
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational design and experimental characterization of a photo-controlled mRNA-cap guanine-N7 methyltransferase†
Dennis
Reichert
ab,
Helena
Schepers
a,
Julian
Simke
c,
Horst
Lechner
de,
Wolfgang
Dörner
a,
Birte
Höcker
 d,
Bart Jan
Ravoo
bc and
Andrea
Rentmeister
*ab
d,
Bart Jan
Ravoo
bc and
Andrea
Rentmeister
*ab
aInstitute of Biochemistry, University of Münster, Correnstraße 36, Münster 48149, Germany. E-mail: a.rentmeister@uni-muenster.de
bCells in Motion Interfaculty Center, University of Münster, Münster 48149, Germany
cOrganic Chemistry Institute, University of Münster, Corrensstraße 36, Münster 48149, Germany
dDepartment for Biochemistry, University of Bayreuth, Universitätsstraße 30, Bayreuth 95447, Germany
eInstitute of Biochemistry, Graz University of Technology, Petersgasse 10-12/II, Graz 8010, Austria
First published on 29th June 2021
Abstract
The spatial and temporal control of gene expression at the post-transcriptional level is essential in eukaryotic cells and developing multicellular organisms. In recent years optochemical and optogenetic tools have enabled the manipulation and investigation of many steps in the involved processes. However, examples for light-mediated control of eukaryotic mRNA processing and the responsible enzymes are still rare. In particular, methylation of the 5′ cap of mRNA is required for ribosome assembly, and the responsible guanine-N7 methyltransferase (MTase) from E. cuniculi (Ecm1) proved suitable for activating translation. Here, we report on a photoswitchable MTase obtained by bridging the substrate-binding cleft of Ecm1 with a tetra-ortho-methoxy-azobenzene. This azobenzene derivative is characterized by efficient trans-to-cis isomerization using red light at 615 nm. Starting from a cysteine-free Ecm1 variant (ΔCys), we used a computational approach to identify suitable conjugation sites for the azobenzene moiety. We created and characterized the four best-ranked variants, each featuring two appropriately positioned cysteines close to the substrate-binding cleft. Conjugating and crosslinking the azobenzene between C149/C155 in a designed Ecm1 variant (VAR3-Az) enabled light-dependent modulation of the MTase activity and showed a 50% higher activity for the cis form than the trans-form of the azobenzene conjugated to VAR3-Az.
Introduction
In higher eukaryotes, mRNA is post-transcriptionally processed, including 5′ capping, splicing, and polyadenylation. Capping comprises the consecutive action of three enzymes–a triphosphatase, a guanylyltransferase, and a guanine-N7 methyltransferase (MTase).1 The methylation of the 5′ cap is essential for its recognition by cap-binding proteins.2 This becomes clear by the fact that the eukaryotic translation initiation factor (eIF4E) has a 100-fold higher affinity to the methylated 5′ cap compared to the unmethylated.3,4 Consequently, methylation at the guanine-N7 position is essential for translation initiation by cap-dependent ribosome recruitment.5,6 To understand these processes at the molecular level, tools are needed to dissect individual functions and activities, ideally with spatial and temporal precision. Therefore, optochemical and optogenetic tools emerged for manipulating and investigating many steps involved in gene expression. These include several ones acting on the post-transcriptional level, such as photosensitive proteins coupled to a modified version of the CRISPR–Cas9 system,7 RNA-binding proteins,8 or regulatory RNA molecules.9We and others reported that 5′ cap methylation is critical for translating eukaryotic mRNA using functionalized cap analogs for in vitro translation studies.10–14 Furthermore, we could show that via site-specific incorporation of a photo-caged tyrosine in the active site of a guanine-N7 MTase from E. cuniculi (Ecm1), the final step of 5′ cap formation, provides a way to bring translation under the control of light.15 Nevertheless, this approach requires amber stop codon suppression and high amounts of the photocaged amino acid, which has to be added to the E. coli culture. Also, once the photo-protecting group has been cleaved, there is no way to regulate enzymatic activity. Therefore, we were interested in a clean and site-specific post-synthetic protein conjugation approach to bring the MTase (or an active variant) directly and reversibly under control of light.
The emerging field of photopharmacology has repeatedly shown that azobenzene-derivatives can be conjugated to proteins and used to manipulate their activity with light.16,17 These photoswitches hold tremendous potential because they are not limited to single photoactivation but enable a reversible and gradual modulation of protein structure and activity. They either can be covalently attached or be tightly bound to their target through noncovalent interactions. This strategy was successfully applied to modulate the conformation and the activity of small peptides,18–20 to ion channels,21 NMDA receptors,22 or structural proteins like cadherin.23 However, this strategy faces particular challenges when applied to bring enzymes under the control of light. Although successful examples of light-regulated enzymes were achieved using azobenzene-based amino acids,24 photoswitchable inhibitors,25 or allosteric regulation,26 modulating enzymatic activity by light resulting from direct crosslinking with a photoswitch remains challenging and rare.
Pingoud and coworkers published a first study using an azobenzene derivative to modulate the activity of a DNA restriction enzyme.27 Here, the double cross-linked variants showed a considerable difference in catalytic activity between the cis and trans configurations of the photoswitch compared to single modified variants. In another approach, a fully reversible change of lipase activity was reported by crosslinking residues crucial for controlling the α-helix structure of the lipase lid domain with an azobenzene photoswitch.28 Also, Khare and coworkers used a structure-based computational approach to obtain reversible, light-mediated control of cytosine deaminase activity by covalently attaching azobenzene.29 From these studies, it can be concluded that (1) the most substantial impact on catalytic activity can be achieved when the photoswitch crosslinks residues in structured regions close to the active site and (2) computational approaches can significantly facilitate the identification of suitable crosslinking sites.
Ecm1 is the smallest known guanine-N7 methyltransferase and features an open binding cleft rather than a substrate-binding pocket.30 It achieves catalytic activity by optimally orienting the reaction partners, S-adenosyl-L-methionine (SAM or AdoMet), and the 5′ cap of mRNA (Fig. 1A and B).31 Considering the specific architecture of Ecm1, we anticipated that post-synthetic conjugation of azobenzenes could be a viable strategy to change the enzymatic activity from the on- to the off-state and vice versa. According to the crystal structure, the two rims of the substrate-binding cleft should be at a distance suitable for crosslinking two adjacent cysteine residues with an azobenzene moiety. We hypothesized that such double conjugation close to the substrate-binding cleft would make the enzyme susceptible to the light-induced conformational changes of azobenzene, allowing to actuate the enzymatic activity by light (Fig. 1C and D). The azobenzene attached at specific positions in the extended trans conformation would be expected to distort the substrate-binding cleft and disfavor optimal substrate binding, thus rendering Ecm1 inactive (Fig. 1D). Light-induced isomerization by irradiation at 615 nm from trans to the more compact cis-state would “switch on” the enzyme by allowing it to return to the native conformation of the binding pocket. Starting from the thermodynamically less stable cis-state of the thiol-conjugated azobenzene, it should be possible to “switch off” Ecm1 activity by irradiation at 450 nm or by thermal relaxation over time.
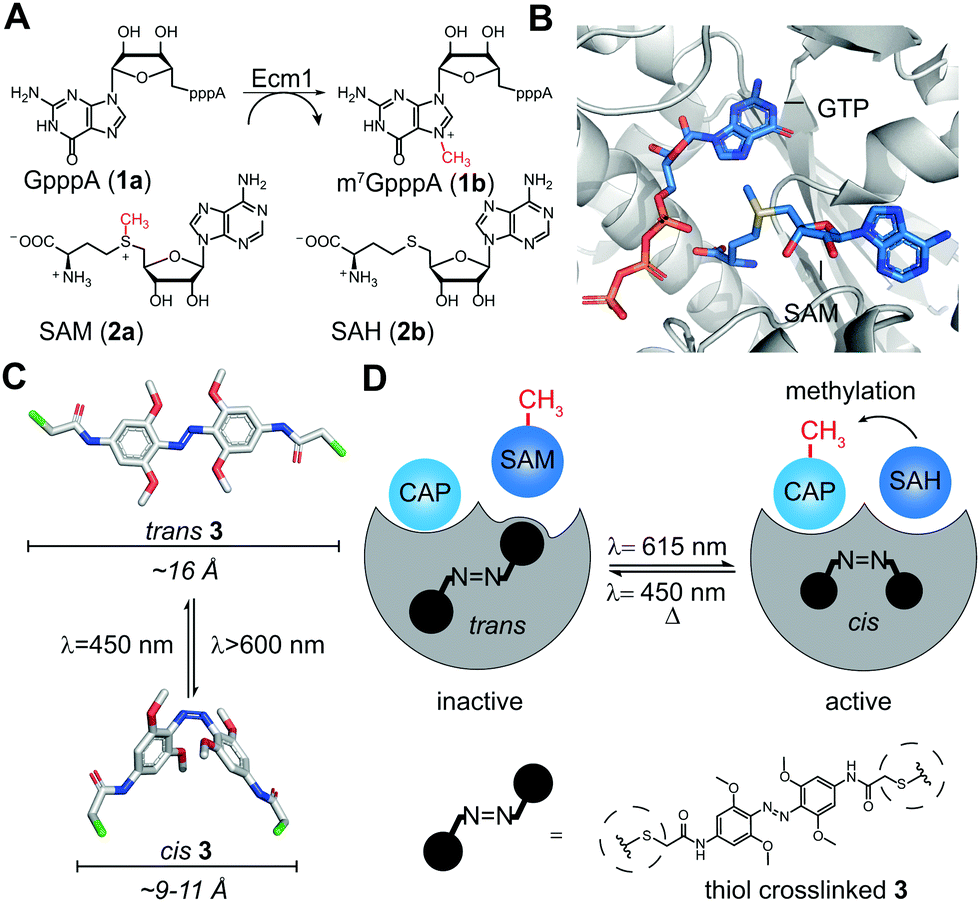 | ||
| Fig. 1 (A) Reaction catalyzed by Ecm1. The 5′ cap analog GpppA (1a) is the minimal substrate, SAM (2a) is the cosubstrate. (B) Overlay of Ecm1 crystal structure in complex with GTP (PDB: 1RI2) and SAM (PDB: 1RI4). (C) Model of trans and cis states of tetra-ortho-methoxyazobenzene-chloroacetamide (3). Size bars indicate the distance between substituents in para position. (D) Schematic illustration of how photo-isomerization of conjugated 3 can influence the methyltransferase activity of Ecm1. | ||
Results
Substituted azobenzene derivatives are well-known to efficiently isomerize using red light.32 The required wavelengths are suitable for biological applications, avoiding harmful effects of UV light and enabling deeper tissue penetration. Here, we used a tetra-ortho-methoxy substituted azobenzene 3 developed by Woolley and coworkers to apply azo compounds in vivo.33,34 The bathochromic shift of the n–π* band allows the selective excitation and conformational change from trans to cis with red light (≥600 nm) and cis to trans with blue light (∼450 nm). An additional advantage of 3 compared to unsubstituted azobenzenes is the prolonged half-life of the cis-conformer in aqueous solution (up to 24 h),34 which leads to a more persistent effect after photoswitching, obviating the need for continuous irradiation during conjugation. To explore the suitability of tetra-ortho-methoxy substituted azobenzene for our application, we synthesized compound 3 and determined its photoswitching properties (Fig. 2).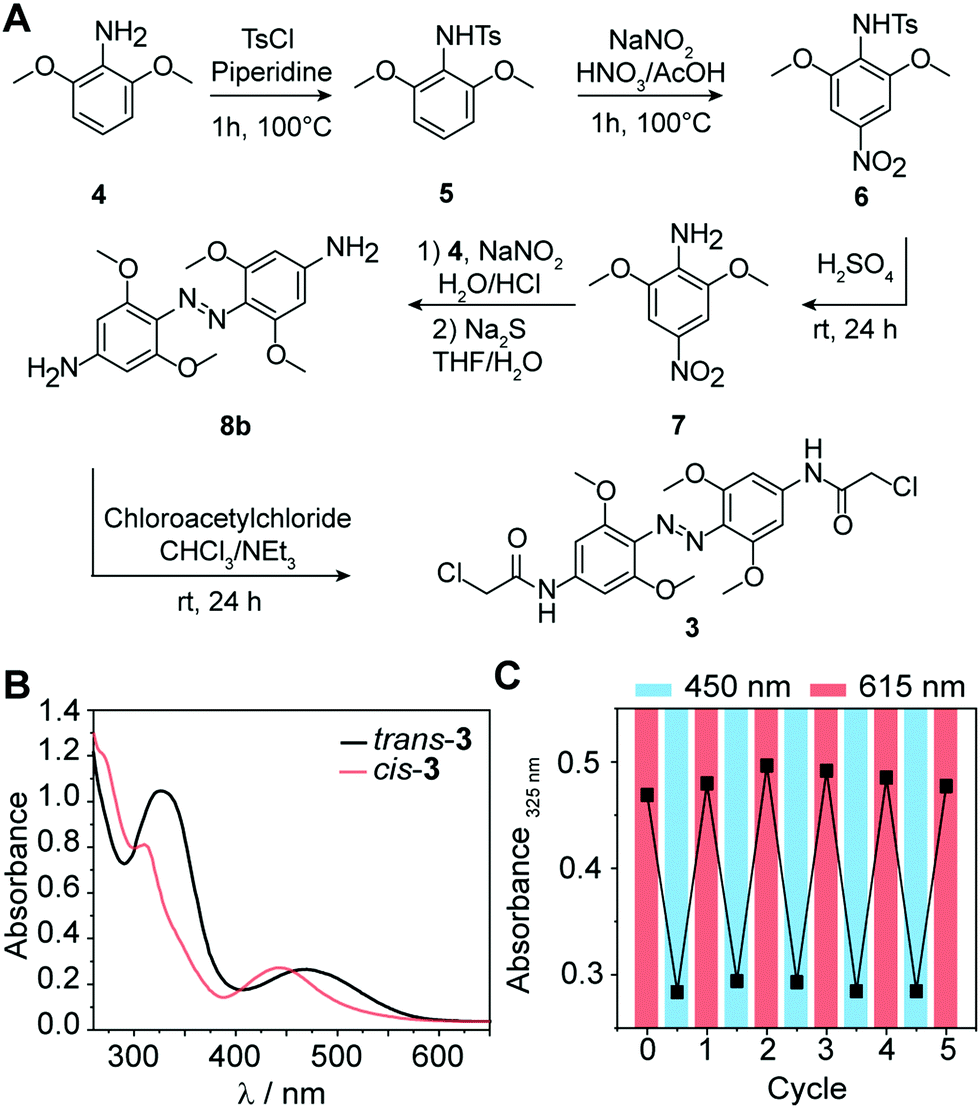 | ||
| Fig. 2 (A) Synthesis scheme of tetra-ortho-methoxy substituted azobenzene. (B) Absorbance spectra of 1 mM 3 in DMSO before and after irradiation with λ = 615 nm for 30 min. (C) Repeated irradiation cycles of 3 using 615 nm for trans-to-cis and 450 nm for cis-to-trans isomerization. | ||
In brief, 3 was synthesized starting from commercially available 3,5-dimethoxyaniline (4) according to the following procedure:34 First, tosylation of the amine (5), followed by nitration (6) with subsequent deprotection, yielded nitroaniline derivative (7). Next, azo-coupling of 7 with 4 led to compound 8a, followed by a reduction of the nitro group with sodium sulfide, which led to compound 8b. Subsequent conversion with chloroacetyl chloride led to 3 (43% yield). Further details about the synthesis and characterization of all compounds are provided in the ESI.†
Next, we recorded the absorbance spectra of the unconjugated compound 3 from 260–650 nm in DMSO (Fig. 2B). We then used red light of λ = 615 nm to induce the photoisomerization from trans to cis. We determined the photostationary state (PSS) and observed near quantitative switching from trans to cis (PSSE→Z 94%) and sufficient switching from cis to trans (PSSZ→E 73%) (Fig. S1A, ESI†). Repeated irradiation cycles with λ = 615 nm to induce the photoisomerization from trans to cis and λ = 450 nm from cis to trans demonstrated the reversible and efficient photoisomerization at all applied wavelengths (Fig. 2C). Furthermore, the generated cis conformer showed a long half-life over 50 h in DMSO (Fig. S1B and C, ESI†). This observation is in line with previous findings that the conformer is stable over several days in DMSO.33 Taken together, these data show that the tetra-ortho-methoxy derivative 3 can be used as an efficient photoswitch because it combines short irradiation intervals with near-complete isomerization (94%) at timescales relevant for biological investigations.
One of the key challenges for bringing protein functions under the control of light is the choice of the conjugation sites. This requires identifying suitable positions in the target protein, which are accessible and appropriately positioned for specific coupling of the photoresponsive moiety. Notably, the light-induced conformational changes of the photoresponsive moiety should be transduced to the protein structure. We aimed to produce an active enzyme conjugated with the azobenzene in the cis-state. This is desirable because it avoids constantly “active” enzymes by thermal relaxation of the azobenzene to the thermodynamically favoured trans-state over time.
Here, we used the crystal structure of Ecm1 combined with state-of-the-art computational methods to guide the choice of conjugation sites. Visual inspection of the crystal structure indicated that amino acids 95–101 (I), 146–156 (II), and 220–228 (III) (Fig. 3A) might be suitable regions, as they are located at the rim of the substrate-binding cleft of Ecm1 and are solvent-exposed, suggesting they will be accessible for conjugation. To identify the best positions to accommodate 3 within these regions in the desired cis conformation, we turned to a computational method based on a set of Rosetta algorithms to predict the impact of light-induced trans to cis conformational change on the protein structure.29,35
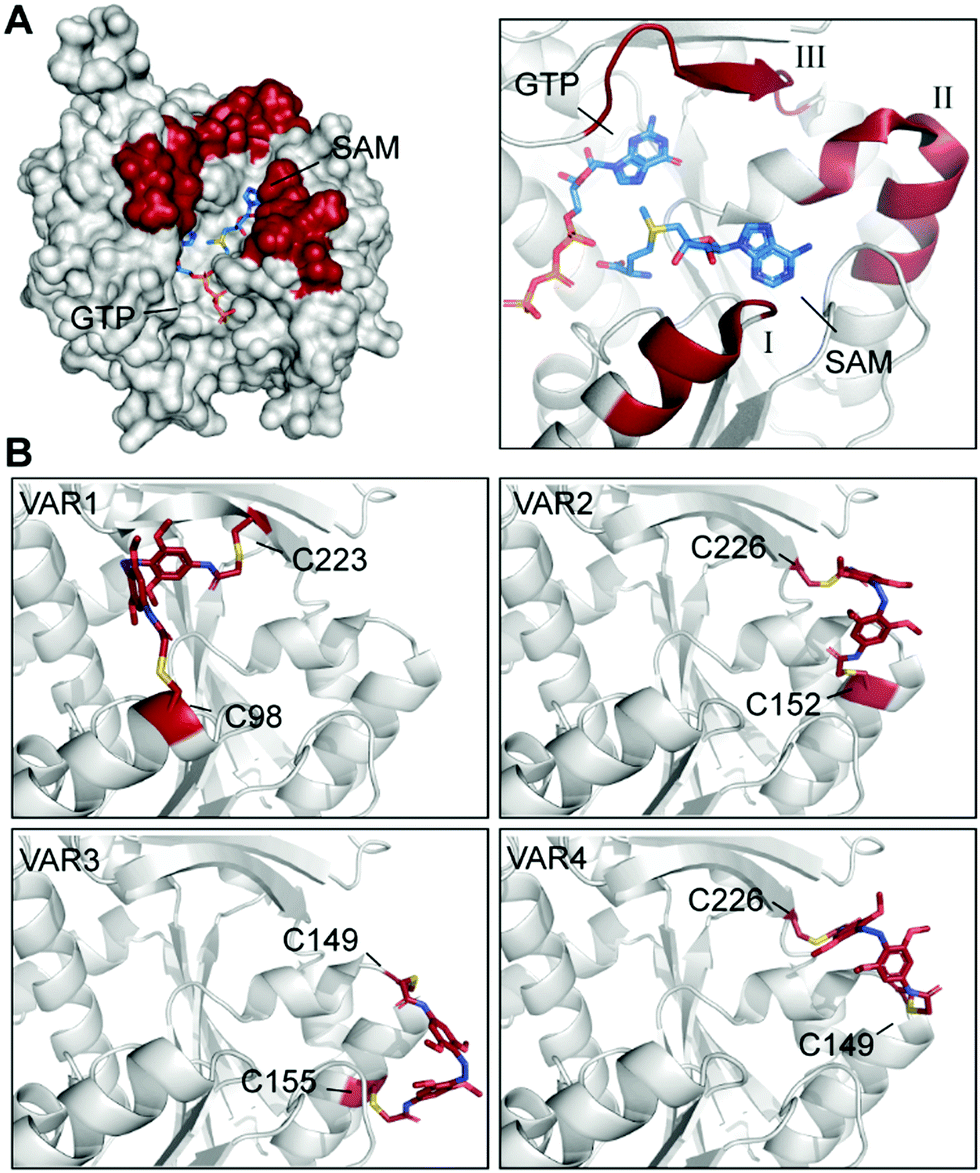 | ||
| Fig. 3 Identification and evaluation of Ecm1 sites for conjugation of 3. (A) Superposition of Ecm1 crystal structures in complex with GTP (PDB: 1RI2) or SAM (PDB: 1RI4). Regions at the rim of the substrate-binding pockets marked in red show amino acids 95–101 (I), 146–156 (II), and 220–228 (III). (B) Ecm1 models of indicated variants with double conjugation to 3 (red) obtained after Rosetta Match and EnzDez application. | ||
In brief, we used the RosettaMatch36 application and a RosettaScripts37 protocol for design and probed all pairs of residues in the ligand-free crystal structure of Ecm1 (PDB 1RI5). For this, the initial structure of 3 in cis conformation and the respective conformer library were assigned to Rosetta parameter files and used with the relaxed structure of cysteine-free variant Ecm1 Δ4C (Table S1, ESI†).
Next, a list of all possible residue positions for azobenzene-derivative conjugation (Fig. 3A), together with a constraint file of the external geometry between the ligand and the protein sidechains, were used as input to the RosettaMatch application. Seven models containing pairs of suitable cysteine substitutions were obtained, accommodating 3 in the desired cis conformation. From these hits, the four models with the lowest total score and constraint energy after the EnzDes application were ranked as VAR1-4 and are shown below (Fig. 3B and Table S2, ESI†). These simulations show that suitable cysteine combinations are VAR1 (C98/C223), C98 located at the end of an α-helix (I, Fig. 3A), and native C223, located in a β-sheet (III), VAR2 (C152/C226) located in an α-helix (II) and a β-sheet (III), VAR3 (C149/C155) located both in the same α-helical region (II), and VAR4 (C149/C226) located in the same α-helix (II) and β-sheet (III) as VAR2. Based on this result, we decided to generate these variants for further experimental characterization.
Before testing variants containing these specific cysteines exclusively, we produced a cysteine-free variant by substituting all four native cysteine residues in Ecm1 (Fig. S2A, ESI†). The resulting variant Ecm1 ΔC4 contained four substitutions, C56S, C73S, C223V, and C272S (Table S1, ESI†), and retained 96% of the wild-type activity (Fig. S2B, ESI†). Ecm1 ΔC4 was then used as starting point to generate VAR2 (S152C/Y226C), VAR3 (T149C/I155C), and VAR4 (T149C/Y226C). VAR1 was generated from Ecm1 ΔC3 by introducing substitution V98C, as it contains the native C223. To evaluate the design strategy, we produced the Ecm1 variants VAR1–4 recombinantly in E. coli Tuner cells. After purification, analysis by PAGE (Fig. S3A, ESI†) and LC/MS (Fig. S3B, ESI†) was used to confirm purity and the identity of the variants. To determine whether the newly introduced cysteines in the native and active enzyme were accessible for efficient bioconjugation, we probed the variants with fluorescein-5-maleimide and detected conjugated cysteines by in-gel fluorescence (Fig. S4, ESI†). As the maleimide residue reacts predominantly with free thiol-groups,38 the obtained fluorescence signal corresponds to the degree of labeling for the respective Ecm1 variants. A fluorescent band at the expected length of 37 kDa was observed for all variants (Fig. S4, ESI†). Although the fluorescence intensities varied, showing the most intense signal for VAR2 (C152/C226) and VAR3 (C149/C155), labeling was successful in all cases. Based on this result, we concluded that the introduced cysteines are accessible and meet the criteria for efficient labeling.
To assess the catalytic activity of the produced variants, we performed biotransformations with the 5′ cap analog GpppA (1a) and SAM (2a) at 25 °C (Fig. 4A). All four variants retained high activity, yielding more than 70% conversion after 24 h. Ecm1 VAR3 showed the highest MTase activity, yielding 93% conversion after 3 h. Still, compared to the wild-type enzyme, the activity was reduced, as previously 91% conversion of 1a were observed after 2 h at 37 °C.15 Nevertheless, these data indicate that our combination of rational and computational design provided active Ecm1 variants suitable to attempt azobenzene conjugation. Overall, we could show that the design strategy yielded functional enzyme variants, with cysteine residues accessible for modification. We chose VAR1 (best computational model) and VAR3 (most active) for subsequent studies.
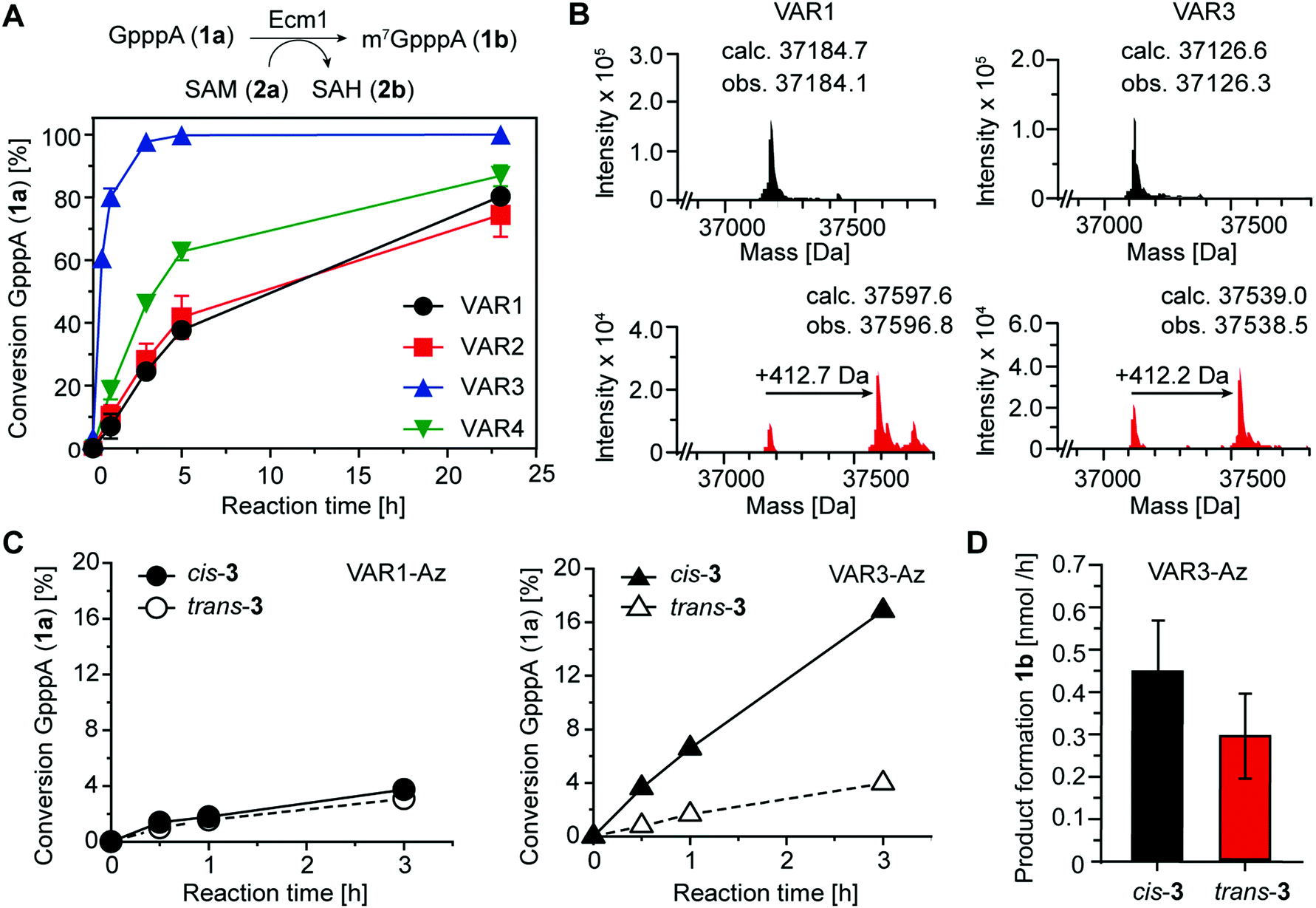 | ||
| Fig. 4 (A) Biotransformations of the 5′ cap analog GpppA (1a) and SAM (2a) catalyzed by indicated Ecm1 variants. (B) LC/MS analysis showing the average mass of VAR1 and VAR3 before (black) and after (red) conjugation with azobenzene 3. The observed mass shift (∼412 Da) corresponds to the molecular weight of 3 when cross-linked. (C) Biotransformations of 1a using cross-linked VAR1-Az or VAR3-Az, respectively. Reaction mixtures were irradiated with either λ = 615 nm or λ = 450 nm to obtain 3 in cis (black) or trans (white) conformation. (D) Average product formation of m7GpppA (1b) for cis-3 and trans-3 using Var3-Az, starting from 0.9 nmol of enzyme. Bars and error bars show the average and s.e.m. of four independent experiments. | ||
As the shape of the binding pocket of Ecm1 is essential for MTase activity, we assumed that isomerization of a cross-linked azobenzene (connecting two cysteine residues) would significantly affect its MTase activity. We first investigated and optimized the labeling efficiency of VAR1 and VAR3 with the azobenzene 3 in the cis conformation. To achieve high degrees of cross-linked protein, 3 was brought to the cis-form by irradiation for 30 min at 615 nm and then added step-wise over 6 h to the protein at pH 8.5. LC/MS analysis showed that the labeling yields were high. For VAR1, 76% of the desired cross-linked species were obtained. For VAR3, the crosslinking yield was 65% (Fig. 4B and Fig. S5, ESI†). These results show that efficient modification of both variants was achieved after careful optimization of the reaction conditions.
As the next step, we wanted to test whether Ecm1 activity can be actuated by light in response to azobenzene-based isomerization. To this end, Ecm1 variants obtained after crosslinking (VAR1-Az, VAR3-Az) were used in methylation reactions of the 5′ cap analog GpppA (1a) and analyzed by HPLC (Fig. 1A, 4C and Fig. S6, ESI†). In the case of Ecm1 VAR1-Az, little to no activity was observed, independent of the conformation of the azobenzene moiety (Fig. 4C). To our delight, VAR3-Az proved active when the azobenzene was in the cis state. When irradiated by light of 450 nm for 2 min, which causes the azobenzene derivative to switch to the trans-state, we observed only minimal conversion. The apparent difference observed after 3 h of incubation, depending on whether 3 was present in the cis (17%) or trans state (4%), shows that the design strategy was successful for VAR3-Az.
To quantify the effect on activity, we repeated the crosslinking experiments and monitored the methylation of 1a over 3 h VAR3-Az in the cis or trans-state (Fig. S7A, ESI†). Importantly, reaction samples were also analyzed by SDS–PAGE to ensure identical enzyme concentrations (Fig. S7B, ESI†). From these data, we calculated an average product formation of 0.3 nmol h−1 for trans VAR3-Az and 0.45 nmol h−1 for cis-VAR3-Az, starting from 0.9 nmol of Ecm1 (Fig. 4D). Thus, light-induced modulation of the active site enzymatic activity increased the activity up to 50%.
Aiming to understand the effects observed during experimental characterization, we turned again to computational modeling. We used the Rosetta FastRelax protocol to study how much flexibility in the protein backbone and side chains is needed to accommodate the azobenzene in the cis conformation. Specifically, we examined the cross-linked regions for Ecm1 VAR1-Az and VAR3-Az using the same geometric constraints for the thiol bonds as in the Rosetta Match application.
The model for VAR1-Az (Fig. S8A, ESI†) indicates that the slight shift in the α-helix in area I (Fig. 3A) is sufficient for efficient conjugation. However, in this relaxed conformation with optimized geometry, the kinked azobenzene moiety of 3 directly points into the binding pocket of Ecm1 (Fig. S8B and C, ESI†), thereby impeding substrate binding and inhibiting the enzyme. These simulations support the observed absent activity-difference for VAR1 in the cis- or trans-form (Fig. 4C).
Next, we used the model of VAR3-Az with the lowest energy and lowest constraint score with the azobenzene cross-linked in cis conformation after the FastRelax protocol. Starting from the designed cross-linked Ecm1 VAR3-Az, we performed 100 simulated cis-to-trans isomerizations by “mutating” the azobenzene derivatives to their trans isomers followed by Rosetta FastRelax application to optimize the geometry and energy of the models (Fig. 5A).
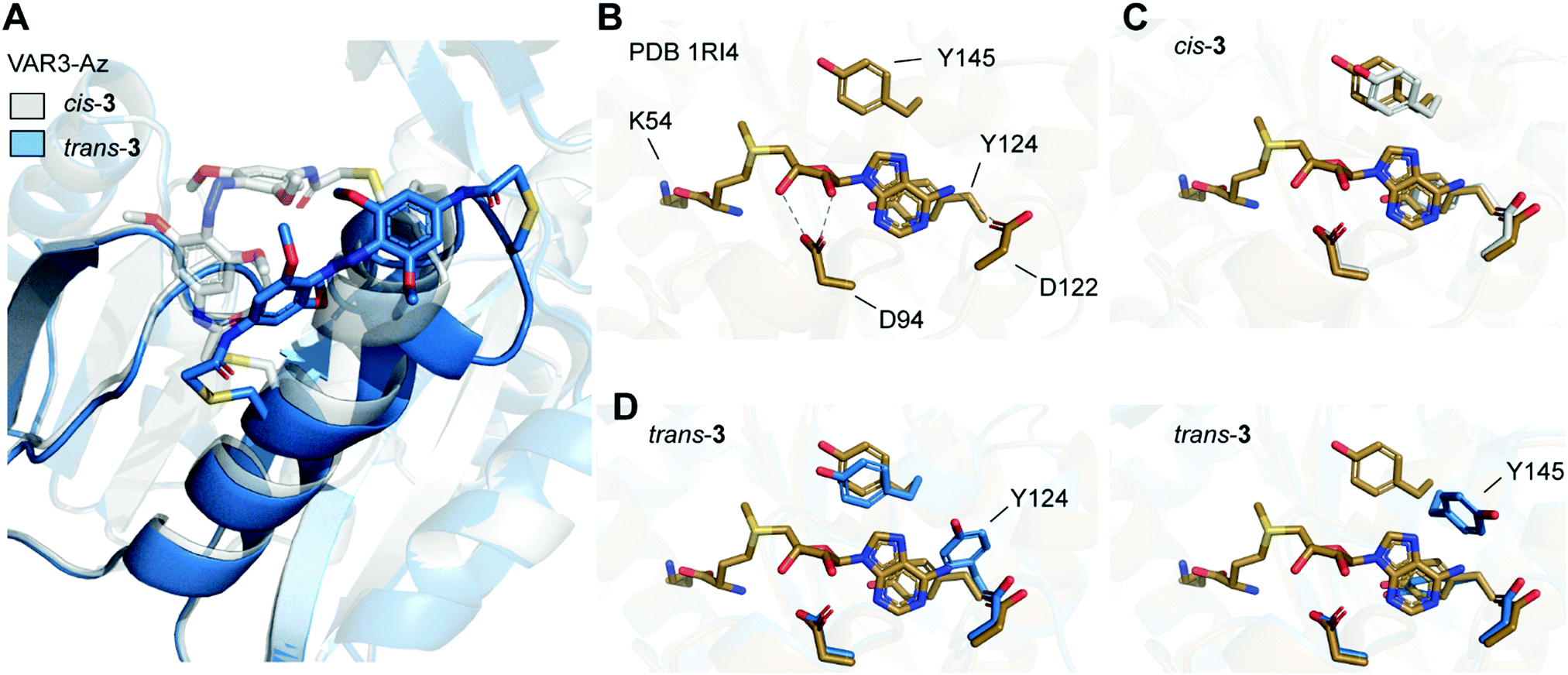 | ||
| Fig. 5 (A) Overlay of Rosetta-based model of Ecm1 VAR3-Az (C149/C155) cross-linked with azobenzene in cis conformation (gray), and model after isomerization from cis to trans (blue). (B) Crystal structure of Ecm1 WT in complex with SAM (brown, PDB 1RI4). SAM and amino acid residues important for SAM coordination are shown as sticks. Hydrogen bonds are indicated as dashed lines. (C) Overlay of WT crystal structure and the Rosetta-based model VAR3-Az conjugated to cis-3 (gray). (D) Overlay of WT crystal structure and two exemplary structures obtained for the Rosetta-based model of VAR3-Az conjugated to trans-3 (blue). Y124 or Y145 are displaced after trans isomerization of the azobenzene impeding SAM binding. | ||
To evaluate the conformational changes, we compared the computational model with the crystal structure of Ecm1 WT in complex with SAM (brown, Fig. 5B–D) and indicated the amino acid residues important for SAM coordination (Fig. 5B).31 Most importantly, Y124 provides stacking interactions with the adenine ring of SAM, and D122 forms hydrogen bonds to the exocyclic amino group at the N6 position of the adenine. Furthermore, D94 interacts with the 2′-OH and 3′-OH of the ribose moiety. The overlay with the initial Rosetta-based model of VAR3-Az conjugated to cis-3 shows that the residues important for coordination are not moved and do not impair SAM binding (gray, Fig. 5C). Next, we evaluated the Rosetta-based model of VAR3-Az conjugated to trans-3 (blue, Fig. 5D), comparing it to the Ecm1 WT bound with SAM (brown, Fig. 5D). The overlay shows two exemplary structures obtained after the simulations. Our models indicate that the isomerization of the photoswitch might induce a slight unraveling of the α-helix in area II and concomitantly a change in the orientation of residue Y124 or Y145. The different rotamers occurred 24 times (Y124) and 29 times (Y145), respectively, in the 100 models we calculated.
Notably, both rotamers never co-occurred together, indicating that both forms interfere with each other. Here, in 53 of the 100 models obtained by cis-to-trans simulations, an effect on the SAM binding pocket was observed. D94, however, is not affected by cis-to-trans isomerization of the azobenzene. This finding could explain our observed experimental outcome since previous mutational characterizations of Ecm1 showed that alanine substitutions of Y124 and Y145 caused an almost complete loss of MTase activity,39 indicating the importance of the stacking interactions for SAM coordination.
Conclusion
In summary, we developed an interdisciplinary approach combining computational design and experimental characterization of a light-dependent 5′ cap guanine-N7 methyltransferase. We created and characterized the best four predicted variants, each featuring two appropriately positioned cysteine residues close to the substrate-binding cleft. Conjugating and crosslinking the azobenzene between C149/C155 in a designed Ecm1 variant (VAR3-Az) enabled modulation of the MTase activity by light.Although azobenzene-based photoswitches hold tremendous potential for a reversible modulation of the protein structure and activity, their application in actuating enzymatic activity by light is rare. It is particularly challenging to predict the effect of the conjugated photoswitch on enzymatic activity. Therefore, this study provided another step to improve design rules leading to photoswitchable enzymes using azobenzene crosslinking.
We could show that the labelling sites must be placed close enough to the active site to affect substrate binding or catalytic turnover as in VAR3-Az, but not so close as to interfere with catalytic activity as shown for VAR1-Az. Our simulations of cis-to-trans isomerization of Ecm1 VAR3-Az showed that not only the labelling sites but also the properties of the secondary structure element used for conjugation must be considered. In our case, local flexibility in the α-helix II (Fig. 3A) was essential to accommodate the conjugated azobenzene, however, at the same time it attenuated the force induced by the photoswitch. Therefore, this study shows that subtle variations in the placement of the azobenzene have a tremendous impact on the photoswitching effect.
In future work, this approach could be readily extended to other small MTases. Promiscuous MTases often feature an open binding pocket potentially accessible for azobenzene conjugation. In addition, the computational method can be applied to other photoswitches making it a general and flexible tool to design light-dependent enzymes.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank S. Wulff for technical assistance in protein LC/MS analyses, Dr F. Höhn and the technical workshop for providing and building custom-made LED boxes. A. R. and B. J. R. gratefully acknowledge funding by the DFG (SFB858). D. R. is a member of CiM-IMPRS, the joint graduate school of the Cells-in-Motion Interfaculty Center (CiM), University of Münster, Germany, and the International Max Planck Research School–Molecular Biomedicine, Münster, Germany.Notes and references
- S. Shuman, Prog. Nucleic Acid Res. Mol. Biol., 2001, 66, 1–40 CAS.
- J. Marcotrigiano, A. C. Gingras, N. Sonenberg and S. K. Burley, Cell, 1997, 89, 951–961 CrossRef CAS.
- A. Niedzwiecka, J. Marcotrigiano, J. Stepinski, M. Jankowska-Anyszka, A. Wyslouch-Cieszynska, M. Dadlez, A. C. Gingras, P. Mak, E. Darzynkiewicz, N. Sonenberg, S. K. Burley and R. Stolarski, J. Mol. Biol., 2002, 319, 615–635 CrossRef CAS.
- C. J. Brown, I. McNae, P. M. Fischer and M. D. Walkinshaw, J. Mol. Biol., 2007, 372, 7–15 CrossRef CAS PubMed.
- N. Sonenberg and A. G. Hinnebusch, Cell, 2009, 136, 731–745 CrossRef CAS PubMed.
- N. E. Shirokikh and T. Preiss, Wiley Interdiscip. Rev.: RNA, 2018, 9, e1473 Search PubMed.
- C. Renzl, A. Kakoti and G. Mayer, Angew. Chem., Int. Ed., 2020, 59, 22414–22418 CrossRef CAS PubMed.
- Y. I. Wu, D. Frey, O. I. Lungu, A. Jaehrig, I. Schlichting, B. Kuhlman and K. M. Hahn, Nature, 2009, 461, 104–108 CrossRef CAS PubMed.
- A. M. Weber, J. Kaiser, T. Ziegler, S. Pilsl, C. Renzl, L. Sixt, G. Pietruschka, S. Moniot, A. Kakoti, M. Juraschitz, S. Schrottke, L. L. Bryant, C. Steegborn, R. Bittl, G. Mayer and A. Moglich, Nat. Chem. Biol., 2019, 15, 1085–1092 CrossRef CAS PubMed.
- E. Darzynkiewicz, J. Stepinski, I. Ekiel, C. Goyer, N. Sonenberg, A. Temeriusz, Y. X. Jin, T. Sijuwade, D. Haber and S. M. Tahara, Biochemistry, 1989, 28, 4771–4778 CrossRef CAS PubMed.
- A. Cai, M. Jankowska-Anyszka, A. Centers, L. Chlebicka, J. Stepinski, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, Biochemistry, 1999, 38, 8538–8547 CrossRef CAS PubMed.
- J. M. Holstein, L. Anhauser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899–10903 CrossRef CAS.
- F. Soukarieh, M. W. Nowicld, A. Bastide, T. Poyry, C. Jones, K. Dudek, G. Patwardhan, F. Meullenet, N. J. Oldham, M. D. Walkinshaw, A. E. Willis and P. M. Fischer, Eur. J. Med. Chem., 2016, 124, 200–217 CrossRef CAS PubMed.
- L. Anhauser, N. Klocker, F. Muttach, F. Masing, P. Spacek, A. Studer and A. Rentmeister, Angew. Chem., Int. Ed., 2020, 59, 3161–3165 CrossRef PubMed.
- D. Reichert, H. D. Mootz and A. Rentmeister, Chem. Sci., 2021, 12, 4383–4388 RSC.
- T. Fehrentz, M. Schonberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156–12182 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS.
- A. M. Caamano, M. E. Vazquez, J. Martinez-Costas, L. Castedo and J. L. Mascarenas, Angew. Chem., Int. Ed., 2000, 39, 3104–3107 CrossRef CAS PubMed.
- D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391–397 CrossRef CAS.
- N. Pozhidaeva, M. E. Cormier, A. Chaudhari and G. A. Woolley, Bioconjugate Chem., 2004, 15, 1297–1303 CrossRef CAS PubMed.
- M. Banghart, K. Borges, E. Isacoff, D. Trauner and R. H. Kramer, Nat. Neurosci., 2004, 7, 1381–1386 CrossRef CAS PubMed.
- S. Berlin, S. Szobota, A. Reiner, E. C. Carroll, M. A. Kienzler, A. Guyon, T. Xiao, D. Trauner and E. Y. Isacoff, eLife, 2016, 5, e12040 CrossRef PubMed.
- R. S. Ritterson, K. M. Kuchenbecker, M. Michalik and T. Kortemme, J. Am. Chem. Soc., 2013, 135, 12516–12519 CrossRef CAS PubMed.
- C. Hoppmann, V. K. Lacey, G. V. Louie, J. Wei, J. P. Noel and L. Wang, Angew. Chem., Int. Ed., 2014, 53, 3932–3936 CrossRef CAS PubMed.
- K. H. DuBay, K. Iwan, L. Osorio-Planes, P. L. Geissler, M. Groll, D. Trauner and J. Broichhagen, ACS Chem. Biol., 2018, 13, 793–800 CrossRef CAS PubMed.
- A. C. Kneuttinger, C. Rajendran, N. A. Simeth, A. Bruckmann, B. Konig and R. Sterner, Biochemistry, 2020, 59, 2729–2742 CrossRef CAS.
- B. Schierling, A. J. Noel, W. Wende, T. Hien le, E. Volkov, E. Kubareva, T. Oretskaya, M. Kokkinidis, A. Rompp, B. Spengler and A. Pingoud, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1361–1366 CrossRef CAS PubMed.
- Y. Liu, X. Gao, D. Z. Wei and Y. H. Ren, ChemPhotoChem, 2017, 1, 393–396 CrossRef CAS.
- K. M. Blacklock, B. J. Yachnin, G. A. Woolley and S. D. Khare, J. Am. Chem. Soc., 2018, 140, 14–17 CrossRef CAS.
- S. Hausmann, C. P. Vivares and S. Shuman, J. Biol. Chem., 2002, 277, 96–103 CrossRef CAS PubMed.
- C. Fabrega, S. Hausmann, V. Shen, S. Shuman and C. D. Lima, Mol. Cell, 2004, 13, 77–89 CrossRef CAS.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS.
- N. Yasuike, K. M. Blacklock, H. X. Lu, A. S. I. Jaikaran, S. McDonald, M. Uppalapati, S. D. Khare and G. A. Woolley, ChemPhotoChem, 2019, 3, 431–440 CrossRef CAS.
- F. Richter, A. Leaver-Fay, S. D. Khare, S. Bjelic and D. Baker, PLoS One, 2011, 6, e19230 CrossRef CAS PubMed.
- S. J. Fleishman, A. Leaver-Fay, J. E. Corn, E. M. Strauch, S. D. Khare, N. Koga, J. Ashworth, P. Murphy, F. Richter, G. Lemmon, J. Meiler and D. Baker, PLoS One, 2011, 6, e20161 CrossRef CAS.
- J. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chemistry, 2019, 25, 43–59 CrossRef CAS.
- S. Zheng, S. Hausmann, Q. Liu, A. Ghosh, B. Schwer, C. D. Lima and S. Shuman, J. Biol. Chem., 2006, 281, 35904–35913 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00109d |
| This journal is © The Royal Society of Chemistry 2021 |