
Broadband spectra with fluorescence and phosphorescence dual emission from bichromophoric platinum metallomesogens containing a 6,12-dihydro-indeno[1,2-b]fluorene linkage†
Yafei Wang*ad,
Jiang Fan‡
a,
Tianyi Li‡b,
Qiong Wanga,
Junwei Shia,
Zuoming Qua,
Hua Tana,
Yu Liua and
Weiguo Zhu*c
aCollege of Chemistry, Key Lab of Environment-Friendly Chemistry and Application of the Ministry of Education, Xiangtan University, Xiangtan 411105, China. E-mail: qiji830404@hotmail.com
bSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, PR China
cSchool of Materials Science & Engineering, Changzhou University, Changzhou 213164, PR China. E-mail: zhuwg18@126.com
dInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
First published on 4th May 2016
Abstract
Two binuclear platinum metallomesogens possessing a 6,12-dihydro-indeno[1,2-b]fluorene bridge, FJ-1 and FJ-2, have been synthesised and characterised by 1H and 13C NMR, TOF-MS and elemental analysis. Their thermal properties were studied by polarised optical microscopy and differential scanning calorimetry. Upon photoexcitation in solution, both complexes exhibited dual fluorescence (1π–π*)/phosphorescence (3π–π*) emission covering most of the visible spectrum (from about 460 nm to 750 nm). This uncommon behaviour has been further investigated by time-resolved optical spectroscopy, including nanosecond time-resolved transient absorption spectroscopy, and further insights were obtained by means of density functional theory (DFT) and time-dependent DFT (TD-DFT). An organic light-emitting diode (OLED) using FJ-1 was prepared. Importantly the two emission bands are retained in electroluminescence. This result suggests that controlling spin–orbit coupling to obtain dual emission can provide an effective way of achieving broadband emission based on a single molecule.
1. Introduction
Cyclometallated platinum complexes exhibit rich and diverse photophysical properties.1,2 Emission can originate from a variety of excited states with different character such as ligand-centred (LC), ligand-to-ligand charge transfer (LLCT), metal-to-ligand charge transfer (MLCT) and metal–metal-to-ligand charge transfer (MMLCT).3–13 In addition, their square-planar geometry is highly suitable for self-assembling processes that can lead to luminescent materials with controlled morphologies.14–20 This unique combination of photophysical and structural properties has made platinum complexes very popular for many applications like photocatalysis,21,22 bioimaging,23–25 sensing,26,27 photodynamic therapy.28,29An important application for cyclometallated Pt complexes is as phosphorescent emitters for organic light-emitting diodes (OLEDs). Over the past two decades, blue, green and red high-efficiency platinum-based phosphorescent materials and OLEDs have been achieved.11,30–35 Due to the importance of lighting efficiency for the future and the complexity of multilayer white OLEDs,36,37 various single-component approaches have been explored to simplify the device architecture.38–41 In this context Pt complexes are particularly appealing as they provide the possibility to prepare white OLEDs simply by increasing the concentration of emitter because of the formation of excimers. As a result, at intermediate concentration white light is obtained by the combination of blue emission from the monomer and orange emission from the excimer.42–48 However, as the formation of excimers is a bimolecular process, the resulting emission can be greatly impacted by small variations of concentration and external condition (e.g. solvent, host, temperature).
An alternative approach to the excimer-based strategy for white light is to develop fluorescent/phosphorescent dual emitters. In general, the presence of heavy atoms such as third row transition metal ions results in very efficient inter-system crossing (ISC) followed by efficient phosphorescence. However, if a steric chromophoric group is introduced into the metal complex, a dual-emission could be observed due to only little electronic interaction between a heavy metal ion and the appended chromophore. This phenomenon has been reported with complexes of osmium,50 iridium,51 gold,52,53 rhenium,54,55 and platinum.49,56–59 In one case, dual fluorescent/phosphorescent emitters have been used as emitter for white OLEDs (Scheme 1).49 External efficiency of 2.6% was obtained with CIE coordinates (0.33, 0.39), very close to a perfect white.
 | ||
| Scheme 1 Previously reported fluorescent/phosphorescent dual emitting Pt complexes for OLEDs.49 | ||
In all aforementioned examples of fluorescent/phosphorescent dual emitters based on third row transition metals, the ratio of heavy atoms to fluorescent units is 1 (mononuclear complexes) or below (polymers). To further investigate this family of dual emitters, we report here binuclear platinum complexes with a single fluorescent unit as the bridge, hence a ratio of heavy atoms to fluorescent units of 2.
Our design is based on naphthyl-pyridine as the cyclometallating unit of the ligand to provide orange/red phosphorescence and 6,12-dihydro-indeno[1,2-b]fluorene as the blue fluorescent bridge. The 6,12-dihydro-indeno[1,2-b]fluorene unit was chosen because of its suitable photophysical properties, namely high fluorescence quantum yield and tunable sky-blue emission, and chemical reactivity, which makes it straightforward to connect to the naphthyl-pyridine unit.60–66 To further explore the relationship between photophysical properties and chemical structures, two ancillary ligands have been used: acetylacetone (acac, complex FJ-1) and dibenzoylmethane (DBM, complex FJ-2). The complexes showed liquid crystalline properties, evidenced by polarized optical microscopy (POM) and differential scanning calorimetry (DSC). The photophysical properties of both complexes were studied by steady-state and time-resolved optical spectroscopy and density function theory (DFT) and time-dependent DFT (TD-DFT) calculations provided additional insights into the properties of the excited state. Importantly, we show that despite the presence of an additional heavy atom, fluorescence/phosphorescence dual emission can be observed both in solution and in electroluminescent devices.
2. Results and discussion
2.1 Synthesis
The synthesis of the binuclear complexes is outlined in Scheme 2. The commercially available compound 1 was borylated by halogen–lithium exchange using n-butyl lithium followed by reaction with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane to afford compound 2 in 90% yield. Compound 5 was obtained in 67% yield from the reaction of precursors 3 and 4 via a Suzuki coupling reaction using Pd(PPh3)4 as catalyst. A Suzuki coupling reaction between the diborane 2 and the halide 5 furnished the cyclometalated ligand 6 in 68% yield.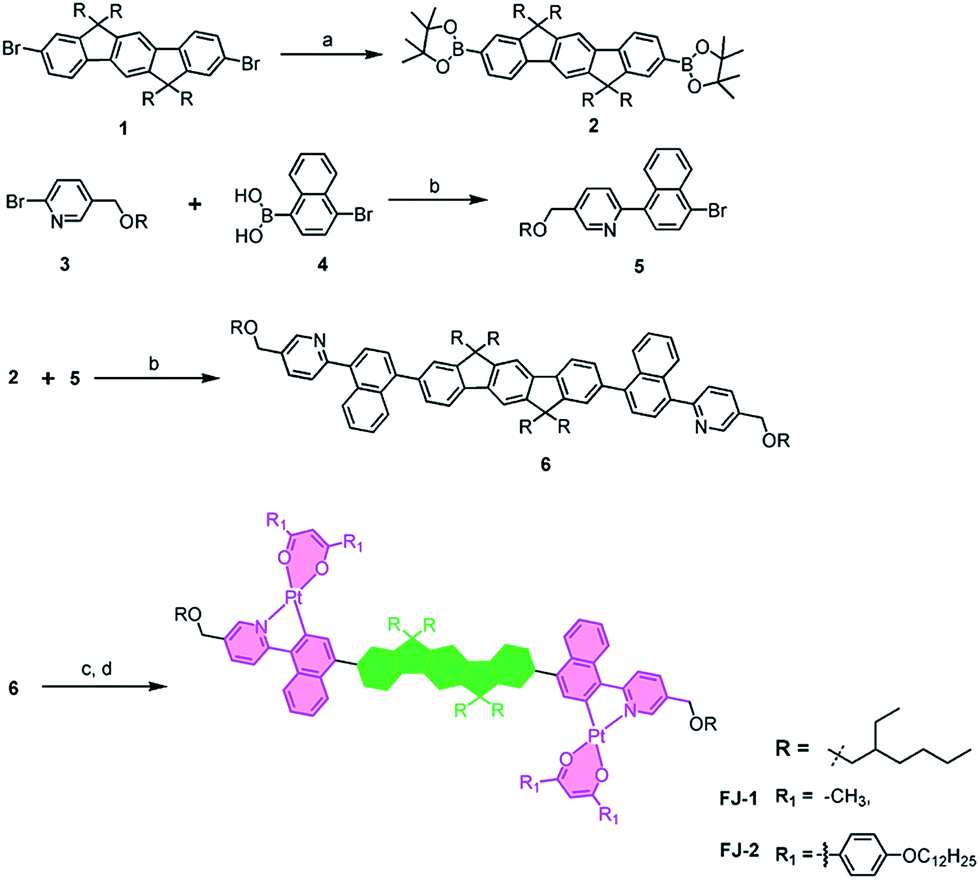 | ||
| Scheme 2 Synthetic route of platinum complexes. Reagents and conditions: (a) 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, n-BuLi, THF, −78 °C, overnight; (b) Pd(PPh3)4, 2 M K2CO3, THF, reflux, 24 h; (c) K2PtCl4, THF, H2O, 80 °C, 24 h; (d) Na2CO3, Na(acac) or Na(DBM), 2-ethoxyethanol, 100 °C, 24 h. | ||
The target binuclear platinum complexes were synthesized in 5–10% yield using our previously reported two-step procedure.26 Both platinum complexes were characterized by 1H NMR, 13C NMR, TOF-MS and elemental analysis (ESI†).
2.2 Thermal properties
The thermal properties of both platinum complexes have been evaluated by thermogravimetric analysis (TGA), and the phase-transition behaviors were studied by differential scanning calorimetry (DSC) and polarized optical microscopy (POM) (Fig. S1–S3†). Both binuclear platinum complexes show good thermal stability (286 °C for FJ-1 and 290 °C for FJ-2) at 5% weight loss. The DSC traces of FJ-1 show two distinct phase transition peaks at 189 °C and 220 °C during the second heating cycle, while no phase transition peak was detected on cooling. On the contrary, FJ-2 shows a crystal–crystal transition in the range of 70–90 °C and the clearing point at 198 °C (on first heating and POM). Compared to FJ-1, FJ-2 exhibits a distinctly decreased clearing point due to the increased number of periphery alkyl chains provided by the ancillary ligand. Interestingly, typical fan-like textures with good fluidity were observed by POM from the isotropic liquid on cooling, pointing to the presence of a smectic phase for FJ-1 and FJ-2.2.3 Electronic absorption spectroscopy
The photophysical data of FJ-1 and FJ-2 are summarised in Table 1. The absorption spectra of the cyclometallating ligand, FJ-1 and FJ-2 measured in CH2Cl2 solutions (10−5 M) at room temperature are depicted in Fig. 1. When the concentration of complex is reduced to 10−6 M, the absorption spectra are identical to those obtained from 10−5 M solutions (Fig. S4†). This shows that there is no interaction in the ground state between the complexes in the range of concentration studied here. Overall, both complexes show similar spectra. By comparison with the absorption spectrum of the ligand, the high-energy absorption band at about 360 nm is attributed to π–π* transition centred on the main ligand. In the case of FJ-2, this LC band is much more intense due to the overlap with the π–π* transition involving the ancillary ligand (the kinks in absorption band at about 350–360 nm are caused by the facility at high concentration). According to previous reports35 and theoretical calculations (vide infra) the low-energy absorption features (400–480 nm) are described as superposition of ligand centred and metal-to-ligand charge transfer (MLCT) transitions. | ||
| Fig. 1 UV-vis absorption spectra of the ligand (black), FJ-1 (red) and FJ-2 (blue) measured in CH2Cl2 (10−5 M) at room temperature. | ||
2.4 Photoluminescence
Upon photoexcitation, both platinum complexes display two well-resolved emission bands in solution, one at high energy (HE, ≈532, 574 nm) and one at low energy (LE, ≈640 nm) (Fig. 2 and Table 1), which is different from our previously reported mononuclear (Piq)Pt(acac) and binuclear (biC12ppy-fl)Pt2(C12DBM)2 complexes.28,29 | ||
| Fig. 2 Emission in CH2Cl2, conc. = 10−5 M, λexc = 460 nm, of (a) FJ-1; (b) FJ-2; (c) FJ-1 at room temperature (red) and 77 K (black) (normalised spectra); (d) FJ-2 at room temperature (red) and 77 K (black) (normalized spectra). | ||
According to the previous reports,49,51 in this case, the HE and LE bands are therefore ascribed to fluorescence and phosphorescence, respectively.
This assignment is supported by degassing the solution of FJ-2 with N2: a large increase of the intensity of the LE band is observed, while the intensity of the HE band remains unchanged (Fig. S5,† the minor intensity increase is attributed to loss of solvent during N2 bubbling). Furthermore, the lifetime of excited state of both HE and LE bands of FJ-1 and FJ-2 were evaluated by time-resolved spectroscopy in the nanosecond to millisecond regimes. As expected, the LE bands of FJ-1 (1.9 μs) and FJ-2 (2.5 μs) are long-lived and on the microsecond time-scale expected for phosphorescence from cyclometalated Pt complexes while the HE bands are very short-lived (<5 ns, limit of the instrument). This fluorescence-phosphorescence dual emission is due to marginal participation of the metal d orbitals to the HOMO of the complexes. Thus the heavy platinum atoms will have only a small effect on the inter-system crossing, which then competes with fluorescence.50 The photoluminescence quantum yield (Φem) is very small at 0.7 and 1.5% for FJ-1 and FJ-2, respectively.
The emission of FJ-2 was measured in solvents of increasing polarity (CH2Cl2, THF, dioxane, MeOH, see Fig. S6†). Only minor solvatochromism was observed pointing to an excited state with large 3π,π* character. Furthermore the shift is hypsochromic as the polarity of the solvent increases, which showed that the dipole moment of the excited state is smaller than the dipole moment of the ground state, as previously reported for similar complexes.67
Fig. 2c and d present the emission spectra of FJ-1 and FJ-2 in degassed CH2Cl2 at 77 K compared to the PL spectra at room temperature. Notably at 77 K, no HE bands are observed, only intense LE phosphorescence bands with a clear vibrational structure. This phenomenon suggests that the rate of intersystem crossing to the triplet state is much faster than the radiative rate of fluorescence.20 This can be explained with the PL of the ligand at 77 K, which displays phosphorescence at ≈440 nm reminiscent of the phosphorescence of the 6,12-dihydro-indeno[1,2-b]fluorene unit (Fig. S7†).63 Such phosphorescence would be easily quenched by the triplet state responsible for the LE emission bands. In addition to an increase of the vibronic structure, blue-shifted LE emission bands (FJ-1: 2 nm; FJ-2: 10 nm) were observed at 77 K compared to that at room temperature because of the rigidochromic effect in the solid matrix. These small shifts further supported the 3π,π* character of the LE emission band in both platinum complexes. The decay lifetimes of FJ-1 (9.6 μs) and FJ-2 (10.9 μs) (Fig. 3) at 77 K are distinctly longer than those at room temperature, which points to the role of thermally activated non-radiative pathways in the low photoluminescence quantum yield of these complexes at room temperature.
 | ||
| Fig. 3 Phosphorescence decays of FJ-1 and FJ-2 measured in CH2Cl2 at 77 K under argon. | ||
In contrast to the emission observed for platinum complexes in solution, only one emission peak (616 nm for FJ-1 and 630 nm for FJ-2) was recorded for platinum complexes in the neat films at the same excitation wavelength (Fig. S8†). There is an aggregation in the neat film, leading to more strong intramolecular interaction.49 Additionally, the profiles in neat films have a blue-shifted emission compared to that in solution, probably that the periphery alkyl chains effectively suppress the intermolecular aggregation in the solid state. It is worth to note that FJ-2 shows a red shift compared to FJ-1, which is attributed to the expanded π-conjugation DBM ligand.
2.5 Transient absorption spectroscopy
To understand further the character of the triplet excited state of the complexes, nanosecond transient absorption (TA) spectroscopy studies were carried out in CH2Cl2 at room temperature (Fig. 4). The decays measured for FJ-1 (2.7 μs) and FJ-2 (5.1 μs) (Fig. S9 and S10†) are single-exponential and similar to their emission lifetimes determined by time-resolved photoluminescence. This confirms that the same excited states are probed with both transient absorption and steady-state emission spectroscopies. | ||
| Fig. 4 Time-resolved transient differential absorption spectra of FJ-1 (a) and FJ-2 (b) in degassed CH2Cl2. The probe wavelength is 525 nm. | ||
As depicted in Fig. 4, both binuclear platinum complexes show the different time-resolved transient absorption spectra. FJ-1 possesses a significant ground-state bleach (GSB) at 362 nm corresponding to its 1π,π* LC transition in the UV-vis absorption spectra. In addition, a broad, positive photoinduced absorption bands between 417 nm and 760 nm was observed for FJ-1. This photoinduced absorption is ascribed to T1–Tn triplet exciton absorption.68,69 In contrast, FJ-2 exhibits the bleach (361 nm and 429 nm) in the region of the π,π* LC transition and charge-transfer absorption band. Also, FJ-2 shows the excited-state absorption bands in the 460–740 nm and 750–790 nm regions. These different spectral features can clearly explain the different lifetimes and the different emission characters of FJ-1 and FJ-2. Furthermore, the triplet excited-state absorption in the 600–700 nm decreases sharply with the expanded π-conjugation in the ancillary ligand.
2.6 Theoretical calculations
To gain insights into the relationship between electronic absorption and molecular structure, density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were carried out for FJ-1. The molecular orbitals around the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap for the geometry optimised in the ground state are depicted in Fig. 5. The HOMO is mainly (94.69% contribution) localized over the indeno[1,2-b]fluorene and naphthalene units, with a small contribution from the pyridine moiety and platinum orbital (4.86% of Pt orbital). The LUMO is predominately based on the entire cyclometalated ligand skeleton and platinum atoms (93.09% contribution of cyclometalated ligand and 5.72% of Pt orbital). Both HOMO−1 and LUMO+1 are mostly centred on the naphthalenylpyridine moiety and platinum atom, with only minor contribution from the indeno[1,2-b]fluorine group. The calculated adiabatic ionization energy (−5.7 eV) and electron affinity (−3.21 eV) of FJ-1 result in an overestimation of the HOMO–LUMO gap evaluated by electrochemistry (see below). The calculated absorption spectrum in CH2Cl2 is given in Fig. S11.† Although blue-shifted, the shape of calculated spectrum shows a good agreement with that of the experimental spectrum with three main bands at 257, 350 and 450 nm. The molecular orbitals involved in the transitions contributing to the absorption bands are listed in Table S2.† The lowest-energy absorption of FJ-1 at about 450 nm is a combination of ILCT involving the cyclometalated ligand and MLCT transition. The high-energy bands involve only the cyclometalated ligand. | ||
| Fig. 5 Contour plots of the frontier molecular orbitals in the ground state of FJ-1. | ||
2.7 Electrochemistry
The redox properties of FJ-1 and FJ-2 were recorded in CH2Cl2 by cyclic voltammetry (Fig. S12†). Both complexes show an irreversible oxidation wave with onset at 0.77 V vs. Fc+/Fc (FJ-1) and 0.72 V vs. Fc+/Fc (FJ-2). Poorly defined quasi-reversible reduction waves are observed at −1.42 V vs. Fc+/Fc and −1.45 V vs. Fc+/Fc for FJ-1 and FJ-2, respectively. According to the empirical formula [EHOMO = −(Eox + 4.8) eV and ELUMO = −(Ered + 4.8) eV],70 the HOMO and LUMO of FJ-1 and FJ-2 were estimated to be at −5.57 eV/−3.38 eV and −5.52 eV and −3.35 eV, respectively, which is in good agreement with the theory calculation. The similarity of the values for the two complexes shows that there is only little effect of changing the ancillary ligand, mostly on the HOMO.2.8 Electroluminescent property
To investigate the electroluminescent (EL) property of these binuclear platinum complexes, OLEDs employing FJ-1 as the emitting dopant were fabricated with the following configuration: ITO/PEDOT:PSS (30 nm)/TFB (20 nm)/PVK![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OXD-7:FJ-1 (70:20:10, 50 nm)/TmPyPB (50 nm)/Liq (2 nm)/Al (100 nm) (Fig. 6 inset). As shown in Fig. 6, the device exhibits a broad EL spectrum in the region of 420–850 nm at 10 V. The Commission International de L'Éclairage (CIE) colour coordinates are (0.39, 0.29), which is in the range of near-white light emission. The emission bands centred at 662 nm and ca. 500 nm are attributed to FJ-1. The emission band at about 438 nm is ascribed to the PVK/OXD-7 host matrix71,72 demonstrating incomplete energy transfer between the host matrix and the emitting dopant. In fact, this phenomenon can be explained by the unmatched spectra between the UV-vis absorption of platinum complex and the emission of host matrix, implying an incomplete Förster energy transfer. Additionally, the OXD-7 is in favor of electron transport, which can increase the exciton energy resulting in the short-wavelength emission.
:OXD-7:FJ-1 (70:20:10, 50 nm)/TmPyPB (50 nm)/Liq (2 nm)/Al (100 nm) (Fig. 6 inset). As shown in Fig. 6, the device exhibits a broad EL spectrum in the region of 420–850 nm at 10 V. The Commission International de L'Éclairage (CIE) colour coordinates are (0.39, 0.29), which is in the range of near-white light emission. The emission bands centred at 662 nm and ca. 500 nm are attributed to FJ-1. The emission band at about 438 nm is ascribed to the PVK/OXD-7 host matrix71,72 demonstrating incomplete energy transfer between the host matrix and the emitting dopant. In fact, this phenomenon can be explained by the unmatched spectra between the UV-vis absorption of platinum complex and the emission of host matrix, implying an incomplete Förster energy transfer. Additionally, the OXD-7 is in favor of electron transport, which can increase the exciton energy resulting in the short-wavelength emission.
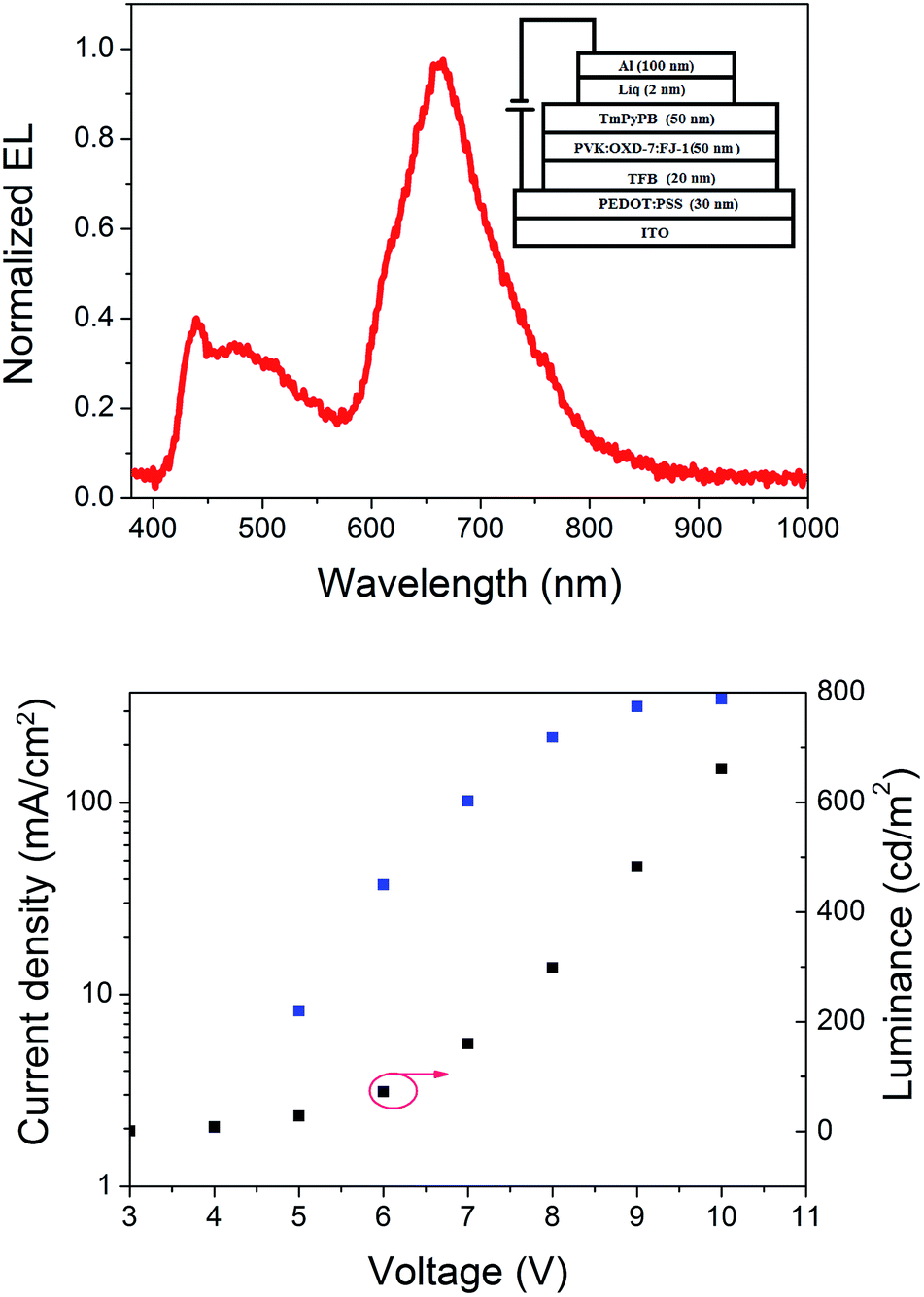 | ||
| Fig. 6 EL spectrum of FJ-1 based device at 10 V (top); I–V–L curves of FJ-1 based devices (bottom). | ||
The current density–voltage–luminance (I–V–L) curves are depicted in Fig. 6. The device shows a low turn on voltage of 3 V. Although the OLED possesses unsatisfying performance with maximum brightness of 661 cd cm−2 at 10 V and a maximum external quantum efficiency of 0.16%, it shows that fluorescent/phosphorescent binuclear platinum complexes can be used for developing broad electroluminescence.
3. Conclusions
Two novel binuclear platinum complexes with a 6,12-dihydro-indeno[1,2-b]fluorene bridging unit were prepared and fully characterized. These binuclear platinum complexes showed a smectic phase and fluorescence/phosphorescence dual emission in solution and EL devices. The relationship between electronics and bichromophoric platinum complexes was explored by both experimental and theoretical studies. The results demonstrate that dual emission can be achieved by incorporation of a large fluorescent organic unit into binuclear platinum complexes. The very small participation of d metal orbitals in the HOMO and LUMO ensure inefficient spin–orbit coupling allowing the fluorescence from the appended chromophore to compete with ISC deactivation. Although the preliminary device performances are not satisfying, this research could pave the road for the design of broad emission materials based on single molecular compound.4. Experimental section
4.1 Materials and measurement
Compounds 1 and 4 are commercial from Energy Chemical Company Ltd. Other reagents were purchased from J&K Chemical and Aladdin companies. All reactions were carried out under N2 atmosphere. Compound 3 has been reported in our previous work.25 1H NMR and 13C NMR spectra were acquired using a Bruker Dex-400 NMR instrument using CDCl3 as a solvent. Mass spectra (MS) were recorded on a Bruker Autoflex MALDI-TOF instrument using dithranol as a matrix. Elemental analysis was determined by Vario EL III. The UV-vis absorption and photoluminescence (PL) spectra were measured with a Varian Cray 50 and Perkin-Elmer LS50B luminescence spectrometer, respectively. Lifetimes were measured with Edinburgh analytical instrument (FLS920 fluorescence spectrometer) in degassed CH2Cl2 at room temperature and 77 K. The time-resolved transient absorption spectra at different delay times after a λ = 525 nm laser pulse excitation were recorded with flash photolysis spectrometer (LP920-K, Edinburgh Instruments). Thermogravimetric analysis (TGA) was carried out with a NETZSCH STA449 from 25 °C to 600 °C at a 20 °C min−1 heating rate under N2 atmosphere. Differential scanning calorimerty (DSC) was measured at the phase transition temperature with a rate of 20 °C min−1 on the first heating circle and 10 °C min−1 on the first cooling and second heating process. Polarized optical microscopy (POM) was recorded the birefringent phenomenon with a rate of 1 °C min−1 on cooling process. Electrochemical property was evaluated by cyclic voltammetry with three typical electrodes in degassed CH2Cl2 solution with a rate of 100 mV s−1. The CV system employed Bu4NPF6 as electrolyte. Platinum disk is used as the working electrode, platinum wire is regarded as the counter electrode and silver wire is used as the reference electrode. Ferrocenium/ferrocene (Fc/Fc+) was used as the internal standard compound.4.2 Computation method
The molecular geometry of the ground state (S0) is fully optimized with the B3LPY exchange–correlation functional using the LANL2DZ basis set for Pt atoms and the 6-31G** basis set for the other atoms. Vibrational frequency calculation was performed to validate that the optimized structure is minima on the potential energy surface. Based on the optimized S0 structure, TD-DFT calculation was conducted, with M062x functional and the same combination of basis sets, to determine the vertical transition characters and simulate the UV-vis absorption spectrum. Solvent effect was always taken under consideration for all the calculations by employing C-PCM model as in CH2Cl2.4.3 Devices fabrication and characterization
The device employing FJ-1 as the dopant were fabricated by solution-processed approach. In these OLEDs, PEDOT:PSS was spin coated onto ITO substrate with 30 nm thick layer, which is utilized as the smooth layer and hole inject layer. The blend of PVK and OXD-7 was used as the host matrix. TmPyPB is the electron transporting layer with 40 nm thick layer. Liq and Al are used as the cathode. Electroluminescent (EL) spectra were recorded using an optical analyzer, Photo Research PR705. The features of current density and brightness versus applied voltage were obtained by a Keithley 2420 and a Konica Minolta Chromameter CS-200, respectively. EQE was calculated from the luminance, current density, and EL spectrum, assuming a Lambertian distribution. The device configurations are as follows: ITO/PEDOT:PSS (30 nm)/TFB (20 nm)/PVK:OXD-7:FJ-1 (70:20:10, 50 nm, 120 °C)/TmPyPB (50 nm)/Liq (2 nm)/Al (100 nm).
4.4 2,8-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6,6′,12,12′-tetraisooctyl-6,12-dihydroindeno[1,2-b]fluorene (2)
n-BuLi (5.6 mL, 2.5 M solution in hexane) was slowly added to a solution of compound 1 (2.0 g, 2.33 mmol) in dry THF (60 mL) at −78 °C under nitrogen. The reaction mixture was stirred for 2 h at −78 °C, and then 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3.5 g, 18.6 mmol) was rapidly added to the solution and the resulting mixture was stirred for 2 h at −78 °C. The reaction solution was allowed to reach room temperature and stirred for 24 h under nitrogen. The mixture was poured into water and extracted with CH2Cl2 (3 × 30 mL). The combined organic layer was dried over anhydrous MgSO4 and filtered. The filtrate was evaporated to remove the solvent and the residue was passed through a flash silica gel column using [CH2Cl2:petroleum ether (PE) (v/v) = 1:3] as the eluent to give a yellow solid (1.0 g, 45.5%). 1H NMR (400 MHz, CDCl3, TMS), δ (ppm): 7.82–7.78 (m, 4H), 7.72–7.69 (m, 4H), 2.04 (m, 8H), 1.37 (s, 24H), 0.89–0.64 (m, 48H), 0.46–0.43 (m, 12H).
4.5 2-(4-Bromonaphthyl)-5-(isooctyloxymethyl)pyridine (5)
To a mixture of compound 4 (189.0 mg, 0.76 mmol), compound 3 (226.0 mg, 0.76 mmol) and Pd(PPh3)4 (45.0 mg, 0.04 mmol) was added THF (15 mL) and K2CO3 aqueous (2 M, 5 mL). The mixture was refluxed for 24 h under nitrogen. After cooling to room temperature, the mixture was poured into water (100 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic layer was dried over anhydrous MgSO4 and filtered. The filtrate was evaporated to remove the solvent and the residue was passed through a flash silica gel column using [CH2Cl2:PE (v/v) = 2:1] as the eluent to give a yellow oily liquid (0.22 g, 67.6%). 1H NMR (CDCl3, 400 MHz, TMS), δ (ppm): 8.74 (s, 1H), 8.34 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.88–7.82 (m, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.56–7.50 (m, 2H), 7.43 (d, J = 7.6 Hz, 1H), 4.61 (s, 2H), 3.46 (d, J = 5.6 Hz, 2H), 1.49–1.11 (m, 12H), 0.93–0.89 (m, 3H).
:CH2Cl2 (v/v) = 1:1] as the eluent to give a white solid (0.6 g, 68.5%). 1H NMR (CDCl3, 400 MHz, TMS), δ (ppm): 8.83 (s, 2H), 8.22 (d, J = 8.4 Hz, 2H), 8.13–8.07 (m, 2H), 7.91 (d, J = 7.6 Hz, 4H), 7.83 (s, 2H), 7.72–7.70 (m, 4H), 7.58–7.47 (m, 10H), 4.69 (s, 4H), 3.53 (d, J = 5.6 Hz, 4H), 2.19–2.11 (m, 8H), 1.69–1.62 (m, 8H), 1.52–1.30 (m, 22H), 1.02–0.74 (m, 48H), 0.62–0.59 (m, 12H). 13C NMR (100 MHz, CDCl3), δ (ppm): 14.15, 14.16, 14.23, 14.27, 22.96, 23.02, 23.17, 23.93, 29.13, 30.69, 34.71, 39.73, 54.74, 70.55, 73.78, 119.02, 124.79, 125.73, 125.87, 126.22, 126.79, 127.07, 128.79, 131.57, 132.56, 135.85, 137.56, 138.64, 141.06, 141.85, 148.83, 150.18, 151.03, 158.62. MALDI-MS (m/z): calcd for C100H132N2O2, 1393.03; found, 1394.063.4.6 General procedures for complexes FJ-1 and FJ-2
To a mixture of K2PtCl4 (161.0 mg, 0.32 mmol), 2-ethoxyethanol (9 mL) and water (3 mL) was added a solution of compound 6 (450.0 mg, 0.32 mmol) in THF (9 mL). The reaction mixture was heated to 80 °C for 24 h under nitrogen. After cooling to room temperature, the mixture was poured into water (5 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was dried over anhydrous MgSO4 and filtered. After removing the solvent, the di-μ-chloro dimer was obtained and used to the next step without any further purification.A mixture of the di-μ-chloro dimer (1 eq.), ancillary ligand (2.5 eq. acac or DBM) and sodium carbonate (10 eq.) in dry 2-ethoxyethanol (15 mL) was stirred at 100 °C for 24 h under nitrogen. After cooling to room temperature, the mixture was poured into water (30 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was dried over anhydrous MgSO4 and filtered. After removing the solvent, the residue was passed through a flash silica gel column using [CH2Cl2:PE (v/v) = 2:1] to give complexes FJ-1 and FJ-2.
Acknowledgements
The authors would like to thank Dr Etienne Baranoff (University of Birmingham, UK) for his help on language. This work is supported by the National Natural Science Foundation of China (21202139, 51473140 and 51273168), the Program for Innovative Research Cultivation Team in University of Ministry of Education of China (1337304), the Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology) (2015-skllmd-08), and the Key Fund Project of Hunan Provincial Education Department (13A133).Notes and references
- J. A. G. Williams, Photochem. Photophys. Coord. Compd., 2007, 281, 205 CAS
.
- J. A. G. Williams, S. Develay, D. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596 CrossRef
- L. Chassot, E. Müller and A. von Zelewsky, Inorg. Chem., 1984, 23, 4249 CrossRef CAS
- C.-M. Che, W.-F. Fu, S.-W. Lai, Y.-J. Hou and Y.-L. Liu, Chem. Commun., 2003, 118 RSC
- H. Jude, J. A. Krause Bauer and W. B. Connick, Inorg. Chem., 2004, 43, 725 CrossRef CAS PubMed
- W. Lu, M. C. W. Chan, K.-K. Cheung and C.-M. Che, Organometallics, 2001, 20, 2477 CrossRef CAS
- D. A. Vezzu, J. C. Deaton, J. S. Jones, L. Bartolotti, C. F. Harris, A. P. Marchetti, M. Kondakova, R. D. Pike and S. Huo, Inorg. Chem., 2010, 49, 5107 CrossRef CAS PubMed
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609 CrossRef CAS PubMed
- A. Chakraborty, J. C. Deaton, A. Haefele and F. N. Castellano, Organometallics, 2013, 32, 3819 CrossRef CAS
- R. M. van der Veen, J. J. Kas, C. J. Milne, V. T. Pham, A. El Nahhas, F. A. Lima, D. A. Vithanage, J. J. Rehr, R. Abela and M. Chergui, Phys. Chem. Chem. Phys., 2010, 12, 5551 RSC
- P. K. Chow, G. Cheng, G. S. Tong, W. P. To, W. L. Kwong, K. H. Low, C. C. Kwok, C. Ma and C. M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084 CrossRef CAS PubMed
- P. Pinter, H. Mangold, I. Stengel, I. Münster and T. Strassner, Organometallics, 2016, 35, 673 CrossRef CAS
- D. Escudero and W. Thiel, Inorg. Chem., 2014, 53, 11015 CrossRef CAS PubMed
- K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 2011, 424 CrossRef PubMed
- N. K. Allampally, C. A. Strassert and L. De Cola, Dalton Trans., 2012, 41, 13132 RSC
- M. G. Debije, M. P. de Haas, J. M. Warman, M. Fontana, N. Stutzmann, M. Kristiansen, W. R. Caseri, P. Smith, S. Hoffmann and T. I. Sølling, Adv. Funct. Mater., 2004, 14, 323 CrossRef CAS
- V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286 CrossRef CAS PubMed
- U. Maran, H. Conley, M. Frank, A. M. Arif, A. M. Orendt, D. Britt, V. Hlady, R. Davis and P. J. Stang, Langmuir, 2008, 24, 5400 CrossRef CAS PubMed
- M. Mauro, A. Aliprandi, C. Cebrian, D. Wang, C. Kubel and L. De Cola, Chem. Commun., 2014, 50, 7269 RSC
- C. Po, A. Y. Tam, K. M. Wong and V. W. Yam, J. Am. Chem. Soc., 2011, 133, 12136 CrossRef CAS PubMed
- D. Zhang, L. Z. Wu, L. Zhou, X. Han, Q. Z. Yang, L. P. Zhang and C. H. Tung, J. Am. Chem. Soc., 2004, 126, 3440 CrossRef CAS PubMed
- Q. Y. Meng, T. Lei, L. M. Zhao, C. J. Wu, J. J. Zhong, X. W. Gao, C. H. Tung and L. Z. Wu, Org. Lett., 2014, 16, 5968 CrossRef CAS PubMed
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144 RSC
- V. M. Manikandamathavan, N. Duraipandy, M. S. Kiran, V. G. Vaidyanathan and B. U. Nair, RSC Adv., 2015, 5, 24877 RSC
- P. Wu, E. L. Wong, D. L. Ma, G. S. Tong, K. M. Ng and C. M. Che, Chem.–Eur. J., 2009, 15, 3652 CrossRef CAS PubMed
- N. J. Demas and B. A. DeGraff, Coord. Chem. Rev., 2001, 211, 317 CrossRef
- K. Li, Y. Chen, W. Lu, N. Zhu and C. M. Che, Chem.–Eur. J., 2011, 17, 4109 CrossRef CAS PubMed
- R. E. Doherty, I. V. Sazanovich, L. K. McKenzie, A. S. Stasheuski, R. Coyle, E. Baggaley, S. Bottomley, J. A. Weinstein and H. E. Bryant, Sci. Rep., 2016, 6, 22668 CrossRef CAS PubMed
- Z. Zhang and X. Dong, Biometals, 2009, 22, 283 CrossRef CAS PubMed
- T. Fleetham, G. Li, L. Wen and J. Li, Adv. Mater., 2014, 26, 7116 CrossRef CAS PubMed
- H. Fukagawa, T. Shimizu, H. Hanashima, Y. Osada, M. Suzuki and H. Fujikake, Adv. Mater., 2012, 24, 5099 CrossRef CAS PubMed
- F. Hua, S. Kinayyigit, J. R. Cable and F. N. Castellano, Inorg. Chem., 2005, 44, 471 CrossRef CAS PubMed
- Z. M. Hudson, C. Sun, M. G. Helander, Y. L. Chang, Z. H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930 CrossRef CAS PubMed
- X. C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753 CrossRef CAS PubMed
- M. Velusamy, C.-H. Chen, Y. S. Wen, J. T. Lin, C.-C. Lin, C.-H. Lai and P.-T. Chou, Organometallics, 2010, 29, 3912 CrossRef CAS
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245 CrossRef CAS
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572 CrossRef CAS PubMed
- Y. Liu, M. Nishiura, Y. Wang and Z. M. Hou, J. Am. Chem. Soc., 2006, 128, 5592 CrossRef CAS PubMed
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2012, 134, 20290 CrossRef CAS PubMed
- H. J. Bolink, F. De Angelis, E. Baranoff, C. Klein, S. Fantacci, E. Coronado, M. Sessolo, K. Kalyanasundaram, M. Gratzel and M. K. Nazeeruddin, Chem. Commun., 2009, 4672 RSC
- S. H. Kim, S. Park, J. E. Kwon and S. Y. Park, Adv. Funct. Mater., 2011, 21, 644 CrossRef CAS
- B. W. D'andrade, J. Brooks, V. Adamovich, M. E. Thompson and S. R. Forrest, Adv. Mater., 2002, 14, 1032 CrossRef
- T. Fleetham, L. Huang and J. Li, Adv. Funct. Mater., 2014, 24, 6066 CrossRef CAS
- B. Ma, P. I. Djurovich, S. Garon, B. Alleyne and M. E. Thompson, Adv. Funct. Mater., 2006, 16, 2438 CrossRef CAS
- E. L. Williams, K. Haavisto, J. Li and G. E. Jabbour, Adv. Mater., 2007, 19, 197 CrossRef CAS
- L. Murphy, P. Brulatti, V. Fattori, M. Cocchi and J. A. Williams, Chem. Commun., 2012, 48, 5817 RSC
- J. Kalinowski, M. Cocchi, D. Virgili, V. Fattori and J. A. G. Williams, Adv. Mater., 2007, 19, 4000 CrossRef CAS
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D'Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171 RSC
- Z. He, W. Y. Wong, X. M. Yu, H. S. Kwok and Z. Y. Lin, Inorg. Chem., 2006, 45, 10922 CrossRef CAS PubMed
- Y. L. Chen, S. W. Li, Y. Chi, Y. M. Cheng, S. C. Pu, Y. S. Yeh and P. T. Chou, ChemPhysChem, 2005, 6, 2012 CrossRef CAS PubMed
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804 CrossRef CAS PubMed
- W. Lu, W. M. Kwok, C. Ma, C. T. Chan, M. X. Zhu and C. M. Che, J. Am. Chem. Soc., 2011, 133, 14120 CrossRef CAS PubMed
- H. Y. Chao, W. Lu, Y. Q. Li, M. C. W. Chan, C. M. Che, K. K. Cheung and N. Y. Zhu, J. Am. Chem. Soc., 2002, 124, 14696 CrossRef CAS PubMed
- D. Bhattacharya, Inorg. Chem. Commun., 2013, 36, 159 CrossRef CAS
- L. Salassa, C. Garino, A. Albertino, G. Volpi, C. Nervi, R. Gobetto and K. I. Hardcastle, Organometallics, 2008, 27, 1427 CrossRef CAS
- F. Geist, A. Jackel and R. F. Winter, Dalton Trans., 2015, 44, 3974 RSC
- F. Geist, A. Jackel and R. F. Winter, Inorg. Chem., 2015, 54, 10946 CrossRef CAS PubMed
- C. J. Lin, C. Y. Chen, S. K. Kundu and J. S. Yang, Inorg. Chem., 2014, 53, 737 CrossRef CAS PubMed
- H. F. Xiang, L. Zhou, Y. Feng, J. H. Cheng, D. Wu and X. G. Zhou, Inorg. Chem., 2012, 51, 5208 CrossRef CAS PubMed
- B. Du, L. Wang, S.-C. Yuan, T. Lei, J. Pei and Y. Cao, Polymer, 2013, 54, 2935 CrossRef CAS
- D. Marsitzky, J. C. Scott, J. P. Chen, V. Y. Lee, R. D. Miller, S. Setayesh and K. Mullen, Adv. Mater., 2001, 13, 1096 CrossRef CAS
- C. Poriel, J. Rault-Berthelot, D. Thirion, F. Barriere and L. Vignau, Chem.–Eur. J., 2011, 17, 14031 CrossRef CAS PubMed
- M. Romain, S. Thiery, A. Shirinskaya, C. Declairieux, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Metivier and C. Poriel, Angew. Chem., Int. Ed., 2015, 54, 1176 CrossRef CAS PubMed
- S. Thiery, B. Heinrich, B. Donnio, C. Poriel and F. Camerel, J. Mater. Chem. C, 2014, 2, 4265 RSC
- D. Thirion, C. Poriel, F. Barriere, R. Metivier, O. Jeannin and J. Rault-Berthelot, Org. Lett., 2009, 11, 4794 CrossRef CAS PubMed
- D. Thirion, C. Poriel, R. Metivier, J. Rault-Berthelot, F. Barriere and O. Jeannin, Chem.–Eur. J., 2011, 17, 10272 CrossRef CAS PubMed
- P. Wang, C. H. Leung, D. L. Ma, W. Lu and C. M. Che, Chem.–Asian J., 2010, 5, 2271 CrossRef CAS PubMed
- S. Goswami, G. Wicks, A. Rebane and K. S. Schanze, Dalton Trans., 2014, 43, 17721 RSC
- Z. J. Li, P. Cui, C. Z. Wang, S. Kilina and W. F. Sun, J. Phys. Chem. C, 2014, 118, 28764 CAS
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed
- V. Jankus, K. Abdullah, G. C. Griffiths, H. Al-Attar, Y. Zheng, M. R. Bryce and A. P. Monkman, Org. Electron., 2015, 20, 97 CrossRef CAS
- M. Yokoyama, T. Tamamura, M. Atsumi, M. Yoshimura, Y. Shirota and H. Mikawa, Macromolecules, 1975, 8, 101 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03391a |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |