
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrace mild acid-catalysed Z → E isomerization of norbornene-fused stilbene derivatives: intelligent chiral molecular photoswitches with controllable self-recovery†
Taotao
Hao
a,
Yongsheng
Yang
a,
Wenting
Liang
 b,
Chunying
Fan
a,
Xin
Wang
a,
Wanhua
Wu
*a,
Xiaochuan
Chen
a,
Haiyan
Fu
*a,
Hua
Chen
a and
Cheng
Yang
*a
b,
Chunying
Fan
a,
Xin
Wang
a,
Wanhua
Wu
*a,
Xiaochuan
Chen
a,
Haiyan
Fu
*a,
Hua
Chen
a and
Cheng
Yang
*a
aKey Laboratory of Green Chemistry & Technology, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, China. E-mail: yangchengyc@scu.edu.cn
bInstitute of Environmental Science, Department of Chemistry, Shanxi University, Taiyuan 030006, China
First published on 28th December 2020
Abstract
Stilbene derivatives have long been known to undergo “acid-catalyzed” Z → E isomerization, where a strong mineral acid at high concentration is practically necessary. Such severe reaction conditions often cause undesired by-reactions and limit their potential application. Herein, we present a trace mild acid-catalyzed Z → E isomerization found with stilbene derivatives fused with a norbornene moiety. By-reactions, such as the migration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and electrophilic addition reactions, were completely inhibited because of the ring strain caused by the fused norbornene component. Direct photolysis of the E isomers at selected wavelengths led to the E → Z photoisomerization of these stilbene derivatives and thus constituted a unique class of molecular switches orthogonally controllable by light and acid. The catalytic amount of acid could be readily removed, and the Z → E isomerization could be controlled by turning on/off the irradiation of a photoacid, which allowed repeated isomerization in a non-invasive manner. Moreover, the Z isomer produced by photoisomerization could spontaneously self-recover to the E isomer in the presence of a catalytic amount of acid. The kinetics of Z → E isomerization were adjustable by manipulating catalytic factors and, therefore, unprecedented molecular photoswitches with adjustable self-recovery were realized.
C double bond and electrophilic addition reactions, were completely inhibited because of the ring strain caused by the fused norbornene component. Direct photolysis of the E isomers at selected wavelengths led to the E → Z photoisomerization of these stilbene derivatives and thus constituted a unique class of molecular switches orthogonally controllable by light and acid. The catalytic amount of acid could be readily removed, and the Z → E isomerization could be controlled by turning on/off the irradiation of a photoacid, which allowed repeated isomerization in a non-invasive manner. Moreover, the Z isomer produced by photoisomerization could spontaneously self-recover to the E isomer in the presence of a catalytic amount of acid. The kinetics of Z → E isomerization were adjustable by manipulating catalytic factors and, therefore, unprecedented molecular photoswitches with adjustable self-recovery were realized.
Introduction
The Z–E isomerization of stilbene derivatives has been known for over one century,1 and it has now become one of the most representative prototype reactions applied to molecular machines,2 such as molecular motors,3 photoswitches,4 molecular ratchets5 and half adders,6 mainly due to the significant geometrical changes that occur during reversible isomerization. The Z–E isomerization of stilbene derivatives has been extensively investigated and is currently known to be triggered by physical (light or temperature) or chemical activators, such as boron fluoride-ether complexes,7 acids8 and bromine, in conjunction with light.9 Photoisomerization is most commonly applied in molecular machines due to its non-invasive and convenient operating features.10On the other hand, protic acids have long been known to promote the Z → E isomerization of stilbene derivatives via an A-SE2 mechanism.11 However, such acid-catalysed isomerization commonly requires the use of strong mineral acids at high concentrations as the solvent, which significantly limits the application of this method due to low functional group tolerance and undesired side reactions caused by the harsh reaction conditions.12 The use of a large excess of strong acids is particularly unfavourable for molecular machines in terms of non-accumulative repeating operations. For an ideal chemically activated molecular machine, the chemical inductor should be mild, work in a catalytic amount, and be readily removable/deactivated.13 To our knowledge, such molecular machines based on stilbene derivatives have not yet been documented.
Although stimulus-responsive molecular switches have been widely constructed based on chemical and physical stimuli, including light,14 electrochemistry,15 temperature,16 pH,17 solvent,18 and additives,19 and we have established rotating switches consisting of molecular universal joints based on thermal20 and redox stimuli,21 the molecule switches obtained by such sophisticated design and synthesis are generally responsive to only one of the above stimuli, and few molecular switches can be manipulated by two independent effectors,2a,f,13 which would offer a higher level of control.22 Herein, we report Z → E isomerization of a novel kind of stilbene derivative unprecedentedly catalysed by even a trace amount of a mild organic acid. Double control of the Z–E isomerization by the two independent external stimuli of light and acid, so-called orthotropic control,22a was realized (Fig. 1a), resulting in intriguing and advanced functions in these molecular photoswitches. The unique self-recovery behavior has been widely presented in nature, e.g. Nymphaea tetragona, which self-recover between OFF (night) and ON (day) with light and chemical stimuli (Fig. 1c). Hence, the overall dynamic properties of the newly prepared stilbene derivative switches mimicked the behaviors of the macroscopic Nymphaea tetragona well.
 | ||
| Fig. 1 (a) Orthotropic control of the Z–E isomerization of norbornene-fused stilbene derivatives by acid and light, (b) the chemical structure of the stilbene derivatives, and (c) schematic representation of the self-recovery process. The overall dynamic properties of the norbornene-fused stilbene derivative switches mimic those of the macroscopic Nymphaea tetragona, which self-recover between OFF (night) and ON (day) under light and chemical stimuli. | ||
Results and discussion
Z-to-E isomerization of norbornene-fused stilbene derivatives catalysed by a trace amount of mild acid
The present study was essentially prompted by a casual observation of spontaneous chemical conversion of 5Z (Fig. 1), which was synthesized by a one-pot stereoselective Pd-catalysed domino reaction (Scheme S6†).23 As illustrated in Fig. 2, a new set of proton signals separate from the original signals of 5Z appeared during the 1H NMR spectral measurements of 5Z in CDCl3 (Fig. 2a and b). This chemical conversion progressed smoothly over time even under strict light-proof conditions, which is unexpected because both the Z and E forms of stilbene derivatives are, in general, thermally stable. This observation prompted us to perform further NMR investigations in different deuterated solvents. Indeed, 5Z has excellent thermal stability, and no chemical conversion was observed in the 1H NMR spectra by heating 5Z in DMSO-d6 at 130 °C for over 24 h (Fig. 2d and e). Further NMR experiments revealed that spontaneous chemical conversion could be observed only in newly opened CDCl3 but not in other deuterated solvents. To examine the possible effects of oxygen, we deoxygenated the solutions by bubbling nitrogen into the CDCl3 solution, which inhibited the reaction. Surprisingly, bubbling oxygen did not facilitate the chemical conversion but instead inhibited it (Fig. S41m†); therefore, we speculated that removal of volatile gas dissolved in CDCl3 by bubbling either N2 or O2 should be responsible.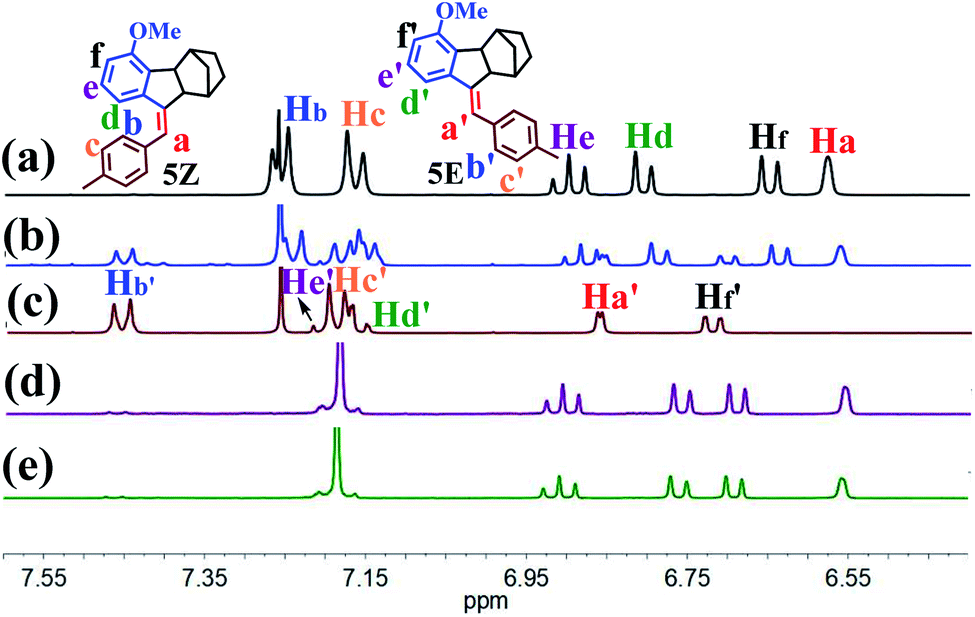 | ||
| Fig. 2 1H NMR spectra of 5Z (a) in CDCl3 sealed with K2CO3, (b) in newly opened CDCl3 (which contains a tiny amount of acid), (c) with addition of 0.5 eq. of TFA to CDCl3, (d) in DMSO-d6 measured soon after preparing the solution, and (e) in DMSO-d6 after heating at 130 °C for 24 h. All measurements were performed at room temperature. | ||
The newly formed compound was then isolated and characterized as the E isomer 5E on the basis of HR mass and 2D NMR spectroscopic studies (Fig. S15–S18†). We then considered that the tiny amount of DCl due to the photodecomposition of CDCl3 may play a role in the spontaneous isomerization of 5Z. To confirm this hypothesis, we added a drop of aqueous DCl to the CDCl3 solution of 5Z at room temperature and observed that isomerization rapidly produced 5E in quantitative yield, as determined by monitoring the 1H NMR spectral changes.
Alkenes are known to undergo electrophilic addition with mineral acids.24 Alternatively, (Z)-stilbene derivatives, mediated by a strong mineral acid,8 isomerize to the E isomers, where proton transfer to the CC double bond is the rate-determining step. The latter was defined as acid-catalysed isomerization, even though a large excess (commonly >50 wt%) of the strong mineral acids was generally required. The above observation is thus unprecedented and prompted us to test the catalytic effect of different kinds of acids. The isomerization of 5Z occurred smoothly in the presence of 0.5 eq. of mineral acid, including HCl, HBr and H2SO4. Surprisingly, when a milder, organic acid, trifluoroacetic acid (TFA), was used instead of DCl to CDCl3, 5Z also underwent isomerization to give 5E within 30 min (Fig. 2c). Catalytic isomerization could be accomplished in the presence of 1% eq. of TFA by increasing the reaction time (Table S1†). The isomerization also proceeded smoothly in the presence of a weaker organic acid, including difluoroacetic acid and dichloroacetic acid.
To understand this exceptional phenomenon, we synthesized a series of stilbene derivatives 4–10 (Fig. 1b) through a reported method,23 the possible absolute configurations of which are shown in Scheme S6,† and we further examined their proton-catalysed isomerization behaviour. The addition of an equal amount of TFA to the CDCl3 solution of 1Z did not cause any isomerization even after standing at room temperature for 1 week (Fig. S37c†). The same was true with 2Z (Fig. S38c†). However, the addition of TFA to the solution of 3Z not only afforded Z → E isomerization product 3E but also led to migration of the CC double bond to give endocyclic alkene 3C. A 3E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3C ratio of 37:63 was finally achieved when equilibrium was reached after four days (Fig. S39e†). In contrast, 4Z–6Z gave the E isomers in quantitative yield under the catalysis of TFA (Fig. S40–S44†), demonstrating a unique chemical reaction behaviour due to the introduction of the norbornene moiety.
:3C ratio of 37:63 was finally achieved when equilibrium was reached after four days (Fig. S39e†). In contrast, 4Z–6Z gave the E isomers in quantitative yield under the catalysis of TFA (Fig. S40–S44†), demonstrating a unique chemical reaction behaviour due to the introduction of the norbornene moiety.
The mechanism of proton-catalysed isomerization and quantitative Z-to-E conversion
To gain insight into the mechanism of such unprecedented proton-catalysed isomerization, we monitored the 1H-NMR spectral change of 5Z in the presence of DCl. As illustrated in Fig. 3b, the addition of DCl into the CDCl3 solution of 5Z afforded 5E quantitatively within 2 h to give a relative integration of Ha′ of 0.35 (Fig. 3a and b), demonstrating that the proton was partially deuterated. On the other hand, the addition of DCl to the CDCl3 solution of 5E did not yield any 5Z, but the integrated intensity of Ha′ also decreased with time (Fig. S41†), indicating that the Ha′ proton of 5E was also exchanged by deuterium. These results implied that both Z and E isomers experienced a carbenium ion intermediate formed by proton transfer. Interestingly, when excess DCl (20 eq.) was added to 5Z, the protons Hd′ and Hf′ were also replaced by deuterium at approximately 82% after 48 h (Fig. 3c–f). Based on these observations, we propose that the proton-catalysed isomerization of 5Z is accomplished through the process shown in Scheme S10.† Thus, D+ transfers to the CC double bond of 5Z first to give intermediate 5I. Carbenium ion 5I is then inverted to the relatively stable species 5II. Elimination of Ha or D from 5II leads to the E isomer. Further deuterium experiments indicated that the deuteration of Hd′ and Hf′ is a slow process and that Hd′ and Hf′ can be completely replaced by D only in the presence of a large excess of DCl.
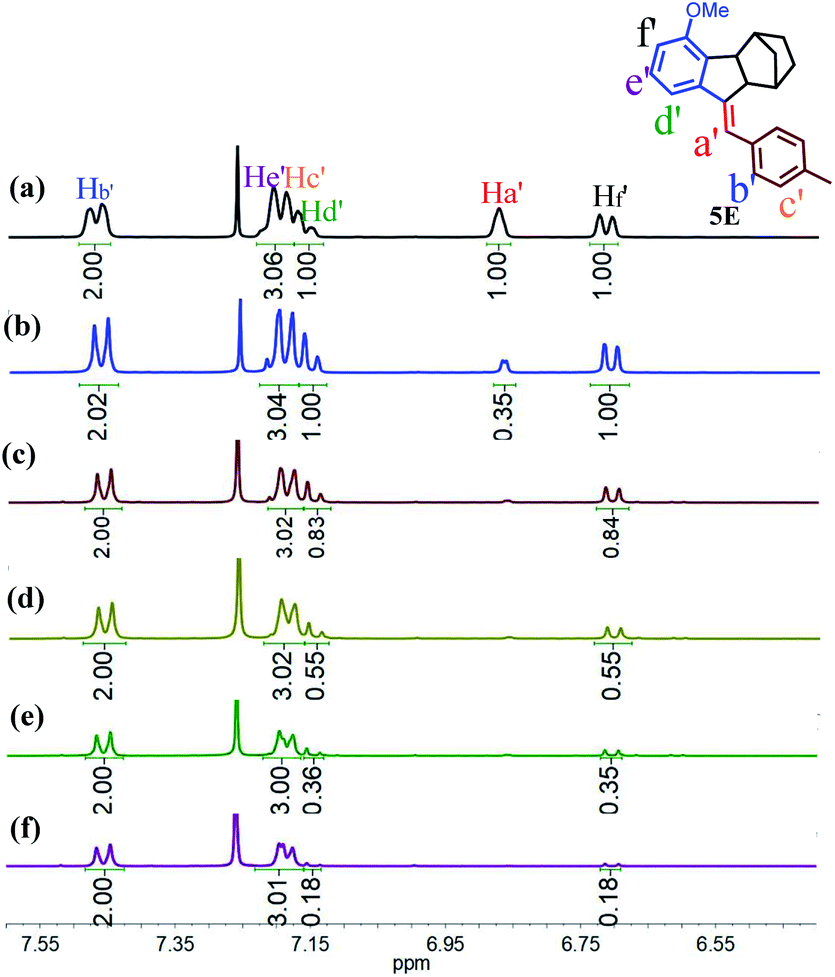 | ||
| Fig. 3 (a) 1H NMR spectra of 5E in CDCl3, (b) 1H NMR spectra of the solution after the addition of 2.0 eq. of DCl to the solution of 5Z in CDCl3 for 2 h, and (c–f) 1H NMR spectra of the solution after the addition of 20 eq. of DCl to the solution of 5Z in CDCl3 for (c) 2 h, (d) 16 h, (e) 24 h, and (f) 48 h. | ||
A comparison between 5Z and 3Z enabled us to better understand the role of the norbornene moiety. Surprisingly, freshly prepared 3Z spontaneously isomerized even in the absence of an acid to give endocyclic alkene 3C and 3E, and a 3C/3E ratio of 89:11 was achieved after six days (Fig. S39c†). In contrast, all norbornene-fused stilbenes 4Z–10Z were stable and did not show spontaneous migration of the CC double bond or isomerization in solution. Even in pure TFA, 5Z led to 5E only, and no formation of 5C or other by-products was observable (Fig. S41k†), which was attributed to the intense ring strain25 in 5C due to the fused norbornene moiety. In contrast, 1Z and 3Z produced complicated unknown products in TFA (Fig. S37e and S39f†).
The quantitative conversion of 5Z to 5E under proton-catalysis conditions was slightly unexpected; in particular, the much larger fused norbornene moiety was expected to cause greater steric repulsion upon rotation of the benzene ring to the norbornene side in the E isomer. To clarify this finding, we grew single crystals of both 5Z and 5E and carefully analysed the corresponding bond and angle parameters through X-ray single-crystal studies (Fig. 4). The α angle (130.53°) of 5Z was larger than the β angle (128.12°) of 5E, implying a smaller steric repulsion caused by the norbornene side than the benzene side.
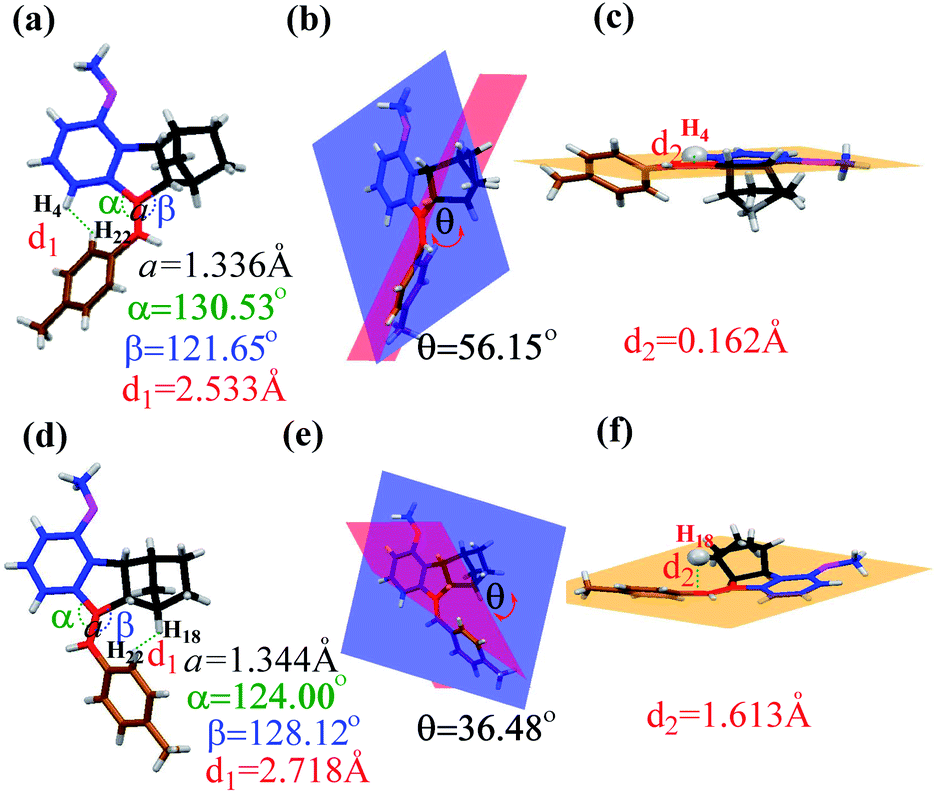 | ||
| Fig. 4 Single-crystal structures of 5Z and 5E (thermal ellipsoid plots at the 50% probability level). The structural parameters for 5Z (a–c) and 5E (d–f). | ||
The dihedral angles θ between the methoxybenzene plane (blue) and the methylbenzene plane (red) were 56.15° for 5Z and 36.48° for 5E (Fig. 4b and e), which clearly indicated that the E isomer had better coplanarity and therefore more effective conjugation than the Z isomer.
Moreover, detailed analyses of the single crystal of 5E revealed that the norbornene moiety leans to one side of the ring. H18 deviated from the CC plane (orange) at a distance of 1.613 Å (Fig. 4f). In contrast, the H4 proton on the methoxybenzene ring lies almost completely in the plane, which unavoidably causes strong steric interaction with the methylbenzene ring with cis configuration (Fig. 4c). These results accounted for the extremely high Z → E conversion in these norbornene-fused stilbenes. Similar differences could be found by comparison of the corresponding parameters obtained from the single-crystal structures of 6Z and 6E (Fig. S49, Table S2†).
On the other hand, 7Z and 8Z, in which the alkylene proton was replaced by a methyl group, also underwent isomerization in the presence of TFA but gave the E isomers in 52% and 10% yields, respectively, when equilibrium was reached (Fig. S45 and S46†). The lower E/Z ratio for the isomerization of 7Z and 8Z could be ascribed to the stronger steric repulsion between the methyl group and the methoxy benzene ring in the E isomer, which makes the E-type carbocation much less stable.
Theoretical study of the proton-catalysed reaction
Styrene is known to undergo an addition reaction with TFA.26 However, no addition products could be detected with the norbornene-fused stilbene derivatives (Fig. S40–48 and S96†). To understand this interesting phenomenon, two possible reaction routes were compared based on density functional theory (DFT) calculations. The computational simulation suggested that 4Z and TFA formed an activated complex com_4Z, which led to the transition state TS-4Z-TFA and then the intermediates 4Z-INT. The bond rotation of 4Z-INT gave 4E-INT-1 and 4E-INT-2, which pass through the transition states TS-4E-TFA and TS-4C-TFA, respectively, to finally give 4C and 4E (Fig. 5). The calculations demonstrated that the Gibbs free energy of 4Z is higher by 2.7 kcal mol−1 than that of 4E but is lower by 9.0 kcal mol−1 than that of 4C, which is entirely consistent with the results of quantitative conversion to 4E under acid catalysis.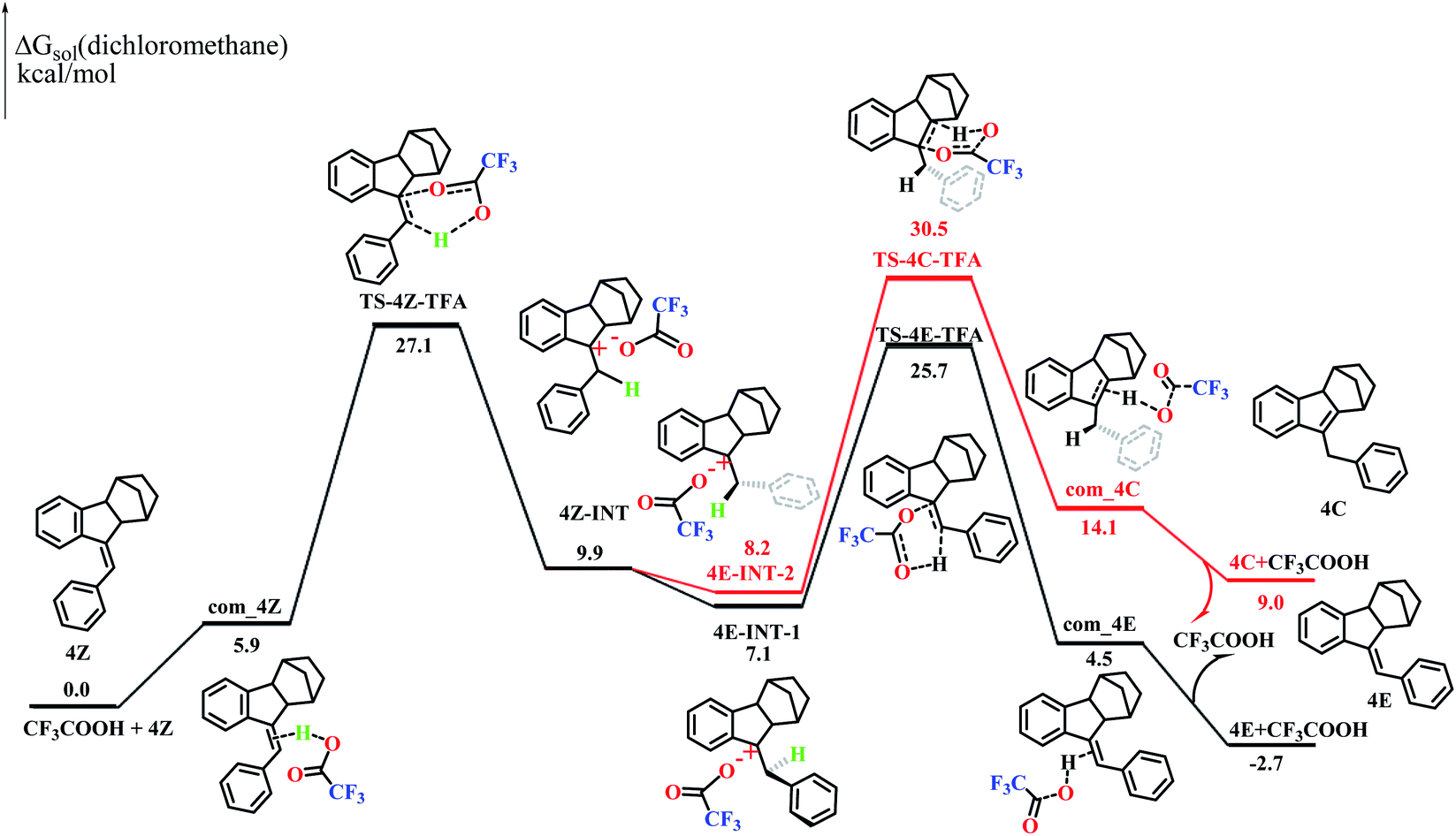 | ||
| Fig. 5 Calculated energy profiles of 4Z to 4E and 4C. | ||
In particular, TS-4C-TFA showed an extremely high Gibbs energy of 30.5 kcal mol−1, further supporting the complete inhibition of the migration of the CC double bond pathway. On the other hand, 3C showed a Gibbs energy lower than that of 3Z by −5.0 kcal mol−1 and lower than that of 3E by −3.1 kcal mol−1 (Fig. S97†), agreeing well with the high ratio of the endocyclic alkene product obtained with 3Z. Thus, the presence of a fused norbornene significantly increased the energy of both 4C and the intermediates affording 4C and hence completely impeded the migration of the CC double bond route. On the other hand, the Gibbs energies of the rate-determining step to the transition state (TS-1Z-TFAvs.TS-3Z-TFA) in the 1Z and 3Z pathways were 41.5 kcal mol−1 and 26.5 kcal mol−1 (Fig. S97†), respectively. The more conjugation of the benzene ring with vinyl in 3Z1d as well as the tension25 resulting from the fusion of five-membered rings should be responsible for the much lower Gibbs energies, and this accounted for the lack of observation of isomerization of 1Z under mild acid conditions.
Factors influencing the proton-catalysed isomerization and non-accumulative cycles
The rate of the proton-catalysed isomerization process was clearly influenced by the solvents used. For example, the isomerization was completed within 2 h in DCM and CHCl3 in the presence of 0.5 eq. of TFA (Table S1, Fig. S67†) but required a longer time in n-hexane and toluene and several days in CH3CN. However, when THF, MeOH, EtOH, or 1,4-dioxane was used as the solvent, no isomerization could be observed, even after 72 h, which was attributed to the stronger solvation of protons in these polar solvents.27The UV-vis spectra of the Z and E isomers of these norbornene-fused stilbene derivatives are distinctively different, and the isomerization could be conventionally detected from UV-vis spectral changes. As illustrated in Fig. 6a, 5Z exhibited one broad absorption band that peaked at λmax = 298 nm. The addition of TFA to the solution of 5Z in DCM led to an apparent increase in absorption to finally give three clear vibrational bands with peaks at 307, 321, and 337 nm, with two isosbestic points (Fig. S64†). It should be noted that, as a very diluted substrate 5Z is required for UV-vis detection, we used superstoichiometric TFA to accelerate the isomerization. However, a catalytic amount of TFA could indeed lead to quantitative isomerization of 5Z at high concentrations as can be seen in Fig. 2c, S41, S92 and Table S1.† The isomerization rate increased with the amount of acid, and the extent of Z → E conversion could be finely manipulated by controlling the amount of acid and the reaction time. In the presence of a large amount of TFA, isomerization was completed within a few seconds (Fig. 6b). The acid-catalysed isomerization proceeded smoothly even at temperatures as low as −40 °C (Fig. S69†). The isomerization rate could be finely tuned by varying the temperature, and increasing the temperature led to an increase in the reaction rate (Fig. S68†). By manipulating the above factors, the isomerization rate could be flexibly tuned from seconds to days.
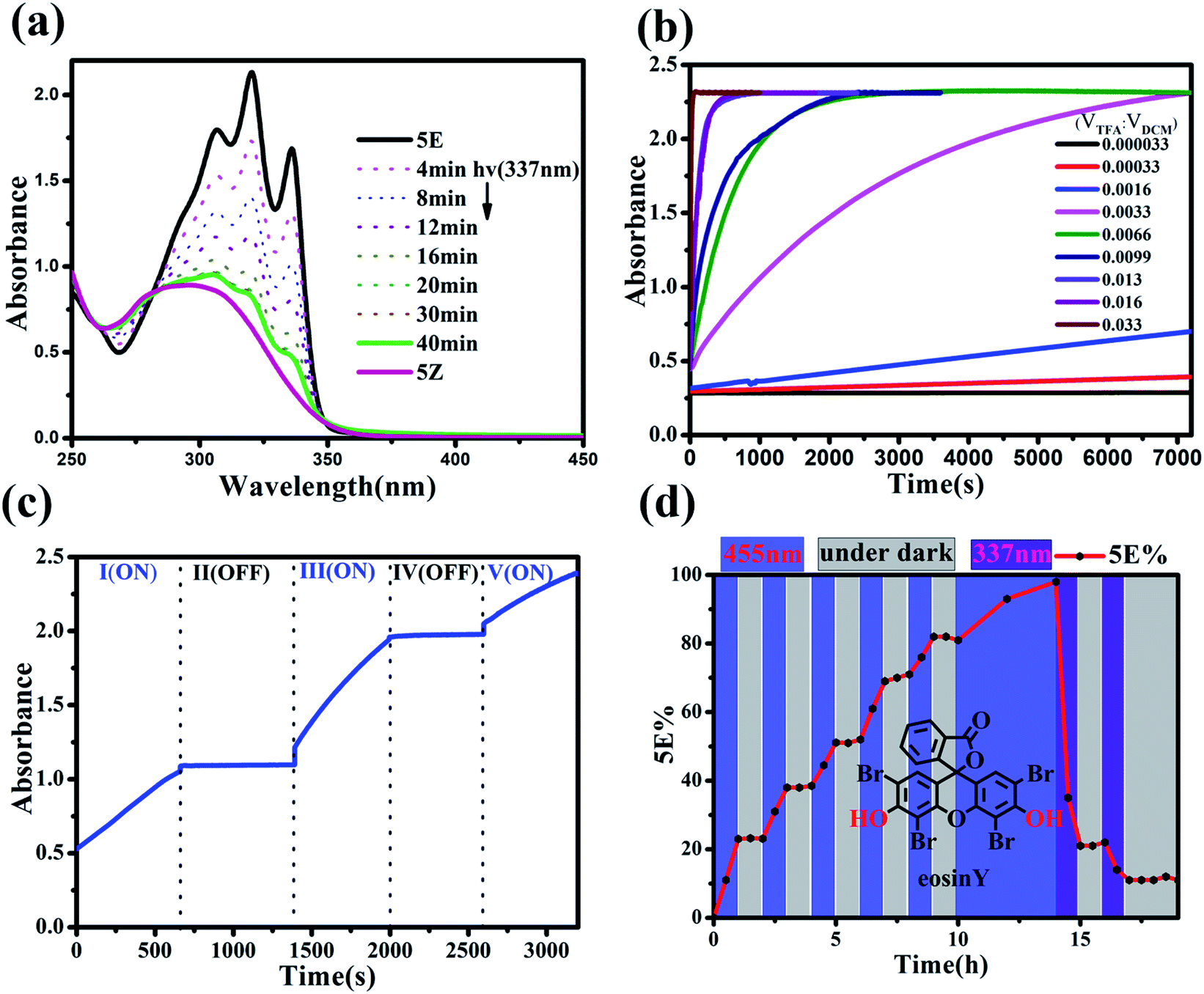 | ||
| Fig. 6 (a) Changes in the UV-vis spectra of 5E (5 × 10−5 M in DCM) upon 337 nm UV irradiation for different time periods, (b) changes in time for the absorbance at 337 nm after the addition of different amounts of TFA to the solution of 5Z (1 × 10−4 M in DCM), and (c) control of the isomerization rate of 5Z by the addition of TFA and Et3N. Conditions: stage I: addition of TFA (VTFA:VDCM = 0.0033); stage II: addition of Et3N; stage III: addition of TFA (VTFA:VDCM = 0.0066); stage IV: addition of Et3N; stage V: addition of TFA (VTFA:VDCM = 0.0099). (d) Changes in the proportion of 5E with eosin Y as the photoacid catalyst (0.1 mmol 5Z and 0.008 mmol eosin Y in 3.0 ml DCM) and the proportion of 5E determined by HPLC analysis. | ||
Seminal pH-activated Z–E switches based on the isomerization of hydrazone28 or enamine29 have recently been explored by using a protic acid as the hydrogen bonding regulator, in which the acid acted as a reactant and the reverse process required the addition of a base to neutralize the acid, which unavoidably led to the generation of waste and side products that eventually hindered the cycle process.30 In the present system, isomerization could be accomplished in the presence of only a catalytic amount of protic acid, which allowed stopping the Z → E isomerization by adding a small amount of alkali. As illustrated in Fig. 6c, the Z → E isomerization of 5Z can be switched on/off by adding TFA and Et3N sequentially. On the other hand, the TFA or HCl could be easily removed from the solution simply by bubbling with nitrogen (Fig. S94 and S95†), thus avoiding the accumulative contamination commonly suffered in chemically induced reversible processes.
Moreover, catalytic isomerization by mild acids allowed another alternative for non-invasive control by the use of a photoacid. The Z → E isomerization of 5Z was examined in the presence of 8% eq. of a commercial photoacid, eosin Y, which releases protons upon photolysis but returns to a neutral species in the dark.31 Indeed, 5Z underwent Z → E isomerization when the system was photoirradiated with a 455 nm OLED, and the isomerization could be stopped by simply turning off the light (Fig. 6d and S91–S93†).
Molecular switch orthogonally controlled by light and acid
The much stronger absorption of 5E in the long-wavelength range is consistent with the better conjugation properties of 5E, and E → Z photoisomerization could be performed by selective excitation at the absorption peak of 5E. As shown in Fig. 6a, when 5E was irradiated with 337 nm UV light in DCM, spectral changes opposite to those of the acid-catalysed isomerization (Fig. S64†) were observed. No further absorption change could be seen after irradiation for 30 min, and a photostationary state (PSS) ratio of 5Z:5E = 88:12 was determined by HPLC analysis. These results revealed that the Z → E and E → Z isomerization of these norbornene-fused stilbenes could be independently manipulated by acid and light, which makes them highly unique molecular switches capable of orthogonal control.22
This orthogonal control makes these molecular switches promising for accomplishing complex functions. For example, they can be used as intelligent molecular switches with controllable self-recovery. Self-recovery switches have wide applications from everyday illumination to microcirculation and are critical in computers and accessory protection.32 A crucial issue for self-recovery switches lies in the precise control of the recovery timescale. The E-to-Z photoisomerization kinetics of the present system can be conveniently controlled by the irradiation light power. Even in the presence of a certain amount of TFA, the E → Z photoisomerization of 5E proceeded smoothly under intensive irradiation, and turning off the light led to spontaneous Z → E isomerization. Without the addition of any base, the on/off switching cycle could be repeated more than 25 times (Fig. S70–S72†). Notably, certain Z forms of molecular switches, such as azobenzene derivatives, can thermally isomerize to the E form, but the thermal isomerization kinetics primarily depend on the inherent molecular properties and are difficult to manipulate.33 In contrast, both the Z → E and E → Z isomerization kinetics of the present molecular switch system are finely controllable. As exemplified in Fig. 7, by changing the concentration of TFA, the recovery time could be conveniently manipulated in a wide time range and therefore should significantly improve the application scenarios of self-recovery.
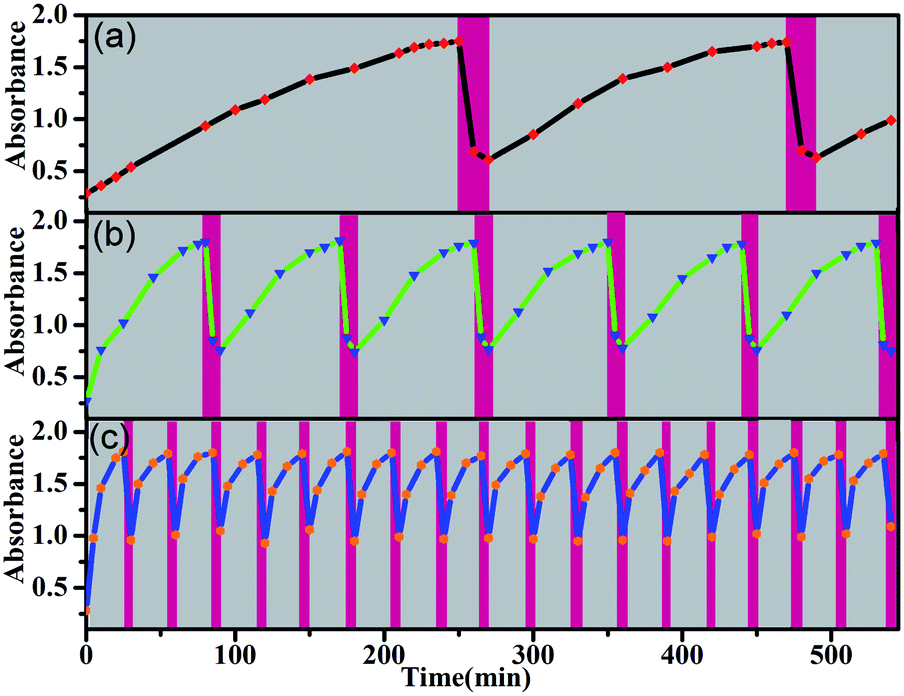 | ||
| Fig. 7 Photoisomerization and recovery cycles of 5Z (5 × 10−5 M in DCM) in the presence of TFA (VTFA:VDCM) (a) 0.0016, (b) 0.0033 and (c) 0.0066 at room temperature. The red and grey areas represent photoisomerization (with 337 nm UV irradiation) and self-recovery processes, respectively. | ||
The potential functional group tolerance was further investigated considering the mild catalysis conditions of the present Z → E isomerization system. For example, compound 10Z, which bears an electron-donating diphenylamino group and an electron-withdrawing cyano group, underwent isomerization smoothly under acid-catalysis conditions. The electronic push–pull effect extends the absorption of 10Z to the visible range, showing an absorption peak at 400 nm, which is a 102 nm bathochromic shift compared with the parent molecule 4Z (Fig. S59†). A blue fluorescence emission was observed for 10Z upon excitation at 380 nm. Upon the addition of TFA to 10Z in DCM, the UV/vis and PL (photoluminescence) spectra showed obvious changes to finally give 10E exclusively (Fig. S87 and S88†). The interconversion between 10E and 10Z could be conducted by acid and visible light for multiple cycles without any loss in performance. The tolerance of additional functional groups, such as 6Z and 10Z, makes it possible to extend the application of this series of compounds through further derivatization or functionalization.
Chiral molecular switch
Norbornene-fused stilbene derivatives are inherently chiral, bearing four stereogenic centers (C7, C8, C11, and C12) in the norbornene component. Our previous studies demonstrated that photochirogenesis is a powerful tool for reading and sensing the supramolecular arrangement of photosubstrates.34 Compared to conventional photochromic systems, chiral molecular switches provide the unique advantage of non-destructive read-out by detecting optical rotation.35 The enantiomeric resolution of 5E was successfully achieved by chiral HPLC (IA column, Fig. S91b†) using a mixed solvent system of n-hexane and DCM (Vn-hexane:VDCM = 80:20). The first eluted fraction 5E1 and the second eluted fraction 5E2 showed perfect mirror-image circular dichroism (CD) spectra, demonstrating an enantiomeric relationship (Fig. 8a). Single crystals of both 5E1 and 5E2 were successfully grown, and the X-ray diffraction analyses demonstrated that the absolute configurations of 5E1 and 5E2 were (7S,8R,11S,12R) and (7R,8S,11R,12S) (Fig. 8c), respectively. The CD spectrum of 5E1 showed clear negative peaks at 236 nm and 300 nm and a positive peak at 252 nm. Irradiation of enantiopure 5E1 at 337 nm led to apparent CD spectral changes (Fig. 8a).
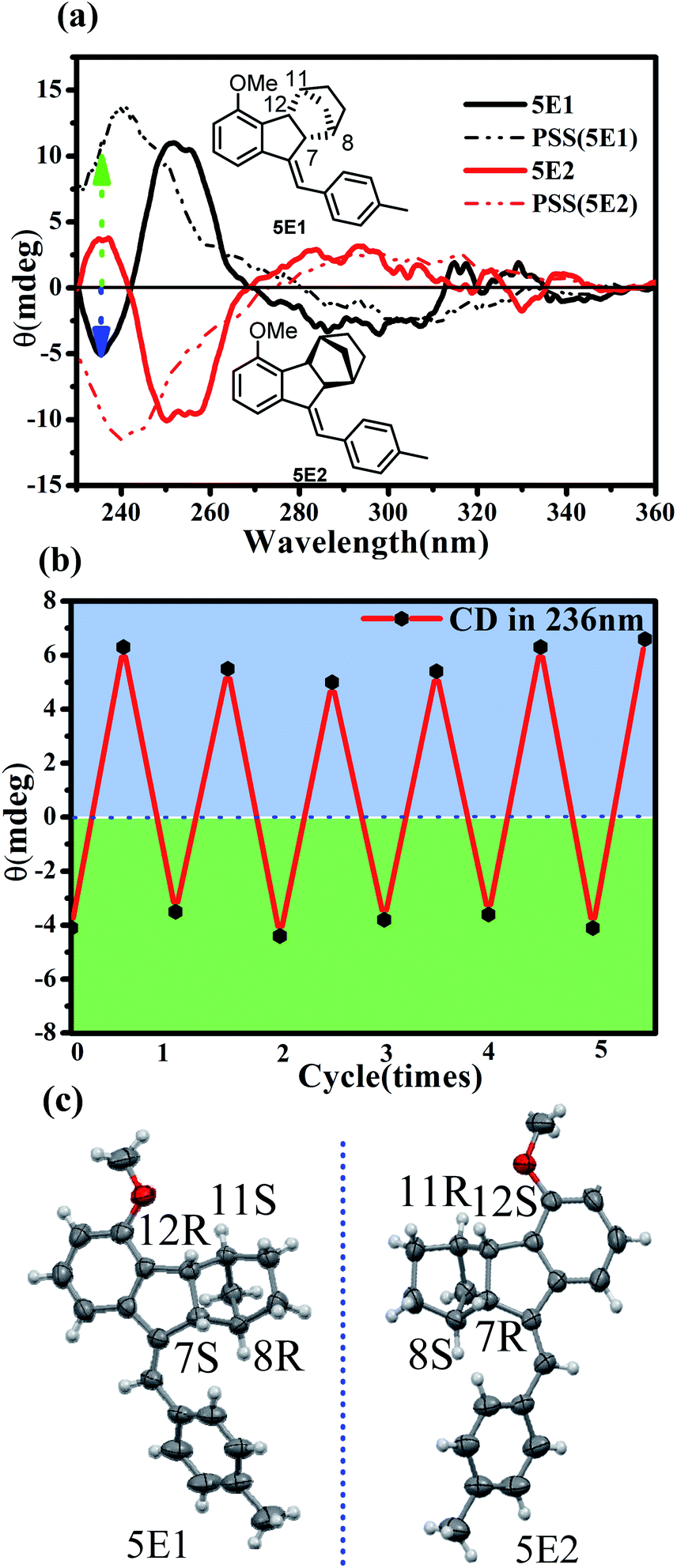 | ||
| Fig. 8 (a) CD spectral changes of 5E1 and 5E2 (5 × 10−5 M) in n-hexane upon irradiation with 337 nm UV light. (b) CD intensity changes at 236 nm for 5E1 (5 × 10−5 M in n-hexane) in the presence of TFA (VTFA:Vn-hexane = 0.0066) after irradiation with 337 nm and then turning off the light until recovery for five cycles. (c) Single-crystal structures of 5E1 and 5E2 (thermal ellipsoid plots at the 50% probability level). | ||
In particular, in the short wavelength region of 230–245 nm, signal inversion from positive to negative was observed. The original negative peak at 236 nm was sharply inverted to a positive peak. This makes it possible to detect E/Z or on/off switching on the basis of the CD sign rather than the spectral intensity of common molecular switches. As illustrated in Fig. 8b, alternately turning on and off the photoirradiation led to switching of the CD signal at 236 nm between −4 and +6, offering an unambiguous on/off signal read-out of the molecular switch. On the other hand, optical rotation measurements indicated that (7R,8S,11R,12S)-5E2 has a specific rotation of 119 deg cm3 dm−1 g−1 at 20 °C with the sodium D line (589 nm) that drastically changed to 226 deg cm3 dm−1 g−1 in the PSS under photoirradiation at 337 nm but completely recovered after adding TFA. Therefore, non-destructive read-out of the switching state of 5 could thus be realized by detecting the specific rotation changes.
Conclusions
In summary, we demonstrated an unprecedented catalytic Z-to-E isomerization of the CC bond in a novel kind of norbornene-fused stilbene derivative triggered by a trace amount of mild organic acids, which is in sharp contrast to the concentrated strong mineral acids required in conventional acid-catalysed isomerization of stilbenes. Such mild catalysis of Z-to-E isomerization occurred quantitatively without any electrophilic addition or migration of the CC double bond by-reaction. The unique properties of these stilbene derivatives make them promising as a novel kind of molecular switch that has the following advantages: the kinetics of Z → E isomerization could be controlled well by the acidity and concentration of acids, temperature and solvents. Photoirradiation at a selected wavelength led to E → Z isomerization in excellent conversion to realize the first molecular switches that are orthogonally manipulated by acid and light, respectively. The Z → E isomerization could be stopped without contamination by using photoacids or removing the acid by bubbling, and therefore, the acid-catalysed Z → E and photo-induced E → Z isomerization switching process could proceed through multiple cycles in a non-accumulative fashion. Molecular switches with controllable self-recovery were thus realized based on these excellent properties. Theoretical studies indicate that the ring strain of the norbornene-fused five-membered ring formed upon stilbene conjugation reduces the activation energy of proton transfer and prevents CC migration, which is responsible for the unprecedented catalysis of isomerization. We believe that similar properties should be achievable through reasonable chemical modification of the fused ring of stilbene, thus opening a new avenue for stilbene derivatives with highly complex functions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of this work by the National Natural Science Foundation of China (No. 21871194, 92056116, 21971169, 21572142 and 22072099), the National Key Research and Development Program of China (No. 2017YFA0505903), the Science & Technology Department of Sichuan Province (2019YJ0160, 2019YJ0090, and 2017SZ0021), and the Fundamental Research Funds for the Central Universities (20826041D4117). Compound characterization was performed with the support of the Comprehensive Training Platform of Specialized Laboratory, College of Chemistry and Professor Peng Wu of the Analytical & Testing Center, Sichuan University.Notes and references
- (a) J. Wislicenus and M. Jahrmarkt, Chem. Zentralbl., 1901, I, 463 Search PubMed; (b) J. Saltiel, S. Gupta, D. W. Eaker, A. M. Kropp and V. K. R. Kumar, Photochem. Photobiol., 2018, 94, 247 CrossRef CAS; (c) W. Fuß, C. Kosmidis, W. E. Schmid and S. A. Trushin, Angew. Chem., Int. Ed., 2004, 43, 4178 CrossRef; (d) J.-S. Yang, C.-K. Lin, A. M. Lahoti, C.-K. Tseng, Y.-H. Liu, G.-H. Lee and S.-M. Peng, J. Phys. Chem. A, 2009, 113, 4868 CrossRef CAS.
- (a) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS; (b) I. Tochitsky, M. A. Kienzler, E. Isacoff and R. H. Kramer, Chem. Rev., 2018, 118, 10748 CrossRef CAS; (c) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174 CrossRef CAS; (d) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS; (e) D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310 CrossRef CAS; (f) M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200 CrossRef CAS; (g) A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117 CrossRef CAS.
- (a) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS; (b) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060 CrossRef CAS; (c) V. García-López, D. Liu and J. M. Tour, Chem. Rev., 2020, 120, 79 CrossRef.
- (a) J.-F. Xu, Y.-Z. Chen, D. Wu, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Angew. Chem., Int. Ed., 2013, 52, 9738 CrossRef CAS; (b) P. Wei, J.-X. Zhang, Z. Zhao, Y. Chen, X. He, M. Chen, J. Gong, H.-H. Sung, I. D. Williams, J. W. Y. Lam and B.-Z. Tang, J. Am. Chem. Soc., 2018, 140, 1966 CrossRef CAS; (c) S. J. Wezenberg, C. M. Croisetu, M. C. A. Stuart and B. L. Feringa, Chem. Sci., 2016, 7, 4341 RSC; (d) D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192 CrossRef; (e) J. Luo, K. Song, F.-L. Gu and Q. Miao, Chem. Sci., 2011, 2, 2029 RSC.
- V. Serreli, C. F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523 CrossRef CAS.
- D.-H. Qu, Q.-C. Wang and H. Tian, Angew. Chem., Int. Ed., 2005, 44, 5296 CrossRef CAS.
- C. C. Price and M. Meister, J. Am. Chem. Soc., 1939, 61, 1595 CrossRef CAS.
- D. S. Noyce, D. R. Hartter and F. B. Miles, J. Am. Chem. Soc., 1968, 90, 4633 CrossRef CAS.
- M. S. Kharasch, J. V. Mansfield and F. R. Mayo, J. Am. Chem. Soc., 1937, 59, 1155 CrossRef CAS.
- (a) X. Guo, B. Shao, S. Zhou, I. Aprahamian and Z. Chen, Chem. Sci., 2020, 11, 3016 RSC; (b) C. L. Fleming, S. Li, M. Grøtli and J. Andréasson, J. Am. Chem. Soc., 2018, 140, 14069 CrossRef CAS; (c) A. Kerckhoffs and M. J. Langton, Chem. Sci., 2020, 11, 6325 RSC.
- C. F. Bernasconi and W. J. Boyle, J. Am. Chem. Soc., 1974, 96, 6070 CrossRef CAS.
- (a) C. Biagini and S. D. Stefano, Angew. Chem., Int. Ed., 2020, 59, 8344 CrossRef CAS; (b) C. Biagini, S. Albano, R. Caruso, L. Mandolini, J. A. Berrocal and S. D. Stefano, Chem. Sci., 2018, 9, 181 RSC.
- I. Cvrtila, H. Fanlo-Virgós, G. Schaeffer, G. Monreal Santiago and S. Otto, J. Am. Chem. Soc., 2017, 139, 12459 CrossRef CAS.
- (a) D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338 CrossRef; (b) A. H. Heindl, J. Becker and H. A. Wegner, Chem. Sci., 2019, 10, 7418 RSC; (c) G. M. Peters and J. D. Tovar, J. Am. Chem. Soc., 2019, 141, 3146 CrossRef CAS; (d) S. Fredrich, A. Bonasera, V. Valderrey and S. Hecht, J. Am. Chem. Soc., 2018, 140, 6432 CrossRef CAS; (e) N. A. Simeth, A. C. Kneuttinger, R. Sterner and B. König, Chem. Sci., 2017, 8, 6474 RSC; (f) D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538 CrossRef CAS; (g) Z. Xu, Q. T. Liu, X. Wang, Q. Liu, D. Hean, K. C. Chou and M. O. Wolf, Chem. Sci., 2020, 11, 2729 RSC.
- (a) I. Hnid, D. Frath, F. Lafolet, X. Sun and J. C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732 CrossRef CAS; (b) J. Yin, A. N. Khalilov, P. Muthupandi, R. Ladd and V. B. Birman, J. Am. Chem. Soc., 2020, 142, 60 CrossRef CAS; (c) M. Herder, M. Utecht, N. Manicke, L. Grubert, M. Pätzel, P. Saalfrank and S. Hecht, Chem. Sci., 2013, 4, 1028 RSC.
- (a) T. Ooi, Science, 2011, 331, 1395 CrossRef CAS; (b) N. Mallo, E. D. Foley, H. Iranmanesh, A. D. W. Kennedy, E. T. Luis, J. Ho, J. B. Harper and J. E. Beves, Chem. Sci., 2018, 9, 8242 RSC; (c) C. Poloni, M. C. A. Stuart, P. Meulen, W. Szymanski and B. L. Feringa, Chem. Sci., 2015, 6, 7311 RSC.
- (a) J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845 CrossRef CAS; (b) X. Fu, H. Han, D. Zhang, H. Yu, Q. He and D. Zhao, Chem. Sci., 2020, 11, 5565 RSC.
- A. Martinez, L. Guy and J.-P. Dutasta, J. Am. Chem. Soc., 2010, 132, 16733 CrossRef CAS.
- (a) S. P. Fletcher, F. Dumur, M. M. Pollard and B. L. Feringa, Science, 2005, 310, 80 CrossRef CAS; (b) T. R. Kelly, H. D. Silva and R. A. Silva, Nature, 1999, 401, 150 CrossRef CAS; (c) J. T. Foy, D. Rayb and I. Aprahamian, Chem. Sci., 2015, 6, 209 RSC; (d) P. C. Knipe, S. Thompson and A. D. Hamilton, Chem. Sci., 2015, 6, 1630 RSC.
- J. Yao, W. Wu, W. Liang, Y. Feng, D. Zhou, J. J. Chruma, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, Angew. Chem., Int. Ed., 2017, 56, 6869 CrossRef CAS.
- C. Xiao, W. Wu, W. Liang, D. Zhou, K. Kanagaraj, G. Cheng, D. Su, Z. Zhong, J. J. Chruma and C. Yang, Angew. Chem., Int. Ed., 2020, 59, 8094 CrossRef CAS.
- (a) M. M. Lerch, M. J. Hansen, W. A. Velema, W. Szymanski and B. L. Feringa, Nat. Commun., 2016, 7, 12054 CrossRef CAS; (b) D. Roke, C. Stuckhardt, W. Danowski, S. J. Wezenberg and B. L. Feringa, Angew. Chem., Int. Ed., 2018, 57, 10515 CrossRef CAS; (c) E. Fuentes, M. Gerth, J. A. Berrocal, C. Matera, P. Gorostiza, I. K. Voets, S. Pujals and L. Albertazzi, J. Am. Chem. Soc., 2020, 142, 10069 CrossRef CAS; (d) A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bleger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335 CrossRef CAS.
- T.-T. Hao, H.-R. Liang, Y.-H. Ou-Yang, C.-Z. Yin, X.-L. Zheng, M.-L. Yuan, R.-X. Li, H.-Y. Fu and H. Chen, J. Org. Chem., 2018, 83, 4441 CrossRef CAS.
- M. B. Smith and J. March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, Wiley, New York, USA, 2006 Search PubMed.
- (a) J. Bredt, J. Houben and P. Levy, Ber. Dtsch. Chem. Ges., 1902, 35, 1286 CrossRef CAS; (b) F. S. Fawcett, Chem. Rev., 1950, 47, 219 CrossRef CAS; (c) K. B. Wiberg, Angew. Chem., Int. Ed., 1986, 25, 312 CrossRef; (d) Z. Huang, Q. Z. Yang, D. Khvostichenko, T. J. Kucharski, J. Chen and R. Boulatov, J. Am. Chem. Soc., 2009, 131, 1407 CrossRef CAS; (e) R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594 CrossRef CAS; (f) Q. Li, H. Qian, B. Shao, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 11829 CrossRef CAS.
- A. D. Allen, M. Resenbaum, N. O. L. Seto and T. T. Tidwell, J. Org. Chem., 1982, 47, 4234 CrossRef CAS.
- A. K. Chew, T. W. Walker, Z. Shen, B. Demir, L. Witteman, J. Euclide, G. W. Huber, J. A. Dumesic and R. C. Van Lehn, ACS Catal., 2020, 10, 1679 CrossRef CAS.
- (a) S. M. Landge and I. Aprahamian, J. Am. Chem. Soc., 2009, 131, 18269 CrossRef CAS; (b) X. Su, T. F. Robbins and I. Aprahamian, Angew. Chem., Int. Ed., 2011, 50, 1841 CrossRef CAS; (c) X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963 RSC; (d) I. Aprahamian, Chem. Commun., 2017, 53, 6674 RSC.
- (a) Y. Ren, P. H. Svensson and O. Ramström, Angew. Chem., Int. Ed., 2018, 57, 6256 CrossRef CAS; (b) Y. Ren, S. Xie, E. Svensson Grape, A. K. Inge and O. Ramström, J. Am. Chem. Soc., 2018, 140, 13640 CrossRef CAS.
- L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 17438 CrossRef CAS.
- G.-Y. Zhao and T. Wang, Angew. Chem., Int. Ed., 2018, 57, 6120 CrossRef CAS.
- (a) M.-X. Wang, Y.-M. Chen, Y. Gao, C. Hu, J. Hu, L. Tan and Z. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 26610 CrossRef CAS; (b) H. J. Sim, H. Kim, Y. Jang, G. M. Spinks, S. Gambhir, D. L. Officer, G. G. Wallace and S. J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 46026 CrossRef CAS.
- (a) N. A. Simeth, S. Crespi, M. Fagnoni and B. König, J. Am. Chem. Soc., 2018, 140, 2940 CrossRef CAS; (b) C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. White and M. J. Fuchter, J. Am. Chem. Soc., 2014, 136, 11878 CrossRef CAS; (c) D. Bleger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597 CrossRef CAS; (d) H. Qian, Y. Y. Wang, D. S. Guo and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 1037 CrossRef CAS; (e) L. Pesce, C. Perego, A. B. Grommet, R. Klajn and G. M. Pavan, J. Am. Chem. Soc., 2020, 142, 9792 CrossRef CAS; (f) Q. Li, H. Qian, B. Shao, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 11829 CrossRef CAS; (g) Z. Chu, Y. Han, T. Bian, S. De, P. Kral and R. Klajn, J. Am. Chem. Soc., 2019, 141, 1949 CrossRef CAS.
- (a) X. Wei, W. Wu, R. Matsushita, Z. Yan, D. Zhou, J. J. Chruma, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Am. Chem. Soc., 2018, 140, 3959 CrossRef CAS; (b) X. Wei, J. Liu, G. J. Xia, J. Deng, P. Sun, J. J. Chruma, W. Wu, C. Yang, Y. G. Wang and Z. Huang, Nat. Chem., 2020, 12, 551 CrossRef CAS; (c) J. Ji, W. Wu, W. Liang, G. Cheng, R. Matsushita, Z. Yan, X. Wei, M. Rao, D. Q. Yuan, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Am. Chem. Soc., 2019, 141, 9225 CrossRef; (d) C. Yang and Y. Inoue, Chem. Soc. Rev., 2014, 43, 4123 RSC.
- (a) B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789 CrossRef CAS; (b) Y. Wang and Q. Li, Adv. Mater., 2012, 24, 1926 CrossRef CAS; (c) J. Li, H. K. Bisoyi, S. Lin, J. Guo and Q. Li, Angew. Chem., Int. Ed., 2019, 58, 16052 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 5Z: 1949916, 5E1/5E2: 1949886 and 6E: 1949917. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05213b |
| This journal is © The Royal Society of Chemistry 2021 |