D. –Y. Hwang and A. M. Mebel, Theoretical study on the reaction mechanism of CO2 with Mg, J. Phys. Chem. A, 2000, 104, 7646–7650 CrossRef CAS.
- K. J. Fricker and A.-H. A. Park, Effect of H2O on Mg(OH)2 carbonation pathways for combined CO2 capture and storage, Chem. Eng. Sci., 2013, 100, 332–341 CrossRef CAS.
- K. Yamauchi, N. Murayama and J. Shibata, Absorption and release of carbon dioxide with various metal oxides and hydroxides, Mater. Trans., 2007, 48, 2739–2742 CrossRef CAS.
- E. Luevano-Hipolito and L. M. T. Martínez, Mg(OH)2 films prepared by ink-jet printing and their photocatalytic activity in CO2 reduction and H2O conversion, Top. Catal., 2018, 61, 1574–1584 CrossRef CAS.
- S. Ogawa, H. Niwa, K. Nakanishi, T. Ohta and S. Yagi, Influence of CO2 and H2O on air oxidation of Mg nanoparticles studied by NEXAFS, J. Surface Anal., 2011, 17, 319–323 CrossRef CAS.
- X. Wu, Y. Li, G. Zhang, H. Chen, J. Li, K. Wang, Y. Pan, Y. Zhao, Y. Sun and Y. Xie, Photocatalytic CO2 conversion of M0.33WO3 directly from the air with high selectivity: insight into full spectrum-induced reaction mechanism, J. Am. Chem. Soc., 2019, 141, 5267–5274 CrossRef CAS PubMed.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Plasmonic control of multi-electron transfer and C-C coupling in visible-light-driven CO2 reduction on Au nanoparticles, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS PubMed.
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, Isolation of Cu atoms in Pd lattice: forming highly selective sites for photocatalytic conversion of CO2 to CH4, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef CAS.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, Turning Au nanoclusters catalytically active for visible-light-driven CO2 reduction through bridging ligands, J. Am. Chem. Soc., 2018, 140, 16514–16520 CrossRef CAS PubMed.
- J. V. Evans and T. L. Whateley, Infra-red study of adsorption of carbon dioxide and water on magnesium oxide, Trans. Faraday Soc., 1967, 63, 2769–2777 RSC.
- Z. Zhang, Y. Zheng, Y. Ni, Z. Liu, J. Chen and X. Liang, Temperature- and pH-dependent morphology and FT−IR analysis of magnesium carbonate hydrates, J. Phys. Chem. B, 2006, 110, 12969–12973 CrossRef CAS PubMed.
- J. Lanas and J. I. Alvarez, Dolomitic lime: thermal decomposition of nesquehonite, Thermochim. Acta, 2004, 421, 123–132 CrossRef CAS.
- V. Rheinheimer, C. Unluer, J. Liu, S. Ruan, J. Pan and P. J. M. Monteiro, XPS study on the stability and transformation of hydrate and carbonate phases within MgO systems, Materials, 2017, 10, 75 CrossRef PubMed.
- S. Feliu Jr, M. C. Merino, R. Arrabal, A. E. Coy and E. Matykina, XPS study of the effect of aluminium on the atmospheric corrosion of the AZ31magnesium alloy, Surf. Interface Anal., 2009, 41, 143–150 CrossRef.
- J. M. Montero, M. A. Isaacs, A. F. Lee, J. M. Lynam and K. Wilson, The surface chemistry of nanocrystalline MgO catalysts for FAME production: An in situ XPS study of H2O, CH3OH and CH3OAc adsorption, Surf. Sci., 2016, 646, 170–178 CrossRef CAS.
- S. Ardizzone, C. L. Bianchi, M. Fadoni and B. Vercelli, Magnesium salts and oxide: an XPS overview, Appl. Surf. Sci., 1997, 119, 253–259 CrossRef CAS.
- S. Feliu Jr, C. Maffiotte, J. C. Galvan, A. Pardo, M. C. Merino and R. Arrabal, The Application of X-ray photoelectron spectroscopy in understanding corrosion mechanisms of magnesium and Mg-Al alloys, Open Surf. Sci. J., 2011, 3, 1–14 CrossRef.
- J. S. Corneille, J. W. He and D. W. Goodman, XPS characterization of ultra-thin MgO films on a Mo(100) surface, Surf. Sci., 1994, 306, 269–278 CrossRef CAS.
- G. Song, X. Zhu, R. Chen, Q. Liao, Y. –D. Ding and L. Chen, Influence of the precursor on the porous structure and CO2 adsorption characteristics of MgO, RSC Adv., 2016, 6, 19069–19077 RSC.
- G. Busca and V. Lorenzelli, Infrared spectroscopic identification of species arising from reactive adsorption of carbon oxides on metal oxide surfaces, Mater. Chem., 1982, 7, 89–126 CrossRef CAS.
- J. A. Lercher, C. Colombier and H. Noller, Acid–base properties of alumina–magnesia mixed oxides. Part 4-Infrared study of adsorption of carbon dioxide, J. Chem. Soc., Faraday Trans., 1984, 1(80), 949–959 RSC.
- A. Lubezky, L. Chechelnitsky and M. Folman, IR spectra of CH4, CD4, C2H4, C2H2, CH3OH and CH3OD adsorbed on C60 films, J. Chem. Soc., Faraday Trans., 1996, 92, 2269–2274 RSC.
- B. G. Oliver and A. R. Davis, Vibrational spectroscopic studies of aqueous alkali metal bicarbonate and carbonate solution, Can. J. Chem., 1973, 51, 698–702 CrossRef CAS.
- K. T. Ranjit and K. J. Klabunde, Solvent effects in the hydrolysis of magnesium methoxide, and the production of nanocrystalline magnesium hydroxide. An aid in understanding the formation of porous inorganic materials, Chem. Mater., 2005, 17, 65–73 CrossRef CAS.
- D. Knapp, B. S. Brunschwig and N. S. Lewis, Transmission infrared spectra of CH3-, CD3-, and C10H21-Ge(111) surfaces, J. Phys. Chem. C, 2011, 115, 16389–16397 CrossRef CAS.
- T. Yabe, Y. Suzuki and Y. Santoh, Renewable energy cycle with magnesium and solar-energy-pumped lasers, Renew. Energy Power Qual. J., 2014, 1, 76–80 CrossRef.
- T. Yabe and T. Yamaji, The magnesium civilization: an alternate new source of energy to oil, Pan Stanford Publishing, 2010, Science, pp. 1–147 Search PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B, 1993, 47, 558 CrossRef CAS PubMed.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, CO2 activation over catalytic surfaces, ChemPhysChem, 2017, 18, 3135–3141 CrossRef PubMed.
- M. Zhang, M. Dou and Y. Yu, DFT study of CO2 conversion on InZr3(110) surface, Phys. Chem. Chem. Phys., 2017, 19, 28917–28927 RSC.
- A. K. Mishra, A. Roldan and N. H. Leeuw, CuO surfaces and CO2 activation: A dispersion corrected DFT+U study, J. Phys. Chem. C, 2016, 120, 2198–2214 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- R. F. W. Bader, Atoms in molecules: A quantum theory, Oxford University Press, Oxford, 1990 Search PubMed.
- I. J. Polmear, Magnesium and Magnesium Alloys, ed. M. M. Avedesian and H. Baker, ASM International, OH, USA, 1999, pp. 3–6 Search PubMed.
- P. Nuss and M. J. Eckelman, Life Cycle Assessment of Metals: A Scientific Synthesis, PLoS ONE, 2014, 9, e101298 CrossRef PubMed.
- G. Demirci and I. Karakaya, Magnesium Technology 2012, ed. S. N. Mathaudhu, W. H. Sillekens, N. R. Neelameggham and N. Hort, John Wiley & Sons, Hoboken, NJ, USA, 2012, pp. 59–62 Search PubMed.
- S. A. Walling and J. L. Provis, Magnesia-based cements: A journey of 150 years, and cements for the future?, Chem. Rev., 2016, 116, 4170–4204 CrossRef CAS PubMed.
- D. T. Vaniman, D. L. Bish, S. J. Chipera, C. I. Fialips, J. W. Carey and W. C. Feldman, Magnesium sulphate salts and the history of water on Mars, Nature, 2004, 431, 663–665 CrossRef CAS PubMed.
- W. Rapin, B. L. Ehlmann, G. Dromart, J. Schieber, N. H. Thomas, W. W. Fischer, V. K. Fox, N. T. Stein, M. Nachon, B. C. Clark, L. C. Kah, L. Thompson, H. A. Meyer, T. S. J. Gabriel, C. Hardgrove, N. Mangold, F. Rivera-Hernandez, R. C. Wiens and A. R. Vasavada, An interval of high salinity in ancient Gale crater lake on Mars, Nature Geosci, 2019, 12, 889–895 CrossRef CAS.
- J. M. Grebowsky, M. Benna, J. M. C. Plane, G. A. Collinson, P. R. Mahaffy and B. M. Jakosky, Unique, non-Earthlike, meteoritic ion behavior in upper atmosphere of Mars, Geophys. Res. Lett., 2017, 44, 3066–3072 CrossRef CAS.
- G. Rupprechter, Operando surface spectroscopy and microscopy during catalytic reactions: from clusters via nanoparticles to meso-scale aggregates, Small, 2021, 2004289 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, revision A.1, Gaussian, Inc., Wallingford CT, 2009, vol. 121, pp. 150–166 Search PubMed.
- G. Henkelman and H. Jónsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional XPS, IR, conversion & DFT data, sustainability and preliminary life cycle assessment (LCA) data. See DOI: 10.1039/d1sc01113h |
| This journal is © The Royal Society of Chemistry 2021 |