DOI:
10.1039/C6RA05413G
(Paper)
RSC Adv., 2016,
6, 45873-45883
D–A conjugated polymers based on thieno[3,2-b]indole (TI) and 2,1,3-benzodiathiazole (BT) derivatives: synthesis, characterization and side-chain influence on photovoltaic properties†
Received
1st March 2016
, Accepted 26th April 2016
First published on 27th April 2016
Abstract
A facile synthetic strategy toward thieno[3,2-b]indole (TI) derivatives was developed by the Cadogan annulation method. Three donor–acceptor (D–A) conjugated polymers P1, P2 and P3 containing N-alkyl-TI derivatives and 4,7-dithien-5-yl-2,1,3-benzodiathiazole (DTBT) derivatives were successfully synthesized and applied to bulk heterojunction (BHJ) polymer solar cells (PSCs). Different side chains were introduced to TI units (for P2) or DTBT units (for P3), the results indicate that the bandgaps, energy levels and photovoltaic performance were finely tuned by the side chains in the TI-DTBT copolymer. Power conversion efficiencies (PCEs) based on the device structure of ITO/PEDOT:PSS/polymer:PC71BM/Ca/Al exhibit a large distinction (1.61% for P1, 0.53% for P2 and 2.73% for P3) under optimal device fabrication conditions. The optimized devices based on P3:PC71BM blends with a relatively higher mobility (2.84 × 10−5 cm2 V−1 s−1) show the best PCE under air mass 1.5 global (AM 1.5 G) irradiation of 100 mW cm−2, which is in good agreement with its high current density and light absorption property. Accordingly, the TI unit can be used as the efficient donor units for D–A conjugated donor materials for PSCs application. In addition, this work suggests that the side chains on low bandgap polymers significantly impact their molecular energy levels and the observed PCEs of the corresponding BHJ solar cells.
Introduction
Bulk heterojunction (BHJ) polymer solar cells (PSCs) based on a blend of conjugated polymers as the electron donors and fullerene derivatives as the acceptors have shown great promise in low processing cost, light weight, mechanically flexible and large-area fabrication in the past decades.1–5 Tremendous efforts have been devoted to modulating photon harvesting materials and developing the device fabrication technology. To date, the best reported power conversion efficiencies (PCEs) in PSCs have commonly approached or exceeded 10%, yet most are lower than 10%, especially in single-junction PSCs.6–10 However, they still cannot achieve the targeted 15% PCEs and satisfying lifetime, which are crucial for future commercialization. According to rapid progress in the field of PSCs, many rational molecular design principles have been deduced.5–11 Theoretically, an ideal conjugated polymer should simultaneously have the following characteristics: broad and strong absorption in the visible region (to harvest enough solar light),12 high hole mobility (to ensure hole transport),13,14 suitable energy levels which well fitted with that of its fullerene acceptor counterpart15–18 and appropriated compatibility with the fullerene acceptor (to form nano-scale bicontinuous interpenetrating networks for efficient exciton dissociation).19,20 In accordance with above-mentioned requirements, a large pool of donor materials has been developed. Donor–acceptor (D–A) conjugated polymers have dominated the library of donor materials for PSCs because their intrinsic optical and electronic properties can be readily tuned by controlling the intramolecular charge transfer (ICT) from donor unit to acceptor unit. The combination of these D and A units has significant influence on the final photovoltaic properties of the resulting copolymers.21–23
Tricyclic 2,7-carbazole unit, as the electron-rich unit with a symmetrical, rigidly fused, coplanar structure, intrinsic electron donating property and prominent hole transportation, has attracted great research interest in recent years for optoelectronic applications. Typically, poly-(2,7-carbazole-alt-dithienylbenzothiadiazole) (PCDTBT)24a (Scheme 1a) derived from carbazole and 4,7-dithien-5-yl-2,1,3-benzodiathiazole (DTBT) unit is considered to be one of the most promising conjugated polymers for the application in PSCs owing to better air stability, high charge carrier mobility and the deep-lying HOMO level (−5.5 eV) resulting in a higher Voc for PSCs, which was first prepared by Leclerc et al., and the PSCs based on a blend of PCDTBT and PCBM delivered a PCE of 3.6%.24 Since then, the PCEs increased to about 7.5% by optimizing PCDTBT:PC71BM blend morphology25 and incorporating a graphene oxide/titanium oxide (GO/TiOx) layer as the electron-transport layer.26,27 In spite of this, PCDTBT also suffers from large optical band gap that limits its light-harvesting ability and thus resulting in the medium Jsc, and many PCDTBT analogues were developed for PSC application. HXS-1 (Scheme 1b) with introducing the octyloxy groups on the electron-withdrawing DTBT unit, exhibits a PCE of 5.4%, but possesses a poor solubility.28 Thus, PCDTBT-8, (Scheme 1c) a more soluble PCDTBT derivative, was synthesized by the branched heptadecyl side chain replacing the straight N-octyl chain on the carbazole unit, and displayed a PCE of 4.22% despite the fact that PCDTBT-8 has a wider band gap than PCDTBT.29 Recently, Yang and Heeney respectively reported a variation on PCDTBT with F atoms (Scheme 1d)30 and incorporating thioalkyl (–SR) (Scheme 1e)31 on the DTBT unit, leading to a high Voc yet a moderate PCE. It was reported that poly{N-[1-(2′-ethylhexyl)-3-ethylheptanyl]dithieno[3,2-b:2′,3′-d]pyrrole-2,6-diyl-alt-4,7-di(2-thienyl)-2,1,3-benzothiadiazole-5′,5′′-diyl} (PDTPDTBT), based on dithienopyrrole (DTP), a carbazole-like unit with N-atom bridged dithiophene, showed a high-lying HOMO (−5.0 eV), a narrow bandgap of 1.46 eV and strong absorption from 300 nm to 850 nm with low Voc (0.52 V) in similar PSC devices.32–36 However, the copolymers based on the DTP possess the relative high planarity, light-harvesting property and high mobility. Inspired by the advantages of carbazole-based PCDTBT analogues and DTP-based polymers, it should be reasonable to exploit the application of thieno[3,2-b]indole (TI), structurally merged by a carbazole unit and a DTP unit, for novel D–A conjugated polymers.
 |
| | Scheme 1 Chemical structures of PCDTBT and its analogues. | |
In present work, we report three polymers based on TI derivatives and DTBT derivatives as the structural analogues of PCDTBT (Scheme 2). Contrary to earlier reports,37,38 the TI unit was synthesized via a simple method of Cadogan annulation.39 In fact, there are few literature that reported TI unit in application of PSCs.40 In this work, the N-alkyl-TI unit was copolymerized with electron-deficient DTBT to afford a new alternating D–A polymer P1, but with a poor solubility that led to low Mn. To further improve solubility and the molecular weights, hexyl side chain (alkoxyoctyl) was attached onto the TI unit and the electron-deficient DTBT unit in conjugated main chain, then P2 and P3 were obtained, respectively. As a result, it is found that the modification has a profound effect on the physical properties and device performance. The optimized device based on P3:PC71BM with a relatively higher mobility (2.84 × 10−5 cm2 V−1 s−1) gave the best photovoltaic performance with a PCE of 2.73% under air mass 1.5 global (AM 1.5 G) irradiation of 100 mW cm−2. These results suggest that the electron-rich unit (TI) can be used as the promising donor unit for designing the D–A conjugated polymers and applied to PSCs.
 |
| | Scheme 2 Chemical structures of P1, P2 and P3. | |
Experimental section
Materials
Pd(PPh3)4, Pd2(dba)3, P(o-tol)3, PdCl2(dppf), anhydrous N,N-dimethylformamide (DMF), 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole, 4,7-bis(5-bromothiophen-2-yl)benzo[c][1,2,5]thiadiazole and [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) were purchased from Energy Chemical, J&K and Suna Tech Inc (China). 2,5-Dibromo-nitrobenzene 1 was synthesized on the basis of the procedures described in the literature.41 Toluene and tetrahydrofuran (THF) were freshly distilled prior to use after processed with Na and benzophenone. All other reagents were used as we received.
Measurements and general methods
1H NMR and 13C NMR data were measured by a Bruker Ultra Shield Plus AV400 spectrometer in deuterated chloroform solution at 298 K with tetramethylsilane (TMS; d = 0 ppm) as the internal standard (1H NMR: 400 MHz, 13C NMR: 100 MHz) Matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) were recorded using a Shimadzu AXIMA-CFR mass spectrometer. Gas Chromatography-Mass Spectrometer (GC-MS) measurements were carried out with a Varian GC-MS 3900-2100T. Molecular weight and polydispersity of the polymers were determined by gel permeation chromatography (GPC) analysis with polystyrene as standard THF (HPLC grade) as eluent at a flow rate of 1.0 mL min−1 at 35 °C. Thermogravimetric analysis (TGA) was performed on a Shimadzu DTG-60 thermogravimetric analyzer under a nitrogen atmosphere at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) experiments were performed on a Mettler DSC823e thermal analyzer at a heating rate of 10 °C min−1, under a nitrogen atmosphere. UV-vis absorption spectra were recorded on a Shimadzu UV-3600. In the solid, the polymers were cast on quartz plates with KW-4A spinner from a chloroform solution (ca. 5 mg mL−1). The electrochemical cyclic voltammetry (CV) was tested in a 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) acetonitrile solution with Au disk as working electrode, Pt plate as counter electrode and Ag/AgNO3 as reference electrode. The polymer films were formed by drop-casting from their toluene solution (ca. 2 mg mL−1). The surface morphology was measured via tapping mode atomic force microscopy (AFM) (Thermo. Auto Probe C P Res.).
Solar cell device fabrication and characterization
BHJ PSCs were fabricated by the following process25b,43 Patterned indium tin oxide (ITO) glass substrates were cleaned sequentially with detergent, de-ionized (DI) water, acetone, and isopropanol (IPA). They were then treated with Jelight UV-ozone cleaner for 15 min. A thin layer of poly(3,4-ethylenedioxythiophene):poly-(styrenesulfonate) (PEDOT:PSS) with a thickness of 40 nm was formed on each ITO substrate by spin coating a PEDOT:PSS aqueous solution (Clevios 4083 from H.C. Starck) at 5000 rpm for 1 min and baked at 140 °C for 15 min. The ITO substrates coated with PEDOT:PSS were then transferred into a glove box filled with dried nitrogen to coat the active layer. Polymers were dissolved in o-DCB, and then PC71BM was added. The solution was then heated at 45 °C and stirred overnight at the same temperature. Prior to deposition, the solution was filtered (0.45 μm filters). The solution of copolymer:PC71BM was then spin coated to form the active layer. The cathode made of calcium (2 nm) and aluminum (200 nm thick) was sequentially evaporated through a shadow mask under high vacuum (<10−6 Torr). Each sample consists of 4 independent pixels with an active area of 0.11 cm2. Finally, the devices were encapsulated and characterized in air. The thickness of the active layer was determined by Dektak 150 profilometer. The photovoltaic performance of the devices was measured with a computer-programmed Keithley 2420 sourcemeter and a Newport's Oriel class A solar simulator, which simulated the AM1.5G sunlight (100 mW cm−2) and was certified to the JIS C 8912 standard. The external quantum efficiencies (EQE) of solar cells were analyzed by using a certified Newport incident photon conversion efficiency (IPCE) measurement system.
Syntheses of monomer and polymers
2-(4-Bromo-2-nitrophenyl)thiophene (2). In a two-necked RBF (100 mL), compounds 1 (2.8 g, 10 mmol) and 2-thiophenylboric acid (1.42 g, 11 mmol) were dissolved in THF (30 mL), 2 M K2CO3 solution (20 mL) was added. The mixture was degassed with N2 flow for 20 min, Pd(PPh3)4 (150 mg, 0.14 mmol) was added under N2 flow. The mixture was stirred at 70 °C under N2 in dark for 24 h. The mixture was then allowed to cool to room temperature and extracted with CH2Cl2 and dried with Na2SO4. The product was purified by silica chromatography with petroleum ether as the eluent, affording the product as a bright orange liquid (2.35 g, 83% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.89 (d, J = 2.0 Hz, 1H), 7.70 (dd, J = 8.3, 2.0 Hz, 1H), 7.48–7.39 (m, 2H), 7.16–7.05 (m, 2H). 13C NMR (100 MHz, CDCl3) δ (ppm) 135.97, 134.98, 133.42, 127.98, 127.64, 127.51, 127.34, 126.84, 121.76. Anal. calcd (%) for C10H6BrNO2S: C, 42.27; H, 2.13; Br, 28.12; N, 4.93; O, 11.26; S, 11.29. Found: C, 42.26; H, 2.16; Br, 28.09; N, 4.94; O, 11.18; S, 11.37. GC-MS: 284.112 for [M]+.
6-Bromo-4H-thieno[3,2-b]indole (3). In a two-necked RBF (100 mL), 2 (9.37 g, 33 mmol) and PPh3 (3.89 g, 14.85 mmol) were dissolved in chlorobenzene (30 mL). The mixture was degassed with N2 flow for 20 min, the mixture was stirred at 140 °C under N2 for 24 h. The mixture was then allowed to cool to room temperature and extracted with CH2Cl2 and dried with Na2SO4. The solvents were removed with reduced vacuum. Pure product of 3 was obtained by column chromatography over silica gel (petroleum ether/ethyl acetate = 10/1, v/v) to give 3 as white solid (7.15 g, yield of 86%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.21 (br, 1H), 7.60 (d, J = 6.7 Hz, 2H), 7.39 (d, J = 5.2 Hz, 1H), 7.29 (dd, J = 8.4, 1.7 Hz, 1H), 7.07 (d, J = 5.2 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm) 143.37, 141.83, 127.73, 123.02, 121.07, 119.98, 117.88, 116.02, 114.81, 111.51. Anal. calcd (%) for C10H6BrNS: C, 47.64; H, 2.40; Br, 31.69; N, 5.56; S, 12.72. Found: C, 47.59; H, 2.43; Br, 31.71; N, 5.58; S, 12.69. GC-MS: 252.862 for [M]+.
6-Bromo-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole (4). In a two-necked RBF (100 mL), to a solution of compound 3 (4.54 g, 18 mmol) in 40 mL of dry DMF was added anhydrous K2CO3 (4 g, 37.8 mmol). The mixture was degassed with N2 flow for 20 min, and then 1-bromo-2-decyltetradecane (8.34 g, 20 mmol) was added. The solution was stirred at 140 °C for 24 h under N2 and then quenched with 30 mL of water. The aqueous layer was extracted with dichloromethane three times. The organic fractions were dried over Na2SO4, and the solvent was removed under reduced pressure. The product was purified by column chromatography over silica gel (petroleum ether as eluent) to give 4 as a colorless liquid (10.2 g, 96%). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.59 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 1.6 Hz, 1H), 7.38 (d, J = 5.2 Hz, 1H), 7.24 (d, J = 1.7 Hz, 1H), 7.02 (d, J = 5.2 Hz, 1H), 4.06 (d, J = 7.5 Hz, 2H), 2.04 (m, 1H), 1.25–1.21 (m, 40H), 0.90–0.87 (t, J = 6.9 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm) 146.15, 142.46, 127.27, 122.03, 120.56, 119.95, 115.82, 115.65, 113.06, 110.61, 49.83, 38.37, 31.96, 31.94, 31.67, 29.91, 29.70, 29.68, 29.63, 29.62, 29.56, 29.40, 29.35, 26.43, 22.72, 14.18. Anal. calcd (%) for C34H54BrNS: C, 69.36; H, 9.24; Br, 13.57; N, 2.38; S, 5.45. Found: C, 69.39; H, 9.27; Br, 13.61; N, 2.41; S, 5.32. GC-MS: 588.758 for [M]+.
2,6-Dibromo-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole (5). The solution of 4 (4.62 g, 10 mmol) in THF (50 mL) was cooled in an ice-water bath. Then, N-bromosuccinimide (NBS, 1.96 g, 11 mmol) was added in three times. The resulting mixture was stirred for 3 h. After the reaction was completed, the mixture was diluted with water and extracted with petroleum ether (100 mL). The organic layer was washed with water (50 mL × 2) and brine (50 mL × 2), then dried over Na2SO4. The solvents were removed with reduced vacuum. The yellow residue was purified by column chromatography over silica gel (petroleum ether as the eluent) to give 5 as a light yellow oil (4.98 g, 92% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.50 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 1.6 Hz, 1H), 7.25 (dd, J = 8.4, 1.6 Hz, 1H), 7.05 (s, 1H), 3.98 (d, J = 7.5 Hz, 2H), 1.99 (m, 1H), 1.44–1.17 (m, 26H), 0.94–0.82 (m, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm) 143.69, 141.18, 122.51, 120.29, 119.75, 115.90, 115.75, 114.06, 113.97, 113.17, 49.79, 38.33, 31.91, 31.80, 31.59, 31.58, 29.91, 29.60, 29.53, 29.31, 26.38, 22.71, 22.61, 14.19, 14.15. Anal. calcd (%) for C26H37Br2NS: C, 56.22; H, 6.71; Br, 28.77; N, 2.52; S, 5.77 Found: C, 56.09; H, 6.72; Br, 28.61; N, 2.36; S, 6.22. GC-MS: 555.112 for [M]+.
4-(2-Hexyldecyl)-2,6-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4H-thieno[3,2-b]indole (M1). In a two-necked RBF (100 mL), compounds 5 (1.11 g, 2 mmol), bis(pinacolato)diboron (1.12 g, 4.4 mmol) and KOAc (1.12 g, 11.4 mmol) were mixed in anhydrous DMF (30 mL). The mixture was then degassed with N2 flow for 20 min, before PdCl2(dppf) (73 mg, 0.1 mmol) was added under N2 flow. The mixture was stirred at 85 °C under N2 in dark for 4 h. The mixture was then allowed to cool to room temperature and extracted with CH2Cl2 and the organic layer was dried with Na2SO4. After purified by silica chromatography with petroleum ether/ethyl acetate as the eluent, M1 was afforded as a bright orange solid (0.56 g, 43% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.85 (s, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.21 (s, 1H), 4.18 (d, J = 7.4 Hz, 2H), 2.15–2.12 (m, 1H), 1.76–1.30 (m, 24H), 1.24 (s, 12H), 0.86 (m, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm) 146.00, 141.21, 140.45, 125.33, 123.93, 117.97, 116.69, 114.80, 106.92, 83.60, 58.53, 49.50, 38.32, 31.92, 31.83, 31.59, 31.58, 29.96, 29.62, 29.56, 29.31, 26.39, 26.35, 24.93, 22.68, 22.64, 18.45, 14.12. Anal. calcd (%) for C38H61B2NO4S: C, 70.26; H, 9.47; B, 3.33; N, 2.16; O, 9.85; S, 4.94. Found: C, 70.23; H, 9.51; B, 3.39; N, 2.14; O, 9.89; S, 4.84. MALDI-MS (m/z): 650.546 for [M]+.
2-(4-Bromo-2-nitrophenyl)-4-hexylthiophene (6). In a two-necked RBF (100 mL), compound 1 (2.8 g, 10 mmol) and 2-(4-hexylthiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3.23 g, 11 mmol) were dissolved in freshly distilled toluene (30 mL). 2 M K2CO3 solution (15 mL) was then added via a syringe. The mixture was degassed with N2 flow for 20 min before Pd(PPh3)4 (50 mg, 0.04 mmol) was added under N2 flow. The mixture was stirred at 95 °C under N2 in dark for 24 h. The mixture was then allowed to cool to room temperature and extracted with CH2Cl2. The crude product was flushed by silica chromatography with petroleum ether as the eluent. 6 was obtained as a waxy solid (2.85 g, yield of 78%). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.85 (d, J = 2.0 Hz, 1H), 7.66 (dd, J = 8.3, 2.0 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.02 (s, 1H), 6.93 (d, J = 1.4 Hz, 1H), 2.62–2.58 (t, J = 7.5 Hz, 2H), 1.61 (m, 2H), 1.34 (m, 6H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm) 149.44, 144.29, 135.52, 134.88, 133.13, 128.69, 127.56, 126.74, 122.22, 121.40, 31.71, 30.46, 30.37, 29.00, 22.66, 14.18. Anal. calcd (%) for C16H18BrNO2S: C, 52.18; H, 4.93; Br, 21.70; N, 3.80; O, 8.69; S, 8.71. Found: C, 52.11; H, 4.98; Br, 21.66; N, 3.81; O, 8.72; S, 8.72. GC-MS: 370.112 for [M]+.
6-Bromo-3-hexyl-4H-thieno[3,2-b]indole (7). Compound 7 was prepared according to the same procedure as that of compound 3, which was used directly for the next step without any further purification.
6-Bromo-3-hexyl-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole (8). Compound 8, a light yellow viscous liquid, was obtained from 7 according to the same procedure as that for compound 4. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.55 (d, J = 8.3 Hz, 1H), 7.49 (d, J = 1.6 Hz, 1H), 7.24 (dd, J = 8.4, 1.7 Hz, 1H), 6.99 (s, 1H), 4.16 (d, J = 7.7 Hz, 2H), 2.87 (m, 2H), 1.99 (m, 1H), 1.75 (m, 2H), 1.52–1.14 (m, 32H), 0.98–0.82 (m, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm) 143.64, 142.93, 127.40, 122.60, 122.03, 121.25, 119.72, 117.13, 115.53, 113.34, 49.21, 39.39, 31.88, 31.77, 31.76, 31.57, 31.56, 30.03, 29.93, 29.62, 29.49, 29.28, 29.27, 28.97, 26.48, 26.46, 22.67, 22.62, 14.13, 14.10, 14.09. Anal. calcd (%) for C32H50BrNS: C, 68.55; H, 8.99; Br, 14.25; N, 2.50; S, 5.72. Found: C, 68.56; H, 9.06; Br, 14.17; N, 2.53; S, 5.68. GC-MS: 561.332 for [M]+.
2,6-Dibromo-3-hexyl-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole (M2). Monomer M2, a colorless viscous oil was prepared according to the same procedure as that for M1 (yield of 58%). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.50 (d, J = 1.4 Hz, 1H), 7.47 (s, 1H), 7.24 (dd, J = 8.3, 1.7 Hz, 1H), 4.13 (d, J = 7.7 Hz, 2H), 2.96 (m, 2H), 2.07–1.92 (m, 1H), 1.72–1.56 (m, 2H), 1.48–1.16 (m, 32H), 0.88 (m, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm) 141.89, 141.02, 127.21, 122.49, 120.17, 119.59, 115.79, 115.39, 113.52, 112.03, 49.06, 39.20, 31.87, 31.74, 31.65, 31.49, 31.46, 30.32, 29.90, 29.59, 29.46, 29.28, 29.25, 27.93, 26.41, 26.39, 22.67, 22.65, 22.61, 14.13, 14.08, 14.07. Anal. calcd (%) for C32H49Br2NS: C, 60.09; H, 7.72; Br, 24.99; N, 2.19; S, 5.01. Found: C, 60.13; H, 7.74; Br, 24.91; N, 2.11; S, 5.11. MALDI-MS (m/z): 640.006 for [M]+.
Synthesis of P1. In a 25 mL Schlenk tube, monomer M1 (214 mg, 0.33 mmol) and 4,7-bis(5-bromo-thiophen-2-yl)benzo[c][1,2,5]thiadiazole (151 mg, 0.33 mmol) were dissolved in a mixture of toluene (12 mL) and 2 M K2CO3 (3 mL). The solution was degassed with N2 flow for 20 min before Pd(PPh3)4 (3 mg) was added under N2 flow. The mixture was stirred at 95 °C under N2 in dark for 36 h. After the reaction system was cooled to room temperature, it was poured into the mixture of methanol (500 mL) and aqueous HCl (1 M, 20 mL). The precipitated solid was collected by filtration. Then the copolymer underwent the Soxhlet extraction with acetone, hexanes, and dichloromethane to remove oligomer and then with chloroform (250 mL) to collect the copolymer P1 fraction. After the chloroform solution was concentrated to 30 mL, it was reprecipitated into methanol (250 mL). And the clean P1 was obtained as a black solid (106 mg, 46%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.12–8.09 (m, 2H), 7.87–7.66 (m, 2H), 7.66–7.54 (m, 2H), 7.54–7.44 (m, 2H), 7.19–6.99 (m, 2H), 4.49–3.29 (m, 2H), 2.06 (s, 1H), 1.77–1.28 (m, 24H), 0.91–0.87 (m, 6H). GPC (THF, polystyrene standard, 35 °C): Mn = 8.6 kDa, Mw = 16.3 kDa, PDI = 1.89.
Synthesis of polymer P2. In a 25 mL Schlenk tube, monomer M2 (320 mg, 0.50 mmol), 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole (313.0 mg, 0.50 mmol), Pd2(dba)3 (4.5 mg) and P(o-tol)3 (6.1 mg) were dissolved in toluene (10 mL). After the solution was degassed with N2 flow for 20 min, the mixture was stirred at 110 °C under N2 in dark for 6 h. When there was much precipitate, subsequent to the addition of tributyl(thiophen-2-yl)stannane, another end-capping reagent, 2-bromothiophene (14 mg, 0.085 mmol) was added, and the reaction continued for another 30 min under otherwise identical conditions. After the reaction mixture was cooled to room temperature and added dropwise to acetone (500 mL) to obtain precipitate. The precipitate was collected by filtration and washed with acetone and dried. The crude polymer was dissolved in CHCl3, filtered through Celite to remove the metal residue, and then precipitated into ethyl acetate and dried under reduced pressure to give P2. P2 underwent the Soxhlet extraction with CH2Cl2 to remove oligomers and then with chloroform to collect the copolymer P2 fraction. After being reprecipitated by methanol, P2 was obtained as a black fiber-like solid (241 mg, 62%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.55–8.46 (m, 1H), 7.72–7.66 (m, 1H), 7.56–7.42 (m, 2H), 7.22–7.18 (m, 2H), 7.00–6.76 (m, 2H), 4.19–3.52 (m, 64H), 2.06–1.96 (m, 1H), 1.68–1.25 (m, 48H), 0.88–0.83 (m, 12H). GPC (THF, polystyrene standard, 35 °C): Mn = 10.2 kDa, Mw = 25.3 kDa, PDI = 2.48.
Synthesis of polymer P3. In a 25 mL Schlenk tube, monomer M1 (214 mg, 0.33 mmol), 4,7-bis(5-bromothiophen-2-yl)-5,6-bis(octyloxy)benzo[c][1,2,5]thiadiazole (236 mg, 0.33 mmol), and Pd(PPh3)4 (3 mg) were dissolved in a mixture of toluene (12 mL) and 2.0 M K2CO3 (2 mL). Then, P3 was synthesized and purified via the similar procedure as that for P1. P1 was obtained as a purple-black solid (210 mg, 67%). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.19–8.15 (m, 1H), 7.90 (s, 1H), 7.73–7.69 (m, 2H), 7.56–7.52 (m, 2H), 7.47–7.41 (m, 1H), 7.34–7.29 (m, 1H), 4.31–4.19 (m, 2H), 3.15–2.25 (m, 1H), 2.10–2.03 (m, 1H), 1.75–1.21 (m, 32H), 0.90–0.83 (m, 9H). GPC (THF, polystyrene standard, 35 °C): Mn = 11.1 kDa, Mw = 26.2 kDa, PDI = 2.36.
Results and discussion
Synthesis and characterization
Synthetic routes of the monomers are depicted in Scheme 3. 1,4-Dibromo-2-nitrobenzene 1 was prepared according to the reported method.41 Then, both 2-(4-bromo-2-nitrophenyl)thiophene 2 and 2-(4-bromo-2-nitrophenyl)-4-hexylthiophene 6 were synthesized via Suzuki coupling reaction using Pd(PPh3)4 as catalyst in high yields of 73% and 83%, respectively. 6-Bromo-4H-thieno[3,2-b]indole 3 and 6-bromo-3-hexyl-4H-thieno[3,2-b]indole 7 were obtained through Cadogan annulation.40 The method is universal and with a high yield around 85%. With KOH as base, KI as catalyst and THF as the solvent, 6-bromo-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole 4 and 6-bromo-3-hexyl-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole 8 were quantitatively obtained by alkylation of compound 3 and 7. Under the exclusion of the light, the bromination of compound 4 and 8 with NBS gives 2,6-dibromo-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole 5 and 2,6-dibromo-3-hexyl-4-(2-hexyldecyl)-4H-thieno[3,2-b]indole M2, quantitatively. 4-(2-Hexyldecyl)-2,6-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4H-thieno[3,2-b]indole M1 was synthesized from compound 5 and bis(pinacolato)diboron by using PdCl2(dppf) as catalyst in 43% yield. Obviously, the synthetic operation and purification of the TI and its derivatives are simple, avoiding the complicated separation and multiple-step synthesis, which is critical for their future commercialization.
 |
| | Scheme 3 Synthetic routes of the monomers. Reagents and conditions: (i) Pd(PPh3)4, 2 M K2CO3, THF/H2O, 70 °C, 24 h, yield > 65%; (ii) PPh3, chlorobenzene, 135 °C, 7–12 h, yield > 85%; (iii) KOH, KI, 1-bromo-2-hexyldecane, THF, reflux, 12 h, quantitatively; (iv) NBS, THF, 0 °C, 12 h, quantitatively; (v) PdCl2(dppf), KOAc, DMF, 85 °C, 24 h, yield > 75%. | |
Synthetic routes of the copolymers are depicted in Scheme 4. The polymer P1 and P3 were synthesized by Suzuki polycondensation using Pd(PPh3)4 as the catalyst in toluene. However, the copolymer P2 was obtained by Stille coupling polymerization using Pd2(dba)3 as catalyst and P(o-tol)3 as ligand in toluene.42 In fact, P1 was also subjected to the Stille methodology, but it gave very low yield and the obtained P1 was almost insoluble. As to P2, higher molecular polymer with good solubility could be obtained under Stille conditions, due to the presence of hexyl group in TI unit, which provided steric hindrance. All copolymers were purified by Soxhlet extraction with hexane, acetone, dichloromethane and chloroform. Adding solubilizing chains to the donor unit or the acceptor unit improves their solubility in commonly employed solvents such as THF and chloroform, and significantly increases the molecular weight of the resulting polymers P2 and P3. The chemical structures of all copolymers were verified by 1H NMR. According to the NMR spectra (Fig. S14–S16†), it can be clearly found that the unsymmetric TI units make the aromatic hydrogen signal very complicated. This result indicates that the TI units are alternating copolymerized with DTBT units in a random fashion. The average molecular weight and polydispersity indices (PDI) of all copolymers were measured by GPC using monodispersed polystyrene as the standard and THF as the eluent. The Mw of P1, P2 and P3 were 16.3, 25.3 and 26.2 kDa, with PDI of 1.89, 2.48 and 2.36, correspondingly. It should be emphasized that the low yield and limited Mn of P1 both resulted from its poor solubility, attributed to strong intermolecular π–π interaction. By introducing hexyl group into TI unit or bisalkyloxyl groups into DTBT units, the solubility of P2 and P3 is both improved with relatively higher molecular weight.
 |
| | Scheme 4 Synthetic routes of polymer P1, P2 and P3. | |
Thermal properties
Thermal stability of all copolymers was investigated by thermogravimetric analysis (TGA). The TGA measurements were carried out at a heating rate of 10 °C min−1 under nitrogen atmosphere. As shown in Fig. S1,† all three copolymers exhibited good thermal stability, and they lost <5% of their weight on the heating to higher than 375 °C. This high thermal stability is favorable for their application in PSCs and other optoelectronic devices. Thermally induced phase-transition behavior of the polymers was investigated using differential scanning calorimetry (DSC) under a nitrogen atmosphere. However, no obvious glass transition temperatures (Tg) were observed during the heating and cooling process. The molecular weights of the polymers were summarized in Table 1.
Table 1 Optical and electrochemical properties of all polymers
| Polymers |
Mn (kDa)/PDIa |
Solutionb |
Filmc |
Eoptg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (eV) d (eV) |
Eoxonset (V)/HOMO (eV) |
Eredonset (V)/LUMO (eV) |
Eecge (eV) |
| λmax (nm) |
λedge (nm) |
λmax (nm) |
λedge (nm) |
| Number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (PDI) of the copolymers were determined by GPC using polystyrene standards in THF at 35 °C. Measured in dilute chloroform solution. Cast from chloroform solution. Bandgap estimated from the onset wavelength (λedge) of the optical absorption: Eoptg = 1240/λedge. Eecg of the polymers were calculated from empirical equation: Eecg = |EHOMO − ELUMO|. |
| P1 |
8.6/1.89 |
408, 570 |
785 |
428, 614 |
791 |
1.57 |
0.37/−5.17 |
−1.08/3.72 |
1.45 |
| P2 |
10.2/2.48 |
383, 544 |
713 |
399, 575 |
730 |
1.70 |
0.42/−5.22 |
−1.13/3.67 |
1.55 |
| P3 |
11.1/2.36 |
446, 523 |
647 |
452, 560 |
697 |
1.77 |
0.52/−5.32 |
−1.11/3.69 |
1.63 |
Optical properties
The normalized ultraviolet-visible (UV-vis) absorption spectra of P1, P2 and P3 in dilute chloroform solutions (ca. 10–5 M) and in film states are shown in Fig. 1. Both in solution and in the solid state, P1 and P2 display two distinct absorption peaks from 350 to 800 nm, which is a common phenomenon of D–A type polymers.43 One absorption peak occurs at the shorter wavelength region (350–500 nm) corresponding to π–π electron transition of the donor units, and the other absorption peak at the longer wavelength region (500–800 nm) can be assigned to the ICT effect from the donor units to the acceptor units. The absorption profiles of all polymers exhibit much red-shifted absorption λmax toward longer wavelengths from the chloroform solution state to the thin film state, demonstrating an effective interchain π–π stacking in thin film. Obviously, since there are alkyl chains substituted to the polymer backbones, the absorption spectra show dramatically different features both in solution and in films. For example, both of the two peaks of P2 are blue-shifted (25–40 nm) compared to those of P1 and the 50–60 nm hypsochromic shift of the absorption edge implies that the hexyl groups on TI units cause steric hindrance, giving rise to large twisting dihedral angles around the interannular single bonds. While the solubilizing octyloxyl groups are attached to DTBT units for P3, the absorption profile of P3 displays one broad peak consisting of one red-shifted peak at short wavelength and one blue-shifted peak at long wavelength, indicating that the solubilizing octyloxyl groups attached to the DTBT units result in weakening electron-withdrawing ability of the BT moiety. The optical band gaps (Eoptg) are 1.57 eV, 1.70 eV and 1.77 eV for P1, P2 and P3, correspondingly, notably, the absorption profiles of TI-containing polymers are different from those of their analogue, PCDTBT, the absorption onset of TI-containing polymers is red-shifted (30–120 nm) in film state, corresponding to a lower optical bandgap than PCDTBT (Eoptg = 1.85 eV). This apparently demonstrates that, in agreement with earlier studies,12 the replacement of benzene ring with thiophene in TI unit results in a reduction of the optical bandgap due to the increase of ICT effect on the polymer backbone. The correlated optical parameters are summarized in Table 1.
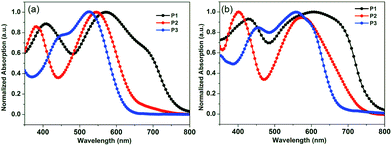 |
| | Fig. 1 Normalized UV-vis absorption spectra of polymers in chloroform solution (a) and in film states (b). | |
Electrochemical properties
The electrochemical properties of all polymers was measured by cyclic voltammetry (CV) in anhydrous acetonitrile (CH3CN) in the presence of 0.1 M Bu4NPF6 with Ag/AgNO3 (0.1 M in CH3CN) as a reference electrode under nitrogen atmosphere at room temperature at a scan rate of 50 mV s−1 (Fig. 2). The energy level of the Ag/AgNO3 reference electrode (calibrated from the Fc/Fc+ redox system) was 4.8 eV below the vacuum level. According to the equation HOMO = −(Eoxonset + 4.8) (eV), LUMO = −(Eredonset + 4.8) (eV),44 HOMO and LUMO energy levels of all polymers are calculated from the onset oxidation potentials (Eoxonset) and the onset reductive potentials (Eredonset), respectively. The Eoxonset of the polymers P1, P2 and P3 are estimated to be 0.37, 0.42 and 0.52 V, corresponding to HOMO energy levels of −5.17, −5.22 and −5.32 eV, individually, which suggests that all polymers have a good air stability for the application in PSCs. The Eredonset of the polymers P1, P2 and P3 were estimated to be −1.08, −1.13 and −1.11 V, corresponding to LUMO energy levels of −3.72, −3.67 and −3.69 eV, respectively (Table 1). Additionally, by introducing hexyl groups into P2 and octyloxyl groups into P3, the HOMO energy levels can be readily tuned, owing to the reduced electronic delocalization along the polymer backbone. Yet, the LUMO energy levels are essentially dominated by the common acceptor unit (DTBT) and all polymers have the similar LUMO levels (about −3.70 eV). The electrochemical band gaps (Eecg) are found to be 1.45 eV for P1, 1.55 eV for P2 and 1.63 eV for P3, which is consistent with their Eoptg.
 |
| | Fig. 2 Cyclic voltammograms of all polymer films on Au disk electrode in 0.1 M Bu4NPF6 acetonitrile solution with a scan rate of 50 mV s−1. | |
Theoretical calculation
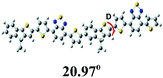
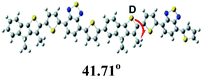
To further understand the molecular geometry, the electron density of states distribution and the fundamentals of the molecular configuration, density functional theory (DFT) theoretical calculations were carried out at the B3LYP/6-31G (d) level.45 To simplify the calculation, only the dimer models of each polymer with all the alkyl side chains replaced by methyl groups were subjected to the calculation. As shown in Table 2, the electron densities of the LUMOs of all polymers are entirely localized on the acceptor unit, while those of the HOMOs of all polymers are fully distributed throughout the whole conjugated backbone. The torsion angles between the electron-rich and the electron-deficient unit of these dimer models are of 20.97° (for P1), 41.71° (for P2) and 21.80° (for P3), respectively. The larger dihedral angle of polymer P2 can be attributed to the hexyl groups on the electron-rich TI units. Additionally, the calculated HOMO/LUMO energy levels and band gaps are in agreement with their experimental values from CV measurements.
Table 2 Dihedral angles, the frontier molecular orbitals and calculated HOMO/LUMO energy levels obtained from DFT calculations on P1, P2 and P3 with a chain length n = 2 at the at the B3LYP/6-31G* level of theory along with (d) optimized geometry
| |
P1 |
P2 |
P3 |
| Dihedral angle |
 |
 |
 |
| LUMO |
 |
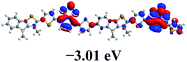 |
 |
| HOMO |
 |
 |
 |
Photovoltaic properties
To investigate the photovoltaic properties of all polymers, bulk heterojunction PSCs devices were fabricated with a conventional configuration of glass/ITO/PEDOT:PSS/polymer:PC71BM/Ca/Al under AM 1.5 G simulated light (Fig. 3a). Herein, PC71BM was chosen as the acceptor due to its stronger light absorption and complemented the absorption valley of the donors in the visible region,46 leading to generating more current to enhance the PCE of device. We optimized the BHJ PSCs device via changing the D/A ratio, the thermal annealing treatment, the additive and the inverted device. J–V characteristics of the optimized devices based on all polymers under illumination of AM 1.5 G simulated solar light (100 mW cm−2) are shown in Fig. 3b and S3–S5† and the optimized device parameters are summarized in Table 3. As a consequence, the optimized weight ratios of polymer to PC71BM were 1:3 for P1, 1:4 for P2 and P3 obtained. P1 showed the best results when devices were annealed at 80 °C while polymers P2 and P3 offered the best power conversion efficiencies when devices were annealed at 150 °C and 25 °C respectively. Solar cells based on P1:PC71BM blend spin-coated from CB give a PCE of 1.79%. When the mixture of CB and CF (9:1, v/v) as the co-solvent used for the same component (Fig. S3†) or by using DIO as additive, the PCE for devices based on P1 have not been improved (Fig. S4†). While after DIO additive treatment, the FF obviously improved a lot, resulting from tuning the morphology for efficient charge dissociation and transport.47 It is assumed that the low solubility and low molecular weight of P1 accounts for the low PCEs. Photovoltaic cells based on P2 show the poor photovoltaic performance with a PCE of only 0.48% and 0.53% by using the optimized co-solvent CB and CF (9:1, v/v), which implies that the hexyl side-chains suppress the interchain stacking and reduce the charge transporting property. Noticeably, the best photovoltaic performance were achieved in P3 solar cells. The PCEs was obtained 2.7% with a Voc of 0.77 V, a Jsc of 7.51 mA cm−2, a FF of 47%. But, in an attempt to further optimize the device based on the polymer P3 by the co-solvent CB and CF (9:1, v/v) or DIO as additive, there is no positive influence.
 |
| | Fig. 3 (a) Device configuration and HOMO and LUMO energy diagram of P1, P2 and P3. (b) J–V curves of PSCs based on polymer:PC71BM blends after thermal annealing at each optimized temperature under standard solar illumination conditions. (c) External quantum efficiency spectra of polymer/PC71BM solar cells. | |
Table 3 Photovoltaic parameters of the polymers/PC71BM-based solar cells at an optimized ratio of D:A (w/w) and different annealing temperature
| Blend |
D/A ratio (w/w) |
Annealing temp. (°C) |
Jsc (mA cm−2) |
Voc (V) |
FF (%) |
PCE (%) |
| DIO as additive. Cosolvent DCB:CF (9:1, v/v). The inverted device (ITO/TiOx/P3:PC71BM/MoO3/Al). |
| P1:PC71BM |
1:3 |
80 |
7.27 |
0.60 |
40.9 |
1.79 |
| 1:3a |
80 |
3.93 |
0.63 |
67.6 |
1.68 |
| 1:3b |
150 |
4.99 |
0.45 |
33.7 |
0.76 |
| P2:PC71BM |
1:4 |
150 |
2.32 |
0.49 |
42.4 |
0.48 |
| 1:4b |
150 |
2.38 |
0.55 |
40.1 |
0.53 |
| P3:PC71BM |
1:4 |
25 |
7.52 |
0.77 |
47.0 |
2.73 |
| 1:4a |
25 |
3.74 |
0.49 |
55.3 |
1.02 |
| 1:4b |
150 |
6.23 |
0.65 |
45.3 |
1.83 |
| 1:4c |
25 |
6.64 |
0.55 |
43.4 |
1.60 |
Comparing the polymer P1, P2 and P3 device performance, it is noted that the variation lies in the Jsc values, with the highest Jsc achieved in P3 solar cells and the lowest in P2 solar cells. The atomic force microscopy (AFM) was performed to study the morphology of blend film P3:PC71BM under optimized conditions. As indicated in Fig. 4, the phase image showed P3:PC71BM blend film forms the interpenetrating structure which would lead to the charge separation and transportation, consequently, the high Jsc and FF are obtained.48–50 Additionally, we made a further optimization on P3 with fabricating the inverted device structure of ITO/TiOx/P3:PC71BM(1:4, w/w)/MoO3/Al, the J–V characteristics of the inverted device are shown in Fig. S5† and Table 3. Surprisingly, the PCE is not improved, demonstrating P3 may not be suitable for the inverted device.
 |
| | Fig. 4 AFM images (4 μm × 4 μm) of blend film P3/PC71BM (1:4, w/w) with thermal annealing at 25 °C (left: height image; right: phase image). | |
To confirm the accuracy of the measurements of the devices and understand the different Jsc values of all polymers, external quantum efficiency (EQE) of the optimized devices was measured (Fig. 3c). It can be seen that the device based on the P2:PC71BM showed photon-to-current responses in the range of 350–700 nm with the lowest EQE response (less than 10%) among the three polymers. However, P3:PC71BM as the active layers can effectively absorb more photons from the solar radiation, consistently, the high Jsc values and an improvement in the device performance.
Space charge limited current (SCLC) measurement
The hole mobility of thin films is measured by the space-charge-limited currents (SCLC) method with a device structure of ITO/PEDOT:PSS/active layer/Al. The SCLC model is fitted according to the Mott–Gurney law:46
where J stands for current density, d is the thickness of the active layer, V (Vappl − Vbi) is the internal voltage of the device, Vappl is the applied potential and Vbi is the built-in voltage due to the relative work function difference of the two electrodes, ε0 is permittivity of free space (1.31 × 10−12) F m−1, εr is the relative permittivity, μh is the hole mobility. The hole mobility for P3-based best devices is of 2.84 × 10−5 cm2 V−1 s−1, which is higher than those of P1 and P2 (1.31 × 10−6 and 4.98 × 10−7 cm2 V−1 s−1, respectively) (Fig. S2 and Table S1†). In fact, if we study the polymer structures further, such results might not be so surprising at all. The optimized geometries for P3 and P1, based on the DFT calculations presented in Table 2. It can be seen that both P3 and P1 have good planarity. Yet, the dihedral angle between hexyl-TI and DTBT is 41.7° for P2, dramatically larger than those for P1 and P3 (ca. 21°). Thus, this difference might result in different UV-vis absorption and π–π stacking distances and then their different PV performance. Once again, the result indicates that the side chain engineering has significant influence on conjugated D–A polymers. And lots of factors should be considered collectively in balance for developing high performance donor materials. Actually, the trend of the hole mobilities of all polymers is consistent with the DFT simulations and their morphological results as discussed above, and the devices based on the polymer P3 also exhibit the best photovoltaic performance among these TI and DTBT based copolymers.
Conclusion
In summary, thieno[3,2-b]indole (TI), structurally analogous to carbazole, a tricyclic unit with rigid and coplanar structure, was successfully prepared via Cadogan annylation. Three new polymers P1, P2 and P3 based on TI derivatives as the electron-rich units and DTBT as electron-deficient units were synthesized and characterized. By the side-chain engineering, the solubility, UV-vis absorption properties, molecular packing behavior and their performance in PSCs were finely tuned. As a result, the modification has a profound effect on the physical properties and device performance. Especially, these TI based copolymers all have strong and broad absorption ranging from 350–800 nm in solid state. These parameters imply they could be ideal copolymers for PSCs. The optimized device based on P3:PC71BM with a relatively higher mobility (2.84 × 10−5 cm2 V−1 s−1) shows the best PCE of 2.73% under air mass 1.5 global (AM 1.5 G) irradiation of 100 mW cm−2. As a consequence, the electron-rich (TI) unit is one candidate and promising donor unit for designing D–A conjugated polymers. To achieve the high performance PSCs, ongoing work is to further tune the ICT transition between TI units and new electron-deficient units, such as BT or F-atoms substituted BT. In addition to this, the optimization of the device is also necessary.
Acknowledgements
This work was financially supported by the National Basic Research Program of China (2012CB933301), the National Natural Science Foundation of China (51103074, 51203077, 81273409, 51173081, 51173199, 51573205, 21204038, 61136003), Natural Science Foundation of Jiangsu Province (BK2012438, BM2012010), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Program for Changjiang Scholars and Innovative Research Team in University (IRT-15R37), Synergetic Innovation Center for Organic Electronics and Information Displays, Specialized Research Fund for the Doctoral Program of Higher Education (20113223110005).
Notes and references
-
(a) N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CAS;
(b) G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
-
(a) H. Y. Chen, J. H. Hou, S. Q. Zhang, Y. Y. Liang, G. W. Yang, Y. Yang, L. P. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS;
(b) R. F. Service, Science, 2011, 332, 293–293 CrossRef CAS PubMed.
-
(a) C. J. Brabec, S. Gowrisanker, J. J. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS PubMed;
(b) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
-
(a) J. Mei and Z. N. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS;
(b) Z. G. Zhang and Y. F. Li, Sci. China: Chem., 2015, 58, 192–209 CrossRef CAS.
-
(a) L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. P. Yu, Chem. Rev., 2015, 115, 12666–12731 Search PubMed;
(b) L. Lu, M. A. Kelly, W. You and L. Yu, Nat. Photonics, 2015, 9, 491–500 CrossRef CAS.
-
(a) Y. Liu, C. C. Chen, Z. Hong, J. Gao, Y. M. Yang, H. Zhou, L. T. Dou, G. Li and Y. Yang, Sci. Rep., 2013, 3, 3356 Search PubMed;
(b) S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS;
(c) D. H. Wang, D. Y. Kim, K. W. Choi, J. H. Seo, S. H. Im, J. H. Park, O. O. Park and A. J. Heeger, Angew. Chem., Int. Ed., 2011, 50, 5519–5523 CrossRef CAS PubMed;
(d) R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657–661 CrossRef CAS PubMed;
(e) R. K. Pai, T. N. Ahipa and B. Hemavathi, RSC Adv., 2016, 6, 23760–23774 Search PubMed.
-
(a) L. J. Huo, J. H. Hou, S. Zhang, H. Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500–1503 CrossRef CAS PubMed;
(b) L. J. Huo, S. Zhang, X. Guo, F. Xu, Y. F. Li and J. H. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS PubMed;
(c) Z. C. He, C. Zhong, X. Huang, W. Y. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636–4643 CrossRef CAS PubMed;
(d) L. T. Dou, J. You, J. Yang, C. C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS.
-
(a) D. H. Wang, J. S. Moon, J. Seifter, J. Jo, J. H. Park, O. O. Park and A. J. Heeger, Nano Lett., 2011, 11, 3163–3168 CrossRef CAS PubMed;
(b) C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S. W. Tsang, T. H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
-
(a) B. Kan, Q. Zhang, M. M. Li, X. J. Wan, W. Ni, G. K. Long, Y. C. Wang, X. Yang, H. R. Feng and Y. S. Chen, J. Am. Chem. Soc., 2014, 136, 15529–15532 CrossRef CAS PubMed;
(b) Q. Zhang, B. Kan, F. Liu, G. K. Long, X. J. Wan, X. Q. Chen, Y. Zuo, W. Ni, H. J. Zhang, M. M. Li, Z. C. Hu, F. Huang, Y. Cao, Z. Q. Liang, M. T. Zhang, T. P. Russell and Y. S. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
-
(a) H. Q. Zhou, Y. Zhang, C. K. Mai, S. D. Collins, G. C. Bazan, T. Q. Nguyen and A. J. Heeger, Adv. Mater, 2015, 27, 1767–1773 CrossRef CAS PubMed;
(b) L. J. Huo, T. Liu, X. B. Sun, Y. H. Cai, A. J. Heeger and Y. M. Sun, Adv. Mater., 2015, 27, 2938–2944 CrossRef CAS PubMed.
- See reviews:
(a) L. Ye, S. Q. Zhang, L. J. Huo, M. J. Zhang and J. H. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS PubMed;
(b) H. X. Zhou, L. Q. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS;
(c) C. Li, M. Y. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed;
(d) Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed;
(e) I. Etxebarria, J. Ajuria and R. Pacios, Org. Electron., 2015, 19, 34–60 CrossRef CAS;
(f) Y. F. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed;
(g) A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed;
(h) Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
-
(a) Z. G. Zhang and J. Z. Wang, J. Mater. Chem., 2012, 22, 4178–4187 RSC;
(b) C. H. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416–10434 RSC.
- Y. Liang, Z. Xu, J. B. Xia, S. Tsai, Y. Wu, G. Li, C. Ray and L. P. Yu, Adv. Mater., 2010, 22, 135–138 CrossRef PubMed.
- T. Chu, J. Lu, S. Beaupr, Y. Zhang, J. R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- B. M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef.
- A. J. Zucchero, P. L. Mcgrier and U. H. F. Bunz, Acc. Chem. Res., 2010, 43, 397–408 CrossRef CAS PubMed.
- H. C. Zhang, E. Q. Guo, Y. L. Zhang, P. H. Ren and W. J. Yang, Chem. Mater., 2009, 21, 5125 CrossRef CAS.
- C. Du, C. Li, W. Li, X. Chen, Z. Bo, C. Veit, Z. Ma, U. Wuerfel, H. Zhu, W. Hu and F. Zhang, Macromolecules, 2011, 44, 7617–7624 CrossRef CAS.
- Y. Wang, X. Xin, Y. Lu, T. Xiao, X. Xu, N. Zhao, X. Xu, B. S. Ong and S. C. Ng, Macromolecules, 2013, 46, 9587–9592 CrossRef CAS.
- C. J. Brabec, C. Winder, N. S. Sariciftci, J. C. Hummelen, A. Dhanabalan, P. A. Van Hal and R. A. J. Janssen, Adv. Funct. Mater., 2002, 12, 709–712 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- L. Ye, S. Zhang, W. Zhao, H. Yao and J. Hou, Chem. Mater., 2014, 26, 3603–3605 CrossRef CAS.
- W. Chen, T. Xu, F. He, W. Wang, C. Wang, J. Strzalka, Y. Liu, J. Wen, D. J. Miller, J. Chen, K. Hong, L. Yu and S. B. Darling, Nano Lett., 2011, 11, 3707–3713 CrossRef CAS PubMed.
-
(a) N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed;
(b) B. N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef.
-
(a) S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–303 CrossRef CAS;
(b) K. Sun, B. M. Zhao, V. Murugesan, A. Kumar, K. Y. Zeng, J. Subbiah, W. W. H. Wang and J. Y. Ouyang, J. Mater. Chem., 2012, 22, 24155–24165 RSC;
(c) T. Y. Chu, S. Alem, S. W. Tsang, S. C. Tse, S. Wakim, J. P. Lu, G. Dennler, D. Waller, R. Gaudiana and Y. Tao, Appl. Phys. Lett., 2011, 98, 253301 CrossRef.
- D. H. Wang, J. K. Kim, J. H. Seo, I. Park, B. H. Hong, J. H. Park and A. J. Heeger, Angew. Chem., Int. Ed., 2013, 52, 2874–2880 CrossRef CAS PubMed.
- W. W. Li, A. Furlan, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 5529–5532 CrossRef CAS PubMed.
- R. P. Qin, W. W. Li, C. H. Li, C. Du, C. Veit, H. F. Schleiermacher, M. Andersson, Z. S. Bo, Z. P. Liu, O. Inganäs, U. Wuerfel and F. L. Zhang, J. Am. Chem. Soc., 2009, 13, 114612–114613 Search PubMed.
- H. Yi, S. Al-Faifi, A. Iraqi, D. C. Watters, J. Kingsley and D. G. Lidzey, J. Mater. Chem., 2011, 21, 13649–13656 RSC.
- J. Kim, M. H. Yun, G. Kim, J. Lee, S. M. Lee, S. Ko, Y. Kim, G. K. Dutta, M. Moon, S. Y. Park, D. S. Kim, J. Y. Kim and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 7523–7534 CAS.
- A. Casey, R. S. Ashraf, Z. Fei and M. Heeney, Macromolecules, 2014, 47, 2279–2288 CrossRef CAS.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS PubMed.
- E. J. Zhou, J. Cong, K. Tajima and K. Hashimoto, Chem. Mater., 2010, 22, 4890–4895 CrossRef CAS.
- J. Jiang, P. Yang, C. Yu, H. Lin, K. Huang and K. Wei, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3960–3969 CrossRef CAS.
- R. G. Brandt, W. Yue, T. R. Andersen, T. T. Larsen-olsen, M. Hinge, E. Bundgaard, C. Krebs and D. H. Yu, J. Mater. Chem. A, 2015, 3, 1633–1639 CAS.
-
(a) W. Yue, X. Huang, J. Yuan, F. C. Krebs and D. H. Yu, J. Mater. Chem. A, 2013, 1, 10116–10119 RSC;
(b) W. Yue, T. T. Larsen-olsen, X. Hu, M. Shi, H. Chen, M. Hinge, P. Fojan, F. C. Krebs and D. Yu, J. Mater. Chem. A, 2013, 1, 1785–1793 RSC.
- R. A. Abramovitch, T. Chellathurai, I. T. Mcmaster, T. Takaya, C. I. Azogu and D. P. Vanderpool, J. Org. Chem., 1977, 42, 2914–2919 CrossRef CAS.
- X. Zhang, Y. Cui, R. Katoh, N. Koumura and K. Hara, J. Phys. Chem. C, 2010, 114, 18283–18290 CAS.
- A. W. Freeman, M. Urvoy and M. E. Criswell, J. Org. Chem., 2005, 70, 5014–5019 CrossRef CAS PubMed.
- J. Y. Shim, J. Y. Baek, J. Kim, S. Y. Park, J. Kim, I. Kim, H. H. Chun, J. Y. Kim and H. S. Suh, Polym. Chem., 2015, 6, 6011–6020 RSC.
- F. Maya and J. M. Tour, Tetrahedron, 2004, 60, 81–92 CrossRef CAS.
- H. H. Huang, G. Y. Jiao, S. L. Liu, Q. Li, X. Shi, N. N. Fu, L. H. Wang, B. M. Zhao and W. Huang, Chem. Commun., 2015, 51, 15846–15849 RSC.
- H. Tan, X. Deng, J. Yu, B. Zhao, Y. Wang, Y. Liu, W. Zhu, H. Wu and Y. Cao, Macromolecules, 2013, 46, 113–118 CrossRef CAS.
- Y. F. Li, Y. Cao, J. Gao, D. L. Wang, G. Yu and A. J. Heeger, Synth. Met., 1999, 99, 243–248 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc, Wallingford, CT, 2010 Search PubMed.
- C. Goh, R. J. Kline and M. D. McGehee, Appl. Phys. Lett., 2005, 86, 122110 CrossRef.
- C. H. Chen, C. H. Hsieh, M. Dubosc, Y. J. Cheng and C. S. Hsu, Macromolecules, 2010, 43, 697–708 CrossRef CAS.
- C. W. Schlenker and M. E. Thompson, Chem. Commun., 2011, 47, 3702–3716 RSC.
- Y. Zhang, S. K. Hau, H. L. Yip, Y. Sun, O. Acton and A. K. Y. Jen, Chem. Mater., 2010, 22, 2696–2698 CrossRef CAS.
- B. Y. Qi and J. Z. Wang, Phys. Chem. Chem. Phys., 2013, 15, 8972–8982 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TGA; SCLC mobility; device optimization and NMR spectra. See DOI: 10.1039/c6ra05413g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
















