
Tuning the electronic energy level of covalent organic frameworks for crafting high-rate Na-ion battery anode†
Sattwick
Haldar
ab,
Dhananjayan
Kaleeswaran
a,
Deepak
Rase
ab,
Kingshuk
Roy
ac,
Satishchandra
Ogale
 bcd and
Ramanathan
Vaidhyanathan
*ab
bcd and
Ramanathan
Vaidhyanathan
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research, Pune, India. E-mail: vaidhya@iiserpune.ac.in
bCentre for Energy Science, Indian Institute of Science Education and Research, Pune, India
cDepartment of Physics, Indian Institute of Science Education and Research, Pune, India
dResearch Institute for Sustainable Energy (RISE), TCG-Centres for Research and Education in Science and Technology (TCG-CREST), Sector V, Salt Lake, Kolkata 700091, India
First published on 25th June 2020
Abstract
Crystalline Covalent Organic Frameworks (COFs) possess ordered accessible nano-channels. When these channels are decorated with redox-active functional groups, they can serve as the anode in metal ion batteries (LIB and SIB). Though sodium's superior relative abundance makes it a better choice over lithium, the energetically unfavourable intercalation of the larger sodium ion makes it incompatible with the commercial graphite anodes used in Li-ion batteries. Also, their sluggish movement inside the electrodes restricts the fast sodiation of SIB. Creating an electronic driving force at the electrodes via chemical manipulation can be a versatile approach to overcome this issue. Herein, we present anodes for SIB drawn on three isostructural COFs with nearly the same Highest Occupied Molecular Orbitals (HOMO) levels but with varying Lowest Unoccupied Molecular Orbitals (LUMO) energy levels. This variation in the LUMO levels has been deliberately obtained by the inclusion of electron-deficient centers (phenyl vs. tetrazine vs. bispyridine-tetrazine) substituents into the modules that make up the COF. With the reduction in the cell-potential, the electrons accumulate in the anti-bonding LUMO. Now, these electron-dosed LUMO levels become efficient anodes for attracting the otherwise sluggish sodium ions from the electrolyte. Also, the intrinsic porosity of the COF favors the lodging and diffusion of the Na+ ions. Cells made with these COFs achieve a high specific capacity (energy density) and rate performance (rapid charging–discharging), something that is not as easy for Na+ compared to the much smaller sized Li+. The bispyridine-tetrazine COF with the lowest LUMO energy shows a specific capacity of 340 mA h g−1 at 1 A g−1 and 128 mA h g−1 at a high current density of 15 A g−1. Only a 24% drop appears on increasing the current density from 0.1 to 1 A g−1, which is the lowest among all the top-performing COF derived Na-ion battery anodes.
New conceptsThough sodium-ion batteries are cost-efficient, the incompatibility of sodium ions with the commercial graphitic anodes and their sluggish ionic mobility limits their usage. Herein, we propose a novel concept of using covalent organic framework pre-coded with pyridine-tetrazine units that deliver low-energy LUMO (anti-bonding) levels. Such electronic levels favour the accumulation of electrons under an applied potential and these electron-dosed LUMO levels provide an extra driving force for the otherwise sluggish Na+ ions to move into the anode from the electrolyte. Also, the strategically positioned heteroatoms offer tailor-made coordination sites to store the Na+ ions. At the same time, the mesoporous structure of the COF provides easy diffusion to these active sites, leading to excellent rate performance. The concept of atomically engineering the electronic levels of the COF to make them superior anodes in Na-ion batteries is quite versatile. The crystalline and modular structure of COF enables a practical experimental–computational approach, leading to futuristic batteries. |
1. Introduction
COFs are crystalline polymers with uniform nanopores.1–5 The out-of-plane π–π stacking of the aromatic rings between the COF layers generates hollow cylindrical channels running along the c-direction.6–10 Their pore size and shape can be tuned by choosing monomers of desired length and geometry. Meanwhile, their organic backbone favors the stoichiometric incorporation of electrochemically active sites into the framework. This molecular-level designability gives a chance to decorate the entire wall of their cylindrical pores with redox-active functional groups. Their crystalline structure ensures a periodic distribution of such active sites, while the sizeable nanopores ensures easy access to such sites. Also, the high surface area of COF helps to store electrical charge via electrical double layer formation.11–16 These redox-active COFs become apt electrode candidates for metal-ion batteries,17–28 particularly in providing the required electronic dynamo for sluggish ions such as Na+. The superior anodic performance of COF in Li-ion batteries with specific capacities surpassing that of commercial graphite has been recently demonstrated.18,20,29–31Nevertheless, graphite is the most used anode in commercial Li-ion batteries. It stores Li+ ions by inserting them into its inter-layer spaces. Unfortunately, graphite does not show such favorability for Na+ ions.32–34 In contrast, even the larger K+ ions can be inserted into the graphite interlayers with relative ease.35 The problem arises due to the lack of thermodynamic favorability for the Na+ ions to reside in the interlayer spaces of graphite.36 This becomes evident from the enthalpy associated with the compounds that form during the intercalation of these different ions into graphite. The enthalpy of formation of LiC6 and KC8 was found to be −16.4 and −27.5 kJ mol−1, respectively. Meanwhile, NaC6 and NaC8 are unstable with positive enthalpies of formation (+20.8 and +19.9 kJ mol−1).37 This is due to the size mismatch. Interestingly, including larger solvated co-ions with Na+ ions improves the latter's size-fit but it does bring down the energy density as many sites get occupied by the “non-contributing” solvated guest.38 Also, since the Na+ ions are larger than Li+ ions, the intercalation of Na+ into small lattice structures (which are desirable for high-energy batteries) might be accompanied by structural and volume changes. In many cases, these changes destroy the contacts between the electrode and the current collector, leading to undesirable capacity fading during the Na-ion battery cycling. However, in nanostructured COFs, there are no such issues, which makes them more suitable porous hosts with diverse active sites and accommodative nano spaces.
On the kinetics front, the mere size and the associated bulk of the Na+ ions compared to Li+ ions certainly make them sluggish (Li+: 0.76 Å vs. Na+: 1.02 Å and the ionic mass).39–41 Hence, introducing a chemical drive to improve the ionic-mobility of Na+ ions would directly impact the battery's rate performance. In fact, with this aim, hard carbon doped with heteroatoms such as B, N, S, and P has been employed as anode in Na-ion batteries with reasonable success (B doped: 278 mA h g−1@0.1 A g−1, N doped: 154 mA h g−1@15 A g−1, S doped: 182 mA h g−1@3.2 A g−1, P doped: 108 mA h g−1@20 A g−1).42–49 Even then, the relative drop in the specific capacity with increasing current density (termed as the rate performance) is a concern in these improved systems. Meanwhile, hard carbons with a 3D mesoporous structure have been far more successful (>280 mA h g−1@100 mA g−1).50,51 Yet, the atomic-manipulation assisted enhancement of anodic performance of such hard carbons, particularly at high current density, is primarily hampered by their amorphous structure.52 Also, they carry a major synthetic disadvantage: the high-temperature pyrolysis-assisted doping of heteroatoms in such carbonaceous materials creates random ordering of the heteroatoms, which makes their reproducibility and scale-up difficult.42,45,48 In contrast, the COF synthesis involves stoichiometric combinations of monomers under mild synthetic conditions, which naturally circumvents this arbitrary doping and a periodic lattice with perfect positioning of active heteroatoms is possible. Recently, carbonyl functionalized COF with noticeable specific capacity at high current density (135 mA h g−1@10 A g−1) in an SIB was reported.22 Independently, the acid-delamination of COF layers was shown to improve the specific capacity in an SIB (200 mA h g−1@5 A g−1).21
Herein, we have developed mesoporous COFs for favourable interactions with Na+ ions and, as a convenient approach, have manipulated their chemical groups so as to gain electronic improvement in driving the Na+ ions into their pores. More specifically, we have considered three large mesoporous COFs (phenyl vs. tetrazine vs. bispyridine-tetrazine) with systematically lowered LUMO levels so as to gain more electron accumulation under an applied potential. This in-operation charged anode forthwith is more attractive for the heavier Na+ ions from the electrolyte through both pseudocapacitance and classical double layer formation. Importantly, the ordered accessible pores make the entire COF participate in the anodic action and not merely its surface. This yields a high specific capacity of 340 mA h g−1@1 A g−1. Its low charge transfer resistance is reflected in the excellent rate performance (only a 24% drop in the specific capacity on increasing from 0.1 to 1 A g−1). Also, the favorable charge degeneracy throughout the nanowalls of the framework enhances the stability even at very high current inputs (128 mA g−1@15 A g−1). It brings excellent cycling stability up to 1400 cycles, with only 8% capacity fading. Interestingly, the phenyl analog that lacks the N-heteroatom in its backbone does not show such a high performance. This notion of atomic-level tuning of the electronic energy level of the COFs for easy Na+ insertion is a promising route to design next-generation anode materials for SIBs.
2. Results and discussion
Synthesis and structural-modelling of IISERP-COF16 (1), IISERP-COF17 (2), and IISERP-COF18 (3)
IISERP-COF16 (1), IISERP-COF17 (2), and IISERP-COF18 (3) have (3+2) frameworks formed by reacting a C3 symmetry trialdehyde [2,4,6-trihydroxybenzene-1,3,5-tricarbaldehyde] with three different C2 symmetry diamine containing terphenyl [(1,1′:4′,1′′-terphenyl)-4,4′′-diamine], s-tetrazine [4,4′-(1,2,4,5-tetrazine-3,6-diyl)dianiline] and s-tetrazine bispyridine [5,5′-(1,2,4,5-tetrazine-3,6-diyl)bis(pyridine-2-amine)] units (Scheme S1 (ESI†) and Fig. 1A(i)–(iii)). The purity of all the monomers was confirmed using solution-state 1H and 13C NMR (Fig. S1–S6 and Tables S1, S2, ESI†). The completion of the polycondensation reactions was confirmed by 13C solid-state NMR studies and IR data analysis of the COFs (Fig. S7–S10 and Table S3, ESI†). With the increase in the nitrogen contents in the framework, the color of the COFs becomes dark brown from golden yellow (Scheme S1, ESI†). While 1 and 2 are known, we are reporting the bispyridine-tetrazine COF (3) for the first time.53,54 These three COFs constitute a family that enables a systematic comparison of the electronic structure with the anodic performance in an SIB.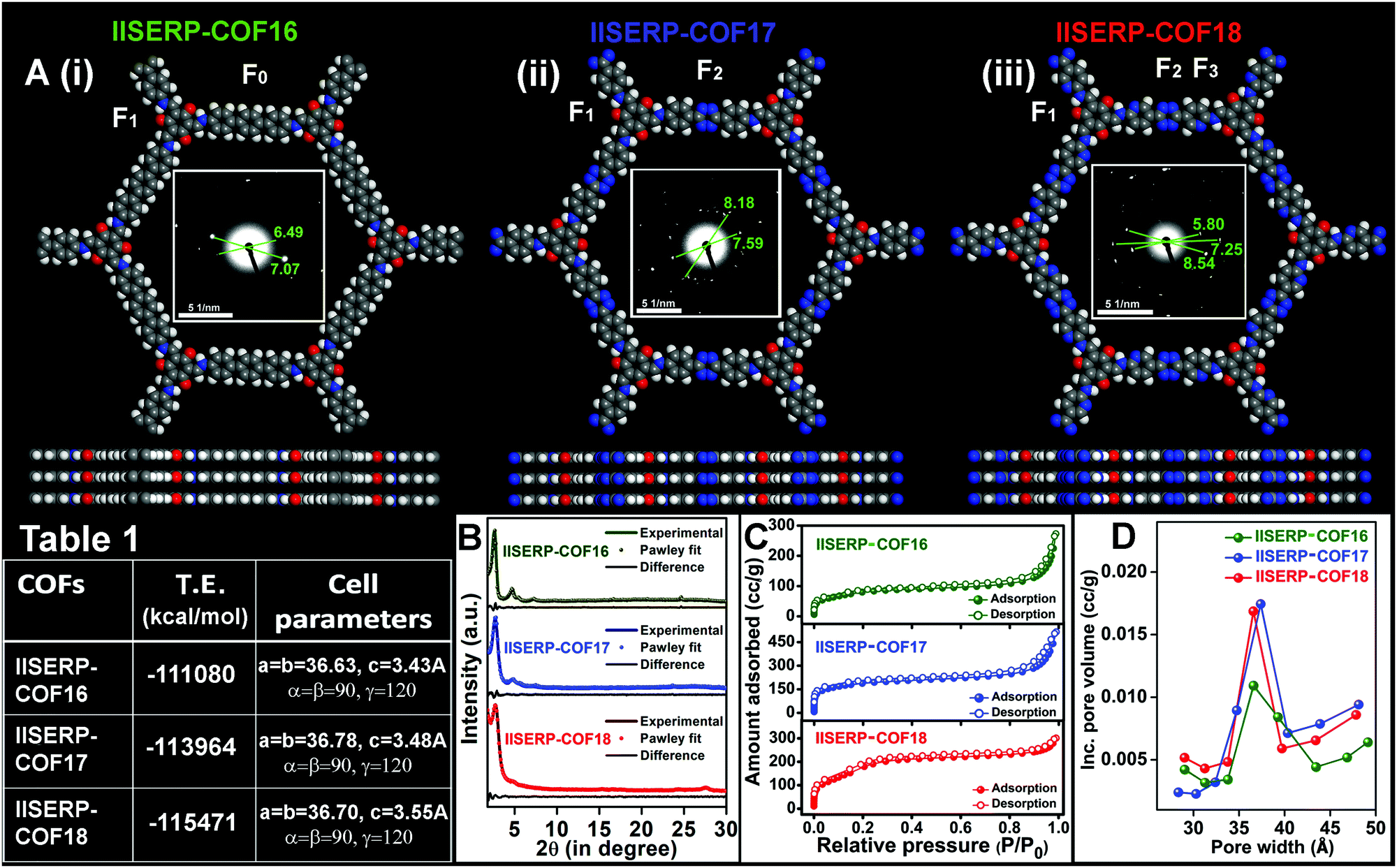 | ||
| Fig. 1 (A) Modeled structures of (i) IISERP-COF16, (ii) IISERP-COF17, and (iii) IISERP-COF18. They prefer an eclipsed configuration with an AA… stacking. (inset) SAED patterns of the COFs observed for higher angle diffraction of the [00l] planes. The units of linear distances between two bright spots of the SAED patterns are in ‘1/nm’. F0, F1, F2, and F3 are the redox-active functional groups present in the IISERP-COFs. Table 1: Total energy and unit cell parameters of geometry-optimized COFs. (B) Pawley fits using the experimental PXRD patterns. (C) Nitrogen (N2) sorption isotherms of the COFs measured at 77 K. (D) Pore size distribution plots of the COFs obtained from BJH fit to the desorption branch. | ||
The structural models for all the three COFs were built using Materials Studio v. 6.0.55–57 An initial indexing and space group search was performed using experimental powder X-ray diffraction (PXRD) employing the Reflex module. All the three PXRD patterns were indexed to a hexagonal cell. A space group search yielded P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) and P6/m, both within a well-acceptable figure of merit (Table 1). Atomic manipulations were carried out in a cell built using the higher symmetry P6/m setting to obtain an initial polymeric model of the COF with apt connectivity. The final structures were optimized with a periodic tight-binding DFT method (DFTB). Total energies were extracted from the DFTB optimizations (1: eclipsed = −111
and P6/m, both within a well-acceptable figure of merit (Table 1). Atomic manipulations were carried out in a cell built using the higher symmetry P6/m setting to obtain an initial polymeric model of the COF with apt connectivity. The final structures were optimized with a periodic tight-binding DFT method (DFTB). Total energies were extracted from the DFTB optimizations (1: eclipsed = −111![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 080; 2: eclipsed = −113964; 3: eclipsed = −115471 kcal per mol per unit-cell). The Pawley refinements of the experimental PXRDs against their optimized models yield excellent fits for all the COFs (Fig. 1B). The presence of strategically positioned keto groups of the phloroglucinol units enables strong O⋯H–N⋯intra-layer hydrogen bonds with the enamine form of the connecting Schiff bonds along the ab-plane (Fig. S11, ESI†).58–60 The three-dimensional structures of the IISERP-COFs have π-stacked columns of resorcinol units and the columns of benzene (for 1), s-tetrazine (for 2), and bispyridine s-tetrazine rings (for 3) covalently linked by Schiff bonds (Fig. 1A). This creates uniform one dimensional (1D) nano-channels with pores of size ∼38 Å (factoring the van der Waals radii of the atoms) running along the c-axis. Experimental PXRD patterns show high intensity peaks located at 2θ of 2.65° (for 1), 2.55° (for 2), and 2.6° (for 3) for the (100) reflections (Fig. 1B). The (001) reflection corresponding to layer-stacking is observed at a 2θ of ∼26.5°. From the selected area electron diffraction (SAED) patterns, the higher angle reflections are observed (Fig. 1A, insets). The SAED ring diameter (2R) ∼ 6.0 1/nm corresponds to inter-planar separation distances (3.4 Å) of the eclipsed configuration of the refined structure. This further confirms the crystallinity of this family of polycrystalline covalent organic frameworks.61–63 Adsorption–desorption isotherms of N2 at 77 K yielded a completely reversible type-2 isotherm for 1, 2, and 3, which proves their expected mesoporous structure (Fig. 1C and Fig. S12–S14, ESI†).64 A Barrett–Joyner–Halenda (BJH) fit to the desorption branch reveals the presence of uniform ∼36.6, 36.9, and 36.5 Å sized pores in 1, 2, and 3, respectively (Fig. 1D). These COFs have a higher Langmuir surface area (920 m2 g−1 for 1; 1452 m2 g−1 for 2; 1745 m2 g−1 for 3) than the Brunauer–Emmett–Teller (BET) surface area (Fig. S12–S14 and Table S4, ESI†).53,55,57 All the powdered samples were subjected to Soxhlet washing using a boiling THF/DMF mixture (48 h) to get rid of any soluble oligomers. The PXRD and porosity data were reproduced well across different batches, confirming that the samples do not have any significant impurity phases.
080; 2: eclipsed = −113964; 3: eclipsed = −115471 kcal per mol per unit-cell). The Pawley refinements of the experimental PXRDs against their optimized models yield excellent fits for all the COFs (Fig. 1B). The presence of strategically positioned keto groups of the phloroglucinol units enables strong O⋯H–N⋯intra-layer hydrogen bonds with the enamine form of the connecting Schiff bonds along the ab-plane (Fig. S11, ESI†).58–60 The three-dimensional structures of the IISERP-COFs have π-stacked columns of resorcinol units and the columns of benzene (for 1), s-tetrazine (for 2), and bispyridine s-tetrazine rings (for 3) covalently linked by Schiff bonds (Fig. 1A). This creates uniform one dimensional (1D) nano-channels with pores of size ∼38 Å (factoring the van der Waals radii of the atoms) running along the c-axis. Experimental PXRD patterns show high intensity peaks located at 2θ of 2.65° (for 1), 2.55° (for 2), and 2.6° (for 3) for the (100) reflections (Fig. 1B). The (001) reflection corresponding to layer-stacking is observed at a 2θ of ∼26.5°. From the selected area electron diffraction (SAED) patterns, the higher angle reflections are observed (Fig. 1A, insets). The SAED ring diameter (2R) ∼ 6.0 1/nm corresponds to inter-planar separation distances (3.4 Å) of the eclipsed configuration of the refined structure. This further confirms the crystallinity of this family of polycrystalline covalent organic frameworks.61–63 Adsorption–desorption isotherms of N2 at 77 K yielded a completely reversible type-2 isotherm for 1, 2, and 3, which proves their expected mesoporous structure (Fig. 1C and Fig. S12–S14, ESI†).64 A Barrett–Joyner–Halenda (BJH) fit to the desorption branch reveals the presence of uniform ∼36.6, 36.9, and 36.5 Å sized pores in 1, 2, and 3, respectively (Fig. 1D). These COFs have a higher Langmuir surface area (920 m2 g−1 for 1; 1452 m2 g−1 for 2; 1745 m2 g−1 for 3) than the Brunauer–Emmett–Teller (BET) surface area (Fig. S12–S14 and Table S4, ESI†).53,55,57 All the powdered samples were subjected to Soxhlet washing using a boiling THF/DMF mixture (48 h) to get rid of any soluble oligomers. The PXRD and porosity data were reproduced well across different batches, confirming that the samples do not have any significant impurity phases.
The characteristic carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching frequency (1718 cm−1) of the triformyl-phloroglucinol was red-shifted (1630 cm−1) and the N–H stretching modes (3388, 3317, 3196 cm−1) of the primary amine disappeared with the formation of the COFs (Fig. S10 and Table S3, ESI†). We notice from the IR spectra that the solid powders of the as-synthesized 1 exists predominantly in the β-ketoenamine form, which arises from the tautomerism between the Schiff bonds (–CN–) and the carbonyl (–CO) units but 2 and 3 also show the presence of the enolic forms.59,60 The presence of appropriate peaks in the 13C solid-state NMR spectra of the COFs corresponding to the keto group of β-keto enamine form (185–190 ppm), pyridine (143–148 ppm), and tetrazine (168 ppm) reveals the functional group integrity maintained during the poly-condensed polymeric structure formation (Fig. S7–S9, ESI†).12,55 The strong interlayer H-bond formation of the β-ketoenamine form enhances the chemical and thermal stability of 1 (stable up to 410 °C). However, the tetrazine containing COFs 2 and 3 exhibit relatively lower thermal stability (gradual weight loss in the thermogravimetric analysis (TGA) curve commences at 280 °C) (Fig. S15, ESI†). All the COFs show ample chemical stability, as confirmed by the PXRD of the samples that were boiled in DMF and treated with acid and base (6 M) (Fig. S16, ESI†).
O) stretching frequency (1718 cm−1) of the triformyl-phloroglucinol was red-shifted (1630 cm−1) and the N–H stretching modes (3388, 3317, 3196 cm−1) of the primary amine disappeared with the formation of the COFs (Fig. S10 and Table S3, ESI†). We notice from the IR spectra that the solid powders of the as-synthesized 1 exists predominantly in the β-ketoenamine form, which arises from the tautomerism between the Schiff bonds (–CN–) and the carbonyl (–CO) units but 2 and 3 also show the presence of the enolic forms.59,60 The presence of appropriate peaks in the 13C solid-state NMR spectra of the COFs corresponding to the keto group of β-keto enamine form (185–190 ppm), pyridine (143–148 ppm), and tetrazine (168 ppm) reveals the functional group integrity maintained during the poly-condensed polymeric structure formation (Fig. S7–S9, ESI†).12,55 The strong interlayer H-bond formation of the β-ketoenamine form enhances the chemical and thermal stability of 1 (stable up to 410 °C). However, the tetrazine containing COFs 2 and 3 exhibit relatively lower thermal stability (gradual weight loss in the thermogravimetric analysis (TGA) curve commences at 280 °C) (Fig. S15, ESI†). All the COFs show ample chemical stability, as confirmed by the PXRD of the samples that were boiled in DMF and treated with acid and base (6 M) (Fig. S16, ESI†).
Under a Field Emission Scanning Electron Microscope (FE-SEM), 1 appears as large smooth-surfaced flakes that form a stacked microstructure (Fig. S17, ESI†). On the other hand, 2 has hexagonal flakes, which further aggregate into microstructures resembling petals. 3 has a thick fibrous morphology. In all the cases, the SEM images corroborate with the morphologies observed under the High Resolution-Transmission Electron Microscope (HR-TEM) (Fig. S18, ESI†). The stacking of the layers becomes visible when viewed at the edges or the thinner portion of the sheets. High resolution images from the HR-TEM showed the presence of lattice fringes, indicating high crystallinity of these COFs (Fig. S19–S21 and Table S5, ESI†). At higher magnifications, sub-micron sized pores could be observed across the whole surface of the COF flakes (Fig. S22, ESI†).
The interlayer spacing (3.3 Å) was determined from the cross-sectional view observed for a few of the crystallites drop-casted on the TEM grid. It matched well with the layer separation distances determined from the energy and geometry-optimized structure. Moreover, distinct SAED patterns of COFs at the 5 1/nm scale confirm the presence of diffraction of the [00l] planes at a higher angle. The lower angle reflections are merged in the smaller diameter range of the SAED, closer to the bright center of the SAED image (Fig. 1A (insets) and Fig. S19–S21, ESI†).61–63,65
Electronic energy levels of the COFs
With the increase in the nitrogen content in the COF backbone, the color of the isostructural COFs changes from golden yellow to brown (Fig. 2A and B). Concomitantly, the Ultraviolet (UV)-visible absorption maxima shifts from lower wavelength to higher wavelength as we go from 1 to 3 (Fig. 2C). Each of the UV band has a long tail in the higher wavelength region, which usually contributes majorly to the color of the COFs. To gain more evidence about the color change with the introduction of the nitrogenous aromatic ring, we estimated the bandgaps using Tauc plots (Fig. 2D).18,66 A continuous decrease in the bandgap from 2.75 to 2.51 to 2.20 eV has been observed with the increase in the color intensity of the COFs. To add further, the band-structure/energy-levels were calculated from the electrochemical methods, namely, cyclic voltammetry (CV). To avoid any interference, the CV measurements were performed in a non-aqueous electrolyte medium (t-butyl-ammonium-hexafluorophosphate dissolved in acetonitrile) using non-aqueous Ag/Ag+ reference and platinum flag counter electrodes (Fig. 2E and Fig. S23, S24, ESI†). Slow scan rate (50 mV s−1) in the potential window of −1.8 to +2.2 V was employed to scrutinize the electrochemical oxidation–reduction of the COFs. The highest oxidation potential provides the energy required to take out one electron from the HOMO, whereas the lowest reduction potential corresponds to the energy needed to provide one electron to the LUMO.67,68 These frontier orbitals precisely define the HOMO–LUMO energy levels of the COFs with respect to NHE (Normal Hydrogen Electrode), which is calculated by converting the potential obtained with respect to Ag–AgCl (Fig. 2F and Fig. S24, Table S6, ESI†). | ||
| Fig. 2 (A) Building blocks of polymeric COFs showing the presence of electron-rich and electron-deficient centers. (B) A photograph showing the color of the COFs under visible light. (C) UV-visible spectra of the COFs showing the absorption maxima. (D) Bandgaps of the COFs evaluated from the Tauc plot using UV-visible absorption spectra. (E) CVs in a non-aqueous three-electrode system showing the oxidation and reduction potentials of the COFs. (F) HOMO–LUMO energy levels of the COFs and their respective bandgaps evaluated from the CV measurements. | ||
Thus, the electrochemically determined bandgaps follow the same trend as the optical bandgap with some differences in their absolute values. Interestingly, the oxidation potentials of these COFs were nearly the same but the reduction potentials continuously go to a more negative value with the introduction of the s-tetrazine and the bispyridine-s-tetrazine ring. Thus, without many alterations in the condensed HOMO levels, the LUMO energy levels get more stabilized to lower energy levels with the inclusion of nitrogen atoms in the COF framework. The lowering of the LUMO energy levels brings out the possibility of facile reduction of the relatively electron-deficient tetrazine and pyridine moieties. In 3, the conjugation of the lone-pair on the pyridine ring with the tetrazine units assists the smooth electron transfer in between electron-deficient tetrazine and carbonyl units of the phloroglucinol units (Fig. 2A). This makes 3 assume the lowest LUMO levels among the three COFs.
Importantly, the position of the pyridine nitrogen (β-position w.r.t. the hydroxyl moiety) is crucial for gaining the maximum conjugation advantage. The relative lowering of the LUMO energy as we move from COF 1 to 3 is quantitatively expressed by how far the reduction potential shifts on the negative axis of the CV (Fig. 2E and Fig. S23, ESI†). Thus, 3, having the electron-accepting LUMO levels sitting at substantially lowered energy, carries the true potential to be the anode for any ion battery.69,70
General principle of an SIB half-cell
In a typical SIB half-cell, the Na-metal plate is employed as an Na+ ion source, which gives an OCV for Na/Na+ of 2.75 V (Fig. 3). When a negative potential is applied to the anode, it lowers the overall potential of the cell from the OCV. This potential difference favors Na → Na+ oxidation. Thus, the generated Na+ ions from the electrolyte combine with the electrons at the anode surface. However, the success lies in making this operation occur at a lower potential and in making the Na+ ions diffuse rapidly towards and into the anode. This can be achieved by making the anodic surface accumulate electrons rapidly and such a negatively biased anode becomes a swift attractor of the incoming Na+ ions. This is where the redox-functionalized COF could bring an advantage. | ||
| Fig. 3 (A) The pictorial representation shows the discharging or sodiation mechanism of the COF derived half-cell (SIB). The Na+ ions reside near the sodium metal interphase at OCV. (B) Under the applied potential, the Na+ ions move towards the negatively-charged COF. The flow of the Na+ ions towards the anode induced by the accumulation of externally-supplied electrons on the anode. | ||
Anodic reduction and concurrent sodiation process of an SIB half-cell
The abundance of the redox-active functional groups (keto groups, pyridine nitrogen) all along the walls of the nano-channel and the presence of highly electron-deficient s-tetrazine ring shows ample potential to use these COFs as an anode in half-cell SIB (Fig. 3). A slurry made by mixing 65% COF (1/2/3), 25% conducting carbon, and 10% polyvinylidene difluoride in N-methylpyrrolidone (NMP) solution was coated on carbon-coated aluminium foil and cut in the size of 2032 coin cell to use as the anode. The half-cell devices were fabricated using Na metal as the reference and 1 M NaPF6 in 1:1 EC–DMC (2% FEC) soaked Whatman paper as the separator. The OCVs of the coin-cells came near about 2.65 V due to the Na/Na+ interface formation on the Na metal electrode.39,71 To understand the sodiation and de-sodiation mechanisms, the CVs of the coin-cells were recorded within the potential window from 0.05 to 3 V@0.5 mV s−1 sweep rate (Fig. 4A). The half-cell CVs of three different COF-derived electrodes have been compared in Fig. 4A. For each case, the CV of the sample subjected to 10 redox cycles was plotted. This was done to avoid the interference from the SEI formation, which happens during the initial cycles. This comparison reveals the insertion of sodium during discharging occurring through a two-step process for 2 and 3 at 0.1 V (R1/O1) and 0.5 V (R2/O2), respectively. But 1 displays very little current output even at a very low potential of 0.1 V (R1/O1) (Fig. 4A). The only molecular-level functional dissimilarity of 1 with respect to 2 and 3 is the absence of π-stacked s-tetrazine ring throughout the nano-channel. This leaves a marked impact on the sodiation process, making 1 the slowest with the most sluggish insertion of Na+ into the nano-pores.21 The participation of the tetrazine rings in the redox-assisted sodiation process is evident from the CV peak at 0.5 V (R2/O2). During the discharge process (Fig. 4D), the anode becomes negatively charged with the applied potential; the incoming electrons are favourably accommodated by the electron-deficient s-tetrazine units of the 2 and 3. The electronic reduction of 2 and 3 goes via two closely-spaced electron transfer steps. Thus, finally, each s-tetrazine unit accommodates 2e− (Fig. 4B).72 Then, two Na+ move from the electrolyte towards the negatively charged tetrazine segment to balance the charge on the COF surface/pores. We believe that the ease of reduction of the anodic COFs, hence the current output, depends on the stabilization of the LUMO energy level. In 3, since the tetrazine units are conjugated with a pyridine ring, the electron-deficient LUMO level are further lowered (Fig. 2B and C), thus accumulating the electrons with extra ease. The high surface area of COF has a role in uniformly dispersing these accumulating electrons on the COF-coated anodic surface. The highest specific capacity of about 410 mA h g−1@100 mA g−1 was achieved by 3 among these three COFs. On the other hand, 2 and 1 show 195 mA h g−1 and 90 mA h g−1, respectively (Fig. 4D(i)–(iii)). Moreover, the potentiostatic charge–discharge profiles of the COFs also corroborate with the characteristic voltage plateau from 0.8 to 0.05 V observed in the CVs. 2 and 3 possess a prominent reversible redox activity with comparable voltage plateau in the identical potential region, which is unlike that of 1.
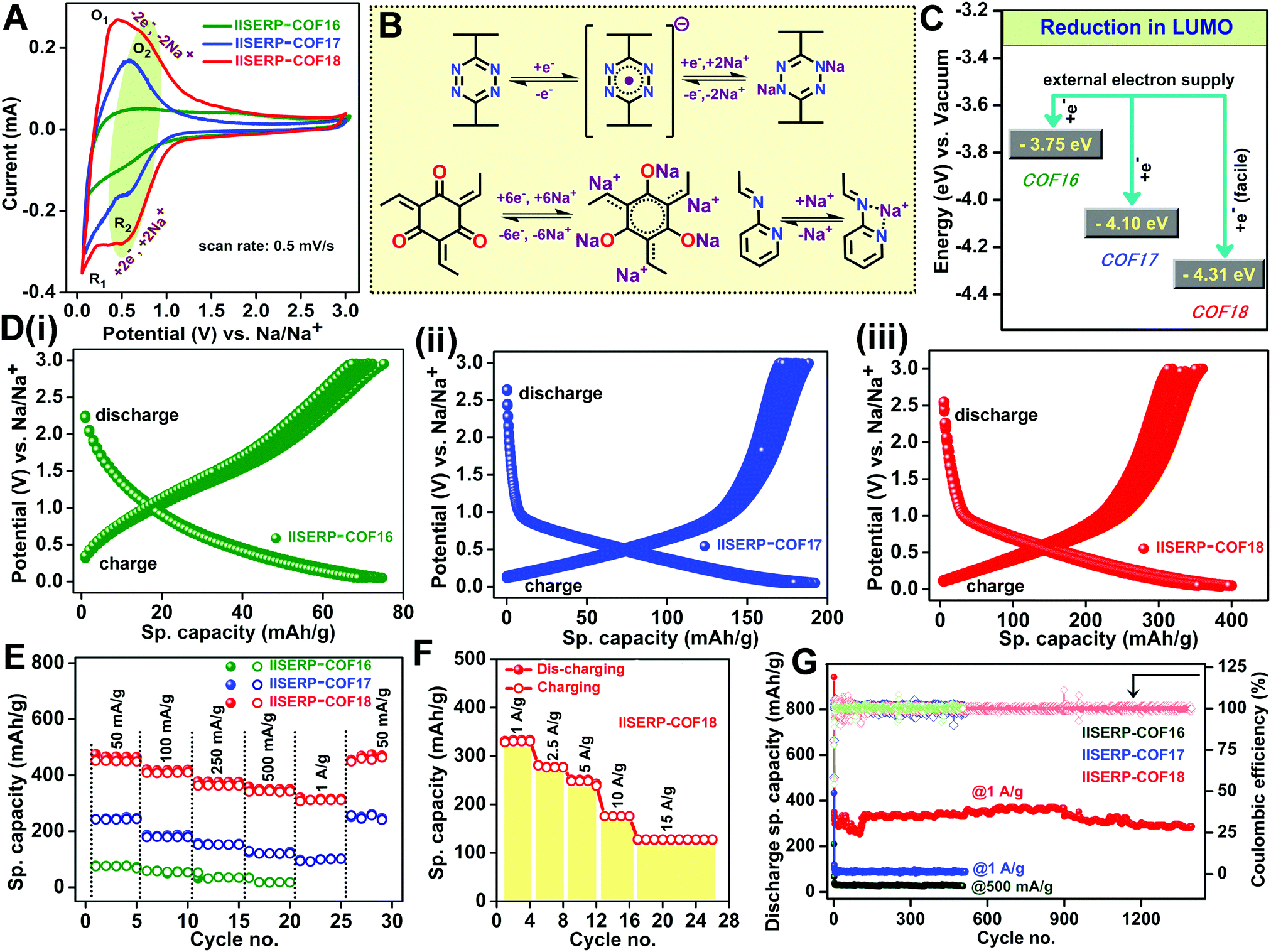 | ||
| Fig. 4 (A) CV curves from the samples of 1, 2, and 3 subjected to 10 redox cycles at 0.5 mV s−1 in the half-cell configuration. The comparison shows the redox-active sodiation–desodiation process (note: the initial cycles were not considered to avoid interference from the SEI formation). (B) Mechanistic pathway of electrochemical reduction of tetrazine and phloroglucinol units, followed by sodiation under a withdrawing potential. Pyridine-β-ketoenamine core provides the chelation site for sodium. (C) The graphical representation of LUMO energy levels shows the energetically favorable electrochemical reduction. (D)(i)–(iii) Charge–discharge profiles of COFs for 250 cycles @100 mA g−1 current density (excluding the initial SEI formations); capacity retention at high currents. (E) Rate performance of the COFs from lower to higher current density excluding the first cycle data points (solid spheres denote discharging or sodiation, hollow spheres denote charging or desodiation). (F) Rate performance of IISERP-COF18 at high current density (from 1 to 15 A g−1). (G) Cycling stability-retention of the discharge capacity @1 A g−1 current density (including the first cycle discharging) and coulombic efficiencies are plotted. Hollow squares denote the coulombic efficiencies of each of the COF-based coin-cells. Note: the specific capacity was calculated considering the mass loading of COF in each of the electrode. | ||
The stable voltage plateau in the charge–discharge curve signifies the ability of the battery to provide a stable potential over a wide window of specific capacities (Fig. 4D). A perfect match of the reduction peak in the CV curve with the sodiation capacity of the COFs helped to estimate the number of sodium ions inserted during the sodiation process (Fig. S25 and Table S7, ESI†). The results showed an almost five-fold enhancement in the sodium acceptance in 3 compared to 1 and two-fold compared to 2. This augmentation is due to the presence of bispyridine-s-tetrazine backbone in the nano-channel of 3. The redox activity at the oxygen-rich phloroglucinol ring also contributes. In 3, there is the possibility of better chelation of sodium ions in between the phloroglucinol oxygen, pyridine-nitrogen, and β-keto enamine nitrogen (Fig. 4B and Scheme S2, ESI†). This sets up chemically-compelled adsorptive sites in 3; in contrast, in 1, most of the Na+ insertion is due to physisorption. Also, the phloroglucinol ring could have some redox activity towards Na+ (peak at 1.7 V) (Fig. S25A, ESI†).21
All the three COFs can also show capacitive storage owing to their good surface areas but the diffusion-controlled storage is influenced by mass transfer of sodium. To compartmentalize these contributions, the CV peak currents at variable scan rates were measured and fitted to the Power law (Fig. S26 and S27, ESI†).12,18,193 obeys Cottrell's equation with a ‘b’ value of 0.95 at 0.5 V (R2/O2) and 0.75 at 0.1 V (R1/O1). This suggests that the reduction of the tetrazine ring, sodiation of the pyridine nitrogen, and phloroglucinol ring all happen via a surface-assisted pathway. Nevertheless, at a very low potential, some contribution comes from sodium insertion into the pores, most likely via a diffusion-controlled pathway.73,74
The above-described rapid diffusion of the Na+ ions at the anodes helps to achieve excellent rate performance. Even at a current density of 1 A g−1, the COFs (2 and 3) retain about ∼80% of the specific capacity obtained at 100 mA g−1, whereas 1 fails at high current inputs (Fig. 4E). Notably, 3 can deliver 127 mA h g−1 specific capacity even at an extremely high scan rate of 15 A g−1 (Fig. 4F). It is impressive to see the stability of COF 3 towards high electron accumulation and a rapid redox process at these high current densities.21,22,73 The electrochemical cyclic stability of 3 was confirmed from the complete retention of its redox activity even after 250 charge–discharge cycles (@100 mA g−1) without any distortion of the voltage plateau and 92% retention of its capacity (340 mA h g−1) even after 1400 charge–discharge cycles at 1 A g−1 (Fig. 4G). The cell with 3 starts with a good specific capacity, which drops from the 30th to 125th cycle, then builds up to the original value within a few cycles, and stays steady at 98%. A plausible reason for this could be the good initial uptake of the Na+ ions by the electrode (after the SEI layer formation), followed by the clogging at the pores, which causes the drop. Over the cycles, the accumulated Na+ ions, under an applied potential, disperse slowly across the electrode, thus improving access to all the sites. This is associated with an improvement in the wetting of the electrode by the electrolyte over time.19 Such an anomalous behaviour appears to be prevailing in other COF systems as well.21 Likewise, 2 also possesses excellent stability. Meanwhile, 1 loses most of its specific capacity even @500 mA g−1. This suggests that the presence of the tetrazine backbone is crucial for accommodating a high electron density at higher current input. Each of the cells was completely discharged before being subjected to the galvanostatic charge–discharge cycles at variable current density. This way, the capacity contribution from SEI formation will not interfere in the rate performances (Fig. 4E and F).75–77 The first cycle coulombic efficiency is only ∼50% for these COFs due to the formation of a solid electrolyte interphase (SEI) on the electrode surface (Table S8, ESI†). A lot of sodium consumption happens irreversibly at the first discharge as the electrolytes decompose on the highly functionalized surface of the COFs. After a few cycles, reversibility is achieved in the sodiation–desodiation processes. This stabilizes the specific capacities with ∼98% coulombic efficiency (Fig. S28 and S29, ESI†). This subsequent steadiness of the coulombic efficiency indicates the stability of the COF-derived electrodes towards multiple cycles of charge–discharge at high current densities. In comparison with other COF derived anodes for SIB, our COF 3 stands at the top in terms of very less capacity drop even at 10 and 15 A g−1 current density (Fig. S30 and Table S9, ESI†).
Lowered resistance to charge-transfer in 3 confirmed from AC-impedance and DC-measurements
To evaluate the state of electronic conductivity and resistance during Na+ propagation within these three isostructural COFs, potentiostatic impedance was measured. The AC sweep in the range of 10000 to 0.10 Hz at 10 mV-rms AC amplitude was implemented on the activated coin-cells. Unlike 1 and 2, the presence of relatively electron-rich (pyridine ring) next to electron-deficient (tetrazine ring) centers increases the in-plane electronic conductivity of 3via a strategic push–pull mechanism (Fig. 2A). This is verified by the three-times lowered semicircle diameters of 3 compared to 1 in the Nyquist plots (resistances of 225 for 3vs. 750 for 1vs. 620 Ω for 2 was observed at the OCV itself; Fig. 5A–C and Fig. S31, ESI†). This is indicative of a lowered charge-transfer resistance for 3. The appearance of a second semi-circle (obtained at lower voltages of 0.5 V and 0.1 V) in the Nyquist plots of 2 and 3 is due to the diffusion resistivity of Na+ when it travels through the electrode–electrolyte interphase.78
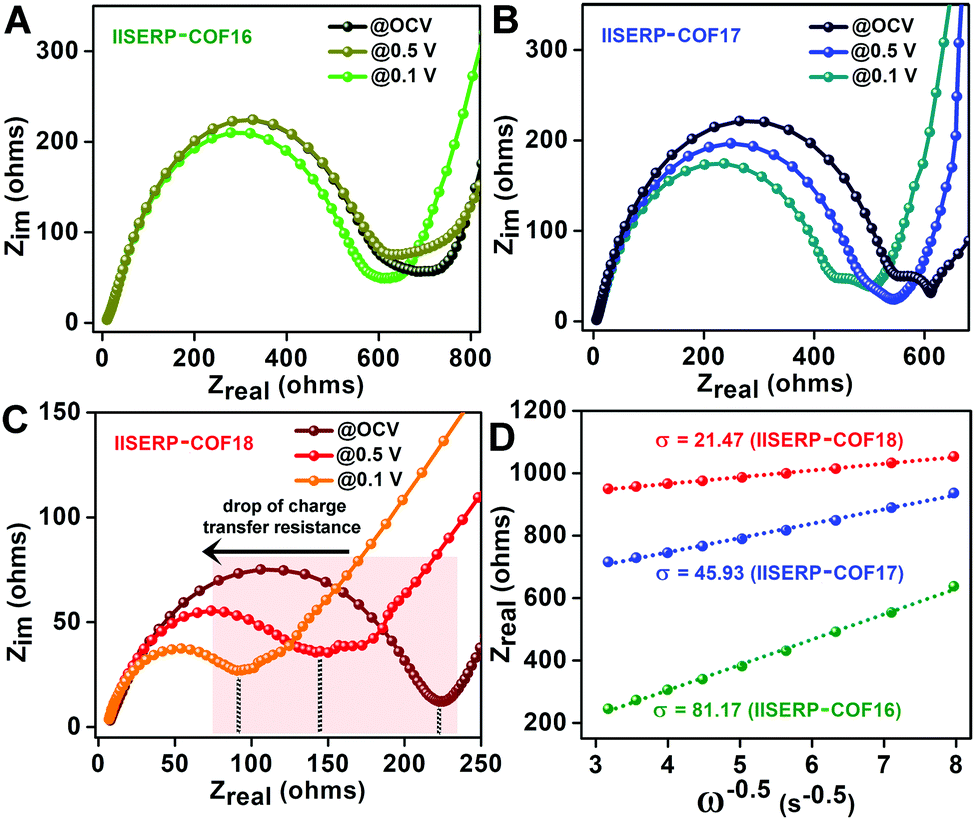 | ||
| Fig. 5 (A–C) Nyquist plots obtained from potentiostatic impedance measurements of COF-derived half-cells @OCV, @0.5 V, and @0.1 V. The shaded area shows the decrease in the charge transfer resistance with the increase in the DC bias. (D) The plot of Zrealvs. the inverse square root of angular frequency (ω) for the COF derived coin-cells (@0.1 V DC voltage). The slopes of the fitted lines represent the Warburg coefficients (σ). | ||
To further understand the advantage of having electron-deficient active sites on the anode, the potentiostatic impedance of the COF derived coin-cells were measured under three different applied DC voltages, i.e., @Na/Na+ = 2.6 V (OCV); @0.5 V (Ered.oftetrazine); @0.1 V (Einsert.ofNa+) (Fig. 5A–C). The decrease in the intrinsic resistances of 2 and 3 with a gradual reduction in the applied potential indicates the excellent responsive charge-transfer lowering of 2 and 3.79 As anticipated, 1 became almost irresponsive to the change in the applied potential. The abrupt decrease in the resistivity of 3 after applying the sodiation potential most likely arises from the easy mass transfer at the electron-rich LUMO levels confined on the tetrazine ring. A higher amount of sodiation makes the structure electronically conducting with time. Moreover, among these COFs, the Warburg resistance (s) at 0.1 V (after the insertion of Na+) is the lowest for 3 (Fig. 5D and Table S10, ESI†). This suggests that the diffusion coefficient of Na+ (DNa+) increases in conjunction with the electron acceptance capability of the COFs (following DNa+α1/σ2). The diffusion coefficient of 3 is four-fold higher than 2 and sixteen-fold higher than 1. Thus, the presence of the bispyridine-tetrazine segment makes the nano-channel of 3 suitable for easy sodium transport during the electronic reduction of the electron-deficient tetrazine ring.
Sodium-COF interactions from atomic modelling
To engage further into the Na-framework interactions, we carried out some modelling studies. For this, an initial configuration was obtained by dispersing 15 sodium ions into an anionic framework of 3 using Grand Canonical Monte Carlo (GCMC) algorithm (number of Na+ ions per unit cell was derived from experimentally determined values) (Fig. 6). The resulting structure was treated as the input for a rigorous DFT optimization carried out on a 3 × 3 × 3 cell using the CASTEP routine embedded in the Materials Studio.80 The details of the modelling can be found in the ESI.† Excellent convergence was achieved and the Na-framework interactions that emerged out of the final model agreed exceptionally well with the experimental findings.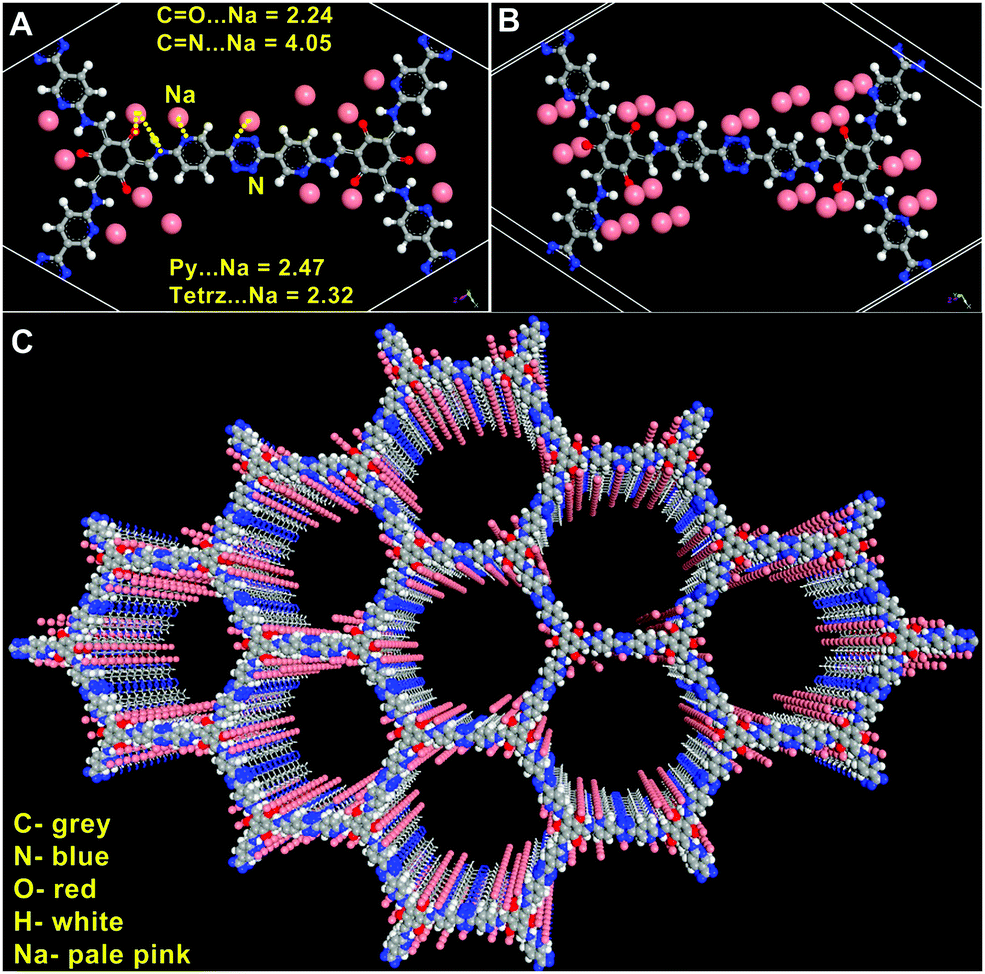 | ||
| Fig. 6 (A) The DFT modeled Na@IISERP-COF18 structure shows the closest interactions between the anionic COF and the Na+ ions. (B) Every active site is sandwiched between two crystallographically equivalent Na+ sites. (C) The 3D framework showing the distribution of Na+ ions around the heteroatoms lining the framework. | ||
Some of the essential findings from this DFT optimized structure of Na@COF are as follows: the distances of the Na+ ions from the N and O atoms of the framework fall within the range of weak coordinate-covalent bonds. The tetrazine, pyridine, and carbonyl atoms seem to be the closest sites while the Schiff nitrogens interact weakly (Fig. 6). Also, Na interacts more precisely with the heteroatoms and faintly with the other C-atoms of the framework compared to Li. In our earlier studies,18,19 Li interacted uniformly with the framework atoms (C, N, and O) and was also dispersed all along the walls of the pores. In contrast, herein, the sodium ions are confined more to the heteroatoms (Fig. 6C).
3. Conclusion
COFs knitting periodically positioned electron-rich and electron-deficient modules using conjugation serve as an excellent push–pull electronics platform. In such frameworks, the electronic activity can be tuned both at the electron-rich and electron-deficient centers. Keeping the electron-rich center fixed, herein, we have utilized tetrazine/pyridine centers as the electron-deficient node to demonstrate the effect that this can bring when used as an anode in a sodium-ion battery. The use of tetrazine/pyridine units lowers the LUMO energy level compared to a phenyl group and this makes the former accumulate electrons with more ease under an applied potential. Such e−-accumulated anti-bonding LUMO levels provide the right driving force for the otherwise sluggish Na+ ions to run into the anode from the electrolyte. This reflects a substantial enhancement in the specific capacity of the SIB. Importantly, the constructed coin-cells show excellent stability even at a current density as high as 15 A g−1. The study establishes a clear-cut correlation between the molecular-level electronic structure and the battery performance. Notably, the demonstrated approach in this family of COF is transferable to many organic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank IISER-Pune for support and the funding by DST for material for energy storage (DST/TMD/MES/2k17/103) program and SERB (EMR/2016/003553). SH thanks DST-Inspire for financial support. DK thanks SERB for NPDF (PDF/2018/000626). We thank MHRD-FAST and the IUSSTF program for the support. SBO and RV also thank DST-Nanomission funding under the Thematic Unit Program (SR/NM/TP-13/2016).References
- K. Geng, T. He, R. Liu, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.9b00550.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- J. Gao and D. Jiang, Chem. Commun., 2016, 52, 1498–1500 RSC.
- C. Yuan, X. Wu, R. Gao, X. Han, Y. Liu, Y. Long and Y. Cui, J. Am. Chem. Soc., 2019, 141, 20187–20197 CrossRef CAS PubMed.
- H. Xu and D. Jiang, Nat. Chem., 2014, 6, 564 CrossRef CAS PubMed.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- Z. Xie, B. Wang, Z. Yang, X. Yang, X. Yu, G. Xing, Y. Zhang and L. Chen, Angew. Chem., 2019, 131, 15889–15893 CrossRef.
- M. Wang, H. Guo, R. Xue, Q. Li, H. Liu, N. Wu, W. Yao and W. Yang, ChemElectroChem, 2019, 6, 2984–2997 CrossRef CAS.
- S. Haldar, R. Kushwaha, R. Maity and R. Vaidhyanathan, ACS Mater. Lett., 2019, 1, 490–497 CrossRef CAS.
- T. Li, X. Yan, Y. Liu, W.-D. Zhang, Q.-T. Fu, H. Zhu, Z. Li and Z.-G. Gu, Polym. Chem., 2020, 11, 47–52 RSC.
- A. Khayum M, V. Vijayakumar, S. Karak, S. Kandambeth, M. Bhadra, K. Suresh, N. Acharambath, S. Kurungot and R. Banerjee, ACS Appl. Mater. Interfaces, 2018, 10, 28139–28146 CrossRef CAS PubMed.
- A. Halder, M. Ghosh, A. Khayum M, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef CAS PubMed.
- M. Wang, H. Guo, R. Xue, Q. Li, H. Liu, N. Wu, W. Yao and W. Yang, ChemElectroChem, 2019, 6, 2984–2997 CrossRef CAS.
- J. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC.
- S. Haldar, K. Roy, S. Nandi, D. Chakraborty, D. Puthusseri, Y. Gawli, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2018, 8, 1702170 CrossRef.
- S. Haldar, K. Roy, R. Kushwaha, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2019, 9, 1902428 CrossRef CAS.
- Z. Zhao, W. Chen, S. Impeng, M. Li, R. Wang, Y. Liu, L. Zhang, L. Dong, J. Unruangsri, C. Peng, C. Wang, S. Namuangruk, S.-Y. Lee, Y. Wang, H. Lu and J. Guo, J. Mater. Chem. A, 2020, 8, 3459–3467 RSC.
- S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li and J. Chen, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- B.-Q. Li, S.-Y. Zhang, B. Wang, Z.-J. Xia, C. Tang and Q. Zhang, Energy Environ. Sci., 2018, 11, 1723–1729 RSC.
- P. Peng, L. Shi, F. Huo, S. Zhang, C. Mi, Y. Cheng and Z. Xiang, ACS Nano, 2019, 13, 878–884 CrossRef CAS PubMed.
- A. Khayum, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 RSC.
- L. Bai, Q. Gao and Y. Zhao, J. Mater. Chem. A, 2016, 4, 14106–14110 RSC.
- H. Yang, S. Zhang, L. Han, Z. Zhang, Z. Xue, J. Gao, Y. Li, C. Huang, Y. Yi and H. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 5366–5375 CrossRef CAS PubMed.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L.-P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang and R. Vajtai, Adv. Mater., 2019, 31, 1901640 CrossRef PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou and X. Feng, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- Z. Lei, X. Chen, W. Sun, Y. Zhang and Y. Wang, Adv. Energy Mater., 2019, 9, 1801010 CrossRef.
- M. Sawicki and L. L. Shaw, RSC Adv., 2015, 5, 53129–53154 RSC.
- L. Seidl, N. Bucher, E. Chu, S. Hartung, S. Martens, O. Schneider and U. Stimming, Energy Environ. Sci., 2017, 10, 1631–1642 RSC.
- Y. Li, Y. Lu, P. Adelhelm, M.-M. Titirici and Y.-S. Hu, Chem. Soc. Rev., 2019, 48, 4655–4687 RSC.
- A. Eftekhari and D.-W. Kim, J. Power Sources, 2018, 395, 336–348 CrossRef CAS.
- O. Lenchuk, P. Adelhelm and D. Mollenhauer, Phys. Chem. Chem. Phys., 2019, 21, 19378–19390 RSC.
- Z. Wang, S. M. Selbach and T. Grande, RSC Adv., 2014, 4, 4069–4079 CAS.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Energy Environ. Sci., 2015, 8, 2963–2969 RSC.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- M.-S. Kim, W.-J. Lee, S.-M. Paek and J. K. Park, ACS Appl. Mater. Interfaces, 2018, 10, 32102–32111 CrossRef CAS PubMed.
- H. Zhang, Y. Huang, H. Ming, G. Cao, W. Zhang, J. Ming and R. Chen, J. Mater. Chem. A, 2020, 8, 1604–1630 RSC.
- N. P. Stadie, E. Billeter, L. Piveteau, K. V. Kravchyk, M. Döbeli and M. V. Kovalenko, Chem. Mater., 2017, 29, 3211 CrossRef CAS.
- S. Wang, L. Xia, L. Yu, L. Zhang, H. Wang and X. W. D. Lou, Adv. Energy Mater., 2016, 6, 1502217 CrossRef.
- G. Zou, C. Wang, H. Hou, C. Wang, X. Qiu and X. Ji, Small, 2017, 13, 1700762 CrossRef PubMed.
- H. Hou, L. Shao, Y. Zhang, G. Zou, J. Chen and X. Ji, Adv. Sci., 2017, 4, 1600243 CrossRef PubMed.
- W. Chen, M. Wan, Q. Liu, X. Xiong, F. Yu and Y. Huang, Small Methods, 2018, 1800323 Search PubMed.
- R. Dan, W. Chen, Z. Xiao, P. Li, M. Liu, Z. Chen and F. Yu, Energy Fuels, 2020, 34, 3923–3930 CrossRef CAS.
- M. Hu, H. Zhou, X. Gan, L. Yang, Z.-H. Huang, D.-W. Wang, F. Kang and R. Lv, J. Mater. Chem. A, 2018, 6, 1582–1589 RSC.
- Q. Jin, K. Wang, P. Feng, Z. Zhang, S. Cheng and K. Jiang, Energy Storage Mater., 2020, 27, 43–50 CrossRef.
- M. Wahid, D. Puthusseri, Y. Gawli, N. Sharma and S. Ogale, ChemSusChem, 2018, 11, 506–526 CrossRef CAS PubMed.
- X. Liu, X. Jiang, Z. Zeng, X. Ai, H. Yang, F. Zhong, Y. Xia and Y. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 38141–38150 CrossRef CAS PubMed.
- W. Song, J. Kan, H. Wang, X. Zhao, Y. Zheng, H. Zhang, L. Tao, M. Huang, W. Liu and J. Shi, ACS Appl. Nano Mater., 2019, 2, 5643–5654 CrossRef.
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed.
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine and R. Abe, ChemRxiv, 2019 DOI:10.26434/chemrxiv.8957039.v2.
- S. Haldar, D. Chakraborty, B. Roy, G. Banappanavar, K. Rinku, D. Mullangi, P. Hazra, D. Kabra and R. Vaidhyanathan, J. Am. Chem. Soc., 2018, 140, 13367–13374 CrossRef CAS PubMed.
- D. Chakraborty, S. Nandi, D. Mullangi, S. Haldar, C. P. Vinod and R. Vaidhyanathan, ACS Appl. Mater. Interfaces, 2019, 11, 15670–15679 CrossRef CAS PubMed.
- D. Mullangi, V. Dhavale, S. Shalini, S. Nandi, S. Collins, T. Woo, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1600110 CrossRef.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS PubMed.
- D. Kaleeswaran, P. Vishnoi and R. Murugavel, J. Mater. Chem. C, 2015, 3, 7159–7171 RSC.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- F. Haase, K. Gottschling, L. Stegbauer, L. S. Germann, R. Gutzler, V. Duppel, V. S. Vyas, K. Kern, R. E. Dinnebier and B. V. Lotsch, Mater. Chem. Front., 2017, 1, 1354–1361 RSC.
- L. Zhang, Y. Zhou, M. Jia, Y. He, W. Hu, Q. Liu, J. Li, X. Xu, C. Wang, A. Carlsson, S. Lazar, A. Meingast, Y. Ma, J. Xu, W. Wen, Z. Liu, J. Cheng and H. Deng, Matter, 2020, 2, 1–15 CrossRef.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dörfler, M. M. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef PubMed.
- F. J. Sotomayor, K. A. Cychosz and M. Thommes, Acc. Mater. Surf. Res., 2018, 3, 34–50 Search PubMed.
- G. A. Bhat, S. Haldar, S. Verma, D. Chakraborty, R. Vaidhyanathan and R. Murugavel, Angew. Chem., Int. Ed., 2019, 58, 16844–16849 CrossRef CAS PubMed.
- S. Nandi, S. K. Singh, D. Mullangi, R. Illathvalappil, L. George, C. P. Vinod, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1601189 CrossRef.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine and Q. Chen, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. Evans, M. Döblinger, S. Herbert and K. Peters, J. Am. Chem. Soc., 2017, 140, 2085–2092 CrossRef PubMed.
- J. Lüder and S. Manzhos, Front. Chem., 2020, 8, 83 CrossRef PubMed.
- S. Wu, W. Wang, M. Li, L. Cao, F. Lyu, M. Yang, Z. Wang, Y. Shi, B. Nan and S. Yu, Nat. Commun., 2016, 7, 13318 CrossRef CAS PubMed.
- K. Li, J. Zhang, D. Lin, D.-W. Wang, B. Li, W. Lv, S. Sun, Y.-B. He, F. Kang and Q.-H. Yang, Nat. Commun., 2019, 10, 725 CrossRef CAS PubMed.
- D. J. Min, F. Miomandre, P. Audebert, J. E. Kwon and S. Y. Park, ChemSusChem, 2019, 12, 503–510 CrossRef CAS PubMed.
- B. C. Patra, S. K. Das, A. Ghosh, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6, 16655–16663 RSC.
- S. Licht, A. Douglas, J. Ren, R. Carter, M. Lefler and C. L. Pint, ACS Cent. Sci., 2016, 2, 162–168 CrossRef CAS PubMed.
- R. Mogensen, D. Brandell and R. Younesi, ACS Energy Lett., 2016, 1, 1173–1178 CrossRef CAS.
- M. Ihsan-Ul-Haq, H. Huang, J. Wu, J. Cui, S. Yao, W. G. Chong, B. Huang and J.-K. Kim, Nano Energy, 2020, 71, 104613 CrossRef CAS.
- J. Jang, Y. Kim, O. B. Chae, T. Yoon, S. M. Kim, H. S. Kim, H. Park, J. H. Ryu and S. M. Oh, Angew. Chem., Int. Ed., 2014, 53, 10654–10657 CrossRef CAS PubMed.
- L. Wang, J. Zhao, J. Gao, J. Li, C. Wan, C. Jiang and X. He, Int. J. Electrochem. Sci., 2012, 7, 345–353 CAS.
- K. Roy, M. Wahid, D. Puthusseri, A. Patrike, S. Muduli, R. Vaidhyanathan and S. Ogale, Sustainable Energy Fuels, 2019, 3, 245–250 RSC.
- L. Ascherl, T. Sick, J. T. Margra, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghiosoff, M. Döblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Nat. Chem., 2016, 8, 310–316 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structure modeling, SAED, solid-state NMR, IR spectra, adsorption, CVs, bandgap calculation, specific capacity comparison, impedance fitting. Materials and methods, characterizations, electrochemistry, and computational details. See DOI: 10.1039/d0nh00187b |
| This journal is © The Royal Society of Chemistry 2020 |