
Tumor-targeted gene therapy with lipid nanoparticles inhibits tumor-associated adipocytes and remodels the immunosuppressive tumor microenvironment in triple-negative breast cancer†
Yun
Liu
 a,
Karthik
Tiruthani
b,
Menglin
Wang
a,
Xuefei
Zhou
a,
Nasha
Qiu
a,
Yang
Xiong
a,
Chad V.
Pecot
c,
Rihe
Liu
*bd and
Leaf
Huang
*a
a,
Karthik
Tiruthani
b,
Menglin
Wang
a,
Xuefei
Zhou
a,
Nasha
Qiu
a,
Yang
Xiong
a,
Chad V.
Pecot
c,
Rihe
Liu
*bd and
Leaf
Huang
*a
aDivision of Pharmacoengineering and Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: leafh@email.unc.edu; Tel: +1-919-843-0736
bDivision of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: rliu@email.unc.edu; Tel: +1-919-843-3635
cUNC Lineberger Comprehensive Cancer Center, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
dCarolina Center for Genome Sciences, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
First published on 15th February 2021
Abstract
Adipocytes are the primary cellular components within the tumor microenvironment (TME) of triple-negative breast cancer (TNBC). Increasing evidence suggests that tumor-associated adipocytes (TAAs) can aggravate tumor progression, exacerbate the immunosuppressive TME and compromise therapeutic efficacy. In this study, the biological effect of TAAs within the breast cancer TME is first investigated, and the C–C Motif Chemokine Ligand 2 (CCL2) which is mainly secreted by TAAs in the extracellular environment is identified as the key mediator. CCL2 recruits immune cells such as monocytes and macrophages that further differentiated into immunosuppressive myeloid-derived suppressor cells (MDSCs) and M2 macrophages. To manipulate CCL2-mediated immune response, a protein trap that binds with CCL2 with high affinity and specificity is designed. The plasmid DNA encoding the CCL2 trap (pCCL2) is specifically delivered to the TME by using targeted lipid-protamine-DNA (LPD) nanoparticles to locally express the CCL2 trap and ameliorate the immunosuppressive TME. Significantly, compared with the commercially available CCL2 antibody, this strategy shows enhanced therapeutic efficacy and appreciable tumor growth inhibition. Furthermore, the pCCL2 trap treatment successfully suppresses TAAs, increases T cell infiltration and decreases the population of immunosuppressive M2 macrophages and MDSCs. Further studies show that the pCCL2 trap could facilitate PD-L1 blockade immunotherapy, demonstrating its translation potential.
New conceptsIn recent years, an improved understanding of cancer biology and the discovery of cellular and molecular mechanisms for innate and adaptive immune responses have significantly revolutionized the fields of cancer immunology and immunotherapy. These have remarkably encouraged researchers to investigate the possibility of restoring the cancer-immunity cycle using nanomaterial-based immunotherapeutics. However, none of them have reached the clinic for patients. One major reason for the lack of clinical translation is the presence of the immunosuppressive tumor microenvironment (TME). In this study, we explored the availability of lipid nanoparticle-based delivery of immuno-regulatory agents to systemically and locally modulate the suppressive milieu within the TME. These works provide a proof of concept for lipid nanoparticle-based TME-modulating methods and illustrate the potential of nanomaterials in the field of cancer immunotherapy. Here, for the first time, we have shown that precise triple-negative breast cancer TME modulation can be achieved by tumor-targeted gene therapy. In addition, our gene therapy strategy significantly outperformed the commercially available monoclonal-antibody in the animal study. We believe that this work represents a promise to revolutionize cancer immunotherapy with non-viral mediated gene therapy and will be of interest to researchers in the fields of drug delivery and cancer immunotherapy. |
Introduction
Triple-negative breast cancer (TNBC) is one of the leading causes of death among women with a high metastasis rate and poor prognosis. Only a few clinical benefits have been observed in TNBC patients receiving chemotherapy and radiotherapy.1 In recent years, the development of checkpoint blockade-based immunotherapy (CTLA4, PD-1 or PD-L1 inhibitors) has greatly reshaped the landscape of cancer therapy. However, a response rate of less than 20% was achieved in TNBC patients owing to the immunosuppressive tumor microenvironment (TME).1,2 Therefore, the development of effective remedies to reverse the immunosuppressive TME is crucial for TNBC treatment.TME is a heterogeneous and dynamic cellular milieu consisting of a variety of resident and infiltrating cells.3 Accumulating evidence has revealed the important role of the TME in tumor initiation, progression, metastasis, therapeutic response and resistance.3 Breast cancer is noted to have abundant and specific resident adipocytes within the TME that differ considerably from other solid tumors.4 Such distinctive cellular component difference in the TME might help to better understand the mechanism that dictates the immunosuppressive TME formation of breast cancer.
Adipocytes have long been considered as a fuel tank to store energy in the form of lipids and triglycerides.5 However, recent findings unraveled the diverse aspects of adipocytes and adipocyte derived factors.4,6 Clinical observations suggested that adipocytes residing in close proximity to cancer cells exhibit tumor-associated functions and phenotypes, including decreased sizes and overexpression of collagen VI and chemokines.6 This specialized type of adipocyte is termed as “tumor associated adipocytes (TAAs)”. TAAs may secrete factors that will promote the immunosuppressive TME.6 Furthermore, TAAs may trans-differentiate into fibroblasts which enhance the desmoplasia of the tumor,6 further promoting the suppressive immune microenvironment. An in-depth understanding of the biology of TAAs might provide new opportunities to modify the immunosuppressive TME in TNBC and facilitate the development of more effective therapies.
C–C Motif Chemokine Ligand 2 (CCL2), which is also referred to as monocyte chemoattractant protein 1 (MCP-1), is a potent inducible chemokine that recruits immune cells, in particular monocytes, to infiltrate into the inflammatory tissue region.7 The plasticity of monocytes enables their phenotype transition and functional changes in response to TME signals. Following infiltration into the TME, monocytes are quickly polarized and differentiated into macrophages and myeloid-derived suppressor cells (MDSCs), accompanied by functional and phenotypical changes.8,9 It is widely documented that tumor-associated macrophages constitute two major subtypes, M1 and M2 macrophages.10 M1 and M2 macrophages are identified based on their surface markers and secrete cytokines/chemokines, which differ considerably in terms of phenotypes and functions.10 M1 macrophages are pro-inflammatory and actively produce high levels of pro-inflammatory cytokines/chemokines to promote Th1 responses. On the contrary, M2 macrophages are anti-inflammatory and actively secrete large amounts of immunosuppressive cytokines such as interleukin 10 (IL-10) and transforming growth factor beta (TGF-β). In addition, M2 macrophages repurpose arginase metabolic pathways to further promote tumor progression.10 Clinical observations suggested that an increased M2 macrophage density in the tumor is closely correlated with poor prognosis, whereas an increased M1 macrophage proportion usually signifies a favorable clinical outcome.11 Moreover, further analysis of gene expression profiles revealed the role of CCL2 as a potent driving factor for M2 polarization and Th2 response. Elevated CCL2 levels positively correlate with enhanced M2 macrophage and MDSC populations, and inhibit T cell infiltration and other immunosuppressive factors that compromise the cancer immunotherapy.9 In this regard, blockade of CCL2 could be a feasible strategy to reverse the immunosuppressive TME and thereby facilitate checkpoint blockade immunotherapy.
There are two common strategies for target-protein based therapy to block the CCL2-specific signaling axis: small molecule inhibitors and monoclonal antibodies (mAbs). Bindarit® is a small molecule inhibitor which exhibits a broad inhibition of CCL2, CCL7 and CCL8, presumably by inhibiting the p65 and p65/p50 mediated MCP-1 promoter activation. A previous study revealed only a moderate inhibitory ability of Bindarit® over lipopolysaccharide (LPS) induced production of CCL2 in human monocytes with IC50 over 170 μM.12 To address the clinical need of a selective inhibitor with enhanced potency and efficacy, CCL2-neutralizing mAb was developed. Preclinical and clinical investigations confirmed the apparent efficacy of a systemic CCL2 mAb in breast cancers with a moderate tumor inhibition effect and a decrease in the population of M2 macrophages and MDSCs in the TME.13 However, discontinuation of CCL2 mAb triggered a rebound effect in CCL2 expression, resulting in a significant increase in the infiltration and accelerated metastasis of M2 macrophages and MDSCs.13 Further mechanistic analysis revealed that the immediate cessation of systemic CCL2 mAb resulted in increased migration and infiltration of monocytes into the primary tumor and lung metastasis.14 Therefore, a local and transient CCL2 blockade may be an effective strategy to modify the immunosuppressive TME with reduced systemic toxicity.
Recent advances in nanotechnology have opened up opportunities for the development of a local and transient in vivo mAb-like blockade with a non-viral gene delivery platform.3 Nanoparticles (NPs) preferentially accumulate within the tumor via the enhanced permeability and retention (EPR) effect that depends on the leaky nature of abnormal vessels in solid tumors.3 To further optimize the targeting efficacy, targeting ligands are often conjugated to NPs to facilitate specific uptake of NPs by cancer cells.15 Plasmid DNA encoding mAb or mAb-like protein can be encapsulated and delivered into the TME in a targeted manner. This approach turns the tumor into a biological factory to produce corresponding mAbs, or the like, followed by extracellular secretion into the TME.16 In this work, we first developed a CCL2-binding single domain antibody, called “trap”, that specifically binds to mouse CCL2. We then constructed and delivered a plasmid encoding CCL2 trap (pCCL2 trap) by using tumor targeted lipid-protamine-pDNA (LPD) NPs to the TME. We hypothesized that locally expressed trap will efficiently decrease the concentration of CCL2, leading to TME remodeling. The surface of LPD NPs was grafted with polyethylene glycol and functionalized with the targeting ligand aminoethyl anisamide (AEAA) to reduce organ accumulation and improve tumor delivery efficacy. AEAA is a high affinity ligand (Kd = 9 nM) for the sigma-1 receptor overexpressed in both tumor cells and tumor associated fibroblasts.17 We demonstrated in a murine orthotopic model of TNBC that the pCCL2 trap delivered by LPD NPs preferentially accumulated within the TME, leading to a decrease in the CCL2 level. In addition, compared with CCL2 mAb, our strategy modified the immunosuppressive TME without any rebound effect, reprogrammed TAAs into normal adipocytes, increased tumor inhibition efficacy and reduced lung metastasis. Furthermore, in combination with pPD-L1 trap-based checkpoint blockade immunotherapy, we observed a significant tumor inhibition effect in the murine TNBC model (Scheme 1).
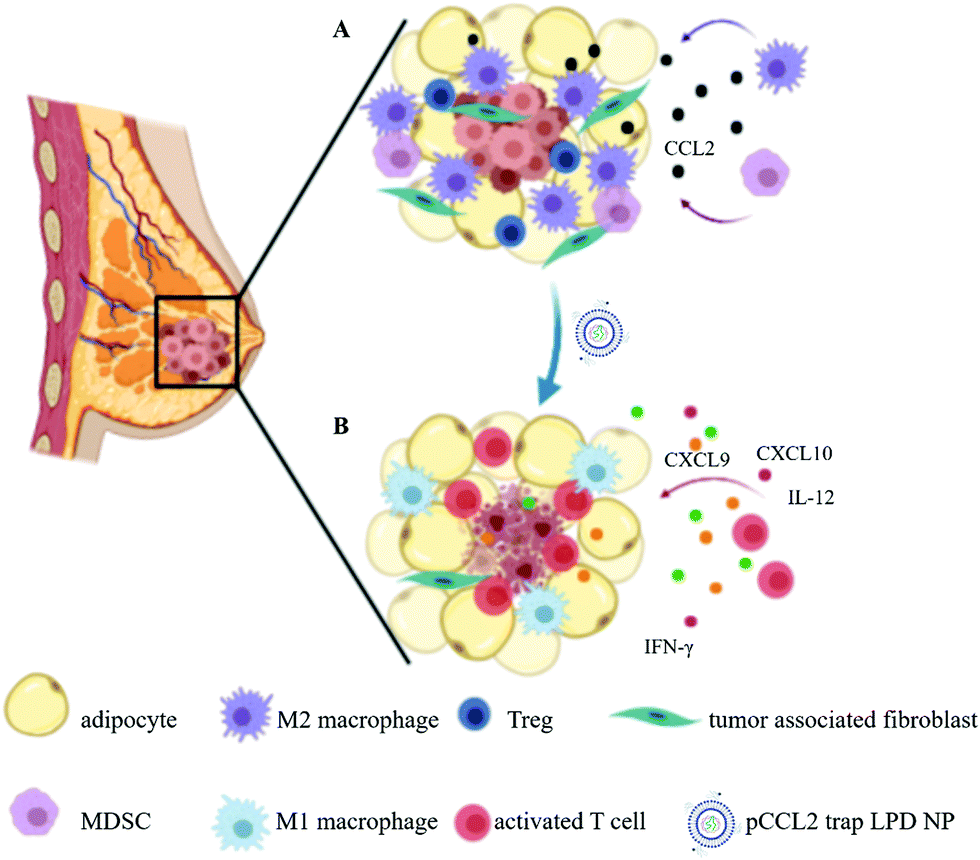 | ||
| Scheme 1 Schematic illustration of the mechanism of pCCL2 trap for the treatment of TNBC in the murine model of TNBC. (A) Primary mammary breast tumor immunosuppressive TME constituting the surrounding tumor associated adipocytes (TAAs) and infiltrated immunocytes. (B) pCCL2 trap successfully remodels the immunosuppressive TME of TNBC in the tumor model. | ||
Results and discussion
Tumor-associated adipocytes facilitate tumor progression and exacerbate the immunosuppressive TME by secreting CCL2
Adipocytes are one of the main resident cells that constitute the TME of TNBC.4 Previous studies suggested the interaction between adipocytes and cancer cells, while the clear role of tumor associated-adipocytes (TAAs) remains undefined.6 To better understand the interaction between mature adipocytes and TNBC cells, mature adipocytes were first differentiated from 3T3-L1 preadipocytes, followed by cultivation in a transwell system in the presence or absence of 4T1 murine TNBC cells (Fig. 1A). Significant morphological changes were observed in adipocytes cocultured with 4T1, which exhibited a significant decrease in lipid droplet size and contents (Fig. 1B). In addition, further RT-PCR analysis revealed altered gene expression in 4T1 cocultured-adipocytes. Fatty acid binding protein 4 (FABP4), a hallmark exclusively expressed in mature adipocytes at high levels,6 was significantly downregulated 2–3-fold in 4T1 cocultured-adipocytes. In contrast, cytokine CCL2, a potent chemokine for the recruitment of monocytes, was significantly upregulated (Fig. 1C).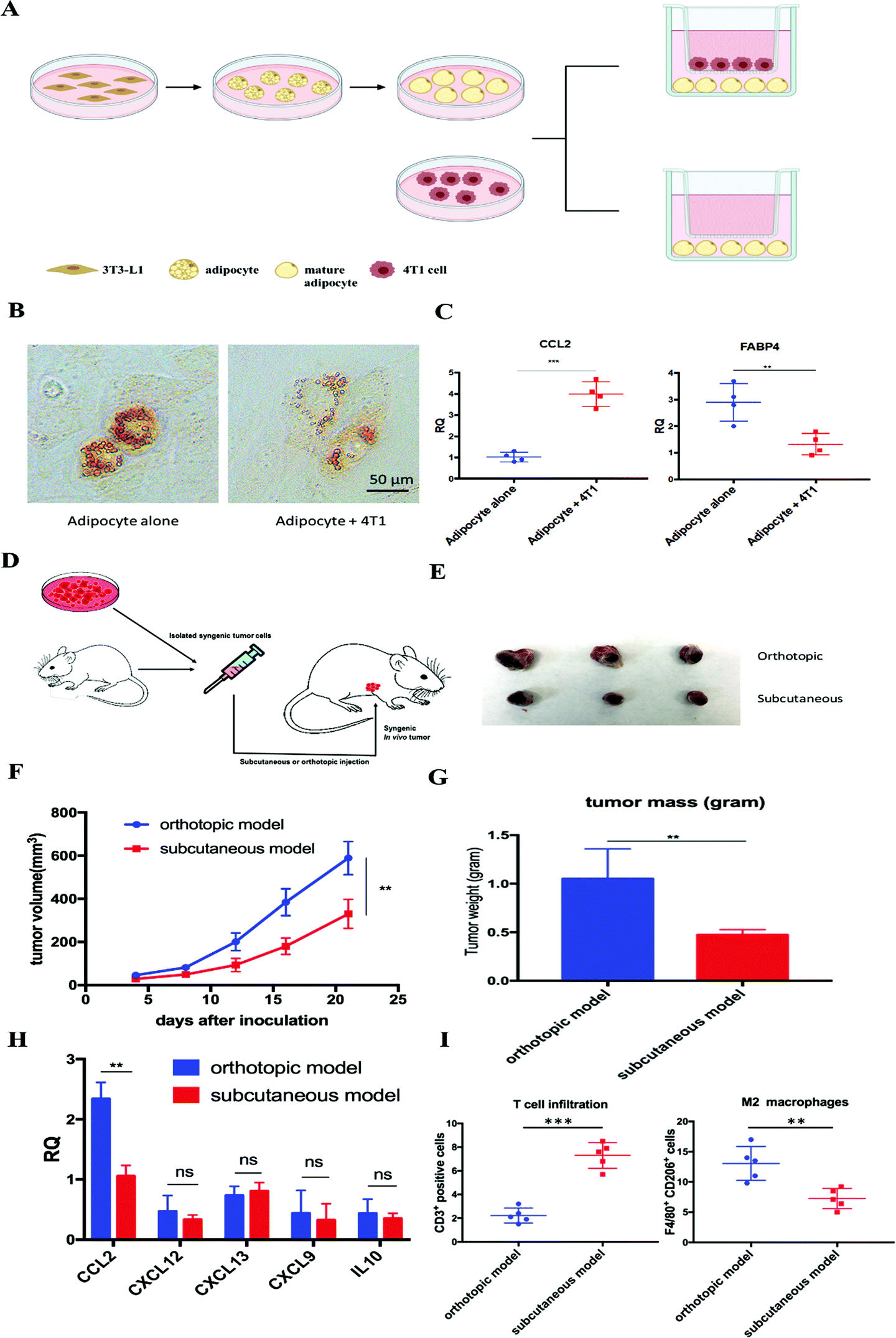 | ||
| Fig. 1 Tumor-associated adipocytes facilitate tumor progression and exacerbate the immunosuppressive TME by secreting CCL2. (A) Cultivation of mature adipocytes in the presence or absence of 4T1 cells. Mature adipocytes were first differentiated from preadipocyte 3T3-L1 and cocultured with or without 4T1 cells in a transwell system. (B) A significant decrease in lipid droplet size and contents was observed in mature adipocytes cocultured with 4T1 cells. Lipid droplets were stained with Oil-Red-O. (C) RT-PCR analysis of cultivated mature adipocytes in the presence or absence of 4T1 cells. CCL2: C–C Motif Chemokine Ligand 2; FABP4: fatty acid binding protein 4. (D) Establishment of the subcutaneous or orthotopic 4T1 mouse model. On day 0, 1 × 106 4T1 cells were inoculated orthotopically on the mammary fat pad or subcutaneously in six-week old female BALB/c mice. (E) Representative picture of tumor burden from orthotopic and subcutaneous 4T1 mouse models on day 21. (F) Tumor progression curve of orthotopic and subcutaneous 4T1 mouse models. **p < 0.01. (G) Quantitative analysis of the tumor weight of orthotopic and subcutaneous 4T1 mouse models on day 21. **p < 0.01. (H) Relative mRNA expression of various chemokines and cytokines in tumors from orthotopic and subcutaneous 4T1 mouse models. CCL2: C–C Motif Chemokine Ligand 2; CXCL12: C–X–C Motif Chemokine Ligand 12; CXCL13: C–X–C Motif Chemokine Ligand 13; CXCL9: C–X–C Motif Chemokine Ligand 9; IL-10: interleukin 10. **p < 0.01; ns, not significant. (I) CD3+ T cell and M2 macrophage population in the tumors from orthotopic and subcutaneous 4T1 mouse models. Cells were identified by using flow cytometry. **p < 0.01, ***p < 0.001. | ||
Enlightened by the in vitro observations, we established in vivo 4T1 tumor models in the presence or absence of adipocytes on BALB/c mice, aiming to investigate the role of TAAs in the 4T1 murine TNBC model. The same amounts of 4T1 cells were inoculated on the mammary fat pad or subcutaneously to establish an orthotopic or subcutaneous 4T1 model, respectively (Fig. 1D). The only significant difference between the two models is the presence or absence of resident adipocytes in the tumor environment. A significant aggressive tumor progression was observed in the orthotopic 4T1 model compared with the subcutaneous 4T1 model (Fig. 1E–G). In addition, consistent with previous in vitro experiments, more detailed gene expression analysis by RT-PCR revealed that CCL2 mRNA was highly upregulated in the orthotopic 4T1 model, whereas no significant differences in the mRNA expression levels of other tumor progression associated chemokines and cytokines between orthotopic and subcutaneous 4T1 models were found (Fig. 1H). Previous studies suggested that tumor vascularization is closely related to tumor growth and metastasis.18 As shown in Fig. S1 (ESI†), there is no significant difference in the tumor vascular density between the two models. Moreover, the orthotopic model revealed an immunosuppressive TME which was characterized by abundant infiltration of M2 macrophages, presumably differentiated from the recruited monocytes via CCL2. Furthermore, activated T cell infiltration was considerably decreased, profiling an immunosuppressive TME in the orthotopic 4T1 model (Fig. 1I).
We next examined the clinical database to further verify our hypothesis. Among all organs and tissues, adipose tissue is the main source of CCL2 in humans. The analysis of clinical specimens also revealed the prevalence of CCL2 expression in breast cancer patients and high expression of CCL2 is positively correlated with shorter survival and poor prognosis in breast cancer patients (Fig. S2, ESI†). Taken together, these data supported our hypothesis that TAAs actively participate in TNBC progression and exacerbate the immunosuppressive TME by secreting excessive CCL2. Therefore, blockade of CCL2 holds great promise to remodel the immunosuppressive TME and facilitate further immunotherapy for TNBC treatment.
Preparation and characterization of in situ CCL2 blockade depot
To develop a CCL2 trap that is ideal for local expression in the TME via nanoparticle-mediated gene delivery, we screened a heavy chain variable domain (VH) library displayed on the yeast cell surface19 and obtained a CCL2 specific trap protein. The trap was engineered with C-terminal c-Myc and Hisx6 tags to facilitate purification and signal detection. The theoretical molecular weight of the CCL2 trap is about 15.8 kDa. The recombinant trap protein was expressed in Expi 293 cells and purified by using Ni-NTA affinity chromatography. The binding affinity to murine CCL2 was measured by using immobilized trap proteins on a Biacore facility. The estimated Kd was ∼229 nM, which is relatively weak. However, the binding appears to be specific. Only a weak binding with C–C Motif Chemokine Ligand 7 (CCL7) was detected, whereas its binding with C–C Motif Chemokine Ligand 8 (CCL8) and other chemokines was not detectable. The CCL2 trap gene was codon-optimized for expression in mammalian cells. A plasmid (pCCL2 Trap) containing the CCL2 trap sequence was constructed in the pcDNA3.1 vector under the control of the CMV promoter.LPD NPs were constructed via a sequential self-assembly process (Fig. 2A). First, blank cationic 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP):cholesterol liposomes were prepared using the thin-film method.20 An anionic complex core was then formulated by mixing cationic protamine with plasmid DNA (pDNA). The anionic complex core was further coated with the blank cationic DOTAP:cholesterol liposome to formulate lipid-protamine-pDNA nanoparticles (LPD NPs). To prolong the circulation time and reduce non-specific uptake by Kupffer cells, polyethylene glycol (PEG) was coated on the surface of LPD NPs. Previous studies demonstrated that the sigma receptor was significantly upregulated in 4T1 murine tumors.1 A specific sigma receptor ligand, aminoethyl anisamide (AEAA), was employed to improve the local gene delivery and expression in 4T1 tumors. DSPE–PEG and the target ligand DSPE–PEG–AEAA were grafted on the surface of liposomes via the post-insertion method.20 The final LPD NPs exhibited a uniform size of ∼100 nm and a low polydispersity index (PDI < 0.2) with neutral surface charge, as suggested by dynamic light scattering (DLS) analysis (Fig. 2A and Fig. S3, ESI†).
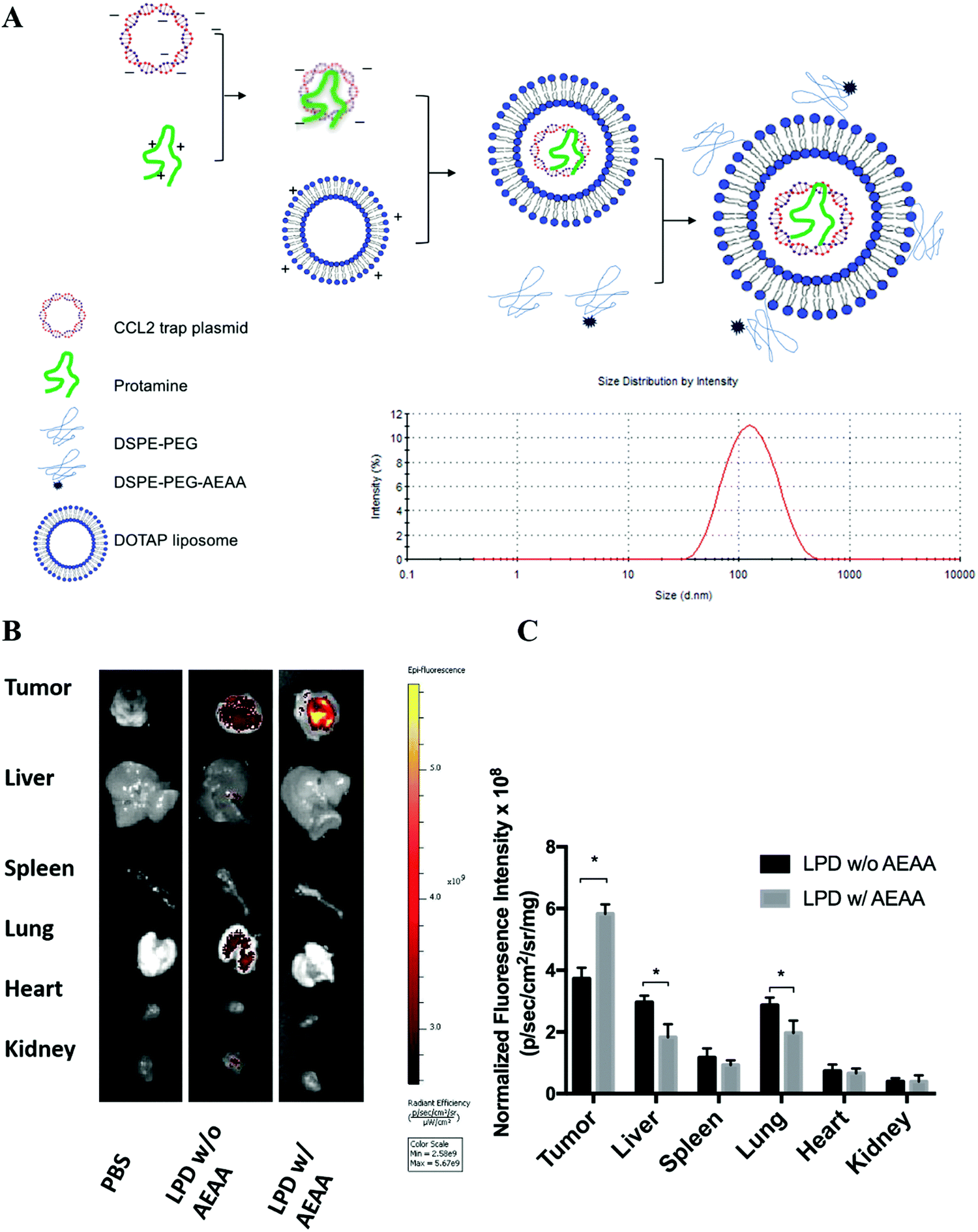 | ||
| Fig. 2 Preparation and characterization of in situ CCL2 blockade depot. (A) Preparation and characterization of pCCL2 LPD NPs. DOTAP, 1,2-dioleoyl-3-trimethylammonium propane chloride salt. AEAA, aminoethyl anisamide. (B) The in vivo biodistribution of LPD NPs with or without AEAA modification in orthotopic 4T1 tumor bearing mice. The hydrophobic dye DiI was incorporated into the outer bilayer of LPD NPs as a fluorescent tracer. Twenty-four hours post intravenous injection of DiI-labeled LPD NPs, mice were sacrificed and the tumors and major organs (liver, spleen, lung, heart, kidney) were harvested and examined under the IVIS imaging system (n = 3). (C) Quantitative analysis of the biodistribution of DiI-labeled LPD NPs in major organs and tumors. *p < 0.05. | ||
The final LPD NPs were labeled with a fluorescent lipophilic dye, DiI, to track its biodistribution in 4T1 orthotopic tumor bearing mice. Twenty-four hours post intravenous injection of DiI-labeled LPD NPs, 4T1 tumor bearing mice were sacrificed and major organs/tumors were harvested and subjected to In Vivo Imaging Systems (IVIS) imaging. LPD NPs modified with the target ligand AEAA predominantly accumulated within the breast tumor site, whereas non-modified LPD NPs showed a non-specific biodistribution in the liver and lungs (Fig. 2B). Based on the normalized fluorescence intensity analysis, a 1.5-fold increase of tumor specific uptake was achieved with AEAA modification, further verifying that successful tumor-targeted delivery was achieved using the AEAA modified LPD NP platform (Fig. 2C).
pCCL2 trap outperformed CCL2 mAb and remodeled the immunosuppressive TME in an orthotopic 4T1 mouse model
Inspired by the exciting target discovery and promising gene delivery platform, we wonder whether the pCCL2 trap would generate a local and transient expression of CCL2 trap protein, remodel the immunosuppressive TME and inhibit TNBC tumor progression as expected. Animal experiments were conducted to evaluate the therapeutic efficacy of the pCCL2 trap in the 4T1 mouse orthotopic model as follows. LPD NPs encapsulating a plasmid encoding the green fluorescence protein (pGFP) were used as the negative control. In addition, CCL2 mAb control was included in the animal study aiming to investigate if the pCCL2 trap could outperform CCL mAb. Mice bearing 4T1 tumors were randomly divided into four groups and treated with different modalities, as shown in Fig. 3A. Longitudinal tumor volume measurements were recorded every other day. In accordance with clinical studies, a partial tumor inhibition effect was observed in the CCL2 mAb treated group within the treatment schedule. Significantly, the CCL2 level in the tumor was the lowest in the group treated with the pCCL2 trap (Fig. 3B). Our gene therapy strategy to deliver the trap protein to the TME outperformed the traditional mAb approach (Fig. 3). It is not surprising that this was the case because the AEAA-targeted NPs specifically delivered the trap gene to the tumor (Fig. 2). In contrast, mAb distributes in the entire body. The trap protein (15.8 kDa), being much smaller than the IgG molecule (150 kDa), can readily diffuse in the TME and neutralize the target chemokine.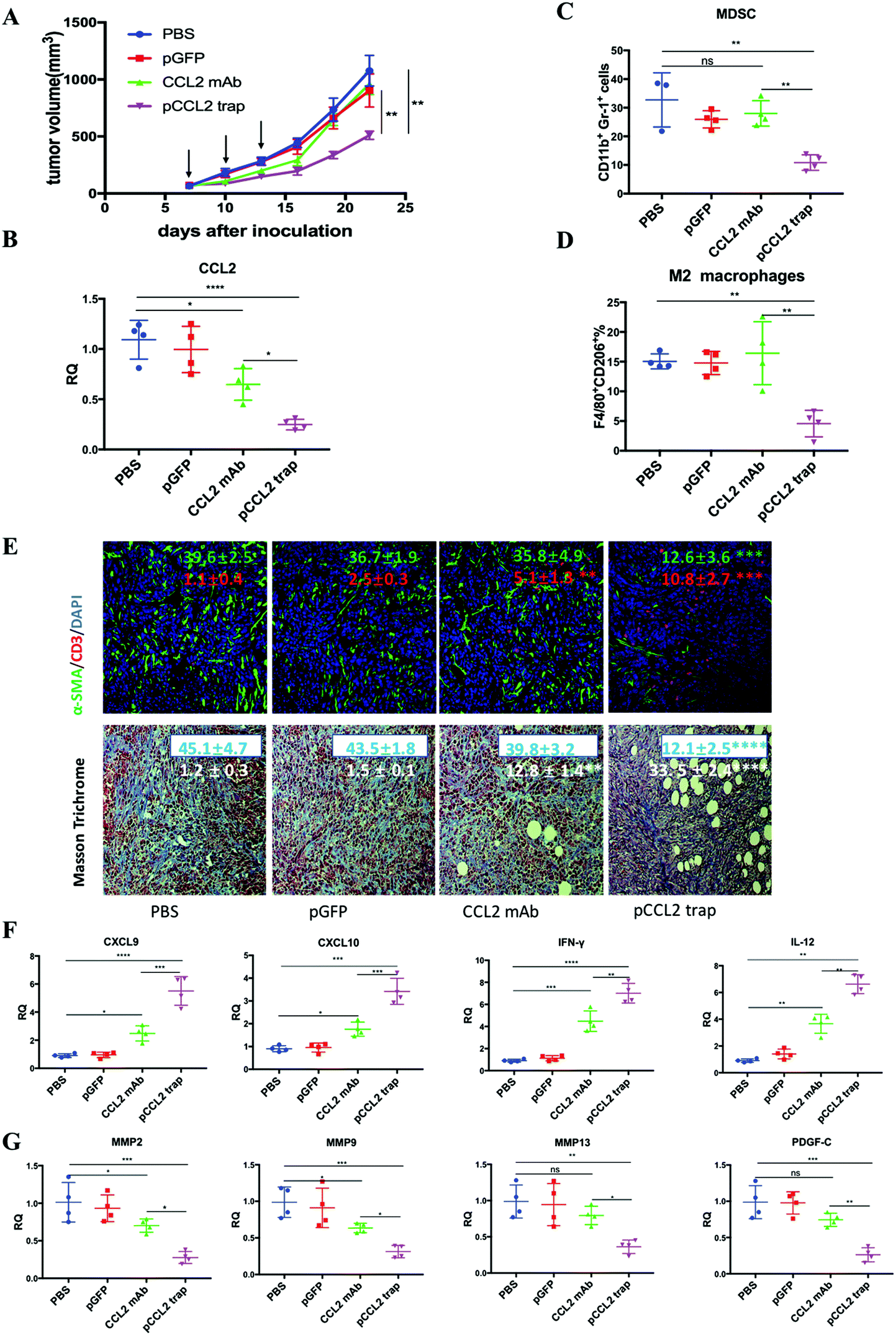 | ||
| Fig. 3 The pCCL2 trap outperformed CCL2 mAb and remodeled the immunosuppressive TME in the 4T1 mouse model. (A) Tumor inhibition effect of PBS, pGFP, pCCL2 trap and CCL2 mAb. 1 × 106 4T1 cells were orthotopically injected into the mammary fat pad of six-week old female BALB/c mice on day 0. Mice were randomly distributed into 4 groups on day 7 (n = 5). Tumor volumes were recorded every other day by caliper measurements. The black arrow indicates the dosing schedule (50 μg pDNA or 200 μg mAb on days 7, 10 and 13). **p < 0.01. (B) The CCL2 mRNA expression in the tumors of mice that received different treatments. Tumors were obtained at the end of the study (day 23, 10 days after the last treatment). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (C and D) The quantitative analysis of MDSC and M2 macrophage populations within the TME on day 23. Cells were measured by flow cytometry. MDSC, myeloid-derived suppressor cells. (E) Upper panel: immunofluorescence staining of tumor samples from different treatment groups using anti-α-SMA antibody (green) and anti-CD3-antibody (red). Cell nuclei were stained blue using DAPI. Lower panel: collagen contents quantified by Masson Trichrome staining. Five random fields were chosen for statistical analysis in each treatment group. Images were analyzed using ImageJ software and quantified using GraphPad 6.0. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The scale bar represents 100 μm. (F) Relative mRNA expression of Th1 chemokines and cytokines in the tumors of mice that received different treatments. Tumors were obtained at the end of the study (day 23, 10 days after the last treatment). CXCL9: C–X–C Motif Chemokine Ligand 9; CXCL10: C–X–C Motif Chemokine Ligand 10; IL-12: interleukin-12; IFN-γ: interferon gamma. *p < 0.05, **p < 0.01, ***p < 0.001. (G) Relative mRNA expression of Th2 chemokines and cytokines in the tumors of mice that received different treatments. Tumors were obtained at the end of the study (day 23, 10 days after the last treatment). MMP, metallopeptidase. PDGF-C, platelet-derived growth factor C. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
In the CCL2 mAb treatment group, the rapid discontinuation of CCL2 mAb resulted in a rebound effect and accelerated tumor progression (Fig. 3A). Further detailed flow cytometry analysis suggested that no significant decrease in the population of M2 macrophages and MDSCs was observed at the end of the study. In contrast, the group treated with the pCCL2 trap significantly inhibited tumor progression, with obviously down-regulated expression of M2 macrophages and MDSCs lasting for two weeks following cessation of pCCL2 trap treatment (Fig. 3C and D). Additionally, immunofluorescence staining revealed a remodeled TME in mice treated with the pCCL2 trap. Alpha-smooth muscle actin (α-SMA), which was highly expressed in TAAs and activated fibroblasts, was reduced 3-fold after pCCL2 trap treatment compared with the pGFP control group (p < 0.001) (Fig. 3E). The collagen content in different treatment groups was measured via Masson Trichrome staining, demonstrating a 4-fold decrease after pCCL2 trap treatment (p < 0.0001) (Fig. 3E). It is also worth mentioning that plenty of normalized adipocytes (cells containing large oil droplets) were observed in the pCCL2 trap treated group, suggesting the restoration of TAAs into normal adipocytes (Fig. 3E). Furthermore, upon CCL2 trap treatment, T cell (CD3+ cell) infiltration within the TME was increased 5-fold compared with the pGFP treated group (p < 0.001), which could be attributed to the remodeled immunosuppressive TME (Fig. 3E).
As an indispensable part of the TME, cytokines and chemokines are important mediators and regulators for immune cell migration, infiltration and action. Highly elevated T cell recruiting cytokines including C–X–C Motif Chemokine Ligand 9 (CXCL9) and C–X–C Motif Chemokine Ligand 10 (CXCL10) are crucial for T cell infiltration. Furthermore, the activation of tumor infiltrating T cells also depends on pro-inflammatory Th1 cytokines, interleukin-12 (IL-12) and interferon gamma (IFN-γ). In accordance with previous T cell profiling, CXCL9, CXCL10, IL-12 and IFN-γ were highly upregulated in the pCCL2 trap treated group (p < 0.01–0.001) (Fig. 3F). Conversely, common matrix metallopeptidases (MMPs) that are involved in extracellular matrix (ECM) remodeling, such as MMP2, MMP9 and MMP13, were significantly downregulated compared with the PBS control group, as was the pro-fibrosis cytokine platelet-derived growth factor (PDGF-C) (p < 0.01–0.001) (Fig. 3G).
pCCL2 trap facilitated the checkpoint blockade therapy
RNA sequencing data derived from the Cancer Genome Atlas (TCGA) database showed up-regulated expression of programmed cell death protein ligand 1 (PD-L1) in TNBC samples in comparison with non-TNBC samples.21 PD-L1 is a checkpoint protein on tumor cells that negatively regulates the immune response by inactivating T cells.22 Monoclonal antibodies that specifically block PD-L1 are referred to as checkpoint blockade therapy. The therapy effectively blocks the inhibition signal for T cell activation and initiates an effective T cell-based anti-tumor response.22 However, TNBC patients do not respond well to checkpoint blockade therapy owing to the immunosuppressive TME.23 Since the data above indicated that the pCCL2 trap successfully remodeled the immunosuppressive TME, we wondered whether pCCL2 trap treatment would enhance the efficacy of checkpoint blockade therapy in our TNBC model.We have previously developed a trimeric protein trap that potently and specifically blocks PD-L1 with an apparent Kd ∼219 pM.24 The plasmid pPD-L1 trap was formulated in LPD NPs and delivered to the orthotopic TNBC tumor in a manner similar to the pCCL2 trap. Our prior studies have shown that the pPD-L1 trap was efficiently expressed in a local and transient manner in the TME of the 4T1 model with better tolerance and lower immune-related adverse effects.25 We next conducted a combination study to investigate the possible synergistic effect of the pCCL2 trap and pPD-L1 trap in the orthotopic 4T1 model. Despite the fact the pCCL2 or pPD-L1 trap by itself only showed a partial tumor inhibitory effect, tumor progression was significantly inhibited in the combined treatment group (Fig. 4A and B). Further terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining studies justified the synergistic effect of the combined treatment group (Fig. 4C).
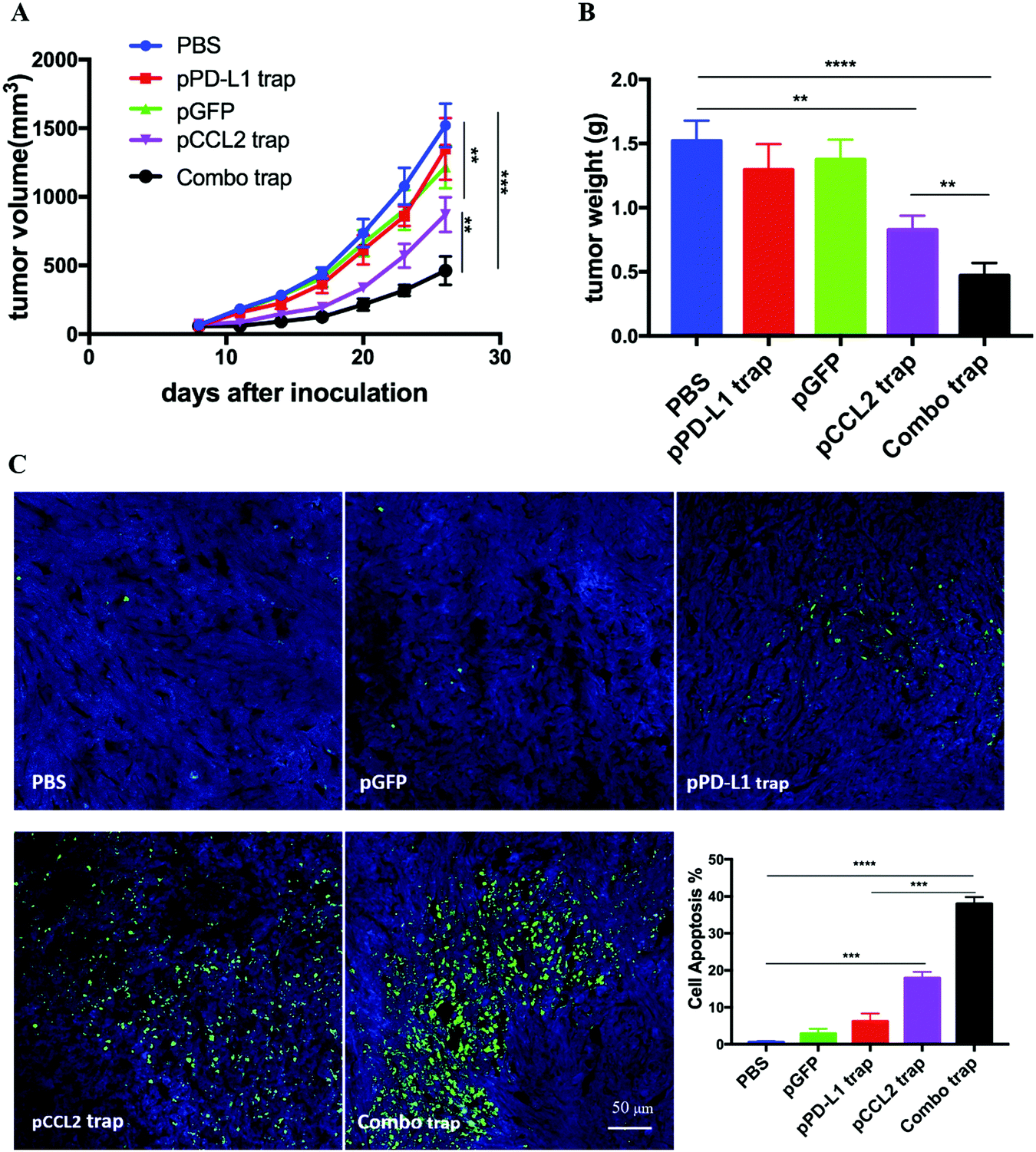 | ||
| Fig. 4 The pCCL2 trap facilitates the checkpoint blockade-based therapy. (A) Tumor inhibition study of various treatments. Combo trap, a combination of the pCCL2 trap and pPD-L1 trap (25 μg pCCL2 combined with 25 μg pPD-L1 on days 7, 10 and 13). *p < 0.05, **p < 0.01, ***p < 0.001. (B) Quantitative analysis of tumor weight of various treatment groups on day 26. *p < 0.05, **p < 0.01, ***p < 0.00, ****p < 0.0001. (C) TUNEL assay and the corresponding quantitative analysis of tumor tissues from various treatment groups. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling. Tumors were harvested on day 26. **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
Safety evaluation of different treatment groups
Major organs, including the heart, liver, spleen, lung and kidney, and blood samples were obtained from different treatment groups at the end of the experiment as shown in Fig. 4. In further safety and toxicity evaluations, we found that the pCCL2/pPD-L1 trap alone and the combo trap treated groups did not exhibit significant morphological damage in any of the major organs (Fig. 5A). In addition, the blood serum and whole blood test suggested that there was no significant difference in the levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and creatinine (CREAT), suggesting the absence of liver- or kidney-associated toxicities (Fig. 5C and Table S1, ESI†). Furthermore, no significant weight loss was observed in different treatment groups (Fig. 5B). Taken together, our therapeutic strategy provided a tumor-targeted, local and transient gene therapy to remodel the immunosuppressive tumor microenvironment in a safe and feasible manner.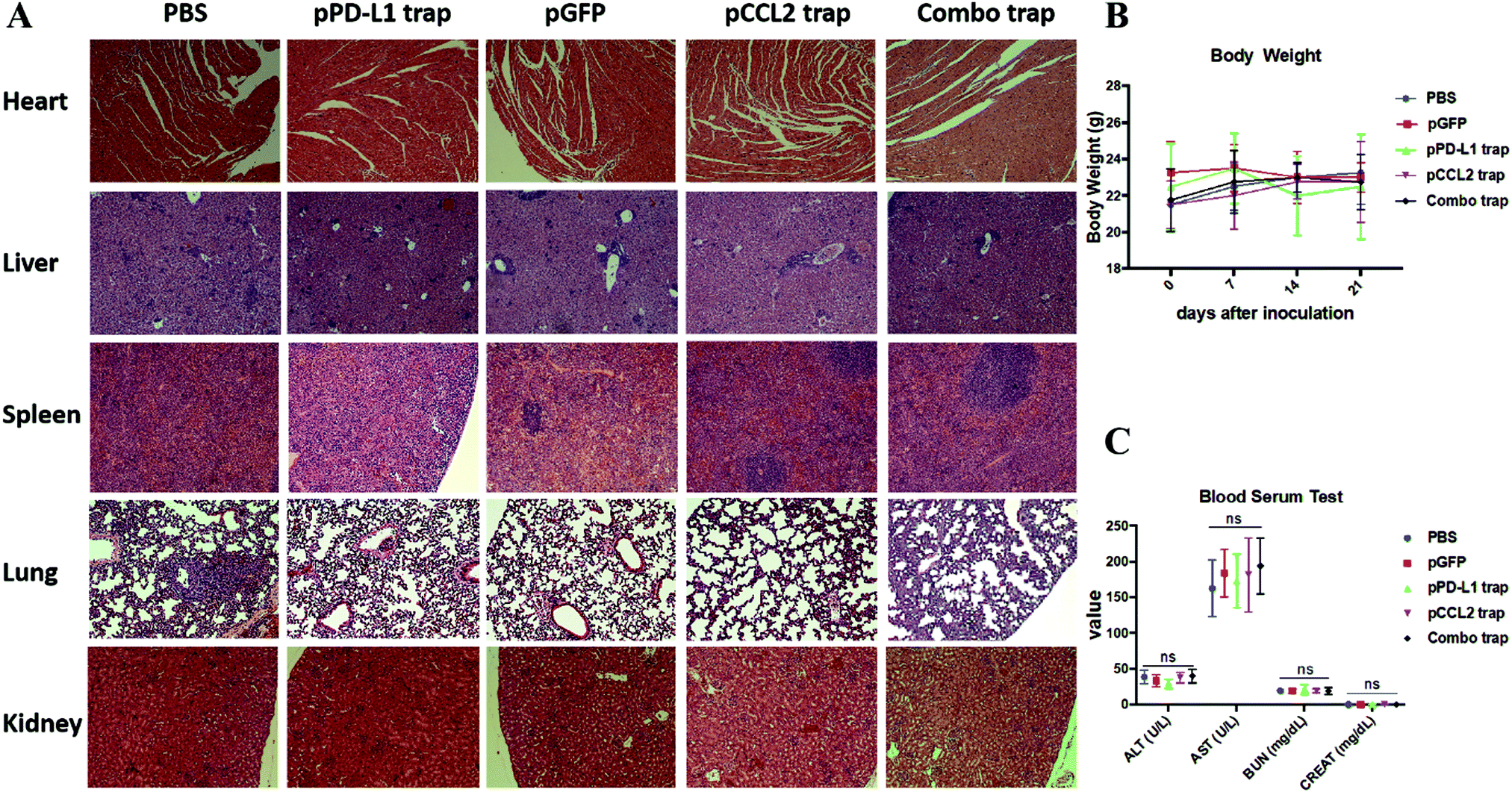 | ||
| Fig. 5 Safety evaluation of different treatment groups. (A) H&E staining of major organs from different treatment groups. At the end of the study, major organs were collected, sectioned and stained with H&E solution and observed under a microscope. (B) Body weights of mice from different treatment groups. (C) Blood serum test of ALT, AST, BUN and CREAT among different treatment groups. ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CREAT, creatinine. ns, not significant. | ||
Conclusion
We have developed a small trap protein with a relatively high binding affinity to mouse CCL2 in a specific manner. This single domain antibody was delivered to the TNBC tumor by using a gene therapy approach, resulting in a persistent decrease of the CCL2 level in the tumor. Local delivery of the trap protein triggered significant remodeling of the immune microenvironment and inhibited tumor growth. Since CCL2 is mainly produced by TAAs in TNBC, our study demonstrated the usefulness of targeting TAAs to improve immunotherapy. In addition, the NPs used in this study are rather simple to prepare, potentially in a large scale, and non-toxic,20 and the approach appears to be translatable into clinics.Materials and methods
Materials
1,2-Dioleoyl-3-trimethylammonium propane chloride salt (DOTAP), 1,2-distearoyl-sn-glycero-3-phosphoethaolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG-2000), 3-(N-succinimidyloxyglutaryl)aminopropyl, and polyethylene glycol–carbamyl distearoylphosphatidyl–ethanolamine (DSPE–PEG–NHS) were purchased from Avanti Polar Lipids (Alabaster, AL). DSPE–PEG–aminoethyl anisamide (DSPE–PEG–AEAA) was synthesized following a previously reported protocol.26Development of the CCL2-binding trap and expression of the recombinant CCL2 trap
The CCL2-binding trap was developed by screening a human single domain antibody library using a yeast surface display against biotinylated mouse CCL2 protein. Yeast cells with the highest binding and expression ratio were selected. The selection was performed until a homogenous population was obtained and no further enrichment was possible. The expression vector encoding the selected CCL2 trap was generated by inserting the synthesized DNAs (codon-optimized for expression in mammalian cells) into the pcDNA3.1 vector. For protein expression, 25 micrograms of plasmid DNA was transfected into 25 mL of ExpiCHO-S cells (1.5 × 108 cells). Ten days post transfection, the supernatant was collected and the protein was purified using an Ni-NTA column. The protein was eluted with 500 mM imidazole in 1× PBS buffer. The eluted protein was dialyzed against PBS buffer and assessed using SDS-PAGE.Affinity measurement and specificity analysis
The binding affinity between the recombinant CCL2 trap and mouse CCL2 was determined using Biacore X100 (Cytiva). Biotinylated mouse CCL2 trap was immobilized on a Biacore Biotin CAPture chip (Cytiva) at 100 nM. Mouse CCL2 analyte (from Shenandoah Biotechnology) was injected into the sample channel at different concentrations in a single-cycle kinetics mode. The data were processed using Biacore analytical software.Synthesis of DSPE–PEG and DSPE–PEG–AEAA
DSPE–PEG–AEAA was synthesized following a previously reported method.26 Briefly, 4-methylbenzoyl chloride and 2-bromoethylamine hydrobromide were mixed for 6 h at room temperature. Then the DSPE–PEG–NH2 was added into the mixture and stirred at 65–70 °C overnight in an oil bath. The obtained product was further precipitated and purified with diethyl ether, lyophilized and dissolved in DI water.Preparation and characterization of LPD NPs
LPD NPs were synthesized via a sequential self-assembly process. Briefly, blank liposomes were first prepared by a thin film dispersion method. DOTAP and cholesterol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) were first dissolved in chloroform and a rotary evaporator was employed to remove the organic solvent. The thin film was further hydrated with distilled water to prepare the final DOTAP/Chol blank liposomes with a concentration of 10 mmol L−1. Unilamellar DOTAP/Chol liposomes were obtained after successive extrusion of the resuspended lipid mixture. 100 μL 0.5 mg mL−1 pDNA and 100 μL 0.2 mg mL−1 protamine were mixed to formulate the LPD polyplex cores. Then, 60 μL of blank DOTAP/Chol liposomes were mixed with 200 μL LPD polyplex cores. 10 μL DSPE–PEG and 10 μL DSPE–PEG–AEAA at a concentration of 10 mg mL−1 were added using a post-insertion method to formulate the final LPD NPs. Glucose solution was added into the system to adjust the osmotic pressure. The size distribution and ζ potential of LPD NPs were determined using Malvern ZetaSizer Nano Series (Westborough, MA).
:1 molar ratio) were first dissolved in chloroform and a rotary evaporator was employed to remove the organic solvent. The thin film was further hydrated with distilled water to prepare the final DOTAP/Chol blank liposomes with a concentration of 10 mmol L−1. Unilamellar DOTAP/Chol liposomes were obtained after successive extrusion of the resuspended lipid mixture. 100 μL 0.5 mg mL−1 pDNA and 100 μL 0.2 mg mL−1 protamine were mixed to formulate the LPD polyplex cores. Then, 60 μL of blank DOTAP/Chol liposomes were mixed with 200 μL LPD polyplex cores. 10 μL DSPE–PEG and 10 μL DSPE–PEG–AEAA at a concentration of 10 mg mL−1 were added using a post-insertion method to formulate the final LPD NPs. Glucose solution was added into the system to adjust the osmotic pressure. The size distribution and ζ potential of LPD NPs were determined using Malvern ZetaSizer Nano Series (Westborough, MA).
Biodistribution of LPD NPs
LPD NPs were labeled with DiI by incorporation of 0.1% lipophilic dye Dil into the blank DOTAP:cholesterol liposomes. Twenty-four hours post intravenous injection of DiI-labeled LPD NPs, the mice were sacrificed and the biodistribution of DiI-labeled liposomes was visualized using the IVIS Kinetics Optical System (PerkinElmer, CA). An excitation wavelength of 520 nm and an emission wavelength of 560 nm were used for IVIS imaging.Cell lines
Murine triple negative breast cancer cells 4T1 were obtained from UNC Tissue Culture Facility. Following ATCC instructions, 4T1 cells were cultured with RPMI 1640 medium supplemented with 10% fetal bovine serum (V/V, Gibco) and 1% antibiotic–antimycotic (V/V, Gibco) in a humified atmosphere at 37 °C under 5% CO2 conditions.Orthotopic animal mouse cancer model
Six-week old female BALB/c mice were purchased from Jackson Laboratories and maintained under germ-free conditions. The orthotopic 4T1 model was established by an orthotopic injection of 1 × 106 4T1 cells in 50 μL PBS into the inguinal mammary fat pad of female BALB/c mice. The subcutaneous 4T1 model was established by a subcutaneous injection of 1 × 106 4T1 cells in 100 μL PBS into the right flank of female BALB/c mice. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. All animal procedures were conducted in compliance with federal regulations (NIH/PHS Animal Welfare Assurance No. D16-00256; USDA Animal Research Facility Registration No. 55-R-0004; AAALAC Institutional No. #329) and ethical regulations approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.Tumor growth inhibition study
The 4T1 tumor bearing mice were randomly divided into different treatment groups when the tumor volume reached 100 mm3. Mice were treated with different modalities according to the treatment schedule (50 μg pDNA or 200 μg mAb on days 7, 10 and 13). Tumor progression was recorded by caliper measurements, and the tumor volume was calculated as V = 0.5 × length × width × width, where the length represents the long axis and the width represents the short axis.Flow cytometry assay
Fresh tissues were obtained and prepared as single cell suspensions. Tumor infiltrating lymphocytes were characterized and quantified using the flow cytometry assay. In brief, fresh isolated tumors were first incubated with collagenase and DNase for 1 h at 37 °C to obtain single cell suspensions. Then the prepared single cell suspensions were stained with a cocktail of fluorescently labeled antibodies for surface marker expression analysis. After 0.5 h staining, the cells were fixed with 4% PFA solution and analyzed using FACS (BD LSR II). The analysis was conducted using FlowJo software (TreeStar, Ashland, OR). The fluorescently labeled antibodies used for flow cytometry are listed in Table S2 (ESI†).Immunofluorescence staining
The mice were sacrificed and fresh tumor tissues were first resected, rinsed in PBS and fixed with 4% PFA solution at 4 °C for 48 h. The tissues were then dehydrated with 15% and 30% sucrose solution. The dehydrated tissues were embedded into O.C.T solution for the preparation of the frozen section. The obtained cryostat sections were permeabilized and blocked with 5% goat serum for 1 h at room temperature. Then the sections were incubated with fluorescently conjugated antibodies for 12 h at 4 °C and mounted with Prolong Diamond Antifade Mountant with DAPI. IF images were taken on a confocal microscope (Zeiss LSM 710) and quantified using ImageJ. Five randomly chosen fields were used for statistical analysis. The antibodies used for immunofluorescence staining are listed in Table S2 (ESI†).TUNEL assay
A TUNEL assay was conducted using a DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) following the manufacturer's protocol. Tumor frozen section slides were prepared and the fragmented DNA of apoptotic cells was stained with a fluorescent dye. Cell nuclei that were stained fluorescent green were defined as TUNEL-positive nuclei. Slides were further mounted with Prolonged Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific) and examined under a confocal microscope (Zeiss, LSM 710). Five microscopic fields were randomly selected for quantification and statistical analysis using ImageJ.Quantitative real-time PCR assay
The RNeasy Microarray Tissue Mini Kit (Qiagen) was used to extract the total RNA from fresh tissues. cDNA was obtained by reverse transcription using the iScript cDNA Synthesis Kit (BIO-RAD) and amplified with TaqMan Gene Expression Master Mix. Mouse specific primers were used and GAPDH was used as the endogenous control. Reactions and the corresponding data analysis were completed using a 7500 Real-Time PCR system. The primers used in the Real-Time PCR assay are listed in Table S3 (ESI†).Safety evaluation
The body weights of mice were measured every other day following the first dose treatment. At the end of the experiment, the mice were sacrificed, and the whole blood, serum and major organs were collected for safety evaluation. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN) and creatinine (CRE) levels in the serum were analyzed as biomarkers for renal and hepatic function. Red blood cells (RBC), white blood cells (WBC), platelets (PLT), hemoglobin (HGB), neutrophils (NEUT), lymphocytes (LYMPH) and monocytes (MONO) in whole blood were measured to quantify myelosuppression.To evaluate the toxicity in major organs, the heart, liver, spleen, lung and kidney, from different treatment groups were harvested, fixed, stained with hematoxylin and eosin (H&E) and examined under a microscope.
Statistical analysis
Results are presented as mean ± SD unless otherwise specified. GraphPad 6.0 was used for statistical analysis. For comparison between two groups, Student's t test was used. For comparison among three or more groups, one-way analysis of variance (ANOVA) was used. The Kaplan–Meier estimator was used to analyse the survival studies. A p value less than 0.05 was considered to be significant. *, **, *** and **** denote p < 0.05, 0.01, 0.001, and 0.0001, respectively.Contributions
Yun Liu and Leaf Huang conceived and designed the research. Yun Liu, Menglin Wang and Xuefei Zhou performed the in vivo mouse experiments. Yun Liu and Nasha Qiu prepared the frozen sections and performed immunofluorescence staining. Yun Liu and Menglin Wang analyzed and quantified the microscopic images. Yun Liu and Yang Xiong performed the flow cytometry assay and analyzed the results. Yun Liu and Nasha Qiu analyzed the clinical data. Karthik Tiruthani and Rihe Liu designed, developed, and characterized the CCL2 trap and PD-L1 trap. Yun Liu performed the statistical analysis. Chad V. Pecot read and edited the manuscript. Yun Liu, Rihe Liu and Leaf Huang analyzed the data and wrote the manuscript.Conflicts of interest
LH is a consultant for PDS Biotechnology, Samyang Biopharmaceuticals, Stemirna and Beijing Inno Medicine. RH is a co-founder of OncoTrap. All other co-authors declare no conflicts.Acknowledgements
This work was supported by the NIH grant CA198999 (to LH) and the Eshelman Institute for Innovation grant RX03812125 (to RL).References
- Y. Liu, N. Qiu, L. Shen, Q. Liu, J. Zhang, Y. Y. Cheng, K. H. Lee and L. Huang, J. Controlled Release, 2020, 323, 431–441 CrossRef CAS.
- H. A. Wahba and H. A. El-Hadaad, Cancer Biol. Med., 2015, 12, 106–116 CAS.
- Y. Liu, J. Guo and L. Huang, Theranostics, 2020, 10, 3099–3117 CrossRef CAS.
- Q. Wu, B. Li, Z. Li, J. Li, S. Sun and S. Sun, J. Hematol. Oncol., 2019, 12, 95 CrossRef.
- P. Cohen and B. M. Spiegelman, Mol. Biol. Cell, 2016, 27, 2523–2527 CrossRef CAS.
- Z. Cai, Y. Liang, C. Xing, H. Wang, P. Hu, J. Li, H. Huang, W. Wang and C. Jiang, Oncol. Rep., 2019, 42, 2537–2549 CAS.
- X. Zhu, M. Fujita, L. A. Snyder and H. Okada, J. Neurooncol., 2011, 104, 83–92 CrossRef CAS.
- K. Pogoda, M. Pyszniak, P. Rybojad and J. Tabarkiewicz, Oncol. Lett., 2016, 12, 4785–4794 CrossRef CAS.
- E. Sierra-Filardi, C. Nieto, A. Dominguez-Soto, R. Barroso, P. Sanchez-Mateos, A. Puig-Kroger, M. Lopez-Bravo, J. Joven, C. Ardavin, J. L. Rodriguez-Fernandez, C. Sanchez-Torres, M. Mellado and A. L. Corbi, J. Immunol., 2014, 192, 3858–3867 CrossRef CAS.
- F. O. Martinez and S. Gordon, F1000Prime Rep., 2014, 6, 13 Search PubMed.
- M. Takeya and Y. Komohara, Pathol. Int., 2016, 66, 491–505 CrossRef.
- W. Chen, S. S. Foo, A. Taylor, A. Lulla, A. Merits, L. Hueston, M. R. Forwood, N. C. Walsh, N. A. Sims, L. J. Herrero and S. Mahalingam, J. Virol., 2015, 89, 581–593 CrossRef.
- L. Bonapace, M. M. Coissieux, J. Wyckoff, K. D. Mertz, Z. Varga, T. Junt and M. Bentires-Alj, Nature, 2014, 515, 130–133 CrossRef CAS.
- T. Kitamura, B. Z. Qian, D. Soong, L. Cassetta, R. Noy, G. Sugano, Y. Kato, J. Li and J. W. Pollard, J. Exp. Med., 2015, 212, 1043–1059 CrossRef CAS.
- Q. Liu, M. Das, Y. Liu and L. Huang, Adv. Drug Delivery Rev., 2018, 127, 208–221 CrossRef CAS.
- L. Shen, J. Li, Q. Liu, W. Song, X. Zhang, K. Tiruthani, H. Hu, M. Das, T. J. Goodwin, R. Liu and L. Huang, ACS Nano, 2018, 12, 9830–9841 CrossRef CAS.
- L. Miao, Q. Liu, C. M. Lin, C. Luo, Y. Wang, L. Liu, W. Yin, S. Hu, W. Y. Kim and L. Huang, Cancer Res., 2017, 77, 719–731 CrossRef CAS.
- R. Lugano, M. Ramachandran and A. Dimberg, Cell. Mol. Life Sci., 2020, 77, 1745–1770 CrossRef CAS.
- G. M. Cherf and J. R. Cochran, Methods Mol. Biol., 2015, 1319, 155–175 CrossRef.
- S. Zhang, D. Zhi and L. Huang, J. Drug Targeting, 2012, 20, 724–735 CrossRef CAS.
- E. A. Mittendorf, A. V. Philips, F. Meric-Bernstam, N. Qiao, Y. Wu, S. Harrington, X. Su, Y. Wang, A. M. Gonzalez-Angulo, A. Akcakanat, A. Chawla, M. Curran, P. Hwu, P. Sharma, J. K. Litton, J. J. Molldrem and G. Alatrash, Cancer Immunol. Res., 2014, 2, 361–370 CrossRef CAS.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS.
- M. J. Kwa and S. Adams, Cancer, 2018, 124, 2086–2103 CrossRef.
- L. Miao, J. Li, Q. Liu, R. Feng, M. Das, C. M. Lin, T. J. Goodwin, O. Dorosheva, R. Liu and L. Huang, ACS Nano, 2017, 11, 8690–8706 CrossRef CAS.
- W. Song, L. Shen, Y. Wang, Q. Liu, T. J. Goodwin, J. Li, O. Dorosheva, T. Liu, R. Liu and L. Huang, Nat. Commun., 2018, 9, 2237 CrossRef.
- R. Banerjee, P. Tyagi, S. Li and L. Huang, Int. J. Cancer, 2004, 112, 693–700 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00588f |
| This journal is © The Royal Society of Chemistry 2021 |