
Tubular assemblies of N-doped carbon nanotubes loaded with NiFe alloy nanoparticles as efficient bifunctional catalysts for rechargeable zinc-air batteries†
Xiaoying
Xie
a,
Lu
Shang
*a,
Run
Shi
a,
Geoffrey I. N.
Waterhouse
b,
Jiaqi
Zhao
ac and
Tierui
Zhang
 *ac
*ac
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: tierui@mail.ipc.ac.cn; lushang@mail.ipc.ac.cn
bSchool of Chemical Sciences, The University of Auckland, Auckland, 1142, New Zealand
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing, 100049, China
First published on 10th June 2020
Abstract
Enormous research effort is presently being directed towards the discovery of low cost bifunctional electrocatalysts capable of efficiently driving the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER), with such bifunctional electrocatalysts being particularly sought after for rechargeable metal-air batteries. Herein, we report the successful synthesis of a highly efficient bifuctional ORR/OER electrocatalyst, comprising tubular assemblies of 20-40 nm N-doped carbon nanotubes containing NiFe alloy nanoparticles (denoted herein as TA-NiFe@NCNT). To synthesize TA-NiFe@NCNT, we first prepared g-C3N4 nanotubes with a diameter ∼200 nm as a sacrificial template and nitrogen source, then decorated the nanotubes with NiFe-layered double hydroxide nanoparticles (NiFe-LDH). The NiFe-LDH/g-C3N4 composite obtained was then coated with a thin layer of glucose (an additional carbon source), then the resulting NiFe-LDH/g-C3N4@Glu composite was pyrolyzed at 900 °C in N2. The obtained TA-NiFe@NCNT product exhibited a low overpotential of only 310 mV at a current density of 10 mA cm−2 during OER in 0.1 M KOH (cf. 401 mV for IrO2) and an ORR activity in 0.1 M KOH (onset potential of 0.93 V and half-wave potential of 0.81 V vs. RHE) comparable to a commercial Pt/C catalyst (onset potential of 0.99 V and half-wave potential of 0.82 V vs. RHE). The remarkable bifunctional performance of TA-NiFe@NCNT can be attributed to the excellent OER and ORR activities of NiFe alloy nanoparticles and NCNTs, respectively, as well as the high porosity and excellent conductivity of the electrocatalyst that benefitted mass and electron transfer processes, respectively. A custom-built rechargeable zinc-air battery constructed using TA-NiFe@NCNT at the air electrode delivered a lower charge-discharge voltage gap (0.92 V) and longer cycling lifetime (170 h at 25 mA cm−2) than a battery fabricated using a mixture of IrO2 and Pt/C as air electrode catalysts.
Introduction
Due to their low cost and high theoretical energy density of 1084 W h kg−1, rechargeable zinc-air batteries are expected to play an important role in future electrical energy storage.1,2 Rechargeable zinc-air batteries comprise a metallic zinc electrode and an air electrode (typically a catalyst on carbon cloth), with an aqueous KOH electrolyte. During discharge, zinc dissolves at the anode and oxygen gas is reduced to hydroxide ions at the air electrode (cathode). During recharge, zinc ions are reduced to Zn metal at the cathode and oxygen is evolved at the air electrode (now the anode). The key to fabricating high performance rechargeable zinc-air batteries are bifunctional electrocatalysts capable of driving the oxygen reduction reaction (ORR) during battery discharge and the oxygen evolution reaction (OER) during battery recharge.3,4 Currently, supported precious metal catalysts are used in the production of rechargeable zinc-air batteries. Pt/C catalysts are particularly efficient in catalyzing ORR, whilst Ru-based or Ir-based catalysts efficiently drive OER. However, their low earth abundance, high cost, modest stability and poor bifunctional activities for both ORR and OER limit the usefulness of these catalysts for large scale rechargeable zinc-air battery production.5,6 This motivates the search for efficient, low cost and stable bifunctional electrocatalysts for ORR and OER based on earth abundant elements.To date, non-precious metal catalyst systems have been pursued for ORR and OER, including nanocarbons,7 metal alloys,8 nitrides,9 and their composites.10 Heteroatom (such as N, S, P)-doped carbon materials are particularly promising for ORR, owing to their electronic structure, good conductivity and surface polarity.3,11–13 N-doped carbon nanotubes (NCNTs) are particularly promising alternatives to Pt-based electrocatalysts for ORR process due to their excellent activities, low cost and chemical/mechanical robustness.4,14–18 However, NCNTs cannot be used directly as bifunctional catalysts in rechargeable zinc-air batteries due to their poor OER activity. Recent studies have shown that certain first row transition metals (Ni, Fe, Co), either as nanoparticles or single atom catalysts, offer suitable active sites for OER.19–21 Amongst them, NiFe alloy nanoparticles have recently emerged as promising replacements for Ru-based/Ir-based catalysts, which can be attributed to their good conductivity, excellent electrocatalytic activity, and outstanding durability in alkaline media.22–24 Density functional theory (DFT) calculations predict that NiFe alloys should afford excellent OER activity relative to Ni or Fe due to their unique surface electronic structure.25 Therefore, depositing NiFe alloy nanoparticles on NCNTs represents a rational approach for the synthesis of bifunctional electrocatalysts capable of driving both OER and ORR.
Various approaches are now being explored to prepare composites of NiFe alloy nanoparticles and NCNTs. Synthetic strategies include the pyrolysis of Prussian Blue analogues26 or alternative precursor mixtures of iron and nickel salts with carbon and nitrogen rich compounds like urea27 and dicyandiamide.28 However, these approaches have inherent limitations since NCNTs formed have a high tendency to agglomerate and entangle with each other, reducing the availability of active sites for ORR and OER, as well as blocking channels for mass transport. Hence, porous NiFe/NCNT nanocomposites with more controllable architectures are needed for rechargeable zinc-air battery applications. Surface functionalisation of NCNTs is a commonly used approach to enhance the nanotube dispersion, though typically involves complex chemical treatments and therefore is undesirable from the viewpoint of practical scalability and cost. Recently, Zelenay's group successfully introduced carbon nanoparticles as a spacer to enhance nanotube dispersion, with the modified nanotubes then being used to create composites with an abundance of available active sites.16 However, amorphous carbon nanoparticles can suppress the excellent conductivity of NCNTs, thus requiring holistic consideration of the benefits of adding carbon nanoparticles. Another strategy is to create composites with a highly porous microstructure, which is commonly achieved via templating strategies. Chen synthesized a mesoporous composite comprising NiFe alloy nanoparticles and carbon nanofibers using a hard SiO2 template technique, with the obtained composite exhibiting excellent activity for both OER and ORR.29 However, the use of hard templates such as SiO2 is becoming less popular as the aggressive chemical treatments often required to remove the templates. In a previous study, we reported that g-C3N4 nanosheets can act as both a template and nitrogen source for the preparation of porous carbon materials with high nitrogen content. By pyrolyzing g-C3N4 nanosheets coated with a thin layer of glucose, porous N-doped carbons were obtained possessing comparable ORR activity to a commercial Pt/C electrocatalyst in alkaline electrolyte.30 By adopting a similar synthetic approach, and introducing NiFe alloy nanoparticles, we hypothesized that efficient bifunctional catalysts could be created capable of efficiently driving OER and ORR.
Herein, we report the successful synthesis of tubular assemblies of N-doped carbon nanotubes internally loaded with NiFe alloy nanoparticles (denoted as TA-NiFe@NCNT). Our approach used 200 nm diameter g-C3N4 nanotubes as a sacrificial template and nitrogen source, glucose as the main carbon source and NiFe-LDH as the NiFe alloy nanoparticles precursor. Pyrolysis of the NiFe-LDH/g-C3N4@Glu precursor at 900 °C in N2 yielded TA-NiFe@NCNT, which in accordance with the hypothesis above offered outstanding ORR and OER performance. A rechargeable zinc-air battery fabricated with TA-NiFe@NCNT as the air electrode catalyst showed a very small voltage gap of 0.92 V and excellent stability (170 h at 25 mA cm−2), indicating that the bifunctional NiFe@NCNT catalyst offered a promising low cost alternative to precious metal-based catalysts for both ORR and OER.
Experimental
Materials preparation
Holey g-C3N4 nanotubes with a diameter of ∼200 nm were synthesized by the calcination of a mixture of melamine and urea in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mass ratio at 550 °C for 4 h under a N2 atmosphere in a quartz tube furnace.31
:10 mass ratio at 550 °C for 4 h under a N2 atmosphere in a quartz tube furnace.31
NiFe-LDH/g-C3N4 nanotubes were prepared by coating the holey g-C3N4 nanotubes with NiFe-layered double hydroxide nanoparticles. Briefly, g-C3N4 nanotubes (500 mg) were dispersed in 40 mL of deionised water. Next, 20 mL of an aqueous solution containing Ni(NO3)2·6H2O (110.25 mg), Fe(NO3)3·9H2O (51 mg) and urea (2700 mg) was added. The resulting dispersion was then heated at 100 °C for 6 h under vigorous stirring. The NiFe-LDH/g-C3N4 product was collected by centrifugation and washed several times with ethanol and deionised water, then finally dried at 60 °C overnight.
NiFe-LDH/g-C3N4@Glu was obtained by coating NiFe-LDH/g-C3N4 nanotubes with glucose using a hydrothermal method. NiFe-LDH/g-C3N4 (505 mg) was dispersed in an aqueous glucose solution (0.3 M, 40 mL) under vigorous stirring for 4 h. The aqueous dispersion was then transferred into a 50 mL Teflon autoclave, which was then heated 120 °C for 10 h. The NiFe-LDH/g-C3N4@Glu product was collected by centrifugation, washed several times with ethanol and deionised water, and finally dried at 60 °C overnight.
TA-NiFe@NCNT were synthesized by direct pyrolysis of NiFe-LDH-C3N4@Glu at 900 °C for 1 h under a N2 atmosphere, using a ramping rate of 5 °C min−1. By controlling the relative concentrations of Ni(NO3)2·6H2O, Fe(NO3)3·9H2O and urea used to prepare NiFe-LDH nanoparticles in the precursor, final products with different NiFe alloy nanoparticle loadings could easily be obtained. Further samples were prepared using the same procedure, but in the absence of NiFe-LDH (product denoted here as tubular N-doped carbon or simply TA-NC), or in the absence of glucose (product denoted here as TA-NiFe@NCNT*), or in the absence of g-C3N4 nanotubes (NiFe/C).
Physicochemical characterization
Scanning electron microscopy (SEM) images were obtained on a Hitachi S4800 SEM. Energy dispersive spectroscopy (EDS) measurements were carried out on a Hitachi S4300. Transmission electron microscopy (TEM) images were measured on HT7700 instrument operating at an accelerating voltage of 100 kV. High resolution TEM (HRTEM) images and element maps were taken on a JEOL-2100F microscope operating at an accelerating voltage of 200 kV. X-ray diffraction (XRD) patterns were collected on a Bruker D8 Focus X-ray diffractometer equipped with a Cu Kα radiation (λ = 1.5405 Å) source. N2 adsorption–desorption isotherms were obtained at 77 K on a Quadrasorb SI MP apparatus. X-ray photoelectron spectroscopy (XPS) data were obtained on a VGESCALABMKII X-ray photo-electron spectrometer using a non-monochromatized Al-Kα X-ray source (hν = 1486.7 eV). Raman spectra were collected on Renishaw in Via Reflex spectrometer system. Spectra were excited using a 532 nm laser. Thermogravimetric analysis (TGA) was obtained by heating samples from room temperature to 900 °C at 5 °C min−1 under a N2 atmosphere on a Rigaku Thermo plus instrument.Electrochemical measurements
The ORR and OER performance of the various catalysts were studied using a standard three-electrode system interfaced with an electrochemical workstation (CHI 760 E, CHI Instrument, China). A Pt plate and a calibrated Hg/HgO electrode served as the counter electrode and the reference electrode, respectively. The Hg/HgO reference electrode was calibrated in hydrogen saturated 0.1 M KOH. The working electrodes were prepared as follows. Catalysts inks were first prepared by adding 8 mg of catalysts to a solution containing 500 μL of deionized water, 436 μL of isopropanol and 64 μL of Nafion solution (5 wt%, Alfa Aesar), followed by ultrasonic treatment for 1 h to achieve a homogeneous dispersion. The resulting dispersions were then pipetted onto freshly polished glassy carbon electrodes (GC, 5 mm in diameter) to achieve an electrocatalyst loading of 240 μg cm−2 (except for IrO2, for which the loading was 500 μg cm−2). The inks were subsequently allowed to air-dry at room temperature. For the OER and ORR tests, 0.1 M KOH was employed as the electrolyte in the three-electrode system. Cyclic voltammetry (CV) curves were recorded at a scan rate of 100 mV s−1. Linear sweep voltammetry (LSV) data for ORR were recorded from 0.2 to −1.0 V (vs. Hg/HgO) at a scan rate of 10 mV s−1 with data collected at rotating electrode speeds from 400 to 2025 rpm. LSV data for OER were recorded from 0 to 0.8 V (vs. Hg/HgO) with a 95%-iR correction at a rotating speed of 1600 rpm.Electron transfer numbers were calculated from the slopes of Koutecky-Levich (K-L) plots, based on the following equations:32,33
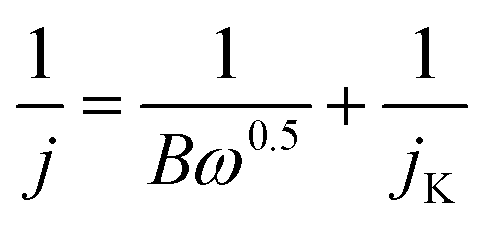
| B = 0.2 nF(DO2)2/3υ−1/6CO2 |
485 C mol−1); CO2, bulk concentration of O2 (1.2 × 10−6 mol cm−1); DO2, diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1); υ, kinetic viscosity of the electrolyte (0.01 cm2 s−1); n, electron transfer number.
Rechargeable zinc-air battery fabrication
Rechargeable zinc-air batteries were constructed, consisting of a zinc plate as the anode, electrocatalyst on carbon paper as the air electrode, and an electrolyte solution containing 6 M KOH and 0.2 M zinc acetate. The air electrodes were prepared by pipetting the catalyst inks described above onto carbon paper over a circular area of 1 cm diameter to achieve a catalyst loading of 1 mg cm−2.Results and discussion
The synthesis of TA-NiFe@NCNT is summarized in Scheme 1. Firstly, g-C3N4 was prepared by thermal decomposition-polymerization of a mixture of melamine and urea (1:10 weight ratio). The obtained g-C3N4 nanotubes had an average diameter around 200 nm with numerous mesopores uniformly dispersed along the tubes confirmed by SEM (Fig. S1a†) and TEM (Fig. S1b and S1c†). Next, NiFe-LDH were grown on the g-C3N4 nanotubes to produce NiFe-LDH/g-C3N4. This was aided by the fact that the Ni2+ and Fe3+ ions were complexed by the pyridine-like nitrogen atoms in the heptazine units of g-C3N4,9,34 which thus served as nucleation sites for NiFe-LDH growth. Fig. S1d–f† show NiFe-LDH nanoparticles homogeneously dispersed over the g-C3N4 nanotubes. Next, a hydrothermal treatment was used to coat NiFe-LDH/g-C3N4 with a thin layer (∼10 nm) of glucose (producing NiFe-LDH/g-C3N4@Glu), which retained the tubular morphology of the g-C3N4 nanotube template (Fig. S1g–j†). Subsequently, NiFe-LDH/g-C3N4@Glu was heated at 900 °C of 1 h in N2 to give tubular assemblies of NCNTs containing NiFe alloy nanoparticles (TA-NiFe@NCNTs). During the pyrolysis process, the glucose was carbonized, with the thermal decomposition of the g-C3N4 nanotubes acting as both a nitrogen and secondary carbon source. TGA showed the decomposition of the g-C3N4 nanotubes was complete at ∼600 °C (Fig. S2†). The evolution of NH3, H2 and CO during the heating of NiFe-LDH/g-C3N4@Glu transformed the NiFe-LDH nanoparticles into NiFe alloy particles supported by NCNTs.14 Heating to 900 °C for 1 h ensured that the NCNTs had a reasonable degree of graphitization and thus good electrical conductivity. The structural evolution of TA-NiFe@NCNT during heating of NiFe-LDH/g-C3N4@Glu was followed by XRD. The g-C3N4 nanotubes showed peaks at 13° and 27°, which could readily be assigned to the (100) and (002) reflections of g-C3N4 (Fig. S3†). The NiFe-LDH/g-C3N4 and NiFe-LDH/g-C3N4@Glu samples showed additional peaks at 34° and 60°, which could readily be assigned to the (012) and (110) reflections of NiFe-LDH (JCPDS Card 51-0463),35 confirming the existence of NiFe-LDH in the two composites. The glucose shell around NiFe-LDH/g-C3N4@Glu was amorphous (Fig. S1j†). XRD data for TA-NiFe@NCNT are discussed below, in conjunction with other characterization data for the composites. The morphology and structure of TA-NiFe@NCNT were studied in detail by SEM and TEM (Fig. 1). As shown in Fig. 1a to c, TA-NiFe@NCNT possessed a similar tubular structure to the g-C3N4 template, with an average tube diameter of about 200 nm. HRTEM revealed that NCNTs with a diameter in a range of 20 to 40 nm have been seeded by the NiFe nanoparticles since each nanotube contained a single metal nanoparticle at one end (Fig. 1d). Metal nanoparticles are well known to act as seeds for carbon nanotube formation in gas mixtures of CH4, acetylene or other C-rich gases at high temperatures. These NCNTs possessed less than 10 layers of N-doped graphitic carbon with interplanar spacing of 0.34 nm, corresponding to the (002) plane of graphitic carbon. The high graphitic content was expected to impart TA-NiFe@NCNT with excellent electrical conductivity. Fig. 1d shows that the metal nanoparticles had a lattice fringe spacing of 0.21 nm, typical for the (111) facets of NiFe alloy.36 The corresponding element maps for TA-NiFe@NCNT (Fig. 1e) confirmed the presence of alloy NiFe nanoparticles on a carbonaceous support. According to EDS results (Fig. S4†), the weight percentages of C, N, O, Ni and Fe in TA-NiFe@NCNT were estimated to be 72.6, 5.6, 12.4, 6.1, and 3.3 wt%, respectively. For comparison, TA-NiFe@NCNT*, TA-NC and NiFe/C were prepared by a similar process to TA-NiFe@NCNT, except that glucose, NiFe-LDH, or g-C3N4, respectively, were not included during the synthesis. Fig. S5† shows characterization data for these additional reference samples. Fig. S5a–c† show that the TA-NiFe@NCNT* product contained NiFe alloy nanoparticles embedded in an entangled NCNT network, indicating that the addition of the glucose shell was beneficial in the synthesis of TA-NiFe/NCNT for preventing excessive nanotube entanglement. In the absence of NiFe-LDH, the direct pyrolysis of g-C3N4@Glu yielded tubular assemblies of N-doped carbon (TA-NC) with a similar morphology to the g-C3N4 nanotube precursor (Fig. S5d–f†). The walls of the submicron-diameter tubes consisted of thin layers. Without the g-C3N4 nanotubular template, pyrolysis of NiFe-LDH@Glu yielded large NiFe alloy nanoparticles supported by semigraphitic carbon (NiFe/C, Fig. S5g–i†).
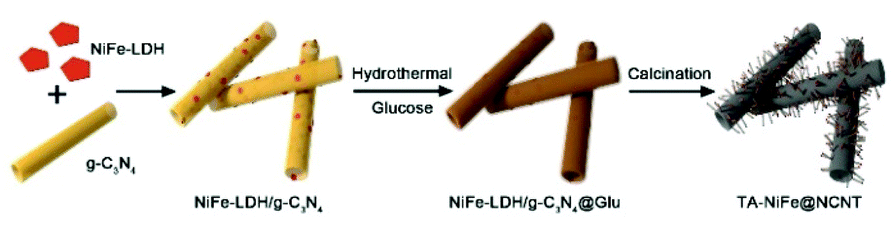 | ||
| Scheme 1 Schematic illustration of the preparation process of TA-NiFe@NCNT. | ||
 | ||
| Fig. 1 (a) SEM image, (b and c) TEM images, (d) HRTEM image, and (e) dark-field image of TA-NiFe@NCNT. Element maps for C, Ni and Fe are also shown in (e). | ||
To investigate the structure and composition of the different final products, XRD was first employed. As shown in Fig. 2a, the XRD patterns of TA-NiFe@NCNT, TA-NiFe@NCNT* (no glucose) and NiFe/C all contained sharp peaks at 44.1, 51.4 and 75.6°, which could readily be indexed to the (111), (200), and (220) reflections of a NiFe alloy (JCPDS Card 38-0419), in good agreement with HRTEM results for TA-NiFe@NCNT (Fig. 1d). These samples, as well as TA-NC, also showed a broad peak centered at 25° due to the (002) plane of graphitic carbon. Raman spectra (Fig. 2b) for all the samples showed two peaks at 1330 and 1580 cm−1, attributed to the D band (disordered sp3 carbon) and G band (graphitic sp2 carbon), respectively, of carbon. The ID/IG ratio for all samples was close to 1, meaning that all catalysts were semigraphitic and containing abundant defective sites. Fig. 2c shows N2 adsorption and desorption isotherms for TA-NiFe@NCNT, TA-NiFe@NCNT*, and TA-NC. All three samples exhibited a type IV isotherm with an obvious hysteresis loop, suggesting the presence of pores with a wide range of sizes in all samples. The BET specific surface areas determined for TA-NiFe@NCNT, TA-NiFe@NCNT*, and TA-NC were 392.6 m2 g−1, 260.9 m2 g−1 and 603.8 m2 g−1, respectively. The porous submicrotubes in TA-NiFe@NCNT accounts for its higher specific surface area relative to TA-NiFe@NCNT*.
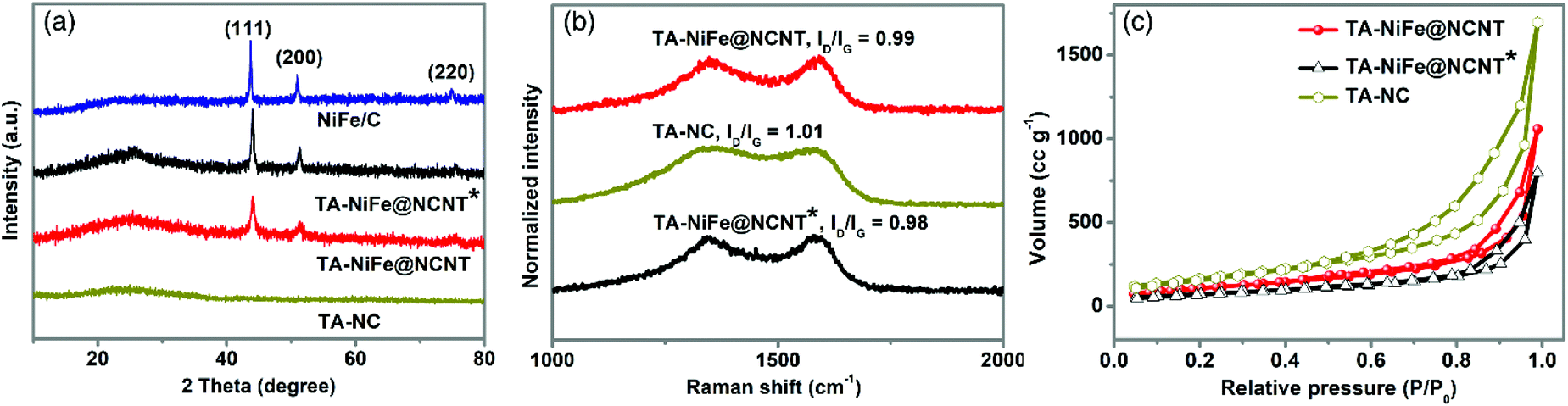 | ||
| Fig. 2 (a) XRD patterns for TA-NiFe@NCNT, TA-NiFe@NCNT*, TA-NC, and NiFe/C. (b) Raman spectra, and (c) N2 adsorption and desorption isotherms of TA-NiFe@NCNT, TA-NiFe@NCNT*, and TA-NC. | ||
XPS was employed to probe the surface chemistry of TA-NiFe@NCNT. The XPS survey spectrum in Fig. S6† revealed the presence of C, N, O, Ni, and Fe. The C 1s spectrum (Fig. 3a) was deconvoluted into three carbon contributions, C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (∼284.7 eV), C–N (∼285.8 eV) and CO (∼287 eV).37,38 The C–N feature, together with the high resolution N 1s spectrum (in Fig. 3b), confirmed that TA-NiFe@NCNT contained N-doped carbon nanotubes. The N 1s spectrum contained peaks due to pyridinic N (398.6 eV) and graphitic N (401.0 eV).39 The presence of pyridinic N has been shown previously to enhance the local electronic structure of carbon materials, thereby improving ORR activity.40 The Ni 2p XPS spectrum of TA-NiFe@NCNT (Fig. 3c), contained 3 sets of peaks, each in a characteristic 2:1 area ratio. Peaks at 853.1 and 870.6 eV correspond to metallic Ni (2p3/2 and 2p1/2, respectively). Peaks at higher binding energies to these features are due to Ni2+ (855.6 and 875.1 eV) and Ni2+ shake up satellites (860.1 and 879.8 eV). Similarly, the Fe 2p XPS spectrum of TA-NiFe@NCNT (Fig. 3d) contained peaks of 707.5 and 720.6 (2:1 area ratio) due to metallic Fe, as well as peaks at higher binding energies due to oxidized forms of iron. The XPS data for TA-NiFe@NCNT is consistent with the presence of metallic NiFe alloy nanoparticles (with partially oxidized surfaces) supported/encapsulated by nitrogen-doped carbon nanotubes.
C (∼284.7 eV), C–N (∼285.8 eV) and CO (∼287 eV).37,38 The C–N feature, together with the high resolution N 1s spectrum (in Fig. 3b), confirmed that TA-NiFe@NCNT contained N-doped carbon nanotubes. The N 1s spectrum contained peaks due to pyridinic N (398.6 eV) and graphitic N (401.0 eV).39 The presence of pyridinic N has been shown previously to enhance the local electronic structure of carbon materials, thereby improving ORR activity.40 The Ni 2p XPS spectrum of TA-NiFe@NCNT (Fig. 3c), contained 3 sets of peaks, each in a characteristic 2:1 area ratio. Peaks at 853.1 and 870.6 eV correspond to metallic Ni (2p3/2 and 2p1/2, respectively). Peaks at higher binding energies to these features are due to Ni2+ (855.6 and 875.1 eV) and Ni2+ shake up satellites (860.1 and 879.8 eV). Similarly, the Fe 2p XPS spectrum of TA-NiFe@NCNT (Fig. 3d) contained peaks of 707.5 and 720.6 (2:1 area ratio) due to metallic Fe, as well as peaks at higher binding energies due to oxidized forms of iron. The XPS data for TA-NiFe@NCNT is consistent with the presence of metallic NiFe alloy nanoparticles (with partially oxidized surfaces) supported/encapsulated by nitrogen-doped carbon nanotubes.
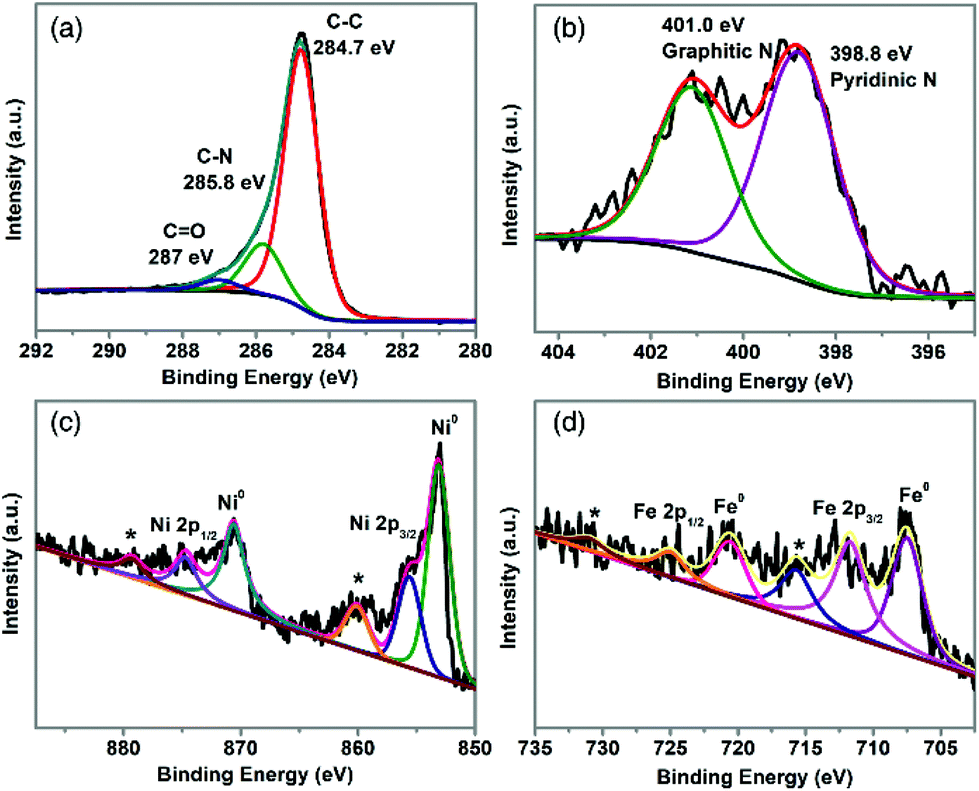 | ||
| Fig. 3 High resolution XPS data for TA-NiFe@NCNT. (a) C 1s region, (b) N 1s region, Ni 2p (c), and Fe 2p (d). | ||
The electrocatalytic performance of TA-NiFe@NCNT and the other electrocatalysts were evaluated in a three-electrode system with 0.1 M KOH as electrolyte. The CV curve (Fig. 4a) of TA-NiFe@NCNT collected at a scan rate of 100 mV s−1 in O2-saturated KOH solution showed an obvious oxygen reduction peak at ∼0.8 V, with the same peak being absent in the CV curve collected in N2-saturated alkaline solution. This demonstrates that TA-NiFe@NCNT was able to drive the oxygen reduction reaction. The ORR activities of the various catalysts were also studied using the LSV technique on a rotating disk electrode. A rotating speed of 1600 rpm and a scan rate of 10 mV s−1 were used for the measurements. As shown in Fig. 4b, the onset potential (0.93 V) and half-wave potential (E1/2, 0.81 V) of TA-NiFe@NCNT were slightly lower than those of the commercial Pt/C catalyst (onset potential = 0.99 V, and E1/2 = 0.82 V, respectively), though TA-NiFe@NCNT offered much better ORR performance than the other catalysts (Fig. 4b). To study the ORR kinetics of TA-NiFe@NCNT, K-L plots (Fig. 4d) were generated from LSV data collected at rotating speeds from 400 to 2025 rpm (in Fig. 4c). The analysis revealed that the electron transfer number during ORR on TA-NiFe@NCNT was 3.81, very close to a perfect four-electron pathway (O2 + 2H2O + 4e− → 4OH−). The excellent ORR performance of TA-NiFe@NCNT can be attributed to appropriate nitrogen doping level, high surface area and excellent conductivity of the NCNTs.
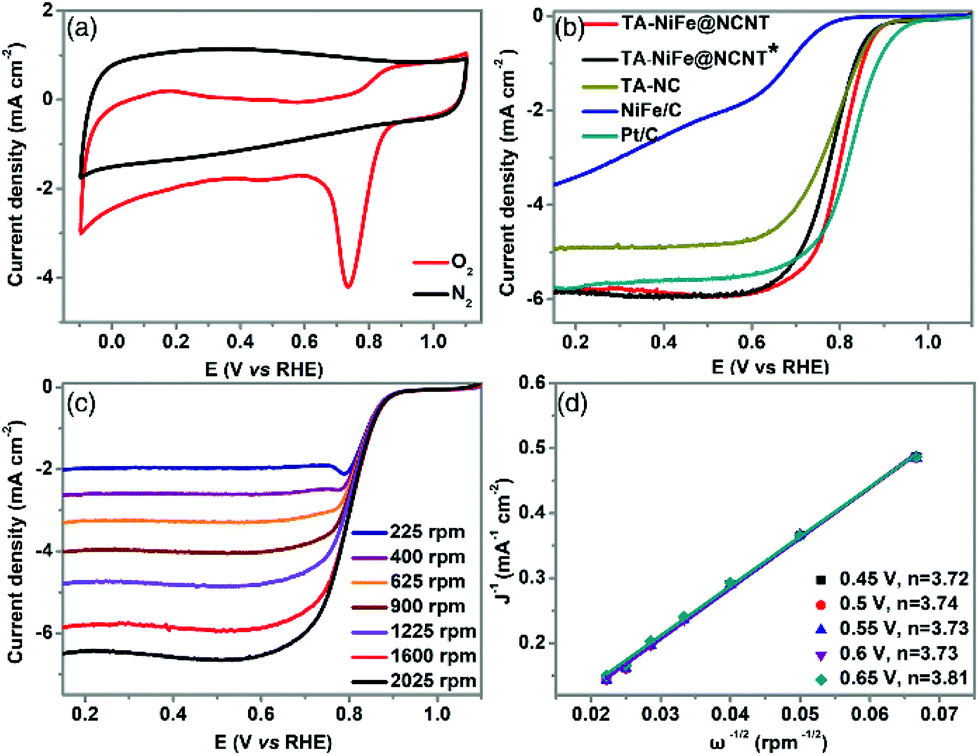 | ||
| Fig. 4 (a) CV curves for TA-NiFe@NCNT in O2 and N2 saturated 0.1 M KOH electrolytes collected at a scan rate of 100 mV s−1. (b) ORR LSV curves for TA-NiFe@NCNT, TA-NiFe@NCNT*, TA-NC, NiFe/C, and Pt/C collected at a rotation speed of 1600 rpm and a scan rate of 10 mV s−1. (c) ORR LSV curves for TA-NiFe@NCNT at various rotation speeds. (d) K-L plots for TA-NiFe@NCNT at different potentials derived from the corresponding ORR LSV curves. | ||
The presence of the NiFe alloy nanoparticles was expected to impart TA-NiFe@NCNT with good OER catalytic properties. The OER performance of TA-NiFe@NCNT and other electrocatalysts were evaluated by 95%-iR corrected LSV measurements at a scan rate of 5 mV s−1 in 0.1 M KOH solution. Fig. 5a shows that TA-NiFe@NCNT delivered an OER overpotential of only 310 mV at a current density of 10 mA cm−2, much lower than corresponding values determined for TA-NiFe@NCNT* (344 mV) and IrO2 (401 mV). The OER kinetics were explored through the Tafel plots in Fig. 5b. TA-NiFe@NCNT gave an OER Tafel slope of only 78 mV dec−1, much lower than that determined for IrO2 (244 mV dec−1), confirming the desirable OER catalytic activity of TA-NiFe@NCNT. The above results indicate that TA-NiFe@NCNT could serve as efficient platinum group metal-free catalyst for both ORR and OER. As shown in Table S1,† the catalytic performance of TA-NiFe@NCNT is comparable to most recently reported bifunctional catalysts. The excellent OER performance of TA-NiFe@NCNT can be attributed primarily to the presence of NiFe nanoparticles, since the control sample TA-NC delivered poor OER activity. The intimate contact between the NCNTs and the NiFe alloy nanoparticles contained within, benefitted electron transfer to the NiFe nanoparticles during OER, whilst the porous tubular assemblies facilitated fast mass transport of O2 away from the NiFe nanoparticles. Nyquist plots in Fig. S7† show that TA-NiFe@NCNT possessed a comparable semicircle to TA-NC, though a much smaller semicircle than TA-NiFe@NCNT* in the high frequency region. The data confirms that TA-NiFe@NCNT had a low charge transfer resistance. Furthermore, TA-NiFe@NCNT delivered a steeper slope than TA-NiFe@NCNT* in low-high frequency, consistent with faster mass transport in TA-NiFe@NCNT. The potential gap (ΔE) between the OER overpotential measured at 10 mA cm−2 and the ORR half-wave potential is an important indicator used to assess the performance of reversible oxygen electrodes. The smaller the potential gap, the closer a bifunctional electrocatalyst is to be an ideal reversible oxygen electrode. Fig. 5c shows that TA-NiFe@NCNT possessed the smallest ΔE (only 0.73 V) amongst the samples tested, outperforming a mixture Pt/C and IrO2 (0.81 V), and TA-NiFe@NCNT* (0.79 V). Therefore, TA-NiFe@NCNT represents an excellent bifunctional electrocatalyst for ORR and OER. This prompted further investigation of the effect of NiFe alloy nanoparticle loading on the ORR and OER activities of TA-NiFe@NCNT. The catalysts with different loading of NiFe alloy nanoparticle loadings were also prepared and tested. XRD patterns (Fig. S8†) for TA-NiFe@NCNT-5%, TA-NiFe@NCNT-3%, TA-NiFe@NCNT, and TA-NiFe@NCNT-0.5% were similar (Taking TA-NiFe@NCNT-5% as an example, 5% means the weight ratio of NiFe-LDH to g-C3N4. For TA-NiFe@NCNT, the weight ratio is 1%), with the NiFe alloy peaks intensifying with loading. ORR and OER results (Fig. S9 and S10†) confirmed that TA-NiFe@NCNT exhibited the best ORR and OER catalytic activities. Either side of 1 wt%, both the ORR and OER activities decreased. As shown in Fig. S11,† TA-NiFe@NCNT-5% suffered from serious aggregation of NiFe alloy nanoparticles. By contrast, TA-NiFe@NCNT-0.5% possessed fewer active sites for OER relative to TA-NiFe@NCNT, hence the lower activity.
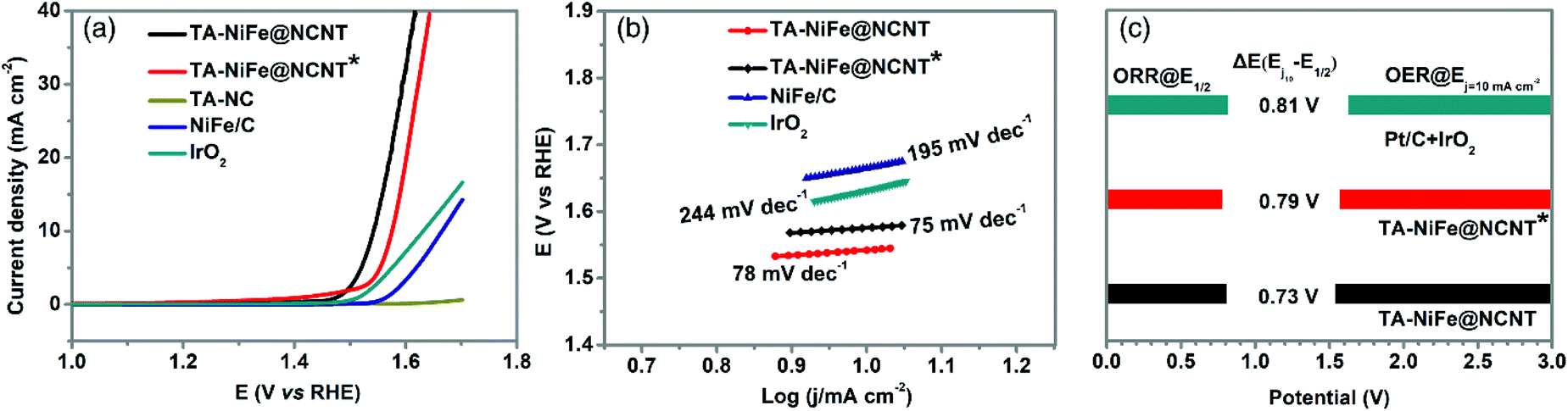 | ||
| Fig. 5 (a) OER LSV curve for various catalysts at a scan rate of 5 mV s−1 and a rotation speed of 1600 rpm after 95%-iR correction. (b) Tafel plots derived from the corresponding OER LSV curves. (c) The potential gap (ΔE) between the OER potential measured at a current density of 10 mA cm−2 and the ORR half-wave potential. | ||
Inspired by the high OER and ORR bifunctional activity and small potential gap of TA-NiFe@NCNT, a custom-built rechargeable zinc--air battery, consisting of a Zn plate (0.1 mm thickness) as one electrode, TA-NiFe@NCNT/carbon paper as other electrode, and an alkali electrolyte (6 M KOH and 0.2 M zinc acetate), was assembled to assess performance and stability of TA-NiFe@NCNT in such battery systems. Another battery was also prepared, using a mixture of Pt/C and IrO2 on carbon paper instead of TA-NiFe@NCNT/carbon paper. The charging-discharging properties of the different batteries were explored through the polarization curves shown in Fig. 6a. Results confirm that the battery constructed using TA-NiFe@NCNT had a comparable discharge voltage to that constructed using Pt/C and IrO2, but a lower charging voltage over a wide range of current densities. Accordingly, TA-NiFe@NCNT is very suitable for use as bifunctional ORR/OER catalyst in rechargeable metal-air batteries. The cycling performance of the batteries constructed using TA-NiFe@NCNT or Pt/C and IrO2 as electrode materials were tested at the current density of 25 mA cm−2. Fig. 6b shows that the rechargeable zincair battery built using TA-NiFe@NCNT demonstrated good stability over 170 h of testing, with no obvious voltage change over the testing period, and outperformed the cycling stabilities of the batteries constructed using the mixture of Pt/C and IrO2, and most of other reported catalysts (Table S2†). The superior cycling stability of TA-NiFe@NCNT likely results from strong interfacial coupling between NiFe alloy nanoparticles and the N-doped carbon nanotubes. The fact that the NiFe alloy nanoparticles were inside the NCNTs, as opposed to on the outer surface, prevents NiFe nanoparticle aggregation and enhances electrocatalyst stability. TEM was used to probe the morphology of TA-NiFe@NCNT after 170 h of charge and discharge cycling. TEM images in Fig. S12† displayed that NiFe alloys were still encapsulated by the graphitic carbon layers and the sizes and the lattice fringe spacing (0.21 nm) of NiFe alloys after cycling did not change obviously.
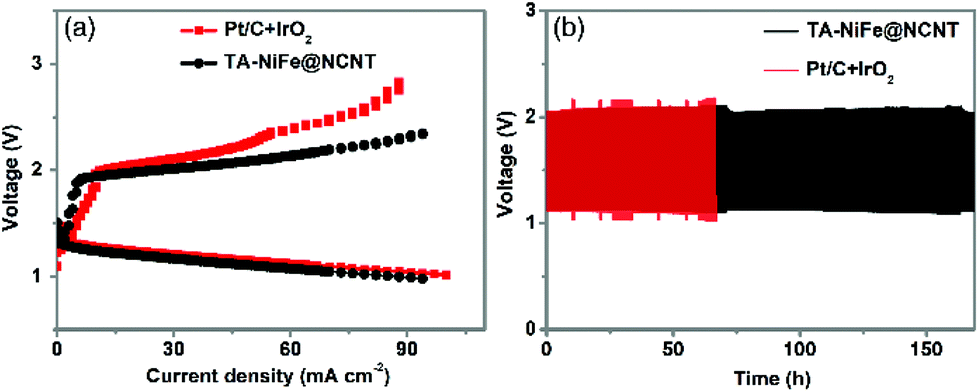 | ||
| Fig. 6 (a) Charge and discharge polarization curves of rechargeable zinc-air batteries. (b) Charge and discharge cycling curves of rechargeable zinc-air batteries at a current density of 25 mA cm−2. | ||
Conclusions
In summary, a novel bifunctional electrocatalyst was developed for ORR and OER. The electrocatalyst, denoted herein as TA-NiFe/NCNT, comprised tubular assemblies of nitrogen-doped carbon nanotubes with NiFe alloy nanoparticles in the individual NCNTs. The hierarchical porosity of TA-NiFe/NCNT, featuring 200 nm macropores from the g-C3N4 nanotube soft template and the mesopores in the NCNTs, ensured facile mass transport during ORR and OER. By adjusting the amount of NiFe alloy nanoparticles in the assembly of TA-NiFe@NCNT, the catalytic performance for ORR and OER can be rationally optimized. Due to synergies realized by combining porosity, NCNT for ORR and NiFe alloy nanoparticles for OER, TA-NiFe/NCNT outperformed its precious metal counterparts (Pt/C and IrO2, respectively) when applied in Zn-air batteries. The battery fabricated using TA-NiFe@NCNT delivered better stability and smaller voltage gap than the counterpart constructed using a mixture of Pt/C and IrO2. Therefore, TA-NiFe@NCNT represents a very promising, low-cost bifunctional catalyst for rechargeable zinc-air batteries. This sacrificial-template strategy adopted here, based on the use of g-C3N4 nanotubes as a soft template and nitrogen source of N-doped carbon catalyst fabrication, is expected to be widely used in the future in electrocatalyst development for reactions involving oxygen.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Key Projects for Fundamental Research and Development of China (2017YFA0206904, 2017YFA0206900, 2018YFB1502002, 2016YFB0600901), the National Natural Science Foundation of China (51825205, 51772305, 51572270, U1662118, 21871279, 21802154, 21902168), the Beijing Natural Science Foundation (2191002, 2182078, 2194089), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17000000), the Royal Society-Newton Advanced Fellowship (NA170422), the International Partnership Program of Chinese Academy of Sciences (GJHZ1819, GJHZ201974), the Beijing Municipal Science and Technology Project (Z181100005118007), the K. C. Wong Education Foundation and the Youth Innovation Promotion Association of the CAS. GINW acknowledges funding support from Greg and Kathryn Trounson (via a generous philanthropic donation), the Energy Education Trust of New Zealand and the MacDiarmid Institute for Advanced Materials and Nanotechnology.Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- P. Sapkota and H. Kim, J. Ind. Eng. Chem., 2009, 15, 445–450 CrossRef CAS.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- B. Xia, Y. Yan, N. Li, H. Wu, X. Lou and X. Wang, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS PubMed.
- W. Wei, Y. Tao, W. Lv, F. Y. Su, L. Ke, J. Li, D. W. Wang, B. Li, F. Kang and Q. H. Yang, Sci. Rep., 2014, 4, 6289 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- L. Shang, H. Yu, X. Huang, T. Bian, R. Shi, Y. Zhao, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 1668–1674 CrossRef CAS PubMed.
- Y. Xiao, Z. Cheng, Y. Zhao and L. Qu, Nanoscale, 2018, 10, 15706–15713 RSC.
- W. Lei, Y.-P. Deng, G. Li, Z. P. Cano, X. Wang, D. Luo, Y. Liu, D. Wang and Z. Chen, ACS Catal., 2018, 8, 2464–2472 CrossRef CAS.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 RSC.
- J. Meng, C. Niu, L. Xu, J. Li, X. Liu, X. Wang, Y. Wu, X. Xu, W. Chen, Q. Li, Z. Zhu, D. Zhao and L. Mai, J. Am. Chem. Soc., 2017, 139, 8212–8221 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- S. Wang, D. Yu and L. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS PubMed.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmekova and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372–4376 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- Q. Wang, L. Shang, R. Shi, X. Zhang, Y. Zhao, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2017, 7, 1700467 CrossRef.
- Q. Wang, L. Shang, R. Shi, X. Zhang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Nano Energy, 2017, 40, 382–389 CrossRef CAS.
- C. L. Huang, X. F. Chuah, C. T. Hsieh and S. Y. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 24096–24106 CrossRef CAS PubMed.
- L. Xu, F.-T. Zhang, J.-H. Chen, X.-Z. Fu, R. Sun and C.-P. Wong, ACS Appl. Energy Mater., 2018, 1, 1210–1217 CrossRef CAS.
- J. Jin, J. Xia, X. Qian, T. Wu, H. Ling, A. Hu, M. Li and T. Hang, Electrochim. Acta, 2019, 299, 567–574 CrossRef CAS.
- Z. Bian, S. Das, M. H. Wai, P. Hongmanorom and S. Kawi, ChemPhysChem, 2017, 18, 3117–3134 CrossRef CAS PubMed.
- M.-S. Wu and Z.-Z. Ceng, Electrochim. Acta, 2016, 191, 895–901 CrossRef CAS.
- X. Zhang, H. Xu, X. Li, Y. Li, T. Yang and Y. Liang, ACS Catal., 2015, 6, 580–588 CrossRef.
- D. Bin, B. Yang, C. Li, Y. Liu, X. Zhang, Y. Wang and Y. Xia, ACS Appl. Mater. Interfaces, 2018, 10, 26178–26187 CrossRef CAS PubMed.
- S. Q. Ci, S. Mao, Y. Hou, S. M. Cui, H. Kim, R. Ren, Z. H. Wen and J. H. Chen, J. Mater. Chem. A, 2015, 3, 7986–7993 RSC.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. Waterhouse, Y. Zhao, C. Zhou, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- X. Wang, C. Zhou, R. Shi, Q. Liu, G. I. N. Waterhouse, L. Wu, C.-H. Tung and T. Zhang, Nano Res., 2019, 12, 2385–2389 CrossRef CAS.
- R. Liu, D. Wu, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- S. Chen, J. Cheng, L. Ma, S. Zhou, X. Xu, C. Zhi, W. Zhang, L. Zhi and J. A. Zapien, Nanoscale, 2018, 10, 10412–10419 RSC.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- L. Qian, Z. Lu, T. Xu, X. Wu, Y. Tian, Y. Li, Z. Huo, X. Sun and X. Duan, Adv. Energy Mater., 2015, 5, 1500245 CrossRef.
- H.-X. Zhong, J. Wang, Q. Zhang, F. Meng, D. Bao, T. Liu, X.-Y. Yang, Z.-W. Chang, J.-M. Yan and X.-B. Zhang, Adv. Sustainable Syst., 2017, 1, 1700020 CrossRef.
- Z. Lin, G. Waller, Y. Liu, M. Liu and C.-P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS.
- Y. Yuan, L. Yang, B. He, E. Pervaiz, Z. Shao and M. Yang, Nanoscale, 2017, 9, 6259–6263 RSC.
- W. Niu, L. Li, X. Liu, N. Wang, J. Liu, W. Zhou, Z. Tang and S. Chen, J. Am. Chem. Soc., 2015, 137, 5555–5562 CrossRef CAS PubMed.
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. M. Lu, Adv. Mater., 2013, 25, 998–1003 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02486d |
| This journal is © The Royal Society of Chemistry 2020 |