
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Masking specific effects of ionic liquid constituents at the solid–liquid interface by surface functionalization†
Bojana
Katana‡
a,
Dóra
Takács‡
a,
Felix D.
Bobbink
 b,
Paul J.
Dyson
b,
Nizar B.
Alsharif
a,
Matija
Tomšič
c and
Istvan
Szilagyi
*a
b,
Paul J.
Dyson
b,
Nizar B.
Alsharif
a,
Matija
Tomšič
c and
Istvan
Szilagyi
*a
aMTA-SZTE Lendület Biocolloids Research Group, Interdisciplinary Excellence Centre, Department of Physical Chemistry and Materials Science, University of Szeged, H-6720 Szeged, Hungary. E-mail: szistvan@chem.u-szeged.hu; Tel: +36 62 343255
bInstitute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
cFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia
First published on 19th October 2020
Abstract
Ion specific effects of ionic liquid (IL) constituents on the surface charge and aggregation properties of two types of particles (positively charged amidine (AL) and polyimidazolium-functionalized sulfate (SL-IP-2) latexes) were investigated in IL solutions containing different anions and the 1-butyl-3-methylimidazolium cation. For the AL systems, the affinity of IL anions to the particle surface followed the sequence chloride < bromide < nitrate < acetate. The critical coagulation concentration values decreased in the same order indicating that ion specific adsorption determines the surface charge density and the extent of the repulsive interparticle forces. In contrast, no tendencies were observed for the SL-IP-2 particles, i.e., both charge and aggregation features were insensitive to the type of anions. This surprising behavior sheds light on that surface functionalization with the polyimidazolium compound effectively masks interfacial ion specific effects. These results indicate new possible routes to the design of processable particle dispersions in ILs irrespective of their composition.
1 Introduction
Ionic liquids (ILs) continue to attract widespread interest due to their advantageous properties such as low vapor pressure, wide electrochemical window and their capacity to be structurally tuned for specific tasks such as for the functionalization of surfaces.1–3 The assembly of the IL cations and anions at solid–liquid interfaces has been extensively studied, since ion specific effects play a major role in the physico-chemical properties of surfaces immersed in ILs or in their solutions.4–6 The interfacial structure is responsible for reduced friction between surfaces,7 exfoliation of layered materials8 and stabilization of various particles in ILs.9–13 In addition, specific interactions of IL ions with synthetic and natural macromolecules influence the stability of enyzmes,14 the rate of amyloid fibrillization15 and phase transition behavior16 through tailoring the inter and intramolecular forces.The origin of ion specific effects on surface forces has been systematically explored by optical tweezer,17 atomic force microscope18 and surface force apparatus19 in aqueous solutions of ILs and one of the main conclusions was that different affinity of the cations and anions for the surface may cause significant variation in the charge density upon ion adsorption. Consequently, this variation results in different extents of electrostatic repulsions between surfaces. Similar hypotheses were also reported on the basis of particle aggregation experiments in IL–water mixtures.9,20,21 Moreover, theoretical studies shed light on the fact that the interfacial assembly of IL ions mainly depends on the composition of the surface, concentration and structure of the ILs.10,22
The IL cations and anions may be arranged in series based on their effects on the surface charge and interaction forces, similar to the Hofmeister series of ions, which was originally constructed for protein destabilization by simple salts.21,23,24 Despite the fact that understanding the interfacial assembly of ILs is of huge importance during development of advanced materials in energy storage,25–27 electrochemistry28 and the preparation of novel catalysts,29 only a few studies dealt with the systematic assessment of the effect of IL constituents on the charge and aggregation features of particles.9,20,21 Another important aspect is that ion specific effects often hinder the application of IL mixtures to disperse and stabilize particles successfully. For instance, different affinity of the ions to the surfaces leads to asymmetric arrangement of the IL constituents near the surface, which may induce destabilization of the dispersions. Therefore, the possibility of masking interfacial ion specific effects would be beneficial in terms of gaining the stability of numerous systems. Nevertheless, to the best of our knowledge, no papers have been published on this topic yet.
In the present study, surface charges and colloidal stability of polymeric particles were investigated in the presence of aqueous IL solutions containing chloride (Cl−), bromide (Br−), nitrate (NO3−) or acetate (Ac−) anions and the 1-butyl-3-methylimidazolium (BMIM+) cation. Two types of positively charged particles were studied, namely, polystyrene-based amidine latex (AL) and SL-IP-2 consisting of a sulfate latex (SL) functionalized with an imidazolium-based polymer (IP-2). In this way, ion specificity was explored with polymeric particles of the same sign of charge, but with different surface functionalities.
2 Experimental
2.1 Materials
Spherical polystyrene latex particles functionalized with sulfate (SL) or amidine (AL) groups were purchased from Thermo Fischer Scientific. The particle size and the charge density of the bare surfaces were determined by the manufacturer using transmission electron microscopy (TEM) and conductometric titration, respectively. The SL has a mean diameter of 430 nm, a polydispersity expressed as coefficient of variation of 1.8%, solid content of 8.1% (w/v%), specific surface area of 1.3 × 105 cm2 g−1 and a charge density of −12 mC m−2. In the case of the AL, the corresponding quantities are 510 nm, 4.6%, 4.1%, 1.1 × 105 cm2 g−1 and +197 mC m−2.The polyimidazolium-based polymer (IP-2) was synthesized using a literature protocol.30 Briefly, 1,4-bis(chloromethyl)benzene and 1-(trimethylsilyl)imidazole were dissolved in acetonitrile in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in a Schlenk-flask. The mixture was heated to reflux for 48 h. The white solid product was removed by filtration, washed with acetonitrile and diethyl ether followed by drying under vacuum for 24 h. All chemicals used for the synthesis are from Sigma-Aldrich. The successful synthesis was confirmed with FT-IR spectroscopy by identifying the characteristic vibration peaks of IP-2 at 1150, 1560 and 1625 cm−1 (for the spectrum see Fig. S1 in the ESI†).30
:1 molar ratio in a Schlenk-flask. The mixture was heated to reflux for 48 h. The white solid product was removed by filtration, washed with acetonitrile and diethyl ether followed by drying under vacuum for 24 h. All chemicals used for the synthesis are from Sigma-Aldrich. The successful synthesis was confirmed with FT-IR spectroscopy by identifying the characteristic vibration peaks of IP-2 at 1150, 1560 and 1625 cm−1 (for the spectrum see Fig. S1 in the ESI†).30
Analytical grade potassium chloride (KCl) was purchased from VWR. The ILs, 1-butyl-3-methylimidazolium nitrate (BMIMNO3, ≥95%), 1-butyl-3-methylimidazolium chloride (BMIMCl, ≥98%), 1-butyl-3-methylimidazolium acetate (BMIMAc, ≥95%) and 1-butyl-3-methylimidazolium bromide (BMIMBr, >97%) were purchased from Sigma-Aldrich. During sample preparation, ultrapure water was obtained from a Puranity TU+ (VWR) apparatus. The water and the salt solutions were filtered with 0.1 μm syringe filters (Millex) to avoid dust contamination. All measurements were carried out at 25 °C and pH value of 4 set by HCl (VWR) stock solutions.
The IP-2-modified SL stock dispersion (denoted as SL-IP-2 thereafter) was prepared by simple mixing of proper volumes of IP-2 of 100 mg L−1 concentration, SL particle stock solution of 10 g L−1 and ultrapure water.
2.2 Characterization of IP-2 and the ILs
The FT-IR spectrum was recorded on a JASCO FT-IR-4700 spectrometer equipped with a DTGS detector in attenuated total reflectance mode. Spectral resolution was 4 cm−1 with 256 scans collected for the spectrum, which was recorded in the wavenumber range of 1800–1000 cm−1.Refractive index and viscosity measurements were carried out with an Anton Paar Abbemat 3200 refractometer and a capillary viscometer, respectively. These data were used for the analysis of the light scattering measurements. For the data treatment, see Fig. S2a, b and Table S1 in the ESI.†
2.3 Electrophoresis
Electrophoretic mobility was measured with a Litesizer 500 instrument (Anton Paar) equipped with a 40 mW semiconductor laser of 658 nm wavelength operating in the backscattering mode at a scattering angle of 175°. For the determination of the electrophoretic mobilities, 2.0 mL samples were prepared. In brief, 0.4 mL particle dispersions of 80 mg L−1 or 0.2 mL of 100 mg L−1 concentration for AL and SL-IP-2, respectively, was added to 1.6 mL or 1.8 mL of solutions composed of ILs or KCl at appropriate concentrations. The samples were left to rest for 2 h at room temperature before measuring the electrophoretic mobilities, which occurred after 1 min equilibration time in the device. The reported values were the average of five individual measurements with a maximum standard deviation of ±5 mV. The experiments were performed in 350 μL volume Ω-shaped plastic cuvettes or with the Univette (Anton Paar) accessory.The measured electrophoretic mobility (u) was converted to zeta potential (ζ) with the Smoluchowski equation as follows31
| ζ = uη/ε0ε | (1) |
| σ = εε0κζ | (2) |
 | (3) |
2.4 Dynamic light scattering
The hydrodynamic radius (Rh) of the particles was measured by dynamic light scattering (DLS) using a NIBS High-Performance Particle Sizer (ALV) equipped with a 3 mW He–Ne laser of 633 nm wavelength. The scattering angle was 173°, the correlation function was collected for 20 s and was fitted with the cumulant method33 to obtain the decay rate constant (Γ). The diffusion coefficient (D) was calculated as| D = Γ/q2 | (4) |
| q = (4πn/λ)sin(Θ/2) | (5) |
| Rh = kBT/6ηπD | (6) |
The viscosity data points were interpolated using eqn (S2) (ESI†). Time-resolved DLS measurements were carried out to assess the possible aggregation processes in the samples. Typical results of these measurements, where 100–250 runs were performed depending on the speed of aggregation, are shown in Fig. S3 (ESI†). During sample preparation, 2 mL dispersions were prepared. The ionic strength or IL dose was adjusted by mixing appropriate amounts of stock solutions and the time-resolved DLS measurements were initiated by adding the desired volume of particle stock dispersions to the solutions containing all other components. The particle concentration was 16 mg L−1 (2.18 × 1014 m−3) for AL and 10 mg L−1 (2.28 × 1014 m−3) for SL-IP-2. These particle doses were good compromises to avoid the formation of higher ranked aggregates and to maintain sufficient scattered intensity for the light scattering experiments. The aggregation rate coefficient (k) was calculated from the time dependent Rh values as33
| (1/R1h)(dRh/dt) = (I2/2I1)(1 − (R1h/R2h))kN0 | (7) |
| I2/2I1 = 1 + (sin(2Rq)/2Rq) | (8) |
| W = kfast/k | (9) |
The critical coagulation concentration (CCC) values were calculated from the W versus cIL plots with the following equation37
| 1/W = 1/(1 + CCC/cIL)β | (10) |
| β = dlog(1/W)/dlogcIL | (11) |
To compare experimental and theoretical CCC data, DLVO theory, which assumes that the total interaction energy can be considered as a superposition of the repulsive electrostatic double layer potential energy and the attractive van der Waals potential energy, was used to estimate the CCC as follows36
| CCC = (0.365/NALB)(Hε0ε)−2/3σ4/3 | (12) |
| LB = e2/4πε0εkBT | (13) |
Using the model above, it was assumed that the energy barrier vanishes at the CCC and attractive interparticle forces predominate beyond this concentration.
3. Results and discussion
3.1 Functionalization of SL with IP-2
The AL particles are commercially available, whereas SL-IP-2 particles were prepared by adsorbing a saturated IP-2 layer on the oppositely charged SL. To determine the polymer dose necessary to fully coat the SL surface, the zeta potential values were determined at different IP-2 doses (Fig. 1). | ||
| Fig. 1 Zeta potential (circles, left axis) and stability ratio (squares, right axis) values of SL particles as a function of the IP-2 dose at pH 4 and 10 mM ionic strength adjusted by KCl. Note that the inverse of the stability ratio is equal to the fraction of particle collisions, which results in dimer formation. The mg/g unit refers to mg IP-2 per one gram of SL. The lines serve to guide the eye. The arrows indicate the axis, to which potential (blue arrow) and stability ratio (red arrow) data points belong. The upper part shows the structure of IP-2. | ||
The SL particles are negatively charged at low IP-2 doses due to the presence of ionized sulfate functional groups. Increasing the amount of IP-2 led to charge neutralization and subsequent overcharging of the SL particles. The zeta potential values remain constant at high IP-2 doses, at which the surface of the particles contains a saturated IP-2 layer. These results resemble to other systems, where polyelectrolytes were adsorbed on oppositely charged latex particles.38–40
The aggregation tendencies were assessed by determining stability ratio values under the same experimental conditions as used for the potential measurements. Note that the stability ratio of unity refers to diffusion controlled rapid particle aggregation, while higher values indicate a decrease in the aggregation rates. The data shown in Fig. 1 suggest that rapid particle aggregation occurred near the charge neutralization point, while stable dispersions were observed far from it. Most importantly, particle aggregation is negligible at high doses.
Very similar observations were taken in systems containing oppositely charged latexes and polyelectrolytes,38–40 but this is the first report, which involves a polyimidazolium compound. Based on the above results, a dose of 500 mg g−1 was chosen (SL-IP-2) for the further studies, since under this condition the particle surface was saturated with IP-2 and stable dispersion was formed.
3.2 Charge and aggregation features of AL and SL-IP-2 particles in the presence of ILs
To investigate the specific interfacial effects of IL constituents, the zeta potentials of AL particles were measured first in aqueous solutions of BMIMAc, BMIMNO3, BMIMBr and BMIMCl in a wide range of IL concentrations. The results are shown in Fig. 2a. Note that the anions are the counterions and the BMIM+ cation acts as co-ion in respect to the positively charged particles. In general, the zeta potential values decreased with the IL concentrations, similar to the case of indifferent KCl electrolyte (Fig. S4a, ESI†).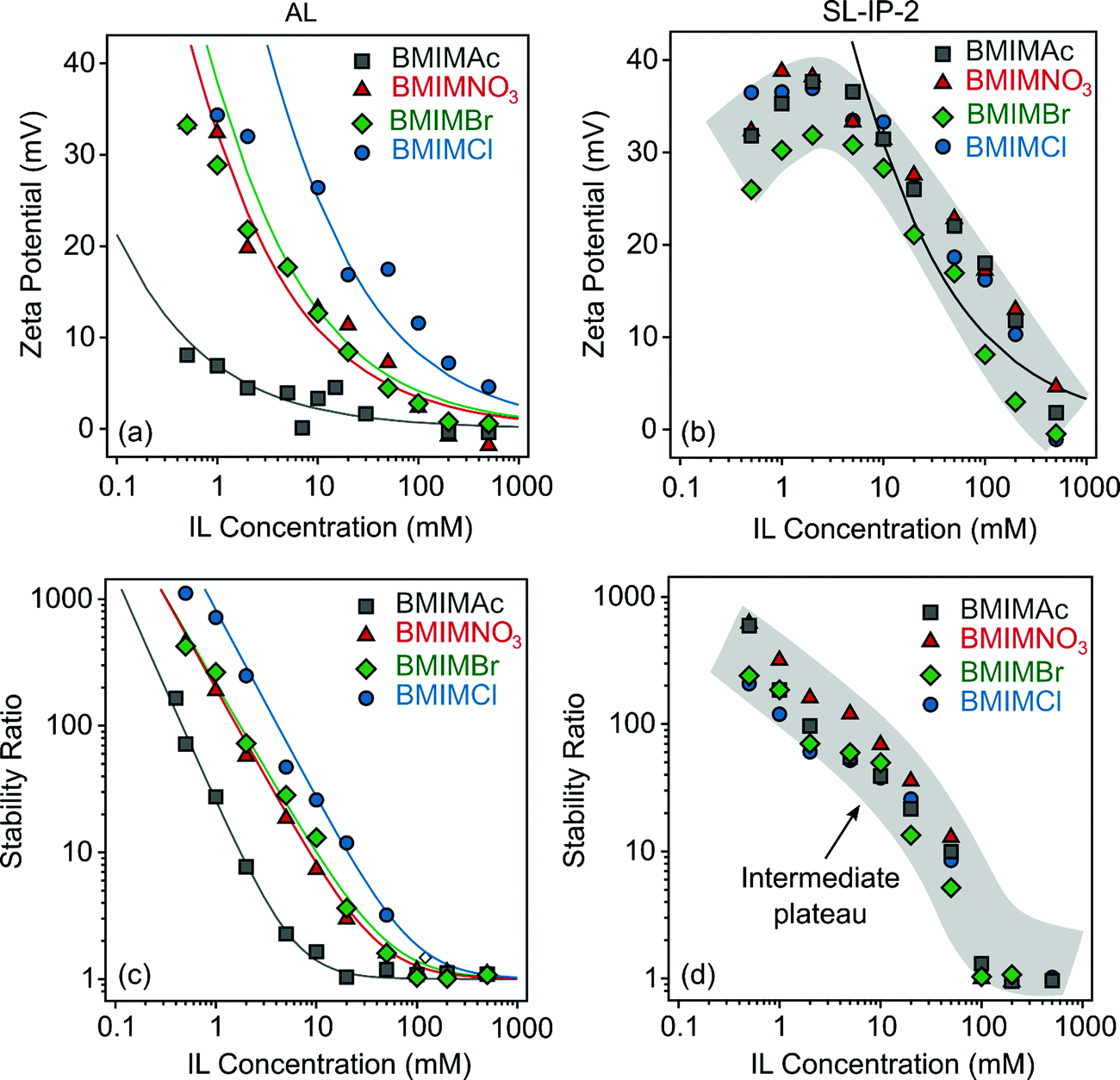 | ||
| Fig. 2 Zeta potentials (AL (a) and SL-IP-2 (b)) as well as stability ratios (AL (c) and SL-IP-2 (d)) as a function of IL concentration at pH 4. The functions represented by lines in (a) and (b) were calculated using eqn (2) and in (c) with eqn (10). | ||
The surface charge densities were calculated from the salt concentration-dependent potential data using the Debye–Hückel model (eqn (2)).41 The values (Table S2, ESI†) decreased in the order Cl− > Br− > NO3− > Ac− indicating specific adsorption of the ions. Note that these charge densities were measured at the slip plane, therefore their magnitudes are much lower than the charge density of the bare surface determined in conductometric measurements by the manufacturer. Such a sequence agrees well with the reversed Hofmeister series, which classifies anions by their affinity towards positively charged hydrophobic surfaces.42 Accordingly, the adsorption of the well-hydrated Cl− is negligible, while Ac− adsorbs strongly on the oppositely charged surface.
For the SL-IP-2 system, nevertheless, the zeta potentials were the same within the experimental error (Fig. 2b). After an intermediate maximum due to the electrokinetic effect,43 the potentials decreased with increasing the IL concentrations in all cases, but no specific adsorption of the IL anions was detected. This result is in a striking contrast to the findings for the AL systems. In addition, specific anion adsorption on positively charged bare latexes21,36 and the ones functionalized with positively charged polyelectrolytes39 was reported in the past. Therefore, the insensitivity of the surface charge to the chemical composition of the surrounding anions in the SL-IP-2 systems is rather surprising. The different affinity of the anions to cationic surfaces was explained by their different level of hydration, i.e., less hydrated anions tend to adsorb on hydrophobic surfaces, while this effect was not observed with the SL-IP-2 particles, most likely due to the less hydrophobic nature of their surface.
The stability ratios were determined by DLS to assess the particle aggregation processes in the IL solutions. The experimental conditions (e.g., pH value, particle and IL concentration range) were the same as in the zeta potential study in order to allow the direct comparison of the results. For the AL systems, the stability ratios follow the same generic trend as a function of the IL concentration, i.e., their values were high (slow aggregation) at low salt levels and decreased with increasing the IL loading and became unity (fast aggregation) at high IL concentrations (Fig. 2c). The threshold value, which separate the slow and fast aggregation regimes, is the so-called CCC, which was calculated by eqn (10).
These results are similar to the ones reported earlier for latex particle dispersions in simple inorganic salt solutions (see the case of KCl in Fig. S4b, ESI†) and they are in line with the prediction of the DLVO theory.35,44 Accordingly, the overall forces acting between charged particles are the sum of the repulsive electrical double layer and the attractive van der Waals forces. Increasing the salt concentration leads to the weakening of the electrostatic repulsion by salt screening and thus, the particles rapidly aggregate after the CCC due to the predominating attractive forces. This explanation is also evident from the very low zeta potential values measured at the CCC (Table S2, ESI†).
Although the generic trend in the stability ratios was very similar in all systems containing AL particles, the CCC values were different (Fig. 3a). This result cannot be explained by the DLVO theory since it predicts the same CCC, once a particle is dispersed in different types of monovalent salt solutions. The CCC values follow the order Cl− > Br− > NO3− > Ac−, in accordance with the surface charge density data determined in the same systems. Again, this order is in line with the suggestion of the reversed Hofmeister series for positively charged hydrophobic particles.36,42
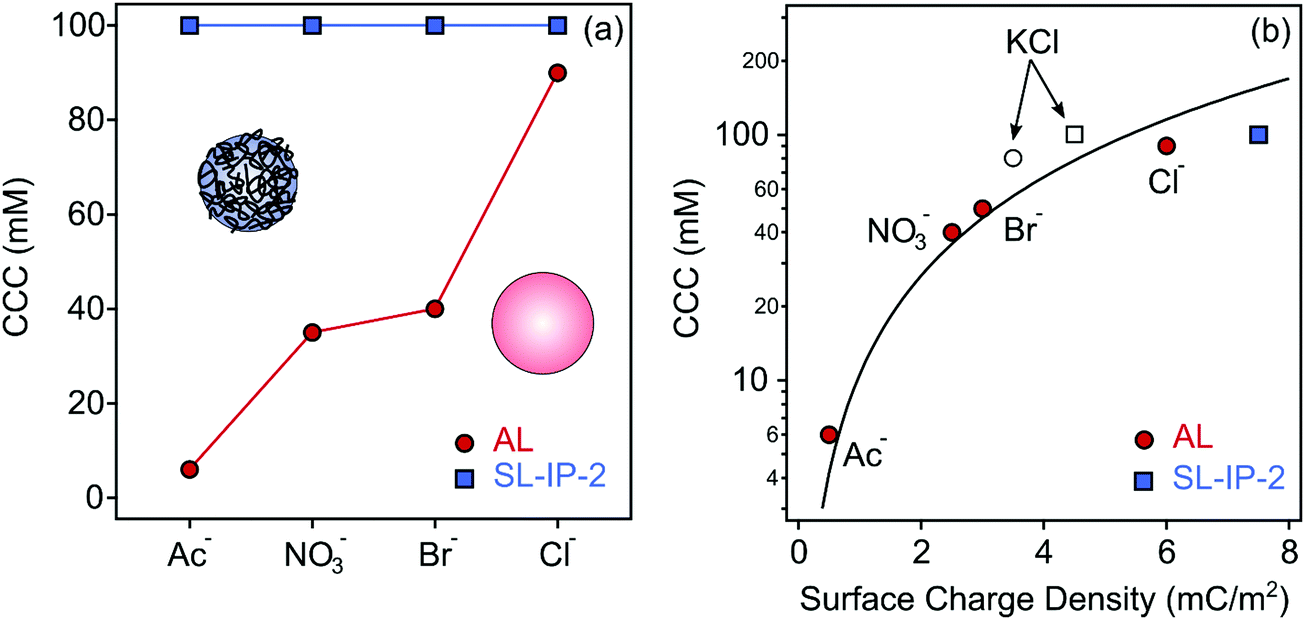 | ||
| Fig. 3 CCC values of AL (red circles) and SL-IP-2 (blue square) determined in the presence of ILs (a) and CCC versus the charge density data (b). The empty symbols indicate the data measured with KCl. The lines in (a) were added merely to guide the eyes, while in (b), it was calculated using eqn (12). | ||
For the SL-IP-2 particles, nevertheless, the measured stability ratio values were identical within the experimental error for BMIMAc, BMIMNO3, BMIMBr and BMIMCl (Fig. 2d) and thus, the onset of rapid particle aggregation was located at the same CCC for all systems. This finding is in striking contrast to earlier results with latex particles functionalized with a positively charged natural polyelectrolyte, where the presence of different inorganic salts led to different CCC values.39 More importantly, we are not aware of findings similar to the present one, which demonstrate that polyelectrolyte coating can be used to abolish ion specific effects on particle aggregation.
Besides, the tendency in the stability ratios was remarkably different from the AL samples and intermediate plateaus were discovered around 10 mM IL concentrations. The term “plateau” refers to the fact that the decrease of the stability ratios was not linear, but curved in the slow aggregation regime. This is rather unusual for latex particles and most likely originates from the swelling of the adsorbed IP-2 layer, which is indeed predicted in the same ionic strength range as the location of the plateaus.45 Such a swelling gives rise to the formation of polyelectrolyte tails and loops on the surface and subsequently, to the rise of stabilizing steric forces12 to a minor extent (the main interparticle forces are still of DLVO origin). The increment in the stability ratios in this concentration range is due to the presence of these additional stabilization forces. An important note is that the height and the location of these plateaus are the same for all IL solutions. Notably, no ion specific effects could be detected for the aggregation of SL-IP-2 particles.
3.3 Origin of interparticle forces
Given the very similar surface charge density and CCC values for the AL and SL-IP-2 particles in inert KCl electrolyte, the striking contrast in the charge and aggregation features in IL solutions is very surprising. Note that deviations in the zeta potentials (Fig. S5a–d, ESI†) and stability ratios (Fig. S6a–d, ESI†) measured in the same IL solution for AL and SL-IP-2 particles are larger in the sequence Cl− > Br− > NO3− > Ac−. These results show that ion specific effects can be abolished by surface functionalization of polyimidazolium.To further clarify the origin of the interparticle forces for both AL and SL-IP-2 particles dispersed in IL solutions, the experimental CCC values were plotted against the surface charge density data and compared to the CCC calculated using the DLVO theory (eqn (12)).36 The data shown in Fig. 3b indicate that the calculated and experimental CCC values are in good agreement, which indicates the presence of DLVO-type forces. However, the ion specific adsorption led to different surface charge densities for AL, which decrease in the order Cl− > Br− > NO3− > Ac− leading to weaker electrical double layer repulsion. Since the van der Waals forces are always present and their strength does not depend on the ionic strength, the CCC values decrease in the above order due to the weakening of the repulsive double layer forces. In other words, DLVO forces govern the aggregation mechanism, while the CCC is determined by specific ion adsorption.
However, such a specific ion effect is absent for SL-IP-2 particles. The basic phenomenon behind this result most probably originates from the hydration level of the surface and the counterions. It was reported in other latex systems that weakly hydrated ions are of higher affinity to hydrophobic surfaces, i.e., they adsorb stronger on hydrophobic latexes,21 similar to the AL particles in the present study. Since the same counterions were applied with the SL-IP-2 particles, the lack of ion specific effects implies that the hydrophobic character of SL weakened upon surface functionalization by IP-2 and thus, the hydration level-based affinity of the counterions is not pronounced. The reason for the decreased hydrophobicity can be that stacking interaction took place between the imidazolium groups of BMIM+ and IP-2 leading to an accumulation of the BMIM+ molecules on the surface and to the formation of a more ionic interfacial environment. Such a stacking interaction was reported earlier based on electronic structure analysis.46
Although the present study focuses on polymeric particles, similar coating can be also possible on other surfaces used in ILs.6,11,13,26,27 This would be a promising strategy to develop ionophobic surfaces, however, the experimental conditions (e.g., pH, polymer concentration and ionic strength) must be precisely optimized before the coating procedure.
4 Conclusions
To conclude, it was successfully demonstrated that anion specific effects on the surface charge and aggregation of AL particles are important and that the tendency can be predicted by the reversed Hofmeister series of anions for positively charged hydrophobic surfaces. The ion specific adsorption led to a decrease in the surface charge density and thus, lower CCC values were measured for strongly adsorbing anions such as Ac−. The ion specificity plays an important role in the adsorption process, while the aggregation mechanism and the major interparticle forces could be described with the DLVO theory. These ion specific interactions, however, can be masked by functionalization of the surface with polyimidazolium, after which the anions behave like indifferent salt constituents giving rise to the same colloidal stability of the SL-IP-2 particles in different ILs. These results are especially important for the design of particle-IL dispersion of desired surface charge and aggregation characteristics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Hungarian National Research, Development and Innovation Office (SNN131558), the Slovenian Research Agency (research core funding No. P1-0201 and the project No. N1-0139 ‘Delamination of Layered Materials and Structure-Dynamics Relationship in Green Solvents’) and the Ministry of Human Capacities of Hungary (20391-3/2018/FEKUSTRAT). The support from the University of Szeged Open Access Fund (5019) is gratefully acknowledged.References
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS.
- Z. P. Zheng, W. H. Fan, S. Roy, K. Mazur, A. Nazet, R. Buchner, M. Bonn and J. Hunger, Angew. Chem., Int. Ed., 2015, 54, 687–690 CAS.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef.
- M. Taskin, A. Cognigni, R. Zirbs, E. Reimhult and K. Bica, RSC Adv., 2017, 7, 41144–41151 RSC.
- K. Z. Donato, L. Matejka, R. S. Mauler and R. K. Donato, Colloids Interfaces, 2017, 1, 5 CrossRef CAS.
- D. A. Beattie, R. M. Espinosa-Marzal, T. T. M. Ho, M. N. Popescu, J. Ralston, C. J. E. Richard, P. M. F. Sellapperumage and M. Krasowska, J. Phys. Chem. C, 2013, 117, 23676–23684 CrossRef CAS.
- R. M. Espinosa-Marzal, A. Arcifa, A. Rossi and N. D. Spencer, J. Phys. Chem. Lett., 2014, 5, 179–184 CrossRef CAS.
- A. Elbourne, B. McLean, K. Voitchovsky, G. G. Warr and R. Atkin, J. Phys. Chem. Lett., 2016, 7, 3118–3122 CrossRef CAS.
- I. Szilagyi, T. Szabo, A. Desert, G. Trefalt, T. Oncsik and M. Borkovec, Phys. Chem. Chem. Phys., 2014, 16, 9515–9524 RSC.
- A. Podgorsek, A. S. Pensado, C. C. Santini, M. F. C. Gomes and A. A. H. Padua, J. Phys. Chem. C, 2013, 117, 3537–3547 CrossRef CAS.
- E. Vanecht, K. Binnemans, S. Patskovsky, M. Meunier, J. W. Seo, L. Stappers and J. Fransaer, Phys. Chem. Chem. Phys., 2012, 14, 5662–5671 RSC.
- K. Ueno, A. Inaba, M. Kondoh and M. Watanabe, Langmuir, 2008, 24, 5253–5259 CrossRef CAS.
- P. P. Arquilliere, I. S. Helgadottir, C. C. Santini, P. H. Haumesser, M. Aouine, L. Massin and J. L. Rousset, Top. Catal., 2013, 56, 1192–1198 CrossRef CAS.
- D. Constantinescu, H. Weingartner and C. Herrmann, Angew. Chem., Int. Ed., 2007, 46, 8887–8889 CrossRef CAS.
- N. Debeljuh, C. J. Barrow and N. Byrne, Phys. Chem. Chem. Phys., 2011, 13, 16534–16536 RSC.
- W. L. Li and P. Y. Wu, Polym. Chem., 2014, 5, 761–770 RSC.
- M. M. Elmahdy, C. Gutsche and F. Kremer, J. Phys. Chem. C, 2010, 114, 19452–19458 CrossRef CAS.
- V. Valmacco, G. Trefalt, P. Maroni and M. Borkovec, Phys. Chem. Chem. Phys., 2015, 17, 16553–16559 RSC.
- M. W. Han and R. M. Espinosa-Marzal, ACS Appl. Mater. Interfaces, 2019, 11, 33465–33477 CrossRef CAS.
- P. Rouster, M. Pavlovic, T. Cao, B. Katana and I. Szilagyi, J. Phys. Chem. C, 2019, 123, 12966–12974 CrossRef CAS.
- T. Oncsik, A. Desert, G. Trefalt, M. Borkovec and I. Szilagyi, Phys. Chem. Chem. Phys., 2016, 18, 7511–7520 RSC.
- T. Kobayashi, A. Kemna, M. Fyta, B. Braunschweig and J. Smiatek, J. Phys. Chem. C, 2019, 123, 13795–13803 CrossRef CAS.
- W. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS.
- A. A. Tietze, F. Bordusa, R. Giernoth, D. Imhof, T. Lenzer, A. Maass, C. Mrestani-Klaus, I. Neundorf, K. Oum, D. Reith and A. Stark, ChemPhysChem, 2013, 14, 4044–4064 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. J. Zheng, S. G. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- S. Kondrat and A. A. Kornyshev, Nanoscale Horiz., 2016, 1, 45–52 RSC.
- S. Kondrat, P. Wu, R. Qiao and A. A. Kornyshev, Nat. Mater., 2014, 13, 387–393 CrossRef CAS.
- X. W. Mao, P. Brown, C. Cervinka, G. Hazell, H. Li, Y. Y. Ren, D. Chen, R. Atkin, J. Eastoe, I. Grillo, A. A. H. Padua, M. F. C. Gomes and T. A. Hatton, Nat. Mater., 2019, 18, 1350–1357 CrossRef CAS.
- X. Xu, Y. Li, Y. T. Gong, P. F. Zhang, H. R. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS.
- W. Zhong, F. D. Bobbink, Z. F. Fei and P. J. Dyson, ChemSusChem, 2017, 10, 2728–2735 CrossRef CAS.
- A. V. Delgado, F. Gonzalez-Caballero, R. J. Hunter, L. K. Koopal and J. Lyklema, J. Colloid Interface Sci., 2007, 309, 194–224 CrossRef CAS.
- G. Trefalt, I. Szilagyi and M. Borkovec, J. Colloid Interface Sci., 2013, 406, 111–120 CrossRef CAS.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- P. A. Hassan, S. Rana and G. Verma, Langmuir, 2015, 31, 3–12 CrossRef CAS.
- B. Derjaguin and L. D. Landau, Acta Phys. Chim., 1941, 14, 633–662 Search PubMed.
- T. Oncsik, G. Trefalt, M. Borkovec and I. Szilagyi, Langmuir, 2015, 31, 3799–3807 CrossRef CAS.
- D. Grolimund, M. Elimelech and M. Borkovec, Colloids Surf., A, 2001, 191, 179–188 CrossRef CAS.
- I. Szilagyi, G. Trefalt, A. Tiraferri, P. Maroni and M. Borkovec, Soft Matter, 2014, 10, 2479–2502 RSC.
- T. Lopez-Leon, A. B. Jodar-Reyes, J. L. Ortega-Vinuesa and D. Bastos-Gonzalez, J. Colloid Interface Sci., 2005, 284, 139–148 CrossRef CAS.
- J. Hierrezuelo, A. Vaccaro and M. Borkovec, J. Colloid Interface Sci., 2010, 347, 202–208 CrossRef CAS.
- W. B. Russel, D. A. Saville and W. R. Schowalter, Colloidal dispersions, Cambridge University Press, Cambridge, 1989 Search PubMed.
- N. Schwierz, D. Horinek and R. R. Netz, Langmuir, 2010, 26, 7370–7379 CrossRef CAS.
- M. Borkovec, S. H. Behrens and M. Semmler, Langmuir, 2000, 16, 5209–5212 CrossRef CAS.
- E. J. W. Verwey and J. T. G. Overbeek, Theory of stability of lyophobic colloids, Elsevier, Amsterdam, 1948 Search PubMed.
- J. Hierrezuelo, I. Szilagyi, A. Vaccaro and M. Borkovec, Macromolecules, 2010, 43, 9108–9116 CrossRef CAS.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp02805c |
| ‡ These authors contributed equally to the work. |
| This journal is © the Owner Societies 2020 |