
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of chiral recyclable catalysts in asymmetric catalysis
GuiPing Hanab,
WenQi Renb,
ShengYong Zhangb,
ZhenYu Zuo*a and
Wei He *b
*b
aDepartment of Pharmacy, Shaanxi University of Chinese Medicine, Xianyang 712046, P. R. China. E-mail: zuozhenyu666@sina.cn
bDepartment of Chemistry, School of Pharmacy, Air Force Medical University, Xi'an 710032, P. R. China. E-mail: weihechem@fmmu.edu.cn
First published on 21st May 2024
Abstract
Chiral drugs hold a significant position within the contemporary pharmaceutical market, and the chiral catalysts play a crucial role in their synthesis. However, current chiral catalysts encounter challenges pertaining to their separation from products and the recycling process. The utilization of chiral recyclable catalysts not only reduces production costs but also aligns with the growing emphasis on environmentally-friendly chiral synthetic chemistry. These recyclable catalysts exhibit diverse carriers and distinct characteristics. Chemists employ the distinctive attributes of individual carriers to render them recyclable, thereby yielding time and cost savings. This review examines the asymmetric recyclable catalytic reactions reported between January 2017 and October 2023, categorizing them based on carrier solubility, and elucidates the loading techniques, catalytic impacts, recovery approaches, and recycling processes associated with these carriers.
1 Introduction
Organic reactions have important significance in daily life, including food and drug manufacturing,1 such as flour fermentation,2 miconazole synthesis,3 and others. Within organic reactions, catalysts assume a crucial function by diminishing activation energy,4 enhancing reaction rates, expediting reaction durations,5 and reducing expenses. Notably, chiral catalysts employed in the production of chiral drugs hold immense potential within the pharmaceutical domain. Nevertheless, the synthesis of chiral catalysts frequently engenders environmental contamination and entails substantial costs. Hence, the significance of synthesizing recyclable catalysts is paramount.6 Not only do they effectively fulfill their intended purpose, but their environmentally friendly and sustainable characteristics have consistently been the ultimate objective in the field of asymmetric catalysis.7 Consequently, the synthesis of catalysts that can be recycled holds significant importance.8The unloaded homogeneous catalytic system presents significant drawbacks, including challenges in separating catalysts from products, difficulties in product purification, high costs associated with catalysts, and complications in catalyst recovery. In comparison to alternative catalysts, both loaded homogeneous and heterogeneous catalysts possess favorable attributes that facilitate their convenient recovery and subsequent reuse. These characteristics are all due to the unique nature of the carrier materials used. Supported catalysts is still an important area worthy of development and attention. Because there are some advantages cannot be ignored, such as improvement of the stability of catalyst, enhancement of the catalytic efficiency, saving costs and environmental protection and energy saving. For example, loading precious metals such as Ag on a carrier such as silicon dioxide, alumina, zirconia can improve its catalytic activity and selectivity, while also slowing down the deactivation rate of precious metals and extending their service life.9 When preparing an organic synthesis reaction catalyst, the active substance can be loaded in micron-scale silica powder, which can improve its catalytic efficiency and reduce the emission of reaction waste. In mass production, this method of saving raw materials can bring great economic benefits. Supported catalysts have been widely used in chemical, pharmaceutical, petroleum, energy and other fields, and are suitable for large scale production. This review also cites industrial cases and emphasizes the importance of supported catalysts. Chiral recyclable catalysts include two parts: carrier and active catalyst. The common ligands in active catalysts are chiral phosphorus ligands, salen ligands and cinchona alkaloids, which are the main skeletons of chiral induction in asymmetric reactions.10 The carriers can be classified into two categories: organic carriers, such as polymers and ionic liquids, and inorganic carriers, which encompass zeolite, molecular sieve, silica, and graphene. Polymers can be classified into three distinct categories according to the configuration of their chain: linear polymers, branched polymers, and cross-linked polymers. Additionally, polymers can be further categorized as either soluble or insoluble based on the solubility of their chain. Soluble polymers encompass linear polymers and branched polymers, exemplified by polyglycerol,11 polyethylene glycol and its derivatives.12,13 Conversely, insoluble polymers comprise highly branched polymers and cross-linked polymers, such as merrifield resin and porous organic polymers,14–16 among others (Fig. 1). Soluble catalysts offer notable benefits such as elevated catalytic efficiency, enhanced stereoselectivity, and convenient design and synthesis featuring uniform active sites. On the other hand, insoluble catalysts possess advantageous traits including robust stability, easy recovery, and, in contrast to soluble catalysts, multiple active sites that enable selective driving of reactions, thereby exclusively yielding the desired products.17 However, the design and synthesis of well-defined uniform active sites pose significant challenges. Researchers have persistently investigated both insoluble and soluble catalysts,18 with the aim of developing molecular catalysts that exhibit high reactivity and selectivity, while also enabling catalyst recovery and recycling.19 Consequently, the development of loaded catalysts has remained a topic of considerable interest.20
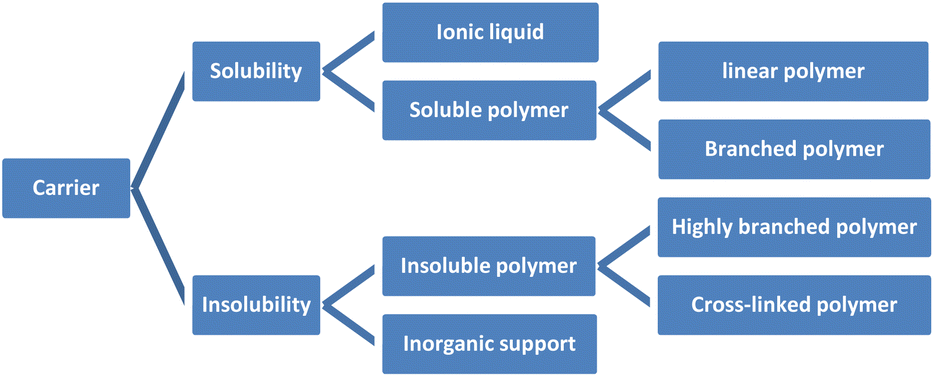 | ||
| Fig. 1 The categorization of catalyst supports. | ||
Currently, there exist two approaches for incorporating catalysts into polymers: physical adsorption and chemical covalent bonding (Fig. 2). The latter method encompasses copolymerization of catalyst monomers with corresponding polymers, as well as subsequent post-modification of the resulting polymers. When formulating a stationary catalyst on a polymer carrier, there are four primary factors to consider: (1) the interconnection properties between the catalytic site and the carrier, (2) the characteristics and length of the spacer between the catalytic site and the polymer network, (3) the density and positioning of the catalytic site along the polymer network, and (4) the physical and chemical attributes of the polymer skeleton.21 Previous research has provided an overview of the utilization of polymer-bound organometallic reagents in organic synthesis, specifically focusing on non-soluble polymer carriers. However, there is a lack of comprehensive analysis regarding the application of soluble carriers.22 Additionally, the utilization of heterogeneous chiral catalysts and soluble chiral catalysts in asymmetric one-pot reactions can be summarized based on the various types of catalysts employed. The absence of a classification and summary of carriers is evident in this review.23 The primary focus of this study is to present the utilization of chiral catalysts, which are supported on both soluble and insoluble carriers, in the context of asymmetric catalytic reactions. We used the Scifinder database to conduct keyword searches on “recyclable catalysts”, resulting in the retrieval of a substantial quantity of research articles published within the period of 2017 to 2023. This data set provides important insights into trends in the field of recyclable catalysts (Fig. 3).
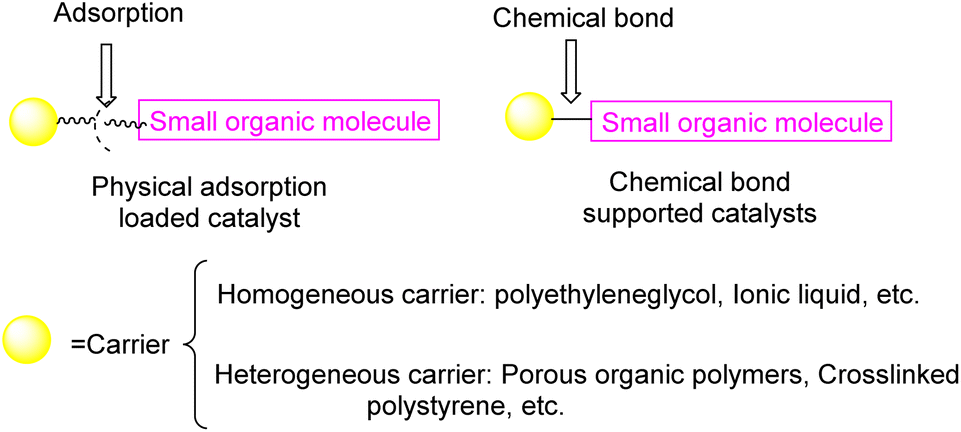 | ||
| Fig. 2 The two ways of catalyst loading. | ||
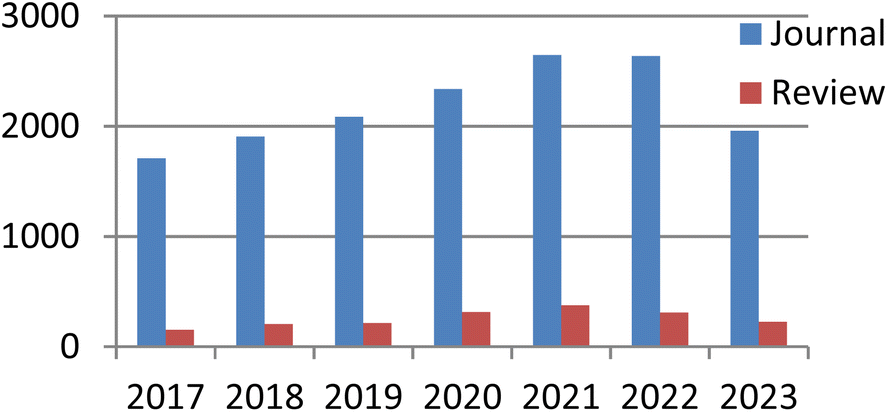 | ||
| Fig. 3 Publications on the topic of “recyclable catalysts” spanning the years 2017 to 2023. | ||
2 Soluble carrier loaded catalyst
The increasing advancement of supported catalysts has led to a growing interest in the investigation of soluble carriers.24,25 “Homogeneous reaction, two-phase separation” is the most vivid description of soluble supported catalyst. This particular type of catalyst facilitates catalytic reactions under homogeneous conditions through the careful selection of an appropriate carrier. Simultaneously, the separation of the product and catalyst can be achieved through solvent precipitation or membrane filtration, leveraging the disparity in solubility between the polymer and the reaction product. For the soluble supported catalyst, the possibility of reducing the catalytic effect due to the leaching of the catalyst is very small, and sometimes it may leach leading to the low catalytic effect, and the reason may due to the select of solvent for precipitation and the impact of soluble supported catalyst on the microenvironment.2.1 Ionic liquid supported catalyst
Ionic liquids, which are molten salts consisting of ions and remain in a liquid state below 100 °C. These liquids possess notable attributes including non-flammability, remarkable thermal stability, minimal toxicity, and adjustable structural properties. Furthermore, they exhibit solubility in polar solvents while remaining insoluble in non-polar solvents,26 such as ether and tert-butyl methyl ether. Consequently, the solvent precipitation method can be employed to recover the catalyst and facilitate the recycling process. The presence of the ionic liquid component not only serves to stabilize the catalytic active center, but also enhances the catalytic activity through its involvement in the reaction as a co-catalyst.27Asymmetric catalytic hydrogenation is considered a highly convenient approach for acquiring chiral alcohols due to its advantageous attributes such as the readily accessible hydrogen source, simple preparation process, cost-effectiveness, and safety.28,29 In 2018, Kinage et al. presented a collection of environmentally friendly amino acid amide chiral ionic liquids 1 (Scheme 1).30 The initial utilization of amino acid amide CILs (0.5 mmol) as a catalyst for the ATH reaction of acetophenone, a small organic molecule, was documented. Research has demonstrated the essential role of amino acid amide CILs and bases as catalysts in this particular reaction. The formation of chiral secondary alcohols through the involvement of cyclic intermediates of CILs compounds resulted in favorable ee and yields. Furthermore, the yields and ee value of chiral ionic liquid 1a remained relatively stable even after undergoing 4 cycles, with yields at 87% and ee at 97%. The utilization of amino acid amide CILs presents a reliable solution for the conventional metal-catalyzed asymmetric ATH reaction of ketones.
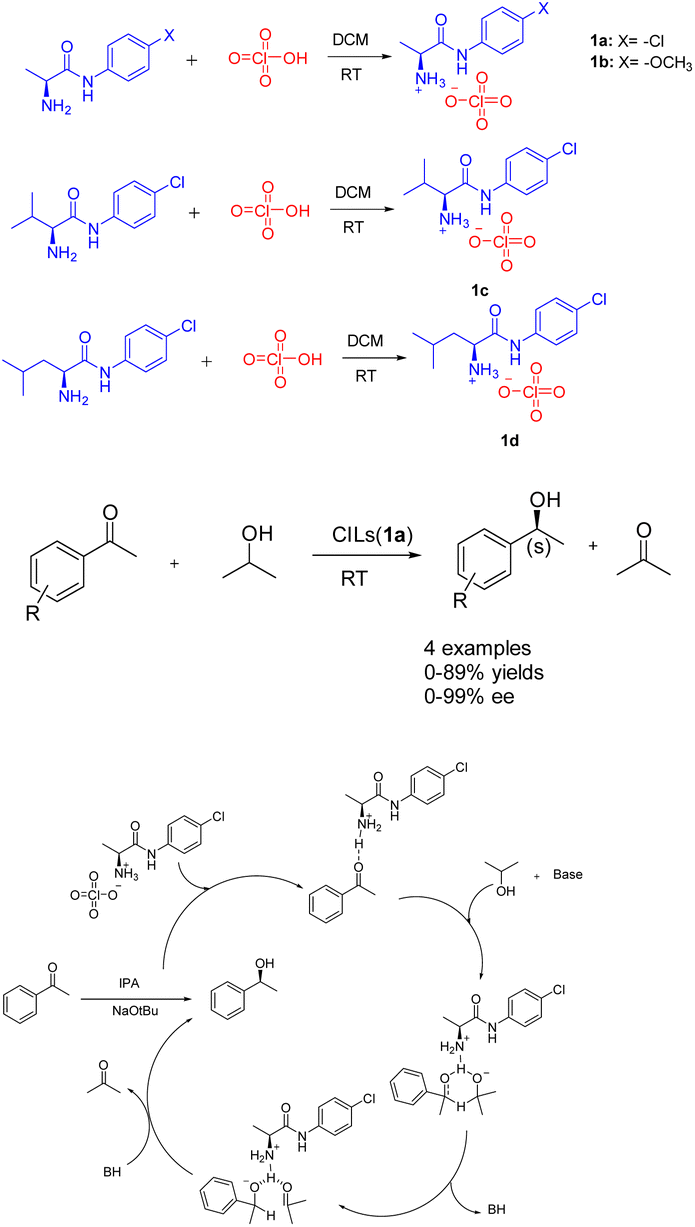 | ||
| Scheme 1 Synthesis of amide-modified chiral ionic liquids and asymmetric ATH reaction of acetophenone and its mechanism. | ||
Water as a green solvent has also been studied.31–34 In their study conducted in 2019, Bica-Schröder et al. employed the characteristics of microemulsions, specifically the precise manipulation of phase behavior and solvent separation through temperature variations. They utilized surface active ionic liquids 2 (0.6 mmol), consisting of surface active anions and cations, in the context of asymmetric catalytic hydrogenation of ketones catalyzed by ruthenium (Scheme 2).35 This transfer hydrogenation is carried out through an ionic mechanism and requires acidic activation of the imine raw material. Therefore, a azeotropic mixture of bisoxazoline and triethylamine (molar ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) instead of sodium formate is used as a powerful and irreversible hydrogen source. The findings indicate that the system exhibits applicability to a diverse range of aromatic ketones, resulting in favorable separation yields (reaching up to 98%) and ee (reaching up to 97%). However, its effectiveness is limited when applied to aliphatic ketones. Despite a decrease in yields after 3 cycles, the ee remains relatively stable without a significant decline.
:2) instead of sodium formate is used as a powerful and irreversible hydrogen source. The findings indicate that the system exhibits applicability to a diverse range of aromatic ketones, resulting in favorable separation yields (reaching up to 98%) and ee (reaching up to 97%). However, its effectiveness is limited when applied to aliphatic ketones. Despite a decrease in yields after 3 cycles, the ee remains relatively stable without a significant decline.
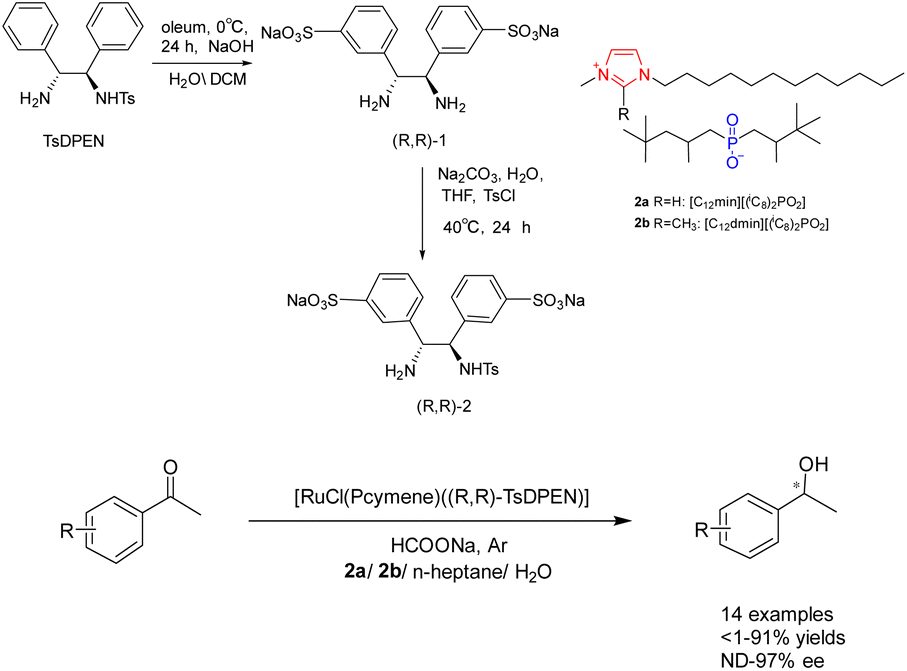 | ||
| Scheme 2 Synthesis of chiral ligands and asymmetric hydrogenation of ketones catalyzed by ionic liquids. | ||
In 2023, Jin et al. modified phosphine ligands and designed and synthesized chiral phosphinxe functionalized polyether ionic liquid 3. This compound was subsequently utilized in the Ru-catalyzed asymmetric hydrogenation of β-ketoesters (Scheme 3).36 The obtained chiral ionic liquid 3 (CIL3) exhibits the characteristics of chiral phosphine ligands and ionic liquids. Moreover, the chiral catalyst can be efficiently retrieved and reused by employing a minimal quantity of CIL3 (0.1 mol%) as a catalyst. The experimental findings demonstrate that, when subjected to the most favorable reaction conditions, the model substrate methyl acetoacetate undergoes complete conversion to β-hydroxybutyric acid methyl ester with an ee of 97%. The chiral catalyst employed in this procedure exhibits a recyclability potential of up to 12 cycles, thereby demonstrating its favorable suitability towards β-ketoesters of diverse structural compositions. This study proposes a new method for using ionic liquids in asymmetric hydrogenation reactions in an environmentally friendly manner.
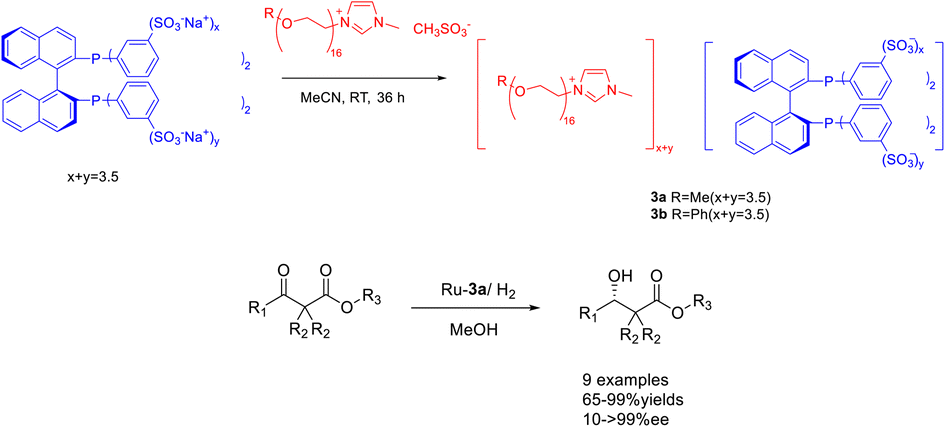 | ||
| Scheme 3 Synthesis and application of phosphine functionalized chiral polyether ionic liquid in asymmetric catalytic hydrogenation reaction. | ||
The catalytic efficacy of double ionic liquids generally surpasses that of single cationic ionic liquids, likely due to the greater number of catalytic sites present in the double ionic liquid catalyst. In a study conducted in 2019, Singh et al. documented a Diels–Alder reaction involving cyclopentadiene and crotonaldehyde. The reaction was carried out using Macmillan catalyst 4 (5 mol%) as an organic catalyst, double ionic liquid (10 mol%) and TFA as co-catalysts. The researchers achieved a yields of 95% and an ee of 87% for the resulting product (Scheme 4).37 The findings indicate that the catalyst demonstrates enhanced reusability when increase of 20 mol% concentration of the double ionic liquid is employed. After undergoing 5 cycles of utilization with the singular ionic liquid catalyst 4c (5 mol%), the resultant product exhibits a yields surpassing 40% and an ee exceeding 50%. Similarly, employing the dual ionic liquid catalyst 4f (5 mol%) for 5 cycles yields a product with a yields exceeding 80% and an ee surpassing 70%. The reactivity of catalysts was depending on the in situ generated Brønsted acids salts of the catalyst. Compared with other reactions in ionic liquids, the MacMillan catalyst modified by BMIm PF6 ionic liquid (4f) is mainly protonated by the hydroxyl group on the catalyst 4 under the action of TFA, forming ion pair with the double ionic liquid 4f, such as the transition state 4′. In addition, the hydrophobicity of the anion PF6 can create a hydrophobic reaction environment, and cyclopentadiene and crotonaldehyde can react better.
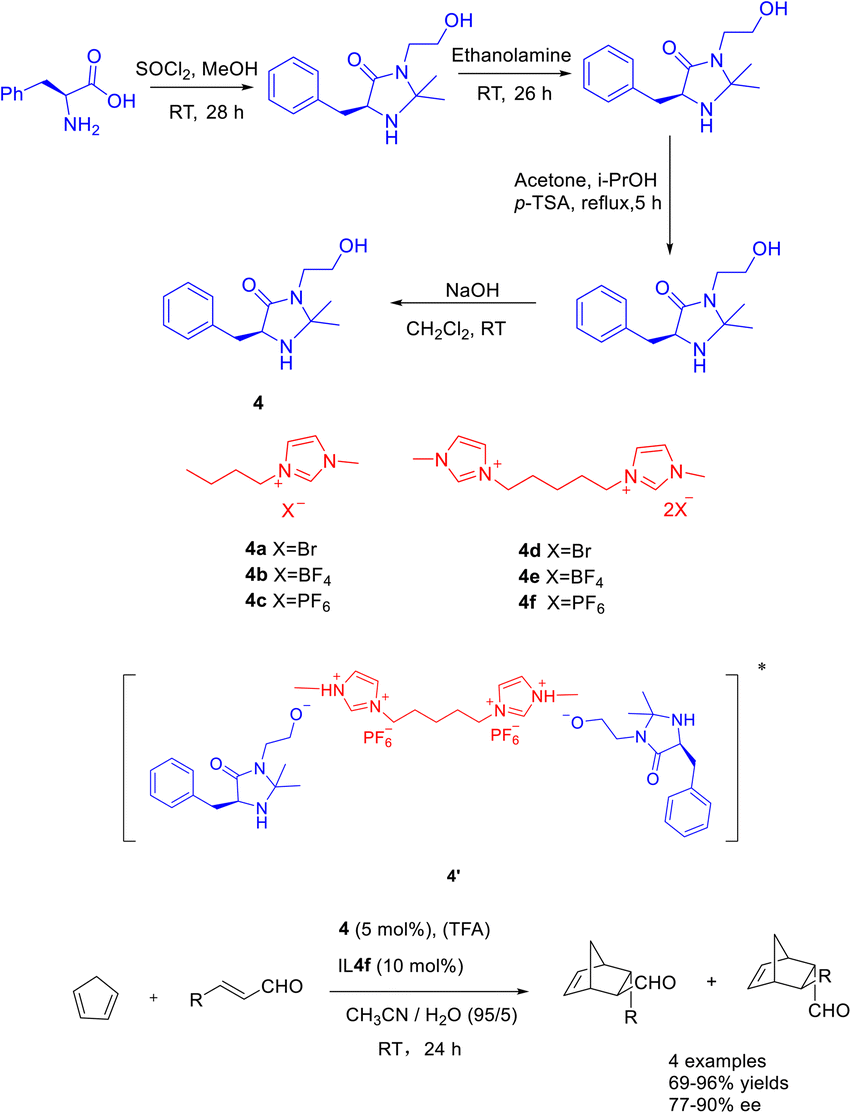 | ||
| Scheme 4 Synthesis of Macmillan ionic liquid and its application in asymmetric Diels–Alder reaction between crotonaldehyde and cyclopentadiene. | ||
In the year 2021, Singh and colleagues published a study wherein they introduced a chiral ionic liquid derived from DABCO modified MacMillan catalyst 5.38 This novel compound was subsequently employed in the asymmetric Diels–Alder reaction involving crotonaldehyde and cyclopentadiene (Scheme 5). The experimental findings revealed that the aforementioned reaction yielded Diels–Alder products with varying yields ranging from 18% to 92%, accompanied by ee ranging from 68% to 93%. The chiral ionic liquid catalyst 5a (5 mol%) exhibits enhanced recyclability. The catalyst demonstrates the ability to undergo 6 cycles of recycling, resulting in a marginal decrease in both ee and yields. Notably, the recycling process yields a product with a 63% yields and an ee value of 79%. Compared with catalyst 4f in the previous case, catalyst 5a is applied to the asymmetric Diels Alder reaction of cyclopentadiene and crotonaldehyde under similar reaction conditions. However, catalyst 5a has better catalytic performance than 4f due to the larger distance between the reaction site secondary amine and the loading site imidazole.
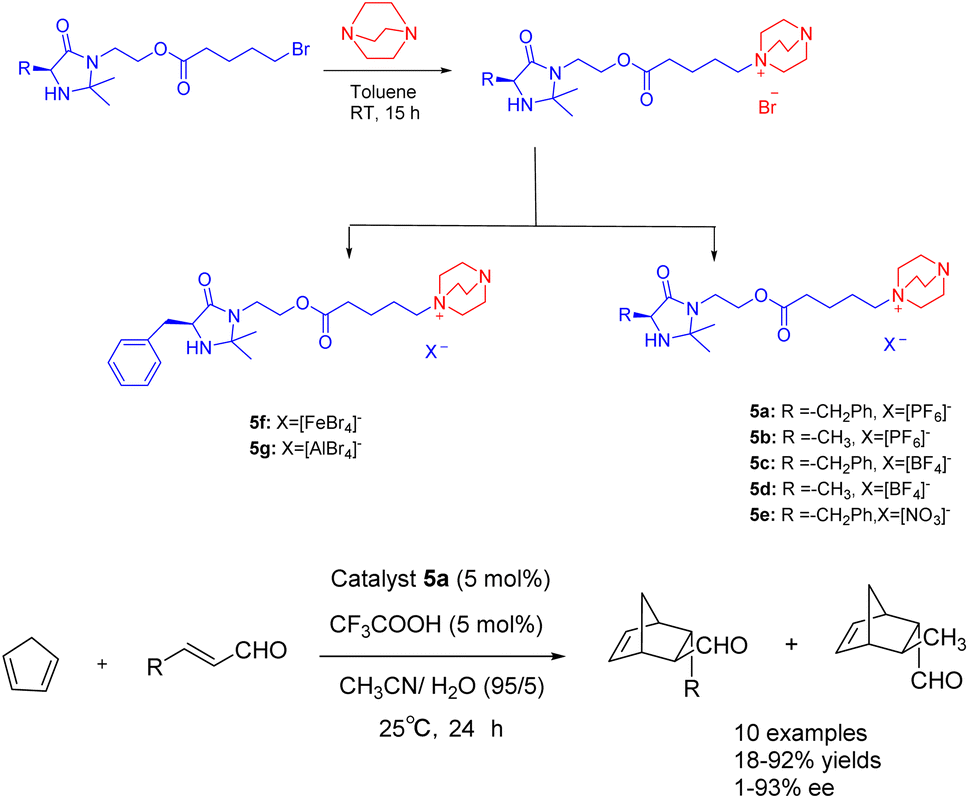 | ||
| Scheme 5 Synthesis of Macmillan chiral ionic liquid catalyst and its application in asymmetric Diels–Alder reaction between crotonaldehyde and cyclopentadiene. | ||
In 2023, Singh et al. published a study on a proline-modified imidazolium-based chiral ionic liquid 6 (20 mol%), which was utilized in the asymmetric Friedel–Crafts reaction of indole with α, β-unsaturated aldehydes (Scheme 6).39 The yields of 62–89% and an ee of 47–88% when triethylamine was used as an additive. Furthermore, it was observed that the catalyst exhibited the ability to undergo recycling up to seven times while retaining a consistent enantioselectivity of 86% ee. Unfortunately, the yield of the product experienced a notable decrease, reaching 75% by the fourth cycle. This phenomenon may attribute to the recovery method of ionic liquid catalyst, which cannot be completely precipitated by solvent precipitation method. The difference in the conformational free energy of imine ions is due to the steric hindrance between the two substituents of the pyrrolidine ring and the orientation of (E)–crotonaldehyde (Table 1). Conformational isomers indicate that the indole attack occurs on the Si surface of the imine ion to avoid the steric hindrance caused by TMS and larger phenyl groups, and to obtain enantioselective (S)-3-alkylated indoles, such as the transition state 6′.
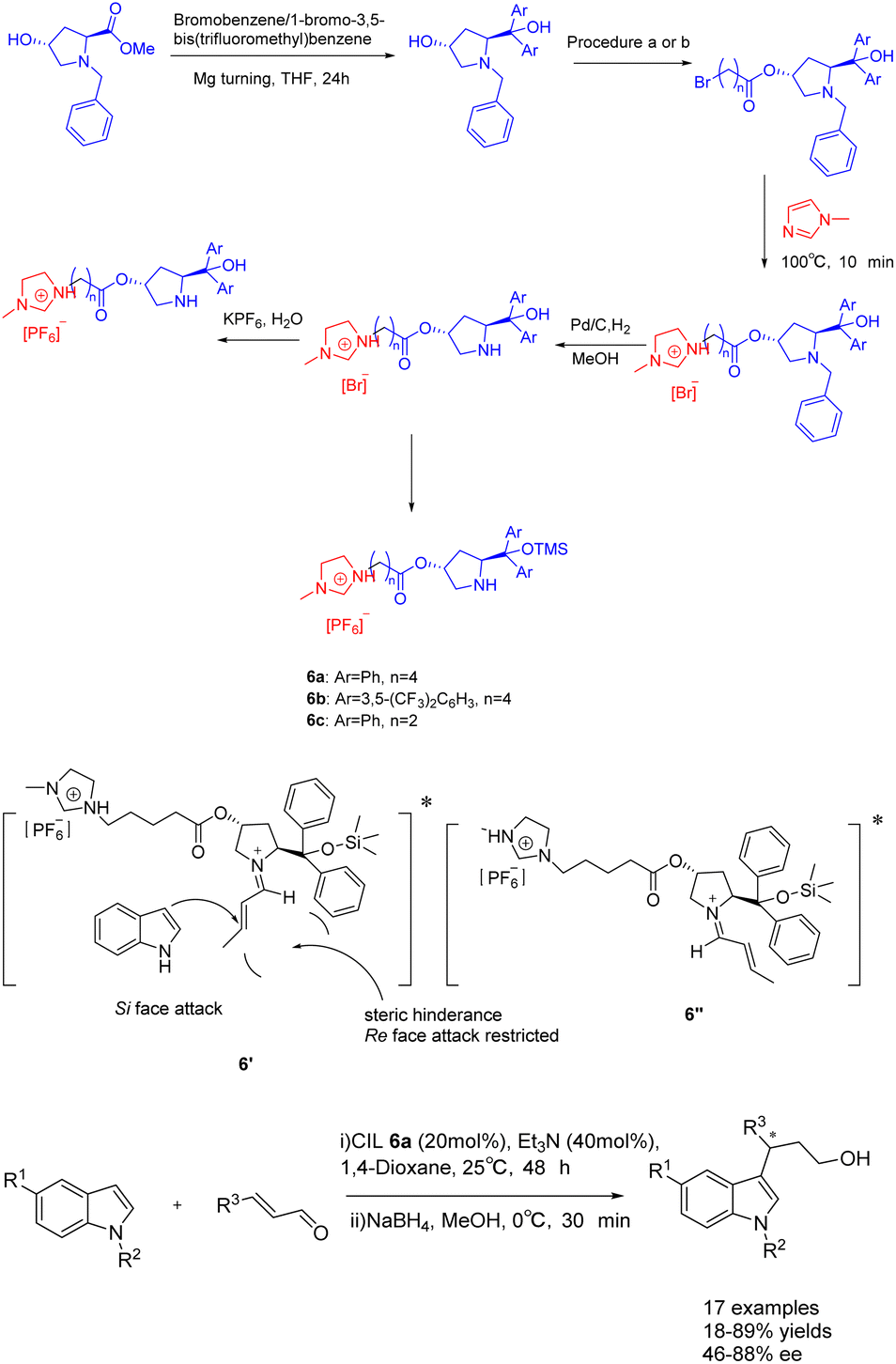 | ||
| Scheme 6 Synthesis of propionylimidazole chiral ionic liquid and its application in asymmetric Friedel–Crafts reactions of indole with α, β-unsaturated aldehydes. | ||
| Structure | E [hartree] | ΔEelec in kcal mol−1 | G [hartree] | ΔG in kcal mol−1 |
|---|---|---|---|---|
| 6′ | −2828.31510740 | 0 | −2827.628521 | 0 |
| 6′′ | −2828.25162066 | 39.8 | −2827.566961 | 38.6 |
Ionic liquids have been investigated not only as catalysts but also as solvents or co-solvents in catalytic reactions.40 In their study, Gonzalo et al. found that room temperature ionic liquids 7, specifically, are extensively employed as co-solvents in various catalytic processes (Scheme 7).41 The addition reaction of tert-butyl hydrazine formaldehyde with various aliphatic α-keto esters was catalyzed by imidazolium-based ionic liquids ([hmim] BF4 and [hmim] PF6) (30 vol%) in the presence of chiral thiourea. The findings indicate that the catalyst exhibits a beneficial influence, and the utilization of low RTILs 7 enhances the catalyst's activity. Chiral tert-butylmethyl tertiary alcohols with yields surpassing 13% and an ee exceeding 60% can be obtained for the majority of the examined substrates. The reaction in the presence of low percentage of RTIL leads to an increase in catalyst activity, making it possible to work at lower temperatures. Therefore, when up to 30 vol% [hmim] PF6 was used as co-solvent in the process carried out in toluene, chiral tert-butylazomethyl tertiary alcohols could be obtained with moderate to good conversion and higher enantioselectivity for most of the substrates studied. Unfortunately, this article does not mention the recycling of the catalyst.
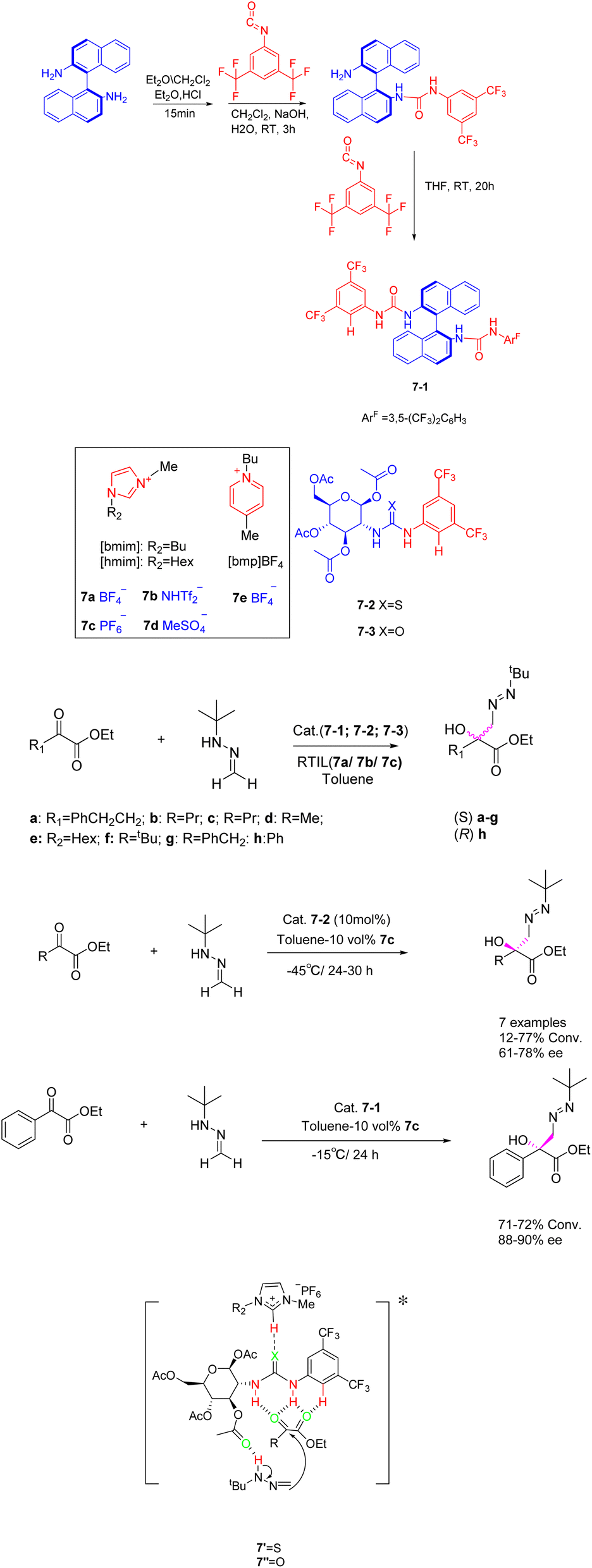 | ||
| Scheme 7 Synthesis of imidazolyl ionic liquid and asymmetric addition reaction of formaldehyde tert-butylhydrazine and α-ketone ester and its mechanism. | ||
The inclusion of ionic liquids in organic catalytic reactions imparts the qualities of homogeneous catalytic effect and facile separation of heterogeneous catalysis, rendering them increasingly prominent in the prospective market. However, it is worth noting that the production expenses associated with ionic liquids are substantial, as are the costs incurred in their preparation and utilization as catalysts. The recovery method of ionic liquids mainly utilizes their insolubility in ether or tert-butyl methyl ether, allowing them to precipitate for recovery.
2.2 Soluble polymer supported catalyst
In their publication in 2019, Shi et al. introduced a pioneering catalyst 8 (5 mol%), supported by polyethylene glycol, for the asymmetric transfer hydrogenation of β-amidonitroolefins (Scheme 8).44 This supported catalyst demonstrated remarkable ee, with an ee reaching up to 95%. Furthermore, it exhibited facile separation through selective precipitation with ether and subsequent filtration from the reaction mixture. Notably, the polymer-bound catalyst 8 was successfully recycled 6 times, yielding the desired product with a 96% yields and a 95% ee value. In general speaking, The symmetric catalyst can reduce the conformation of the transition state and improve the selectivity of the reaction.45 PEG acting as a bridge to construct a C2-symmetric recyclable catalyst, good yield and selectivity were obtained, According to the mechanism, it can be seen that 8b is activated by multiple hydrogen bonds through catalyst 8 to form an intermediate (8c). Subsequently, hydrogen is transferred from the negative hydrogen compound 8d to obtain chiral products 8e and pyridine compounds.
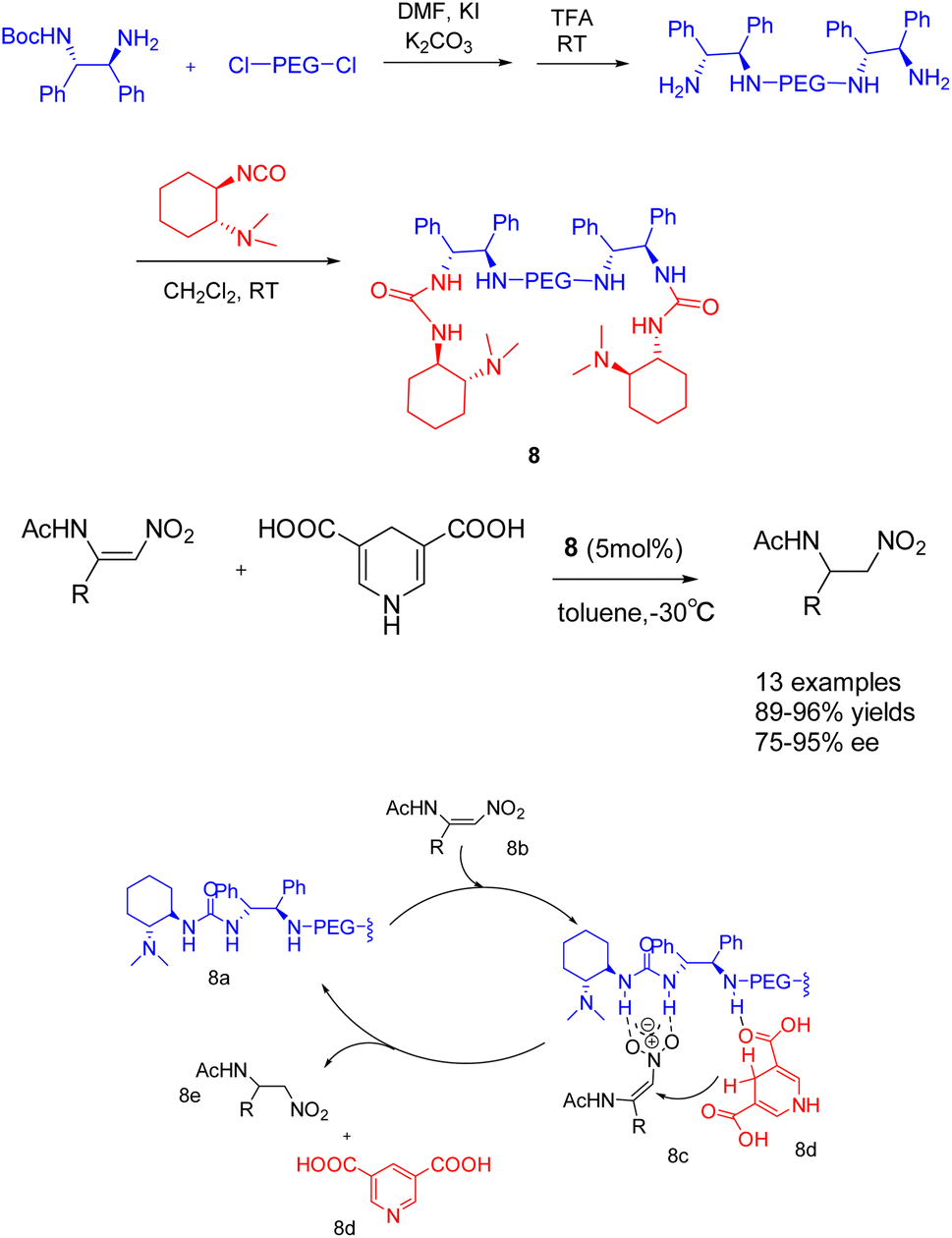 | ||
| Scheme 8 A pegylated supported catalyst with multiple hydrogen bonds was synthesized for asymmetric catalytic hydrogenation of β-amido-nitroolefin and its mechanism. | ||
In their 2020 study, Dai et al. conducted reversible addition fragmentation chain transfer (RAFT) polymerization to synthesize a range of L-proline-functionalized pH-responsive copolymers, utilizing macromolecular RAFT agents as catalysts (Scheme 9).46 The researchers then employed these catalysts in the direct asymmetric aldol reaction of p-nitrobenzaldehyde and cyclohexanone in an aqueous environment, subsequently evaluating the catalytic activity and stereoselectivity. Research findings have indicated that the pH level of the aqueous solution and the composite structure of the copolymer catalyst significantly impact both the catalytic activity and stereoselectivity. Notably, when the pH was set at 7.0, catalyst 9 (10 mol%) exhibited the most favorable catalytic performance, resulting in a yields, anti-syn ratio, and ee of the β-hydroxy ketones product at 92%, 92/8, and 96%, respectively. Furthermore, even after undergoing 3 cycles, catalyst 9 maintained a high ee value of 95%, with only a slight reduction in yields to 61%.
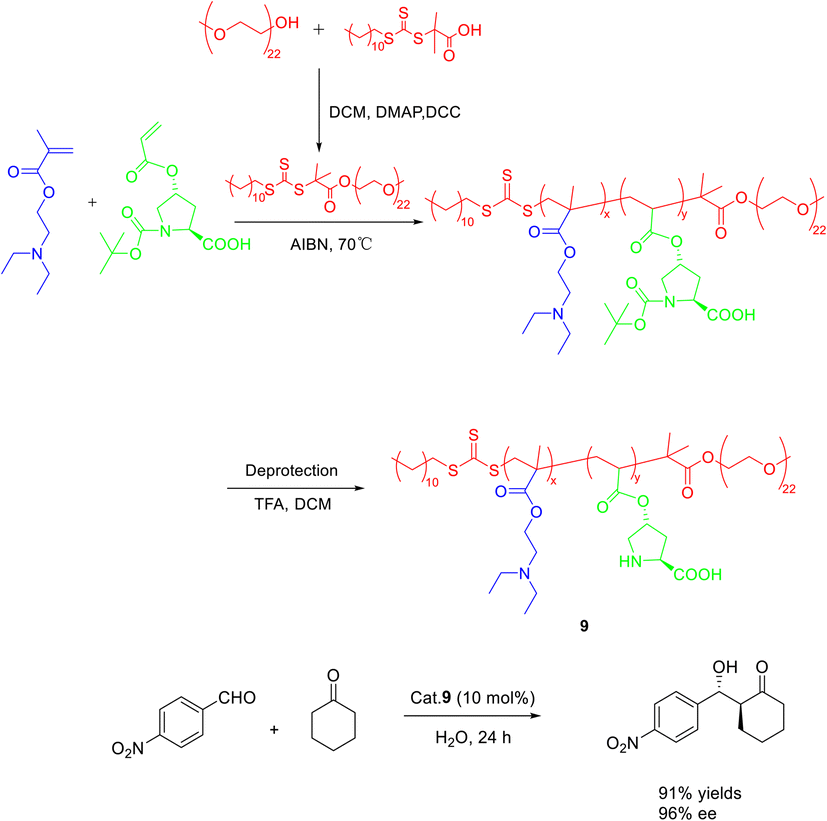 | ||
| Scheme 9 The asymmetric acetal reaction between p-nitrobenzaldehyde and cyclohexanone was catalyzed by the synthesis of L-proline functionalized copolymer. | ||
In their 2023 publication, Chen et al. presented their findings on the utilization of heterogeneous ligand 10 (10 mol%), supported by polyethylene glycol (PEG), in the copper catalyzed asymmetric Henry reaction between nitroethanol and aldehydes (Scheme 10).47 The catalyst exhibited notable levels of both yields and stereoselectivity, while also offering the advantage of facile purification and recovery based on the dissolution properties of polyethylene glycol. Following a single utilization cycle, the inclusion of copper acetate resulted in an 89% increase in yields. Simultaneously, the ee value experienced a slight reduction to 94%. This catalyst's practicality was further substantiated by its successful application in the gram-scale synthesis of crucial intermediates for florfenicol and reboxetine.
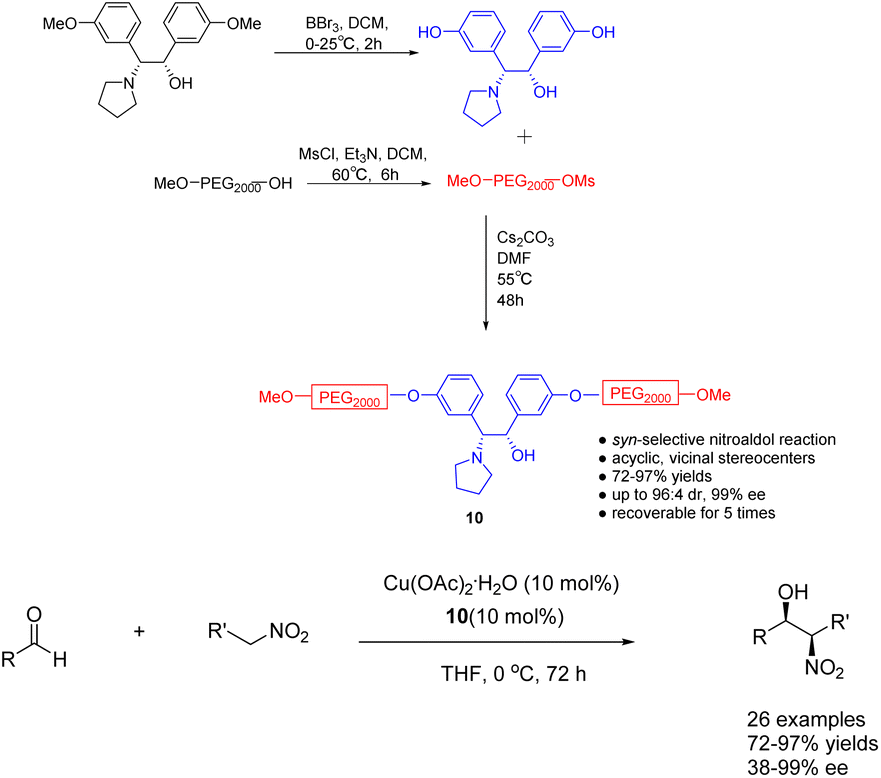 | ||
| Scheme 10 The synthesized PEG-supported chiral amino alcohols were used for the asymmetric Henry reaction of nitroethanol with aldehydes. | ||
Linear polymers possess specific elastic and plastic properties, enabling them to dissolve, undergo melting, and exhibit a significant degree of rotational freedom due to chemical bonding. Among the soluble polymer matrices employed, polyethylene glycol (PEG) stands out as the most successful option due to its solubility in numerous organic solvents, insolubility in select solvents, and ease of functionalization.
The chiral diamine polymer synthesized through RAFT exhibits pronounced hydrophobicity and displays low reactivity. In 2022, Jia et al. successfully synthesized a star-shaped thermal responsive polymer 11 via ATRP polymerization (Scheme 11).49,50 The resulting polymer was immobilized onto a chiral metal–organic catalyst prepared using Ru metal and chiral diamine ligands, with the chiral ligand coordinating with Cu2+ metal. The research revealed that the interaction between the chiral polymer and Cu2+ results in the emergence of a chiral amplification phenomenon. Additionally, the Ru–Cu catalyst (0.85 mol%/0.45 mol%) with a star-shaped structure can be effectively assembled into a single chain polymer in aqueous environments, showcasing superior catalytic performance and selectivity compared to the Ru catalyst during the asymmetric hydrogenation of ketones in water. Furthermore, this catalyst can be reused up to 6 times while maintaining a yields of 90% and an ee value of 95%. Furthermore, this approach obviates the need for supplementary ligands and eliminates the necessity of Cu2+ removal post-reaction, thereby mitigating metal contamination and minimizing cost inefficiencies. β-Cyclodextrin is a hollow cyclic oligosaccharide compound with 21 hydroxyl groups for modification, making it an ideal raw material for synthesizing star shaped polymers. In this paper, it was converted to a 21-Br-β-CD macroinitiator, which can be further modified with TsDPEN to realize the loading of the chiral catalyst.
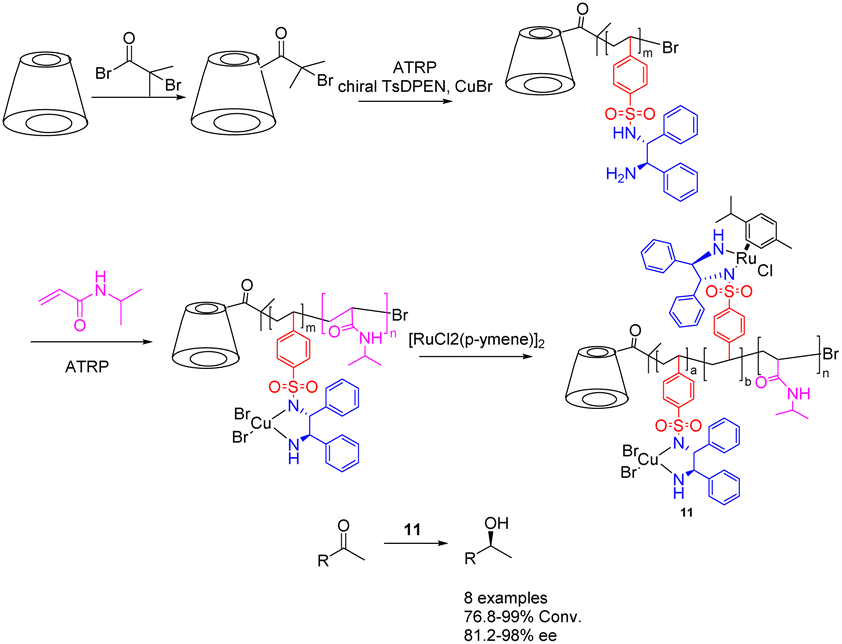 | ||
| Scheme 11 Synthesis of star polymer supported metal Ru and chiral diamines and their application in asymmetric catalytic hydrogenation of ketones. | ||
In 2023, Song et al. conducted a synthesis of bisoxazolinegrafted amino acid polymers 12 (0.03 mmol), which were utilized as recyclable chiral catalyst ligands for copper catalyzed asymmetric Henry reaction (Scheme 12).51 The observed reaction yields ranged from 67% to 95%, while the ee value ranged from 50% to 90%. The polymer-supported catalyst 12 demonstrated a notable recovery efficiency, allowing for at least 7 cycles of recycling without requiring reactivation. Furthermore, it exhibited commendable catalytic performance in the gram-scale reaction.
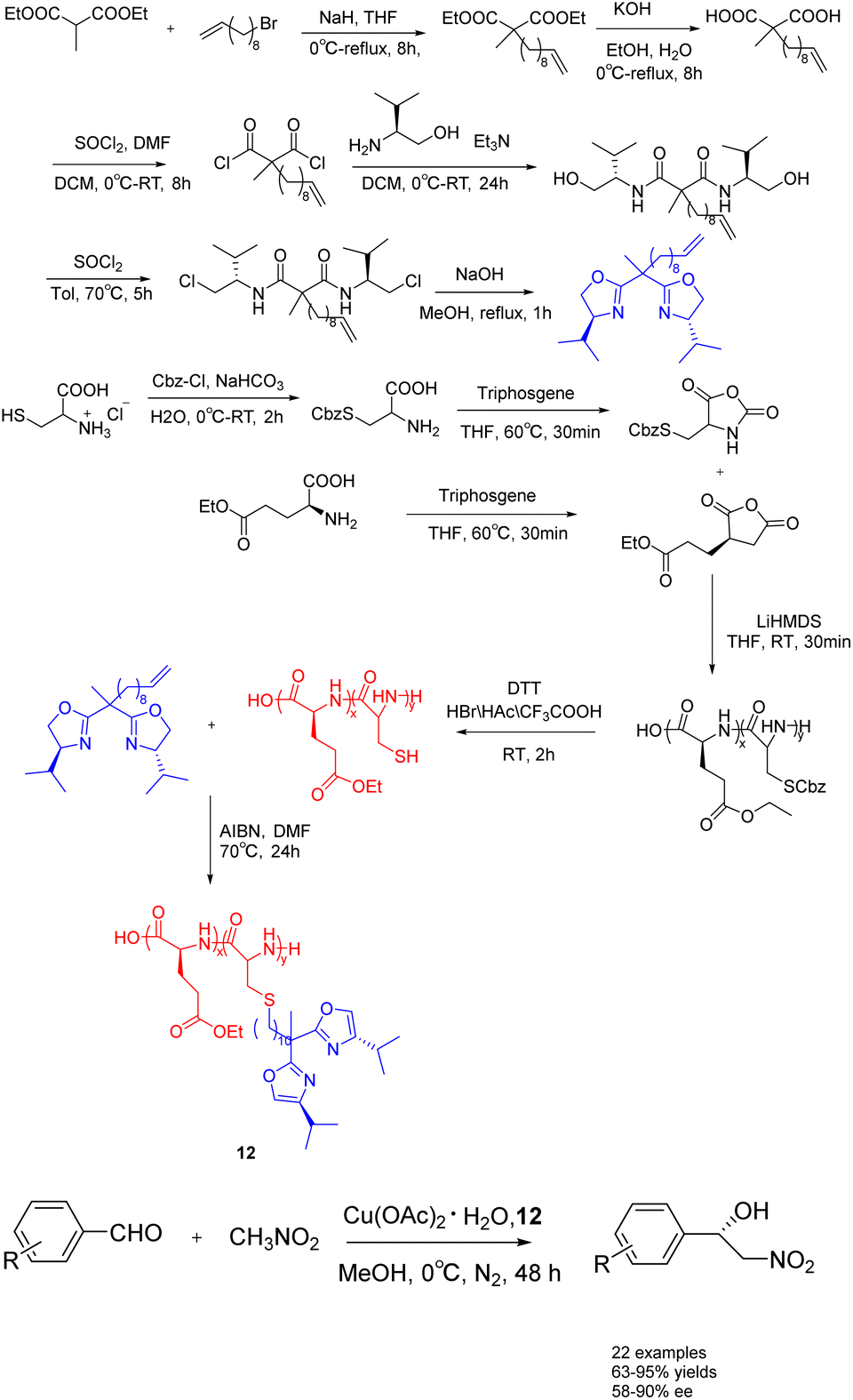 | ||
| Scheme 12 Synthesis of bisoxazoline amino acid polymers and their application in copper catalyzed asymmetric Henry reaction. | ||
In 2022, Wu et al. published a study detailing the utilization of atom transfer radical polymerization to graft diverse proline organic catalysts 13 (0.019 mmol) onto protein scaffolds, resulting in the synthesis of artificial multienzymes (Scheme 13).52 The efficacy of the artificial multienzyme as an aldolase mimic for facilitating asymmetric aldol reactions in the presence of water was confirmed, yielding a yields of 33% and ee of 94%. Unfortunately, this article has not tested the recycling situation.
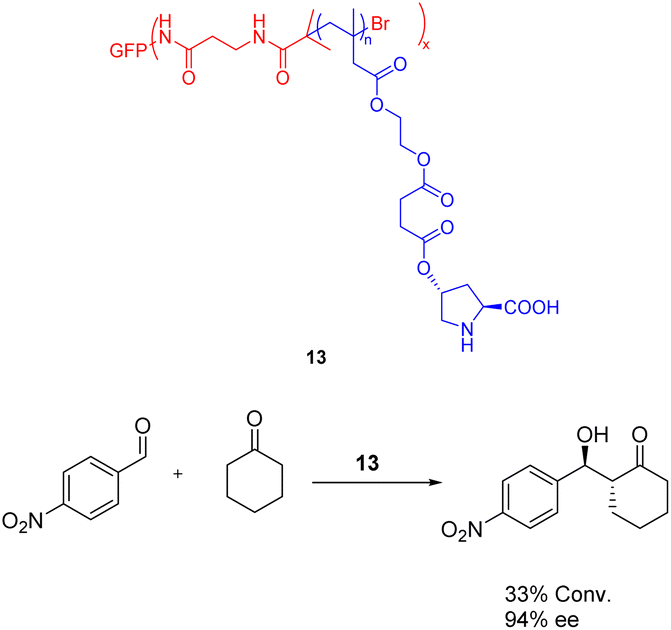 | ||
| Scheme 13 The utilization of proline artificial multienzyme in the asymmetric aldol reaction involving p-nitrobenzaldehyde and cyclohexanone. | ||
The branched polymers exhibit favorable rheological properties, facile processability, low viscosity, desirable solubility, thermal stability, and chemical reactivity. However, their extensive steric hindrance, loose molecular packing, and limited intermolecular chain entanglement result in weak intermolecular forces and hindered crystallization.
3 Insoluble carrier supported catalyst
Insoluble carriers such as Merrifield resin,53 porous organic polymer,54 zeolite,55 SiO2,56,57 and diatomite58 are commonly utilized in multiphase reactions with two-phase separation. This approach allows for easy recovery of the carriers through filtration post-reaction, followed by simple washing and recycling, owing to their solubility characteristics. The successful incorporation of chiral functionalities into porous materials relies on two key factors: (a) the site isolation effects and (b) the constraints imposed by rigid frames.59 For the insoluble supported catalyst, there are two ways of loading the catalyst. One way of loading is to adsorb or coat the catalyst, in which unstable adsorption or incomplete encapsulation may cause catalyst leakage. Another way of loading is the covalent catalysts with polymer. In this case, the covalent may be not stable after multiple cycles of use or high-temperature condition, which may lead to the catalyst leakage.3.1 Insoluble organic polymers
In 2019, Itsuno et al. conducted a study wherein they synthesized chiral hyperbranched polymers 14 (5 mol%) containing cinchona alkaloids through Mizoroki–Heck (MH) coupling polymerization (Scheme 14).61 The resulting polymer demonstrated remarkable catalytic activity in the asymmetric Michael reaction. The reaction between β-ketoester and trans-β-nitros tyrene yielded a product of >99% ee. The chiral HBPs exhibited a robust three-dimensional network structure. Among them, chiral HBP 14a exhibited exceptional reaction efficacy and could be reused up to 6 times, maintaining an 84% yields and >99% ee. After conditional screening, the structural differences between iodides 14-1, 14-2 and 14-3 have little effect on the catalytic activity of their corresponding chiral HBP. The catalyst 14a with a skeleton of 14-3 has the best catalytic effect.
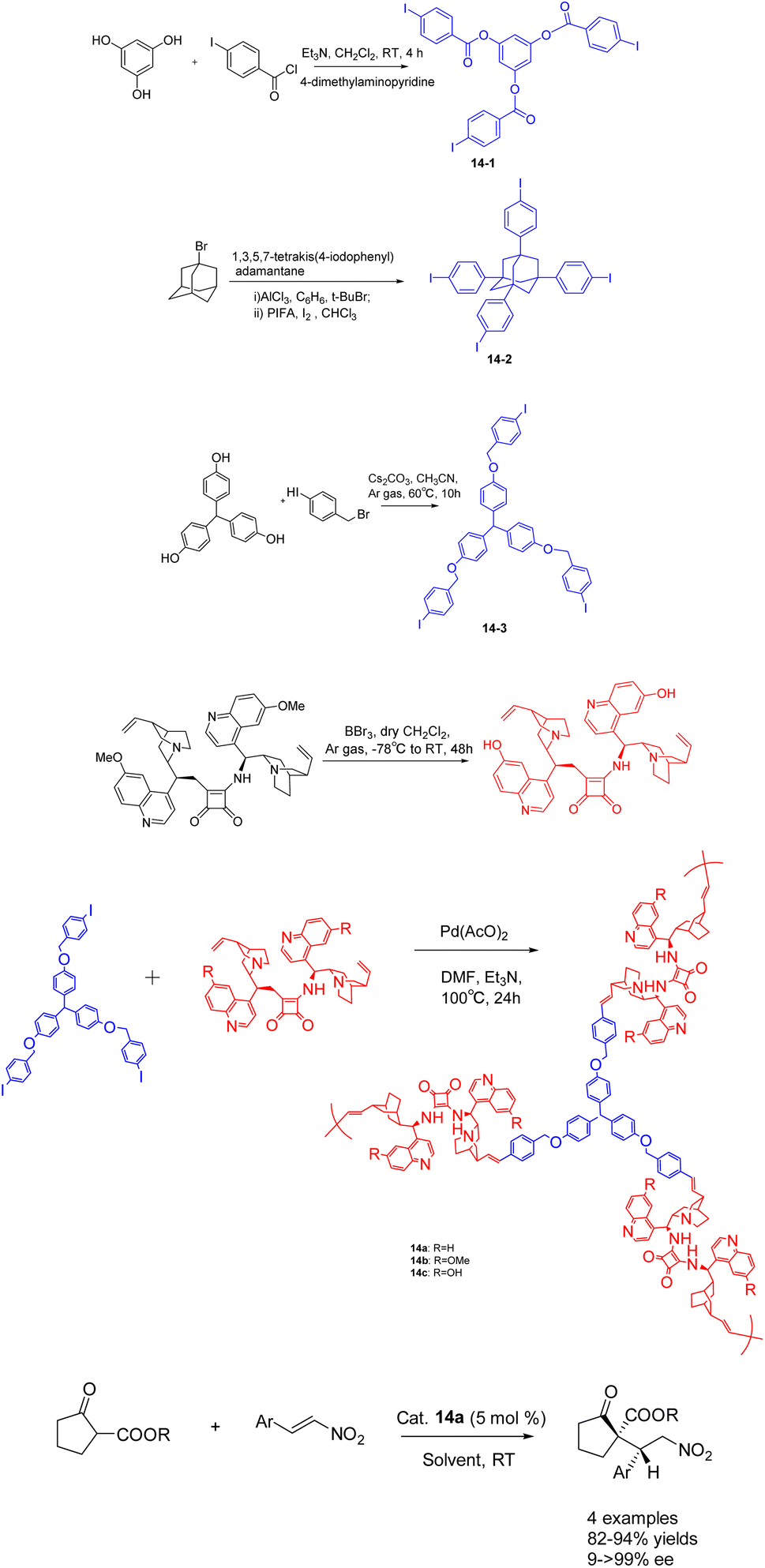 | ||
| Scheme 14 Synthesis of chiral hyperbranched polymers of cinchona alkaloids and their application in asymmetric Michael reaction between β-keto esters and β-nitrostyrene. | ||
In 2020, Itsuno et al. published a study wherein they synthesized chiral HBPs 15 (5 mol%) with cinchonaamide groups using the same method (Scheme 15).62 The researchers successfully employed the heterogeneous catalyst in the asymmetric Michael addition reaction of 2-oxocyclopentanecarboxylic acid methyl ester and trans-β-nitrostyrene, demonstrating remarkable diastereoselectivity and ee. It is worth noting that the catalytic effect of the heterogeneous catalyst is better than that of the unsupported catalyst, because the complex branch structure of the heterogeneous catalyst is well dispersed in the solution. In addition, the HBP catalyst can be easily recovered from the reaction mixture by filtration because of its large molecular weight, difficult to dissolve, and can only swell. After repeated use for 4 times, there is still 86% yields and 99% ee.
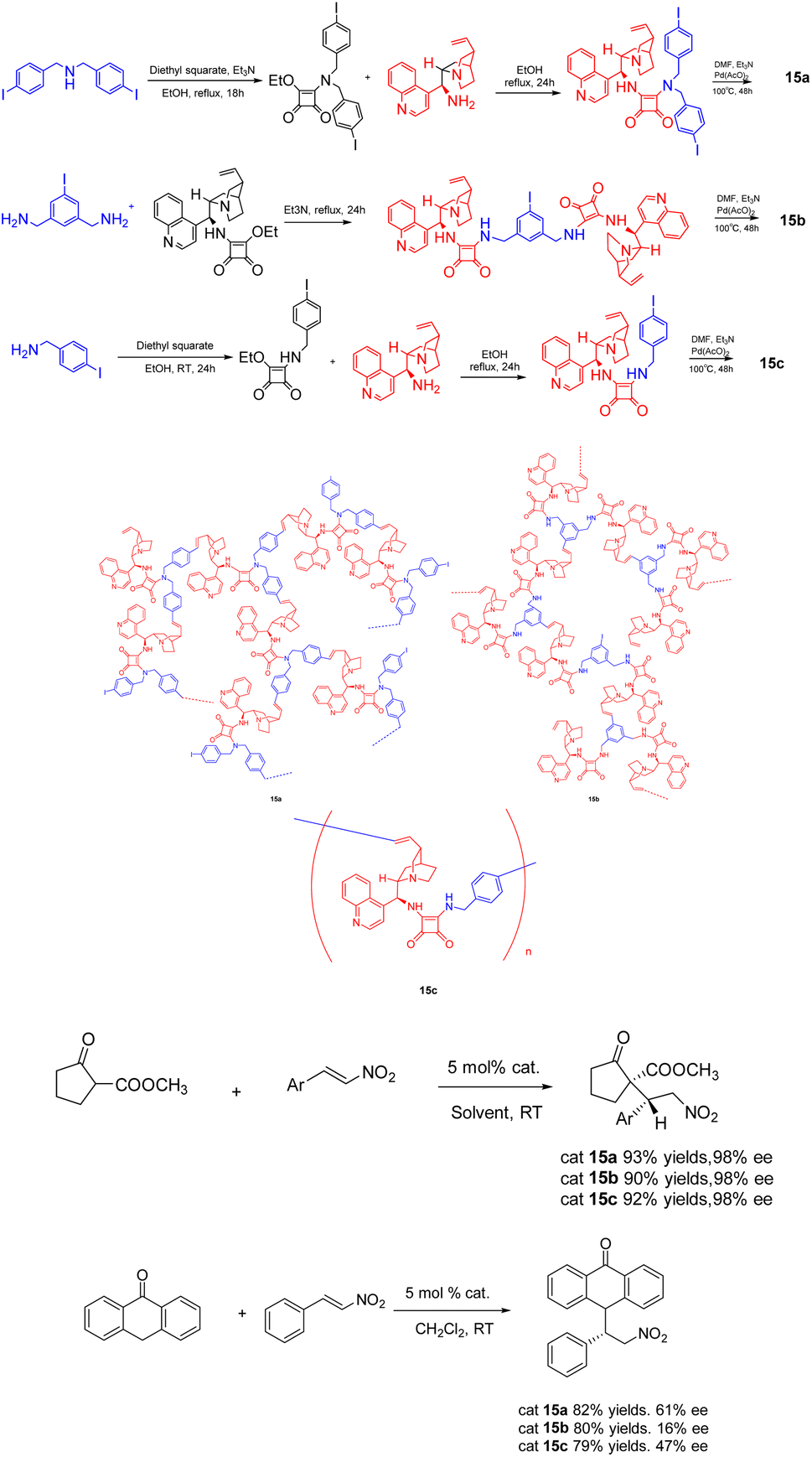 | ||
| Scheme 15 Synthesis of chiral hyperbranched polymers functionalized with cinchona square amide group and their application in asymmetric Michael addition reaction. | ||
Furthermore, in 2023, Öztürk et al. documented the eco-friendly synthesis of a range of asymmetric dihydropyrimidinones through the asymmetric Biginelli reaction, employing sulfo-functionalized hyperbranched polylysine 16 (0.03 g/1 mmol) as a heterogeneous catalyst (Scheme 16).63 The inclusion of minute cavities within the chiral heterogeneous catalyst's structure facilitates efficient asymmetric induction during the production of chiral compounds. The reaction was conducted in the absence of solvent at a temperature of 70 °C, resulting in the synthesis of the desired product of 83–97% yields and 70–98% ee. The catalyst exhibited remarkable stability, as it could be employed for a minimum of five successive reactions without experiencing a substantial decline in activity or chiral induction. Merely a marginal decrease of 7% in yields was observed following 5 repeated reactions. The reaction, facilitated by the catalyst, offers significant benefits such as straightforward post-treatment, high yields and ee.
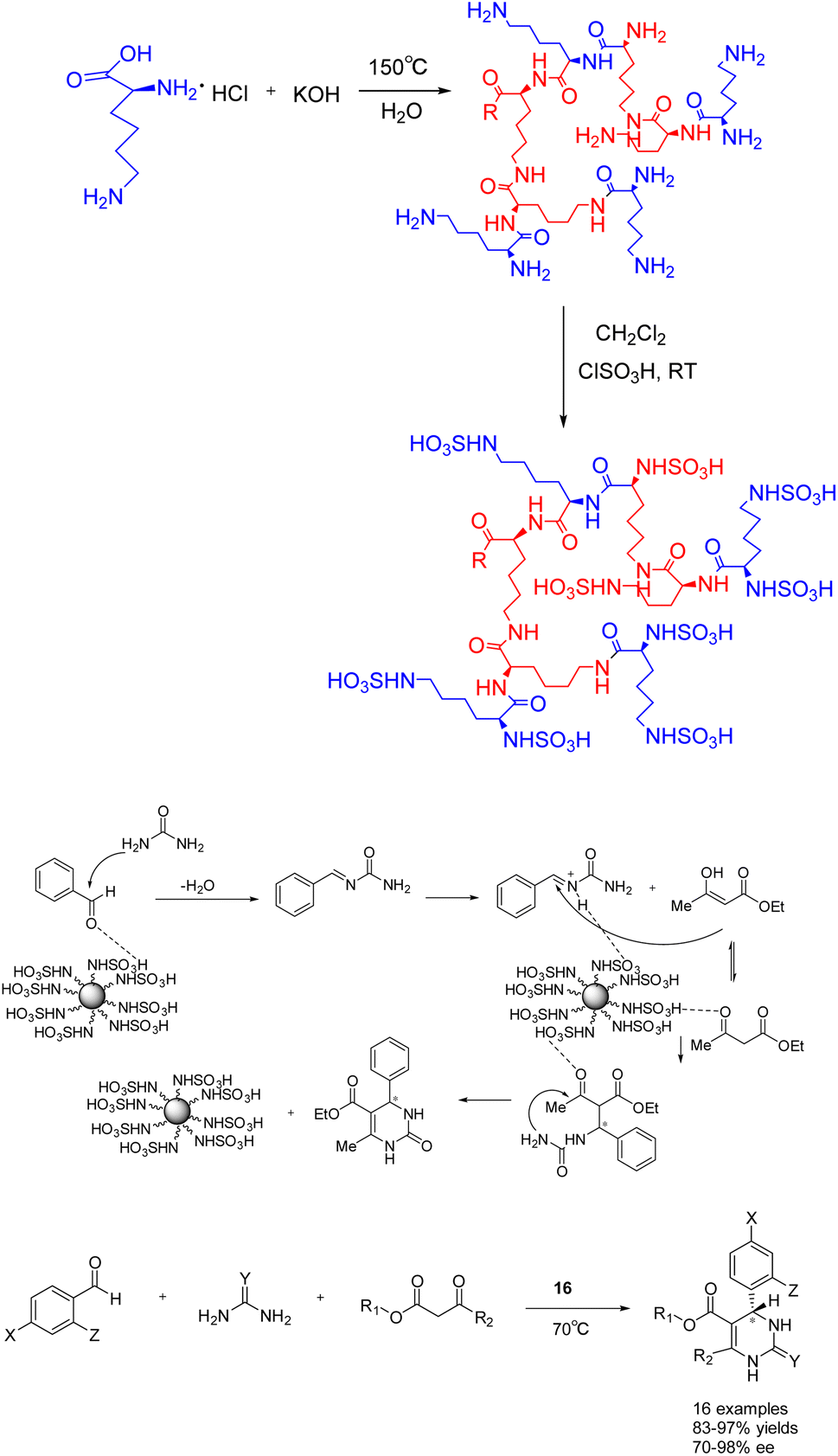 | ||
| Scheme 16 Synthesis of sulfonated hyperbranched polylysine in asymmetric Biginelli reaction and its mechanism. | ||
There exist three primary approaches for the synthesis of hyperbranched polymers: the high temperature synthesis method, which enables the incorporation of branched chains into the polymer, resulting in enhanced molecular weight and structural stability. This method offers the advantage of simplicity and expediency; however, it is accompanied by the drawback of generating a substantial quantity of waste. The second method involves copolymerization, wherein hyperbranched polymers are formed through the copolymerization of two distinct polymers. One advantage of employing this method lies in its ability to achieve a reduced waste yields. However, a corresponding disadvantage is the necessity for additional processes and a prolonged duration. Another method, known as the branched chain synthesis method, involves integrating the branched chain into the primary chain of the polymer to generate a hyperbranched polymer. This method offers the advantage of effectively altering the polymer's performance. Nonetheless, a drawback associated with this approach is the challenge of regulating the increase in molecular weight. Multifunctional hyperbranched polymers possess a multitude of functional groups on their surface. The preservation of these reactive groups results in heightened reactivity and enhanced dispersion in solution, thereby facilitating their utilization in diverse applications. These polymers exhibit a highly branched structure that is often challenging to dissolve, instead undergoing swelling. Synthesis involves the chemical reaction of multiple branched polymers, yielding a complex structure characterized by substantial molecular weight, excellent fluidity, high compressibility, elasticity, and melting point.
3.1.2.1 Covalent organic framework. The COF material, a porous crystalline polymer material formed through covalent bonds and pure organic groups, has garnered significant attention from scientists due to its ordered crystal structure, uniform distribution, exceptional porosity, and adjustable structure. Nevertheless, the construction of functional COFs, particularly chiral covalent organic frameworks (CCOFs), poses considerable challenges. The simultaneous consideration of asymmetry, porosity, and crystallinity is imperative in their construction. The stability, porosity, and crystallinity are significant factors in the development of efficient heterogeneous asymmetric organic catalysts.70,71 Two primary strategies are typically employed for the construction of such catalysts. The first strategy involves integrating asymmetric organic catalysts into the support after synthesis, although this approach often results in decreased loading and uneven distribution of the catalysts. The second strategy entails the direct construction of heterogeneous asymmetric organic catalysts using functional building blocks. Despite the complexity of the preparation process, the precise control of the environment of chiral catalytic sites has been demonstrated.72
In a study published in 2019, Cui et al. documented the synthesis of compounds 17a (10 mol%) and 17b using chiral organic catalysts. The author emphasizes the advantageous advantages of covalent organic frameworks (CCOFs), which have clear one-dimensional channels and periodic chiral organic catalytic sites. These features enable CCOFs to exert effective control over the chiral environment of organic catalytic sites (Scheme 17).73 The heterogeneous catalyst was utilized for the asymmetric Steglich rearrangement and asymmetric Michael addition reactions, resulting in stereoselectivity and diastereoselectivity values of 84% ee and 86% ee, respectively, along with a 17:1 dr. These values are comparable to or even surpass those achieved by homogeneous and amorphous counterparts. Furthermore, even after undergoing 5 cycles, the product still exhibited a yields of >81% and an ee of 80% under the catalysis of 17a, while under the catalysis of 17b, the product maintained a yields of >90% and an ee range of 81–86%. Remarkably, the catalytic activity and ee remained unaltered throughout the cycles.
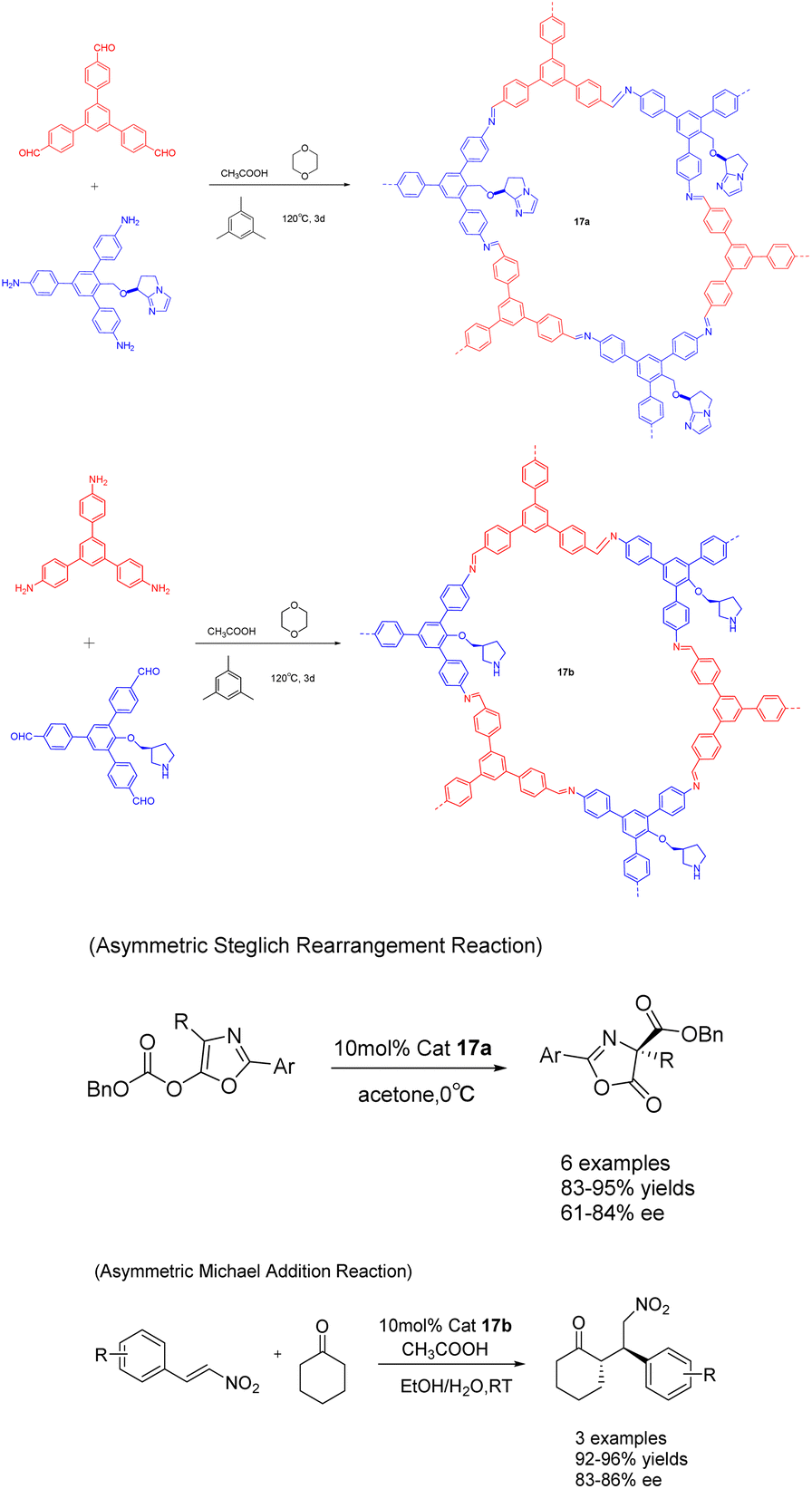 | ||
| Scheme 17 The synthesis of chiral proline covalent organic scaffolds is applied to asymmetric Steglich rearrangement and asymmetric Michael addition reactions. | ||
In 2020, Dong et al. conducted a study wherein they prepared CCOFs 18 through the asymmetric polymerization of prochiral monomers (Scheme 18).74 The resulting propargylamine-linked chiral heterogeneous catalyst 18 (3.3 mol%) was subsequently employed in the asymmetric Michael addition reaction. The researchers observed that the catalyst exhibited favorable surface area, porosity, and stability, enabling effective regulation of the chiral environment within the organic catalytic sites. After subjecting the catalyst 18 to 10 cycles of recycling, the yields and ee value remained at 99%, thereby demonstrating the catalyst's remarkable stability. The introduction of catalytic asymmetric polymerization as a concept may potentially pave the way for the creation of CCOFs that were previously unattainable through conventional CCOF synthesis approaches.
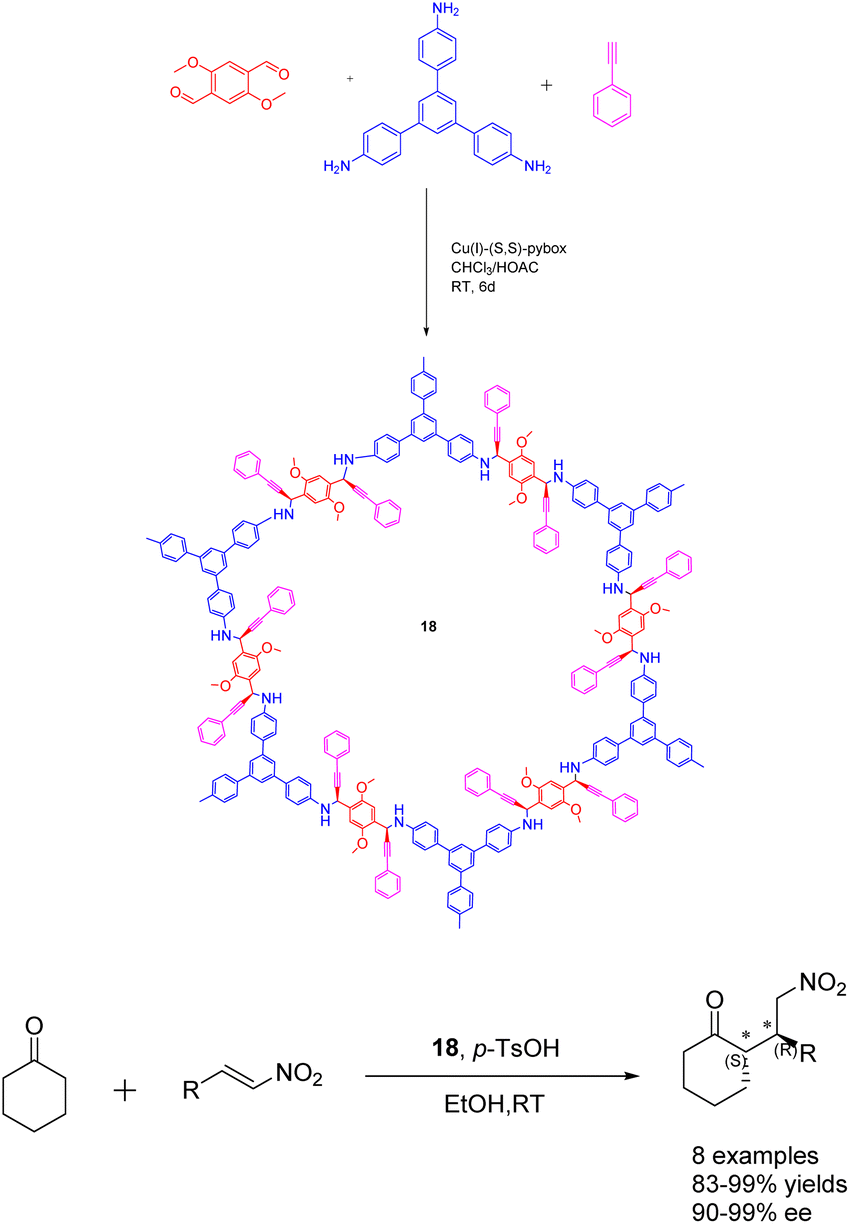 | ||
| Scheme 18 Synthesis of chiral propargylamine covalent organic frameworks and their application in asymmetric Michael addition reaction. | ||
Schiff base condensation, being a common reversible organic reaction, serves as the predominant method for synthesizing both COFs and CCOFs.75,76 Nevertheless, the existing literature demonstrates that CCOFs obtained through Schiff base condensation are typically produced via direct polymerization of chiral or achiral monomers in the presence of chiral additives, utilizing harsh solvothermal reaction conditions.77 The aforementioned stringent and burdensome protocols pose significant obstacles to the efficient mass production of CCOF derived from Schiff base condensation. In 2014, the Cid group introduced a novel amino catalyzed approach, wherein the electrophilic imine intermediate generated by the secondary amine catalyst and the aldehyde substrate can readily undergo reaction with diverse primary amines under mild conditions, yielding the corresponding molecular aldimines with high efficacy.78
In 2022, Dong et al. published a study on the synthesis of a chiral covalent organic framework 19 of β-ketoenamines using an amino catalyzed method, conducted under ambient conditions (Scheme 19).79 This approach demonstrates significant advantages over the conventional acid-catalyzed solvothermal method, particularly in terms of its environmentally friendly synthesis. A series of β-ketoenamine CCOFs 19 (3.0 mol%), derived from tris (N-salicylidene amine), were synthesized through the direct reaction of prochiral aldehydes and primary amine monomers using a chiral 2-methylpyrrolidine catalyst. The resulting β-ketoenamine CCOFs 19 can be further metallized using a solid-state coordination method. Specifically, Cu II 19a exhibits remarkable catalytic activity and ee, effectively promoting the asymmetric A3 coupling reaction. Even after 10 cycles, Cu II 19a maintains its catalytic activity and ee, yielding a 92% yields and 93% ee.
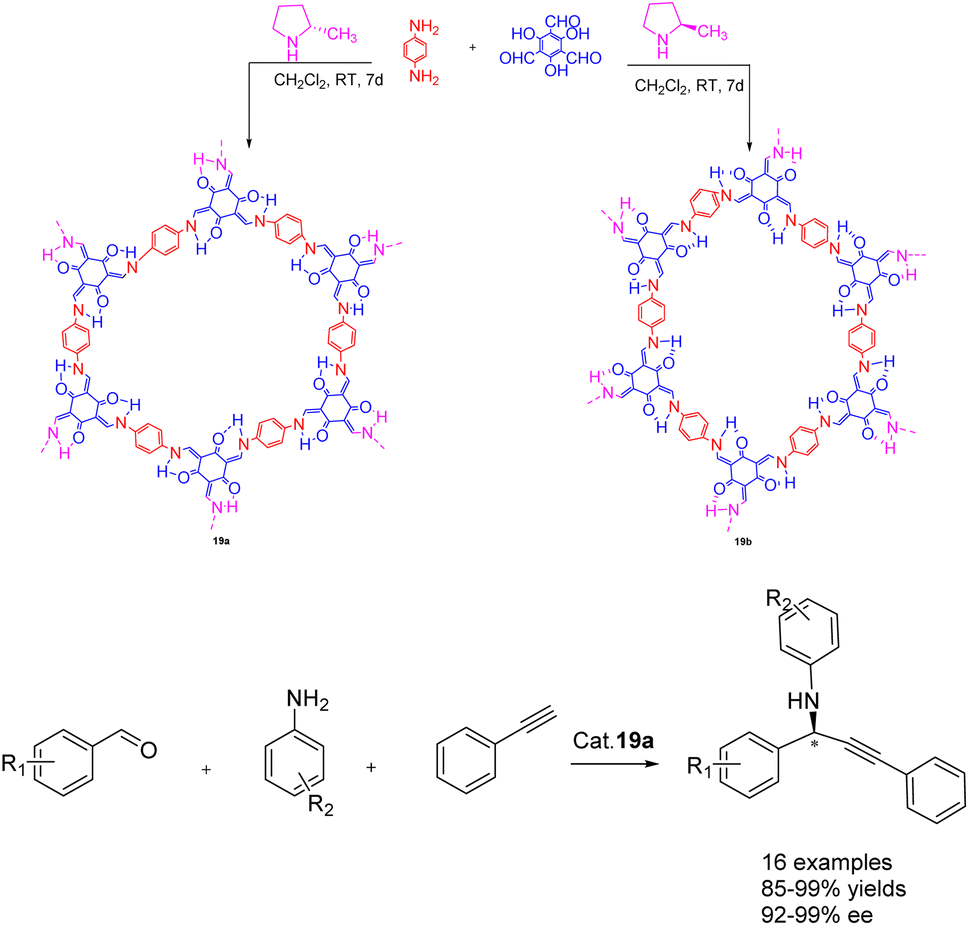 | ||
| Scheme 19 Synthesis of β-ketoamine chiral covalent organic framework and its application in asymmetric A3 coupling. | ||
However, the introduction of sizable functional groups onto the pore wall typically results in a notable reduction in the crystallinity and chemical stability of covalent organic frameworks (COFs), thereby constraining their practical utility. In this scenario, the efficacy of two-dimensional COFs surpasses that of their one-dimensional counterparts. Among the diverse array of two-dimensional COFs, imine-linked COFs exhibit stability across a broad spectrum of solvents and aqueous solutions, while also accommodating a wide pH range.80
In their 2017 publication, Cui et al. presented a multivariable approach for synthesizing chiral covalent organic frameworks (CCOFs) suitable for asymmetric catalysis (Scheme 20).81 The incorporation of an organic catalyst onto the channel wall in a periodic manner significantly impacts the chemical stability and crystallinity of the CCOFs. By manipulating the three-component condensation system, the authors successfully achieved the preparation of three distinct stacking modes in the two-dimensional porous CCOFs series 20 (20 mol%). In comparison to binary CCOFs, ternary CCOFs exhibit enhanced crystallinity and stability. In demanding environments, ternary CCOFs demonstrate efficacy as heterogeneous catalysts for asymmetric aminooxygenation, Aldol reaction, and Diels–Alder reaction, with their stereoselectivity and diastereoselectivity potentially surpassing that of homogeneous analogues. Following 5 cycles, catalyst 20b′ achieved a 68% yields and a 90% ee value in the asymmetric aminooxygenation reaction. The 20c′ catalyst demonstrated a yields of 89% and an ee value of 89% in the asymmetric Aldol reaction, while the catalyst 20d′ achieved a yields of 75% and an ee value of 82% in the asymmetric Diels–Alder reaction. These findings not only present a novel approach for synthesizing CCOFs, but also offer the potential for modulating the crystallinity, stability, and performance of CCOFs.
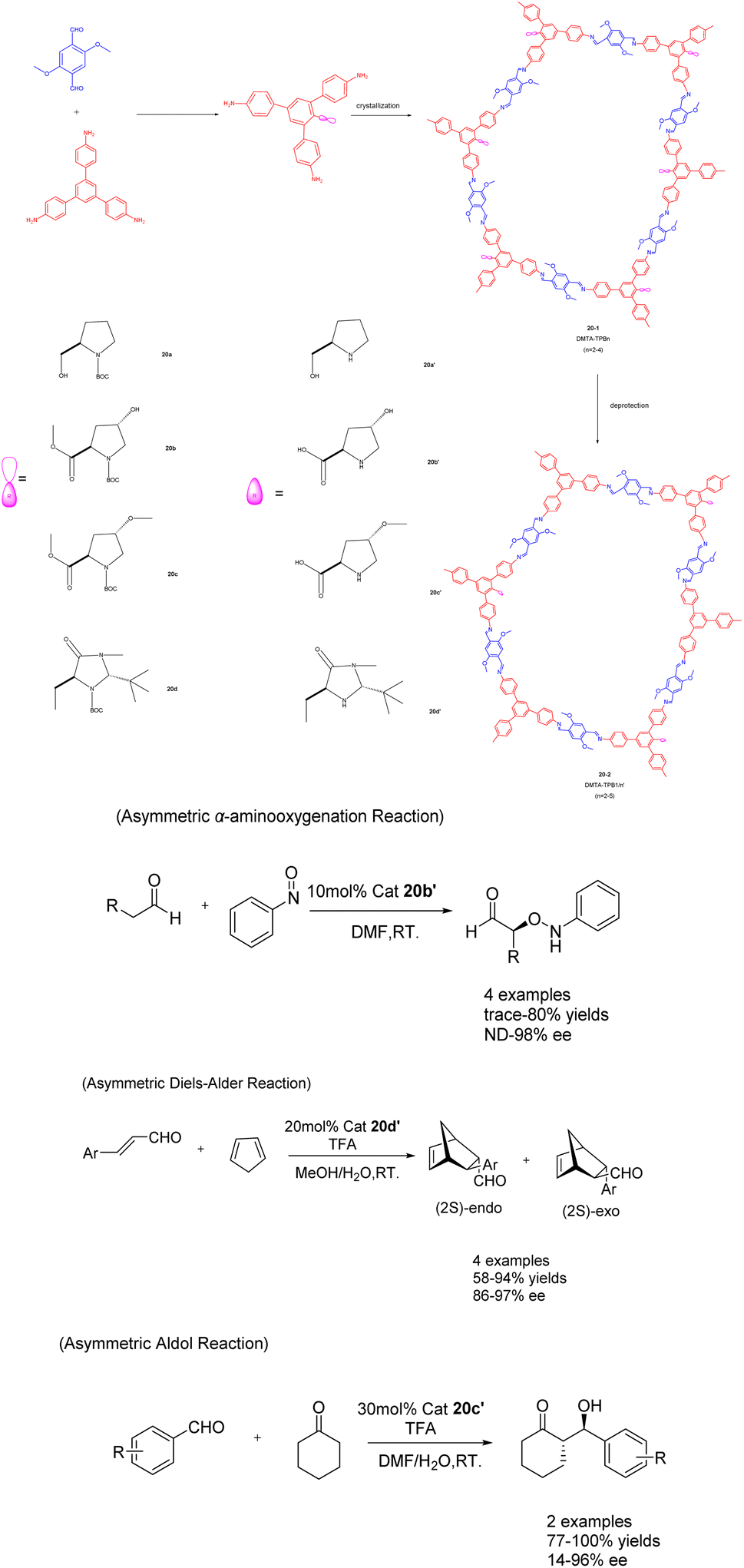 | ||
| Scheme 20 Synthesis and application of ternary covalent organic frameworks (CCOFs) in asymmetric reactions. | ||
The construction of chiral covalent organic frameworks (CCOFs) remains a significant challenge due to the disparity between asymmetry and crystallinity. Additionally, COFs encounter the obstacle of limited stability in harsh conditions, thereby restricting their practical application.82
In 2017, Cui et al. conducted a study on the imine condensation reaction between chiral 1,2-diaminocyclohexane and 3-symmetric trialkyl aldehydes with 1 or 0 3-tert-butyl groups. The purpose of this study was to synthesize two chiral COFs 21 (5 mol%) under metal orientation (Scheme 21).83 Further investigations revealed that Zn (salen)-based CCOFs exhibit a two-dimensional hexagonal grid network with AA stacking. Moreover, the chemical stability of COFs doped with tert-butyl on the pore wall was found to be significantly enhanced under both acidic (1 M HCl) and alkaline (9 M NaOH) conditions, when compared to non-alkylated analogues. The Zn (salen) module within the CCOFs is substituted with the synthesized metal to incorporate the multi-metal into the framework. These substituted CCOFs exhibit notable crystallinity and porosity, rendering them suitable as effective and reusable heterogeneous catalysts for various reactions including aldehyde asymmetric cyanation, Diels–Alder, olefin epoxidation, epoxide ring-opening, and related chain reactions, achieving ee values as high as 97%. After 5 cycles, a conversion rate of at least 69% and an ee value of 81% can still be achieved.
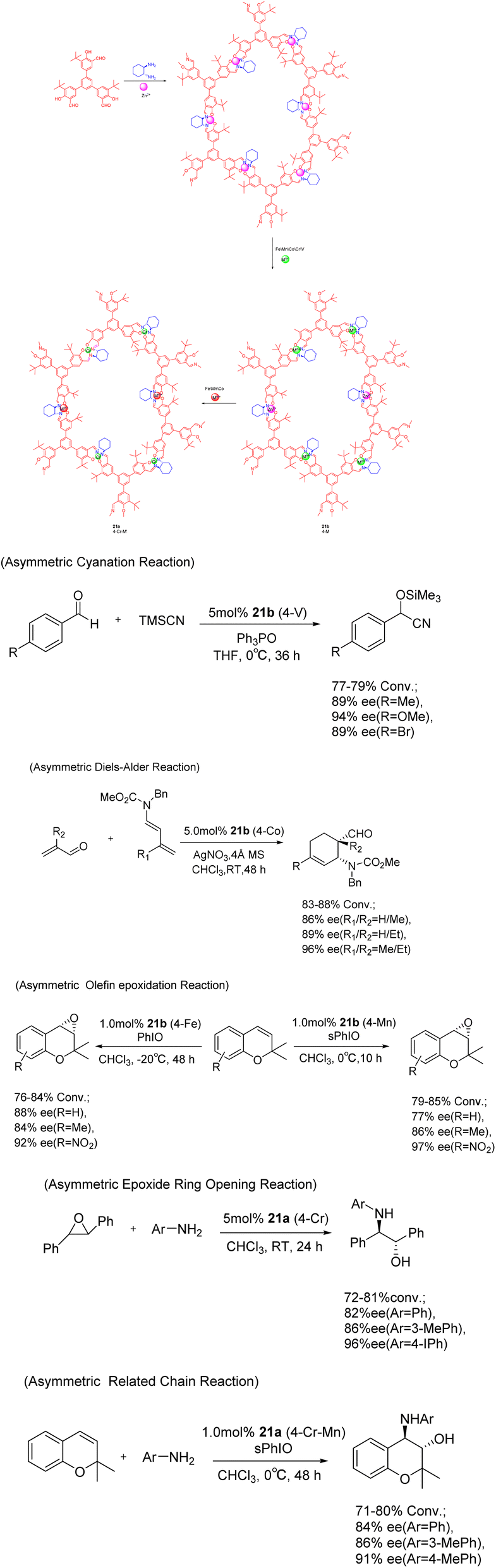 | ||
| Scheme 21 Chiral iminyl covalent organic framework was synthesized and used for asymmetric reactions. | ||
The COF material exhibits a well organized crystal structure over long distances, accompanied by a relatively uniform distribution and size of pores.84 The COF's advantageous attributes, including thermal stability, low density, extensive surface area, multidimensionality, adjustable structure, and exceptional porosity, contribute to its ideal properties. Simultaneously, the integration of a substantial functional group with the COFs material typically induces a notable reduction in both crystallinity and chemical stability, thereby compromising the structural integrity of the COFs framework. Moreover, the solvothermal synthesis technique is intricate and entails harsh conditions, rendering it inadequate for meeting the substantial industrial demand. COF finds extensive application in the realms of energy, drug delivery, and catalysis.
3.1.2.2 Conjugated microporous polymers. The CMP material is a porous organic polymer characterized by a π-conjugated skeleton. Its overall conjugated and highly cross-linked interior confers several advantages, including a large specific surface area, high stability, adjustable pore size, and a diverse range of monomers.85,86 However, it should be noted that this material is amorphous and lacks long-range order. Nonetheless, CMPs hold significant potential for various applications such as catalysis, energy storage, fluorescence sensing, and separation.87
In their seminal work published in 2022, Zang et al. demonstrated the utilization of CCMPs as carriers for the incorporation of chiral amide groups, leading to the synthesis of chiral conjugated microporous polymers containing amide functionalities. The resulting heterogeneous catalysts were subsequently employed in asymmetric catalysis (Scheme 22).88 This approach capitalizes on the remarkable stability of chiral amides and their facile coordination with transition metals, enabling their successful integration into conjugated microporous polymers possessing both stability and porosity. The Pd/CCMPs heterogeneous catalyst was prepared through the coordination of the chiral amide group of CCMPs with Pd. When Pd/22c (10wt%) is used as a catalyst, it exhibits good catalytic activity and enantioselectivity when it was applied to asymmetric Henry reactions under mild conditions. No significant decrease was observed after 3 cycles (yield of 80%, ee of 82%). An increasing the number of cycles, the Pd content of Pd/CCMP decreased from 2.4 atomic% to 1.88 atomic%. The results indicate that the collision between the agitator and the reactor wall during the reaction process may cause the exposed Pd surface to rupture, which may lead to a decrease in product selectivity. However, catalyst is costly, posing challenges for its implementation in large scale production and application. Additionally, the removal of residual transition metals presents a significant difficulty.
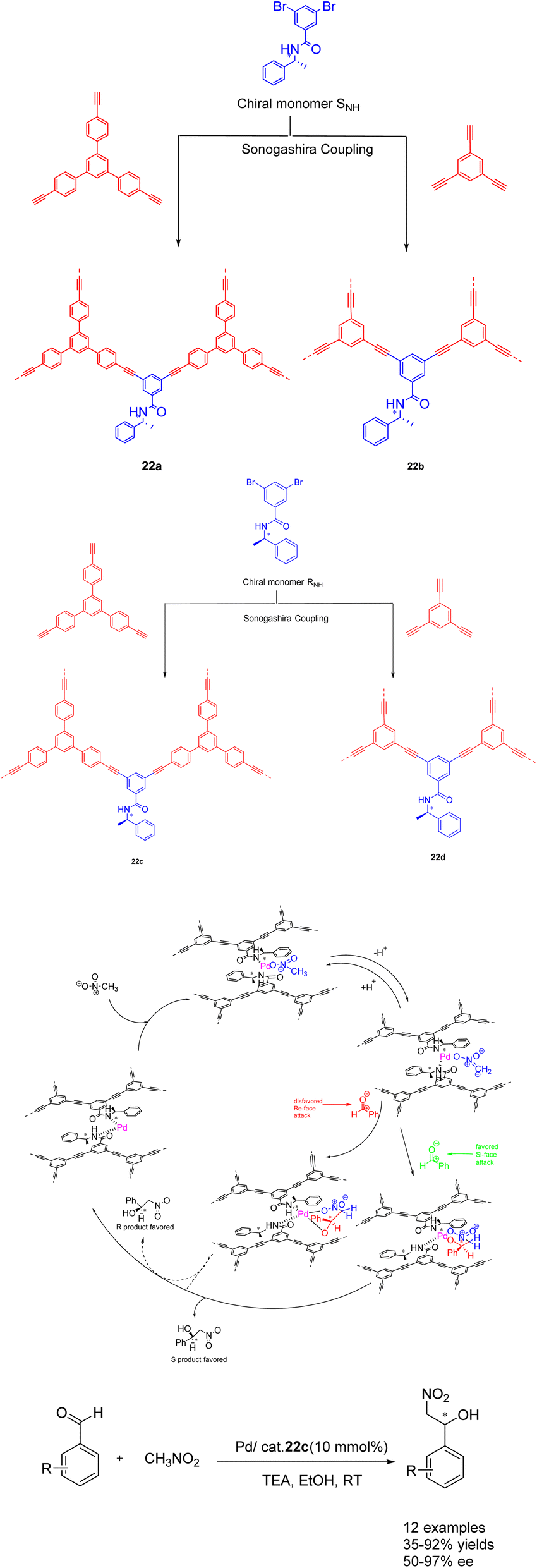 | ||
| Scheme 22 Synthesis of chiral amide conjugated microporous polymers for asymmetric Henry reaction and its mechanism. | ||
The synthesis of conjugated microporous polymers primarily relies on noble metals or rare metals as catalysts for coupling reactions. Nonetheless, the utilization of transition metal-catalyzed coupling polymerization presents certain drawbacks, including costly catalysts, rigorous reaction conditions, and intricate post-treatment procedures. Additionally, the complete elimination of residual metals within the polymer framework poses a challenge, thereby restricting its application in specific domains.
In 2018, Turner mentioned in his review that the concept of HCP material was first proposed by Davankov et al.89 Typically, HCP materials are synthesized through cross-linking reactions, such as the Friedel–Crafts reaction.90 The resultant crosslinked polymer chain exhibits a specific level of stiffness, effectively impeding structural collapse and ensuring the polymer material maintains a permanent pore structure.91 One advantage of HCP material lies in its commendable chemical stability, while a drawback is its tendency to exhibit specific swelling characteristics. The synthesis conditions significantly influence the structure, pore size, and specific surface area of HCP material, posing challenges in their deliberate design and control.92 Moreover, the reaction conditions necessitate the involvement of potent Lewis acids, thereby restricting the incorporation of specific functional groups. The network structure characterized by a shallow cross-linking degree exhibits the ability to undergo softening upon exposure to heat, without undergoing complete melting. Furthermore, the application of an appropriate solvent can induce swelling in the network structure, without causing complete dissolution. On the other hand, the shape structure characterized by a deep cross-linking degree does not exhibit softening upon heating, and it is resistant to swelling by solvents, resulting in the formation of a rigid solid. This structure possesses several advantageous properties, including high porosity, high specific surface area, high adsorption capacity, and high chemical stability.93
The rapid development of hyper-crosslinked polymers (HCPs) can be attributed to their diverse range of monomers, convenient functionalization, favorable synthesis conditions, and cost-effective catalysts and external cross-linking agents.94,95 In a study conducted in 2017, Ding et al. achieved successful synthesis of a novel vinyl-functionalized chiral monomer (S)-5,5′-divinyl-BINAP and incorporated it into two distinct porous organic polymers 23 (Scheme 23).96 In the context of a heterogeneous asymmetric hydroformylation reaction of styrene, a polymer catalyst featuring chiral nano-encapsulated pores was employed. The incorporation of flexible chiral nanocages resulted in a 1.67 fold increase in the ee of Rh/23a (0.0096 mmol) compared to its homogeneous counterparts. Furthermore, the chiral porous organic polymer catalyst, which is based on BINAP, exhibited remarkable swelling capabilities under solvothermal conditions and demonstrated favorable activity and recyclability. After obtaining 94% yields and 59% ee of the product, it was recycled 7 times and achieved a yield of 40% and an ee value of 50%. Consequently, the malleability of active sites within solid catalysts assumes a critical role in enhancing catalytic performance.97 A porous polymer exhibiting exceptional swelling characteristics was synthesized through the polymerization of vinyl-functionalized monomers under solvothermal conditions.98 The expanded polymers possess the attributes of elastic solids rather than liquids, thereby endowing the polymer with remarkably elevated flexibility.99
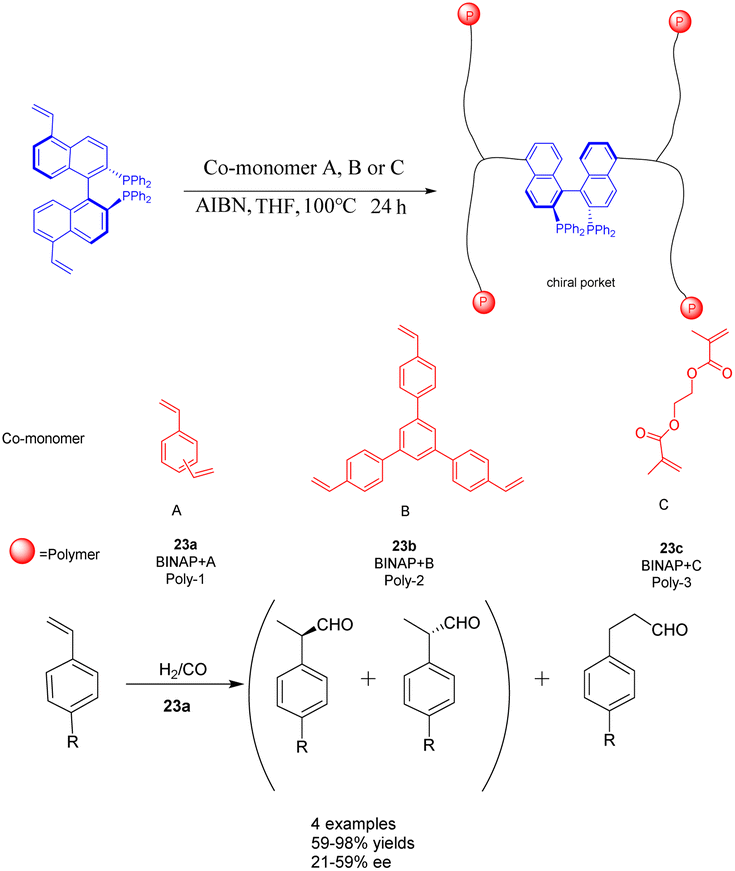 | ||
| Scheme 23 BINAP chiral porous organic polymer was synthesized and applied to the asymmetric hydroformylation of styrene. | ||
In the year 2021, Liu et al. devised a construction approach to directly immobilize axially chiral phosphoric acid within hyper-crosslinked polymers using a one-pot Friedel–Crafts alkylation reaction. Additionally, they fabricated a range of chiral HCPs 24 utilizing BNPPA, which exhibited a substantial surface area, exceptional thermal stability, and chemical stability (Scheme 24).100 The resulting heterogeneous organic catalyst 24e (2 mol%) possessed a certain degree of rigidity, effectively preventing structural collapse, thereby endowing the polymer material with a permanent pore structure, high porosity, good stability, and adjustable catalytic centers. The solid polymer demonstrates remarkable catalytic activity, ee, and recyclability in the asymmetric transfer hydrogenation of benzoxazine, benzothiazine, and benzoxazinone. It possesses durability, ease of separation, and the ability to be recycled multiple times. Following a minimum of 10 recycling cycles, 24e exhibits no substantial decline in catalytic activity (yielding 91–97%) and ee (with an ee of 94–96%). This study presents a novel approach for the fabrication of robust chiral Brønsted acid catalysts with exceptional catalytic efficiency, ee, and reusability. This advancement is advantageous for the development of heterogeneous catalytic porous functional materials at the molecular level.
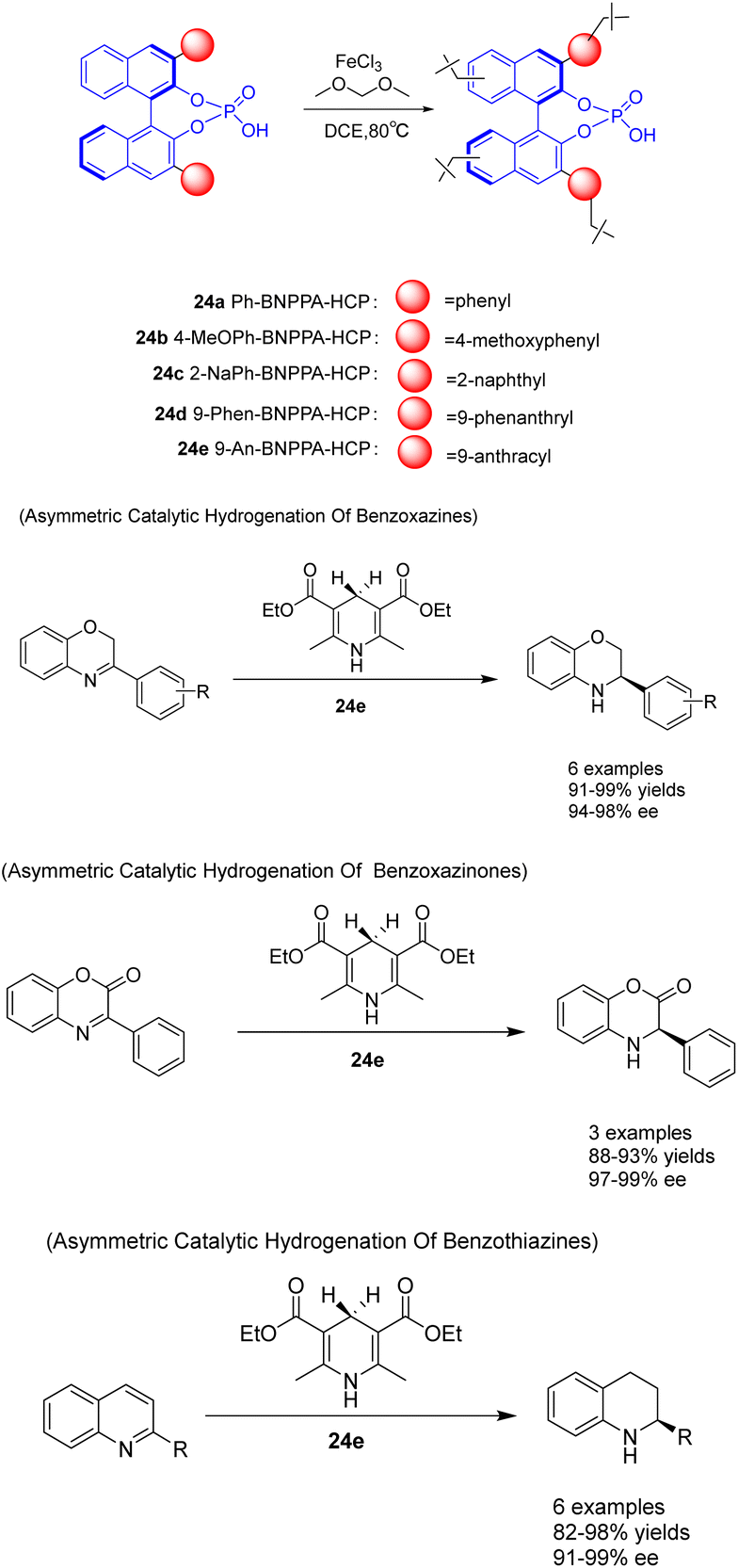 | ||
| Scheme 24 Synthesis and application of chiral phosphate ester hypercrosslinked polymers in asymmetric reactions. | ||
In 2022, You et al. successfully achieved the synthesis of chiral phosphoric acid 25 (5 mol%) through the utilization of a heterogeneous porous BINOL and the ‘weaving’ strategy (Scheme 25).101 By introducing an anthracene group at the 3,3′-position of BINOL, a three-dimensional environment was created, facilitating the formation of highly cross-linked networked PCPAs as controllable connecting units. These PCPAs exhibited remarkable catalytic activity in the asymmetric transfer hydrogenation of 2-phenylquinoline and the dearomatization of β-naphthol with DEAD, resulting in an impressive yields of 98% and a high ee of 99%. The catalyst was successfully employed for 10 consecutive cycles without any substantial decline in its catalytic efficiency. It Can still achieve the product in 98% yields and 96% ee.
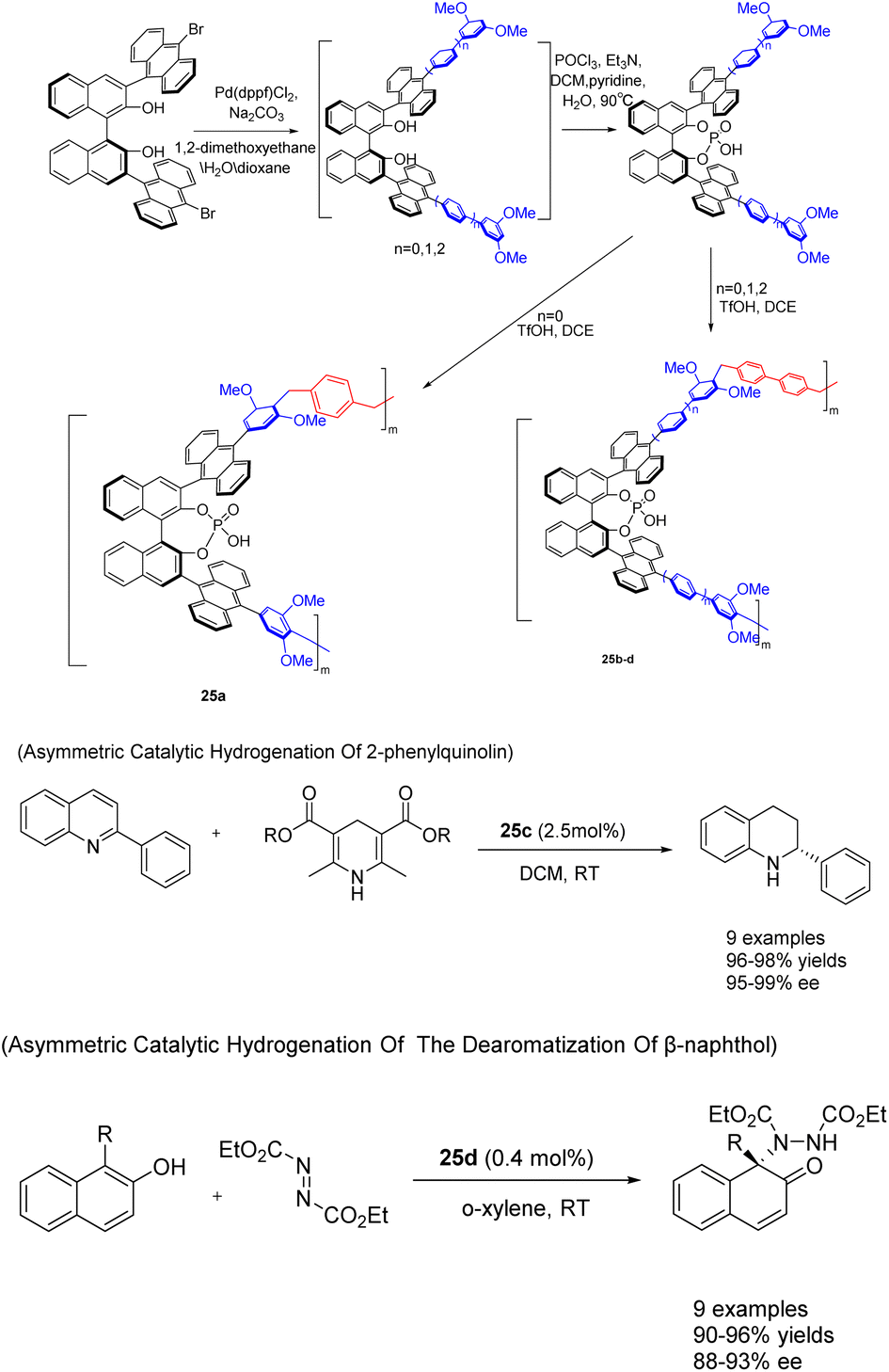 | ||
| Scheme 25 Synthesis and application of porous polymers containing BINOL chiral phosphoric acid in asymmetric reactions. | ||
In the past few years, there has been a growing interest in the construction and development of chiral secondary amine functionalized organic porous networks. These networks have been synthesized using post-synthetic modification strategies or direct incorporation methods.102 In a recent study conducted by Liu et al. in 2023, a heterogeneous chiral porous polymer skeleton, HCP 26 (Scheme 26), was formed by cross-linking L-phenylalanine dipeptide monomer. Subsequently, the HCP 26 catalyst was further modified with L-proline (L-Pro) to achieve a specific surface area of 261.68 m2 g−1.103 The catalytic activity of HCP 26 (6 mg) was demonstrated in the successful facilitation of the asymmetric Michael addition reaction and Mannich reaction. In comparison to the unloaded condition, the reaction time for the Michael addition of cyclohexanone with trans-β-nitrostyrene was reduced from 7 days to 4 days, accompanied by an increase in the ee value from 71% to 97%. This notable enhancement was similarly observed in the asymmetric Mannich reaction. Furthermore, the investigation encompassed an examination of the optimal parameters for the catalytic reaction, including the influence of time, solvent, and catalyst dosage, while also assessing the catalyst's suitability through an analysis of substrate structure. The catalyst exhibited facile recovery through a straightforward filtration process and demonstrated commendable recovery efficacy. Notably, the catalytic activity and ee of the HCP 26 catalyst remained largely unaffected even after undergoing 5 cycles, resulting in yields of 92% and ee of 89%.
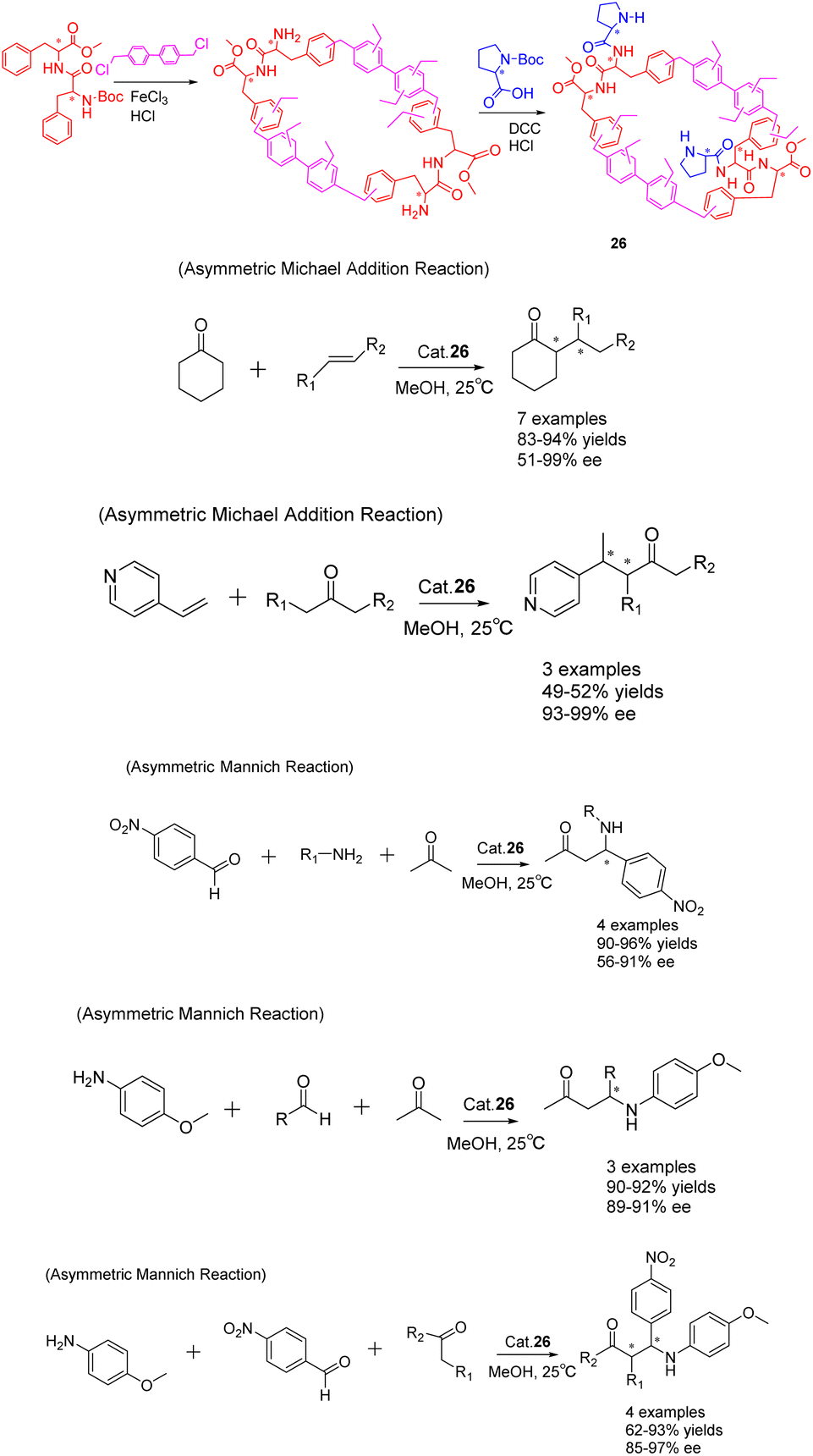 | ||
| Scheme 26 Synthesis of chiral porous polymers containing secondary amines and their applications in asymmetric Michael addition and Mannich reactions. | ||
The formation of hyper-crosslinked polymers (HCPs) does not occur through the compact arrangement of structural units; rather, it involves dissolving or swelling the linear precursor polymer in a benign solvent to achieve full stretching. Subsequently, the polymer is highly cross-linked or extended cross-linked under the influence of a catalyst. Once the solvent is removed, the resulting hyper-crosslinked polymer forms a stable and non-collapsed microporous structure with a significantly high specific surface area. Consequently, the porous characteristics of HCP materials are closely associated with the process of crosslinking, including the choice of crosslinking agent and solvent. There exist two primary approaches for the synthesis of hyper-crosslinked polymers: one involves the crosslinking of linear polymers using crosslinking agents, while the other entails the formation of hyper-crosslinked polymers through self-assembly. Notably, the self-assembly method yields hyper-crosslinked polymers with distinct advantages, including a uniform distribution of pore sizes, adjustable pore dimensions, and exceptional porosity. Consequently, this method exhibits significant potential for diverse applications in adsorption and separation processes.
The composition of the PIM material consists of interconnected covalent bonds between rigid and twisted molecules. The preservation of the inherent pore system is attributed to the ingenious incorporation of curved and rigid links, preventing effective filling of the interior and resulting in the formation of continuous micropores within the depressed region. Notably, the distinctive characteristic of PIM materials lies in their capacity to be transformed into organic microporous membranes for adsorption and separation, as highlighted in ref. 104. One drawback lies in the disordered molecular structure, which results in a broader distribution of pore sizes. Additionally, the presence of ultra micropores and closed pores hinders the increase in specific surface area and poses challenges in accurately controlling pore size.105 Apart from its high porosity and machinability, the versatility of PIM synthesis stands out as a notable advantage, enabling the incorporation of diverse functional groups.106
In the year 2020, Kegnæs et al. presented the initial instance of enantioselective C(sp3)–H functionalization, wherein they synthesized chiral phosphamide porous organic polymers 27 (20 mol%) (Scheme 27).107 The catalyst was generated through the coordination of palladium with polystyrene-supported chiral phosphoramides and was employed for the enantioselective C(sp3)–H functionalization of 3-arylpropionamides, yielding ee values of up to 86%. One advantage of utilizing polystyrene as a carrier lies in its ability to expand within a suitable solvent, thereby creating a homogeneous reaction environment. Simultaneously, the catalyst can be easily separated and reused from the reaction medium, albeit with a gradual decline in ee. After 4 cycles, 73% yields and 54% ee were obtained. One drawback of the catalyst is its limited substrate range, with a more pronounced catalytic effect observed for smaller substrates, while encountering challenges when applied to larger substrates.
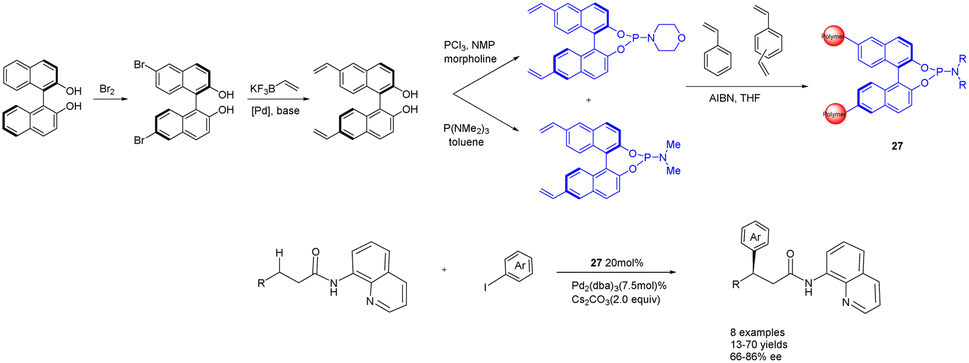 | ||
| Scheme 27 Synthesis of chiral phosphamide porous organic polymer and its application to the asymmetric C(sp3)–H functionalization of 3-aryl propionamide. | ||
PIM exhibits a greater BET surface area,108 thereby resulting in the formation of smaller pores. Consequently, the utilization of PIM as a catalyst yields improved performance with smaller substrates, while encountering challenges with larger substrates. This limitation, identified as a significant drawback,109 can be mitigated by incorporating more adaptable monomers during the synthesis of PIM. This modification enhances the ability of PIM to expand in the presence of solvents, facilitating the creation of larger pores. In the realm of practical applications, inherent microporous polymers have found significant utilization in gas masks, protective clothing, gloves, and air filters. Additionally, appropriately functionalized porous organic polymers (POPs) hold promise in addressing chemical threats. Extensive research has been conducted on persistent organic pollutants, exploring their potential in diverse areas such as energy storage and conversion, chemical catalysis, gas storage and release, and chemical separation.110
3.2 Inorganic polymer
In 2018, Janiak et al. conducted a study wherein they reported the synthesis of a mixed catalyst denoted as catalyst 28 (10 mg). This catalyst was prepared by covalently linking Mn-porphyrin complexes with graphene oxide (GO) nanosheets through propyl bonds. The researchers investigated the catalytic performance of this catalyst in the enantioselective epoxidation of various unfunctionalized olefins using O2 as the oxidant (Scheme 28).115 Notably, when isobutyraldehyde was present, the graphene-supported Mn-porphyrin exhibited significant activity in the epoxidation of unfunctionalized olefins. Furthermore, the catalyst could be easily recovered and reused without any noticeable loss in its catalytic activity and selectivity. After undergoing 5 cycles, the yields remains at approximately 87%, while the ee experiences a reduction to approximately 72%. The optical selectivity of the resulting epoxides ranges from 86% to 100%.
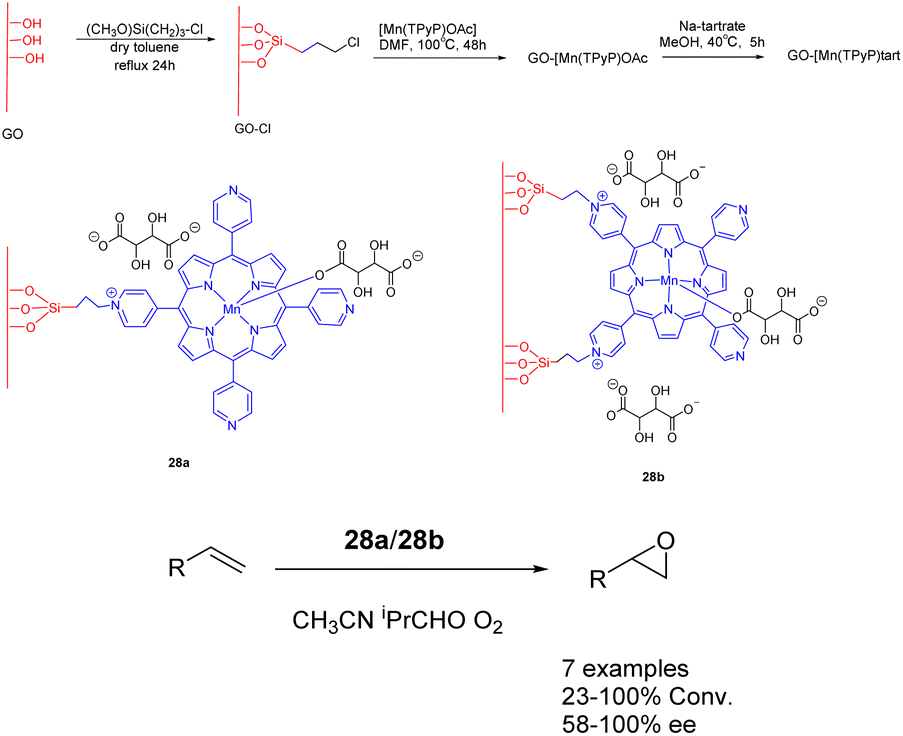 | ||
| Scheme 28 Synthesis of graphene-supported manganese porphyrins and their application to asymmetric epoxidation of olefin. | ||
In 2019, Yan et al. published a study introducing a novel heterogeneous catalyst 29-Ti (Scheme 29), based on graphene oxide.116 The catalyst consists of an amino-terminated BINOL molecule, which is covalently linked to the graphene oxide sheet. The preparation of this graphene oxide catalyst involved the initial oxidation of commercial graphene oxide to nanoporous graphene oxide in an acidic environment. Subsequently, the enantiomer (R) or (S)-NH2-BINOL (covalent) was attached to the nanoporous graphene oxide, followed by treatment of the oxidized 29 (10 mol%) with Ti(OiPr)4. In the asymmetric selective addition reaction of diethylzinc to aromatic aldehydes, the utilization of 29-Ti demonstrated favorable reactivity (99%) and moderate ee (45% ee). Following 4 cycles, the yield of the product is 99%, with an ee of 36%.
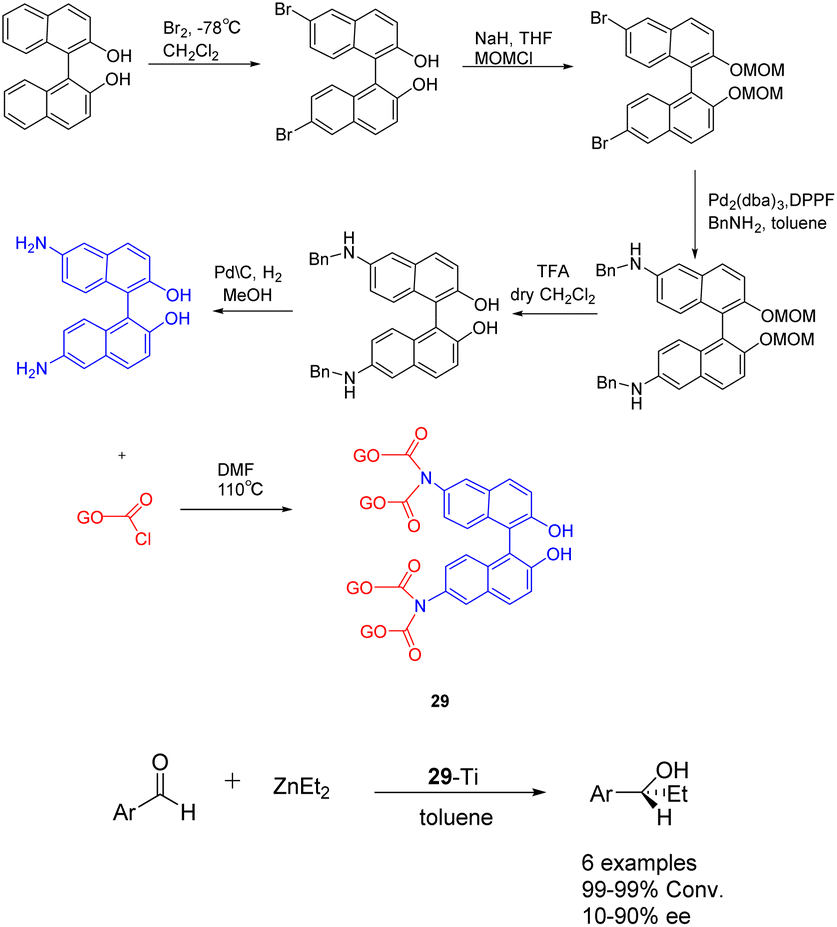 | ||
| Scheme 29 Synthesis of BINOL graphene oxide and its application in asymmetric selective addition of aromatic aldehydes to zinc diethyl. | ||
In their 2021 study, Schulz et al. demonstrated the non-covalent loading of pyridine-labeled Cr-salen complexes onto reduced graphene oxide, resulting in the acquisition of a highly stable heterogeneous catalyst 30 (2 mol%) (Scheme 30).117 This catalyst was successfully employed in the ring-opening reaction of cyclohexene oxide, yielding a yields of 96% and an ee of 68% after 10 cycles. Additionally, the catalyst was utilized in the hetero-Diels–Alder cycloaddition reaction between various aldehydes and Danishefsky diene, achieving a yields of 84% and an ee of 63% after 8 cycles. Notably, the catalyst exhibited recyclability without any discernible loss in activity or ee. This study represents a significant milestone as it marks the inaugural instance of immobilizing a salen derivative onto a carbon surface via a π–π interaction. The successful outcome of this endeavor paves the path for numerous future expansions, owing to the extensive versatility of these complexes in the realm of asymmetric catalysis.
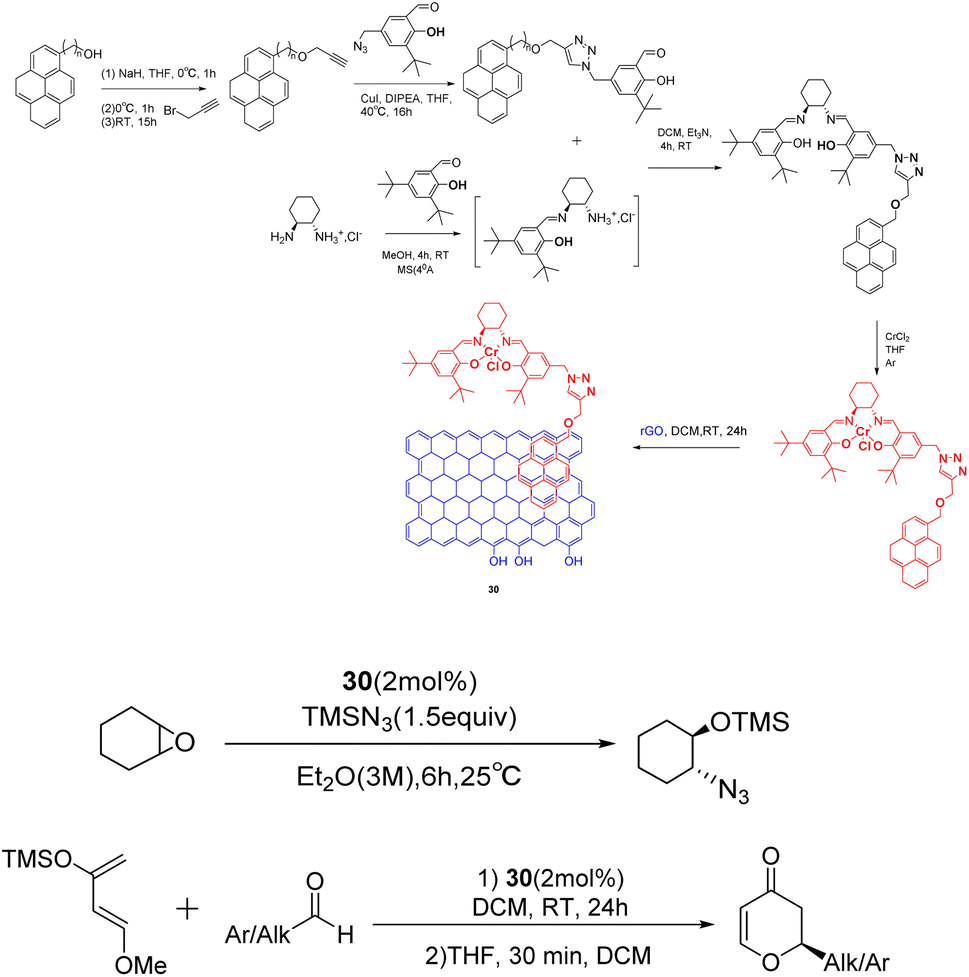 | ||
| Scheme 30 Synthesis and application of pyridine-graphene oxide to ring-opening reactions of cyclohexene oxide and asymmetric Diels–Alder cycloaddition reactions involving aldehydes and Danishefskedienes. | ||
In their 2022 publication, Durmaz et al. documented the synthesis of two newly developed bifunctional carbon catalysts, namely (S, S) 31 and (R, R) 31 (25 mg). These catalysts were created by covalently attaching enantiopure organosilanes containing primary amine and thiourea groups onto the graphene oxide framework through a silane coupling reaction, as depicted in Scheme 31.118 The researchers further investigated the catalytic efficacy of these catalysts in the asymmetric Michael addition reaction involving α, α-disubstituted aldehydes, nitrostyrene, and N-substituted maleimides. Under the influence of L-and D-camphorsulfonic acid, significant yields (reaching up to 85%) and high stereoselectivities (up to 95% ee) were achieved in dichloromethane at ambient temperature. Furthermore, the investigation of catalyst recovery and reutilization proved successful. Even after undergoing 7 cycles of recycling, the product still exhibited a yields of 70% and an ee of 80% ee.
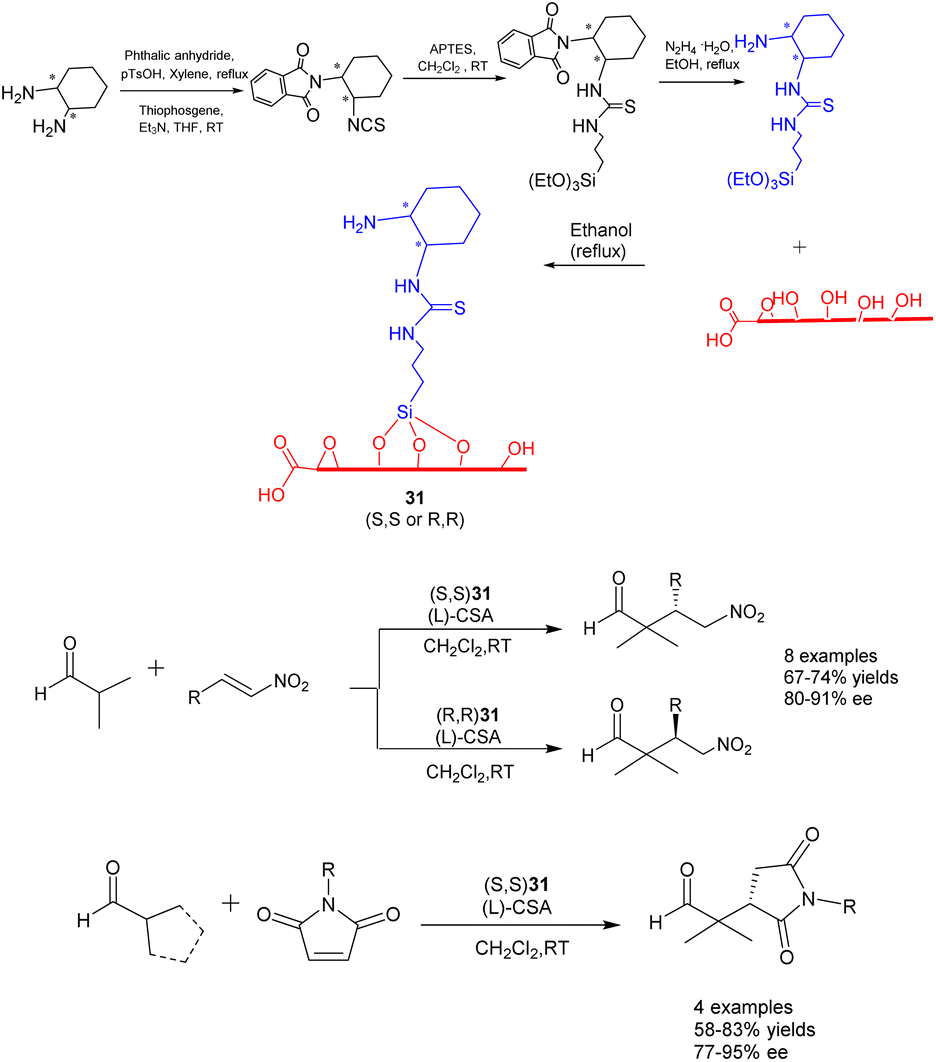 | ||
| Scheme 31 Synthesis and application of graphene-supported primary amine and thiourea bifunctional catalysts to the asymmetric Michael addition of α, α-disubstituted aldehydes to nitrite styrene and N-substituted maleimide. | ||
In the year 2020, Bingol et al. conducted a study wherein they reported the grafting of L-proline onto graphene oxide sheets, resulting in the creation of two innovative graphene-supported proline bifunctional carbon catalysts 32 (50 mg) (Scheme 32).119 These catalysts were subsequently utilized in the asymmetric aldol reaction, leading to the discovery that the enantioselective aldol reaction performed in hexane under mild conditions yielded the corresponding aldol adducts with a separation yields of up to 88% and an ee of up to 85%. Furthermore, the carbon catalyst composed of graphene exhibits a high degree of recoverability and recyclability, facilitated by a straightforward washing procedure. Moreover, its catalytic activity remains largely unaffected even after undergoing multiple cycles, as evidenced by the attainment of 70% yields and 72% ee after the 5 cycles.
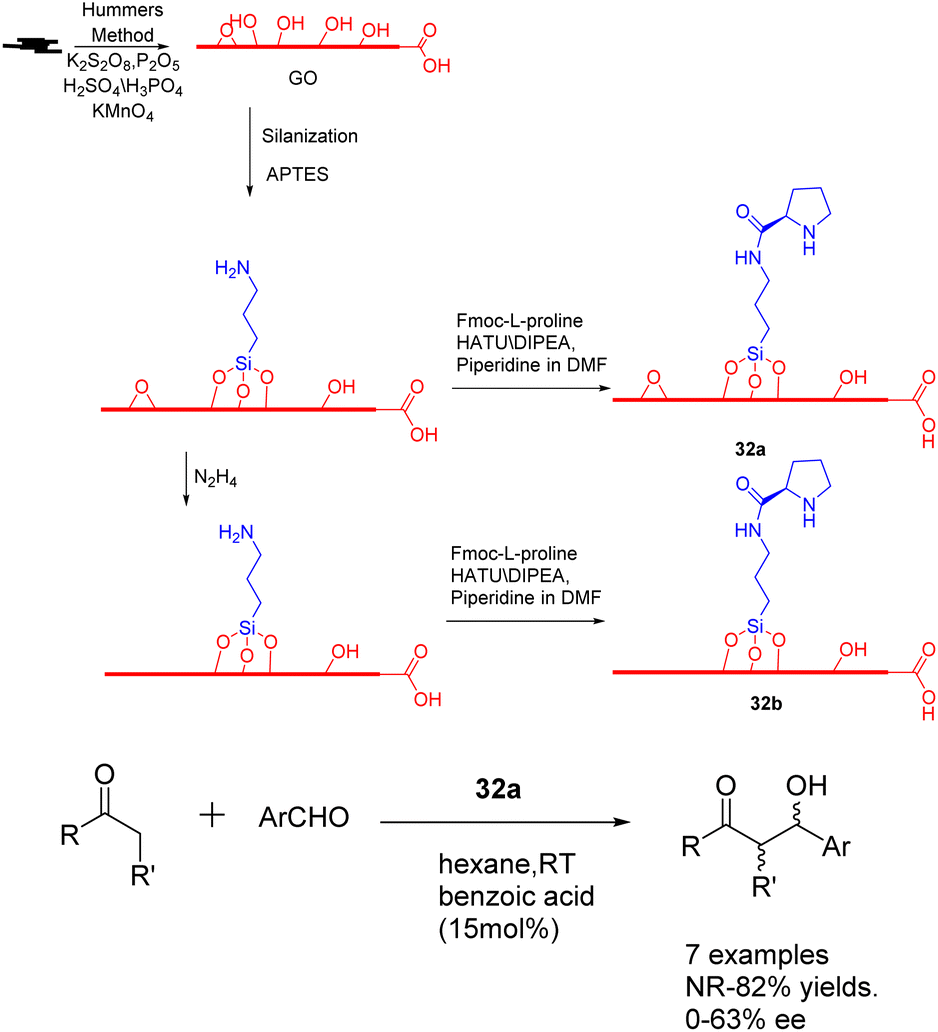 | ||
| Scheme 32 Synthesis and application of graphene-supported proline bifunctional carbon catalyst in asymmetric aldol reaction. | ||
The utilization of graphene oxide in catalytic reactions is advantageous due to its high specific surface area and presence of reactive oxygen functional groups. These properties facilitate an increased contact area between the reactant and catalyst, resulting in enhanced reaction rates and improved overall efficiency. Additionally, graphene oxide exhibits remarkable resistance to oxidative corrosion from environmental factors such as oxygen and water, ensuring its stability in a wide range of reactions. Moreover, its distinctive surface structure and electron transfer effect enable accelerated catalytic reactions. The surface structure of graphene oxide can be manipulated and enhanced through control, thereby enhancing its selectivity towards various reaction substances and facilitating the optimization of the reaction process. However, the intricate chemical properties of graphene oxide pose challenges in accurately controlling its surface structure and functional groups, necessitating improvements in its selectivity towards different reaction substances. Additionally, the high cost associated with its preparation, coupled with the expensive nature of current graphene oxide catalyst production, hinders its widespread adoption in industrial applications. The reaction is susceptible to the presence of oxygen and water in the surrounding air, leading to a decline in its activity and hindrances in its regeneration. Inevitably, the preparation process encounters technical challenges, including the utilization of foaming technology.
In their 2019 study, Villani et al. documented the utilization of bifunctional catalysts comprising chiral primary amine cinchona alkaloids and their achiral acids, which were supported on silica carriers (Scheme 33).121 Notably, the in situ activated acid/base catalysts demonstrated remarkable potential in facilitating the addition of cycloketones to trans-β-nitrostyrene, exhibiting diastereoselectivity and ee comparable to that of homogeneous catalysts (with a dr value reaching 90/10 and an ee value reaching 90%). Furthermore, the catalyst 33 (0.144 mmol) exhibited consistent activity throughout a minimum of 4 cycles. The synthesis of the chiral drug warfarin involved the enantioselective addition of 4-hydroxycoumarin to 4-phenyl-3-buten-2-one, resulting in a total yields of 78% for the precursor 4-hydroxycoumarin. These initial findings present intriguing prospects for the utilization of heterogeneous asymmetric ammonia catalysis in extensive-scale applications and flow reactions.
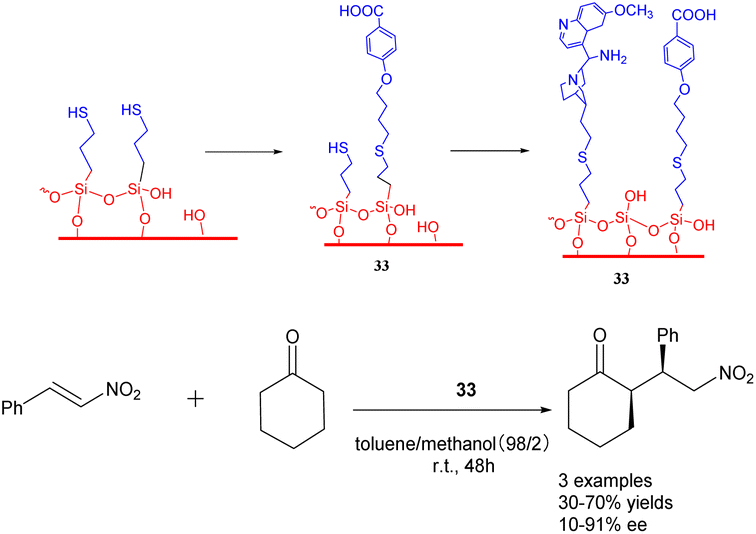 | ||
| Scheme 33 Synthesis of chiral primary amine Cinchona alkaloids and achiral acid bifunctional catalysts supported by silica and their application to the asymmetric addition reaction of cycloketone and trans-β-nitrostyrene. | ||
In 2022, Modi et al. published a study detailing the synthesis of heterogeneous catalyst 34 (0.5 g) through the functionalization of L-proline methyl ester on a silica support, without the need for protective or deprotective groups (Scheme 34).122 This catalyst exhibited remarkable efficacy in promoting the asymmetric aldol reaction under environmentally friendly conditions, eliminating the requirement for excessive organic solvents. The reaction achieved complete conversion with a high ee of >99% (ee) for the S-isomer. Furthermore, the synthesized catalyst exhibits a remarkable ability to be recovered consecutively for 9 cycles, displaying negligible activity loss. Notably, a yields of 95% and a product with an ee of 95% can still be achieved.
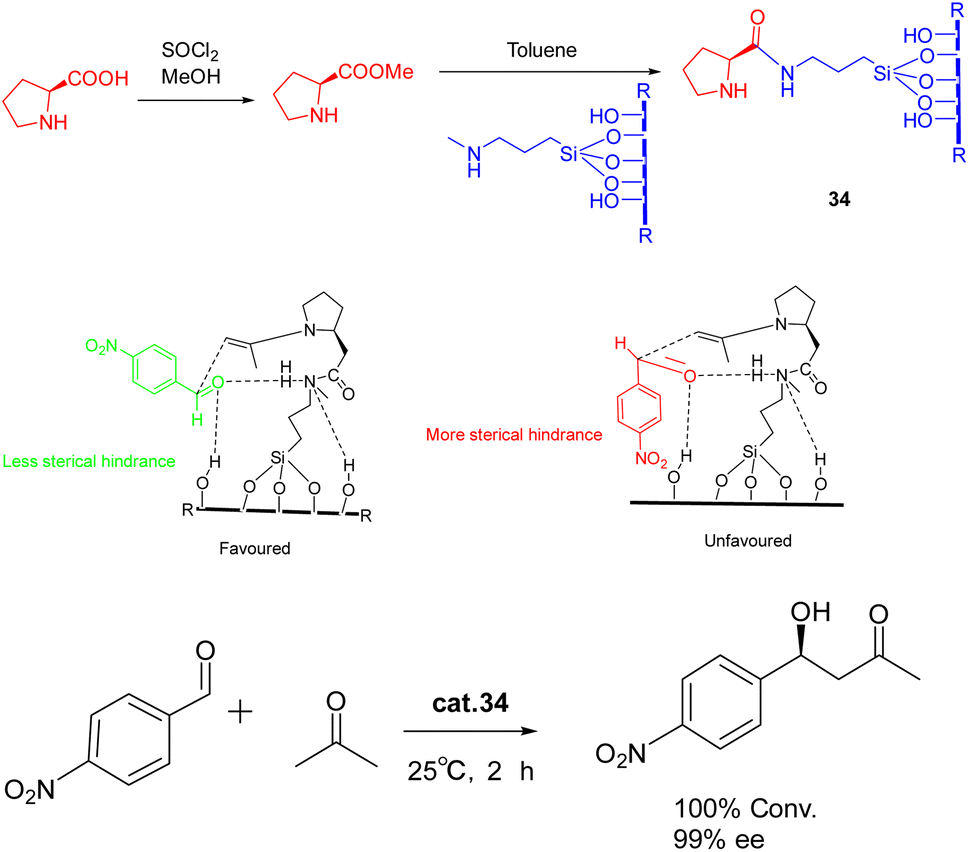 | ||
| Scheme 34 Synthesis of L-proline methyl ester supported by silica and its application in asymmetric aldol reaction and its mechanism. | ||
In 2018, Liu et al. published a study on a bimolecular catalyst 35 (12.35 mg), wherein a yolk–shell-mesostructured silica was utilized to separate the active sites. The silicate yolk contained a single-point chiral ruthenium/diamine functional group, while the basic DABCO functional group was fixed in the shell. This information can be found in Scheme 35 of their publication.123 The bifunctional catalyst exhibited significant catalytic activity and ee in the Michael addition/ATH one-pot enantioselective tandem reaction. This can be attributed to the homogeneous distribution of morphology, isolation of active sites, and the dual-component catalytic properties. These findings offer valuable insights for the development of efficient one-pot enantioselective tandem reactions and the synthesis of valuable chiral aryl substituted γ-secondary amino alcohols. Unfortunately, this article did not provide a detailed introduction to the recycling of catalysts.
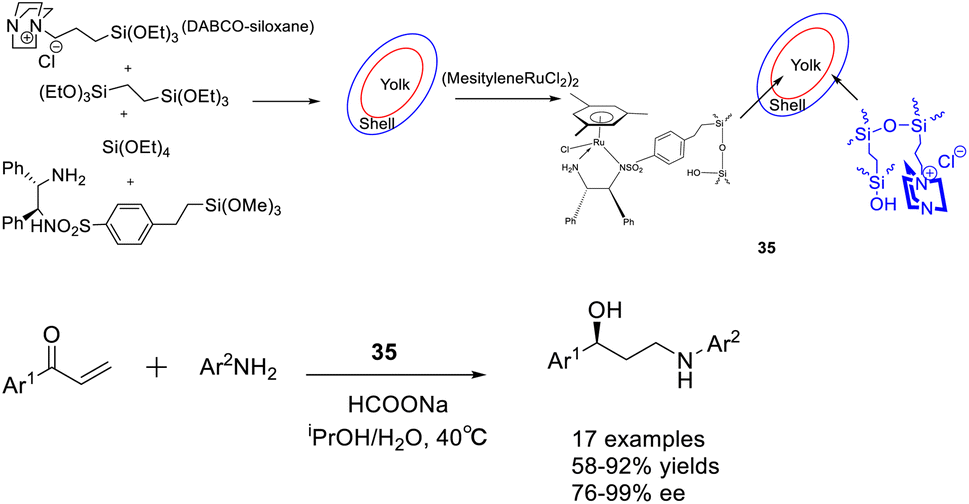 | ||
| Scheme 35 Synthesis of chiral ruthenium/diamine functional groups supported by silica and their application in asymmetric Michael addition/ATH enantioselective tandem reactions. | ||
In 2013, Li et al. conducted a comparative analysis of two loading strategies, namely post-grafting, post-modification, and co-condensation, to obtain chiral heterogeneous rhodium catalysts.124 These catalysts were synthesized using organo–rhodium complexes incorporating sulfonylated diamines within mesoporous silicate networks. The resulting catalysts were then employed in the asymmetric transfer hydrogenation of aromatic ketones in an aqueous environment. The study revealed notable variations in catalytic activity and ee among the different loading strategies. The direct immobilization of chiral organic rhodium complexes onto the external surface of mesoporous silica enables the retention of elevated ee, whereas the co-condensation of chiral organic rhodium complexes onto the internal surface of mesoporous silica results in a homogeneous dispersion of active rhodium sites. In terms of ee, both approaches exhibited superior catalytic efficacy compared to the post-modification approach. The findings of this study unequivocally illustrate the characteristics of these heterogeneous catalysts, which are contingent upon diverse immobilization strategies. Additionally, this study offers a comprehensive approach to enhance the efficacy of prepared strategies in order to fine-tune the catalytic efficiency of heterogeneous catalysts.
In their 2017 study, Celentano et al. documented the utilization of polystyrene and silica-supported chiral imidazolidine organocatalysts, denoted as Scheme 36.125 These supported heterogeneous catalysts, referred to as catalysts 36, were employed in the asymmetric catalytic reduction of imines using trichlorosilane. Polystyrene has demonstrated superior chemical and stereochemical efficiency as a carrier compared to silicon dioxide, as the latter is inert and poses challenges in effectively engaging active components to facilitate catalysis in a synergistic manner. Despite a loading rate as low as 1 mol%, the catalyst with the highest level of support exhibits noteworthy activity and stereocontrol ability, demonstrating a stereoselectivity of 98% ee. The method showcases a broad range of applicability, while also highlighting the efficient reusability of the immobilized catalyst. However, it is worth noting that the catalytic activity and ee of the polystyrene-supported catalyst 36c (0.057 mmol g−1) experienced a slight decrease after undergoing 7 cycles, resulting in a yields of 76% and an ee of 87%.
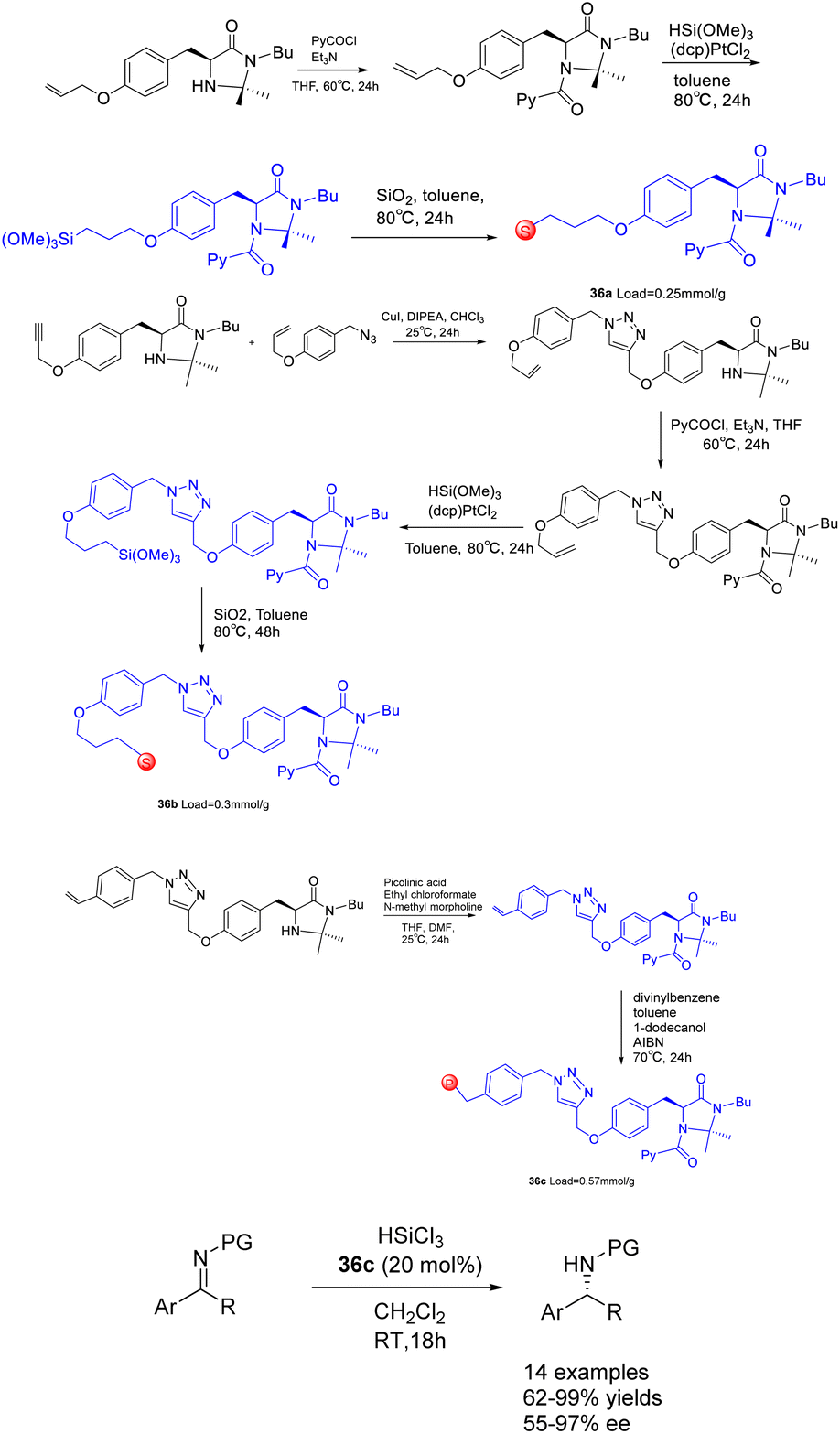 | ||
| Scheme 36 Synthesis of chiral imidazolidins supported by polystyrene and silica and their application in asymmetric catalytic reduction of imines by trichlorosilane. | ||
In 2019, He et al. conducted a study in which they successfully grafted a chiral amine catalyst onto mesoporous silica. This catalyst 37 (20 mol%), consisting of Si-lanol hydroxyl group and chiral amine, was employed to catalyze the tandem reaction of aromatic aldehyde, malononitrile, and phosphite (Scheme 37).126 The resulting product exhibited a yields of 99% and an ee of 99% ee. Furthermore, the researchers investigated the impact of benzaldehyde substituents and phosphite molecular size. Their findings revealed that larger substrates could access the catalytic sites within larger mesopores (6.6 nm), leading to enhanced yields and ee of the final product. After 3 cycles of use, catalyst 37a still maintains 98% yield and ee.
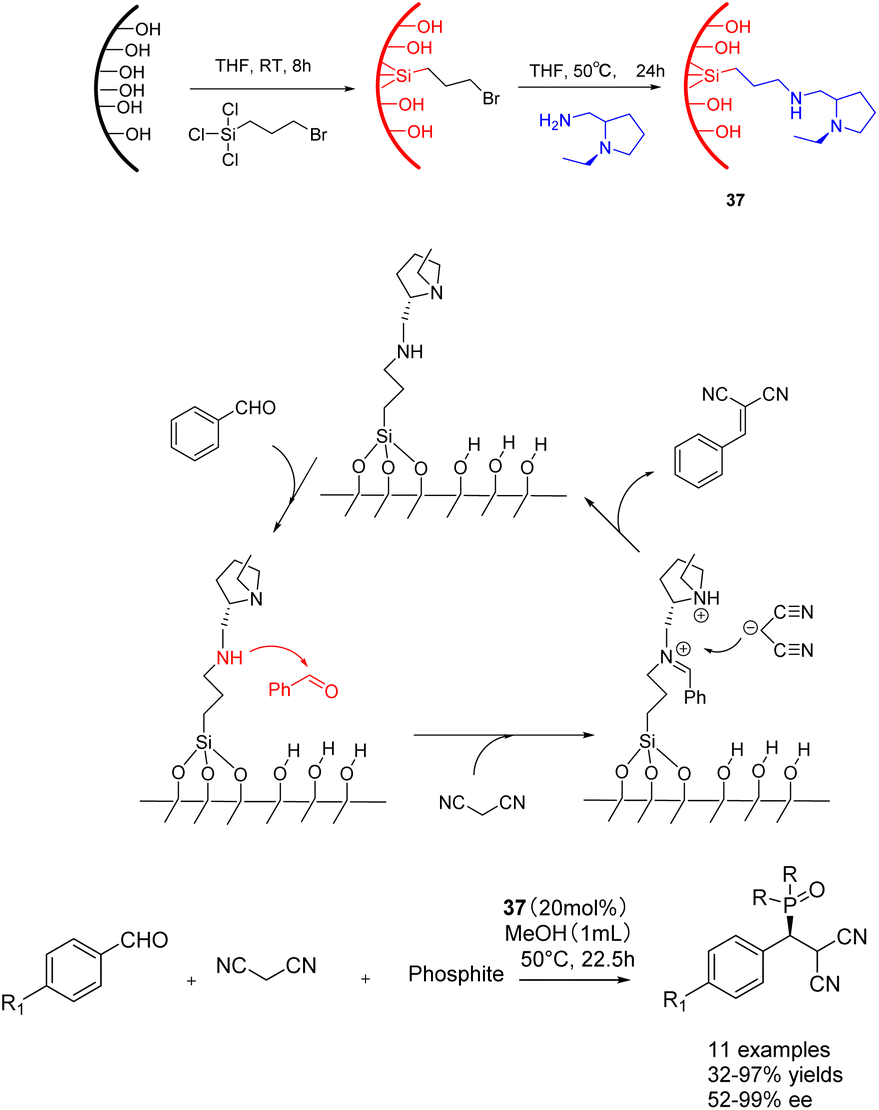 | ||
| Scheme 37 The synthesis and mechanism of chiral amine catalyst supported on silica and its application in the tandem reaction of aromatic aldehydes, malononitrile and phosphite. | ||
In their study published in 2020, Gök et al. successfully synthesized mesoporous silica-supported chiral urea-amine bifunctional catalysts by grafting chiral urea-amine ligands onto SBA-15 and magnetic MCM-41. These catalysts, referred to as 38 (7 mol%) (Scheme 38).127 demonstrated remarkable activity in transfer hydrogenation (achieving a yields of up to 99% and an ee of 65%) and enantioselective Michael reaction (achieving a yields of up to 98% and an ee of 26%). The solid catalyst loaded on SBA-15 (38c/38d) can be easily separated by filtration, while the solid catalyst loaded on magnetic MCM-41 (38e/38f) can be separated and reused for up to 3 cycles through simple magnetic precipitation. Obtained a conversion rate of over 34%, almost losing enantioselectivity.
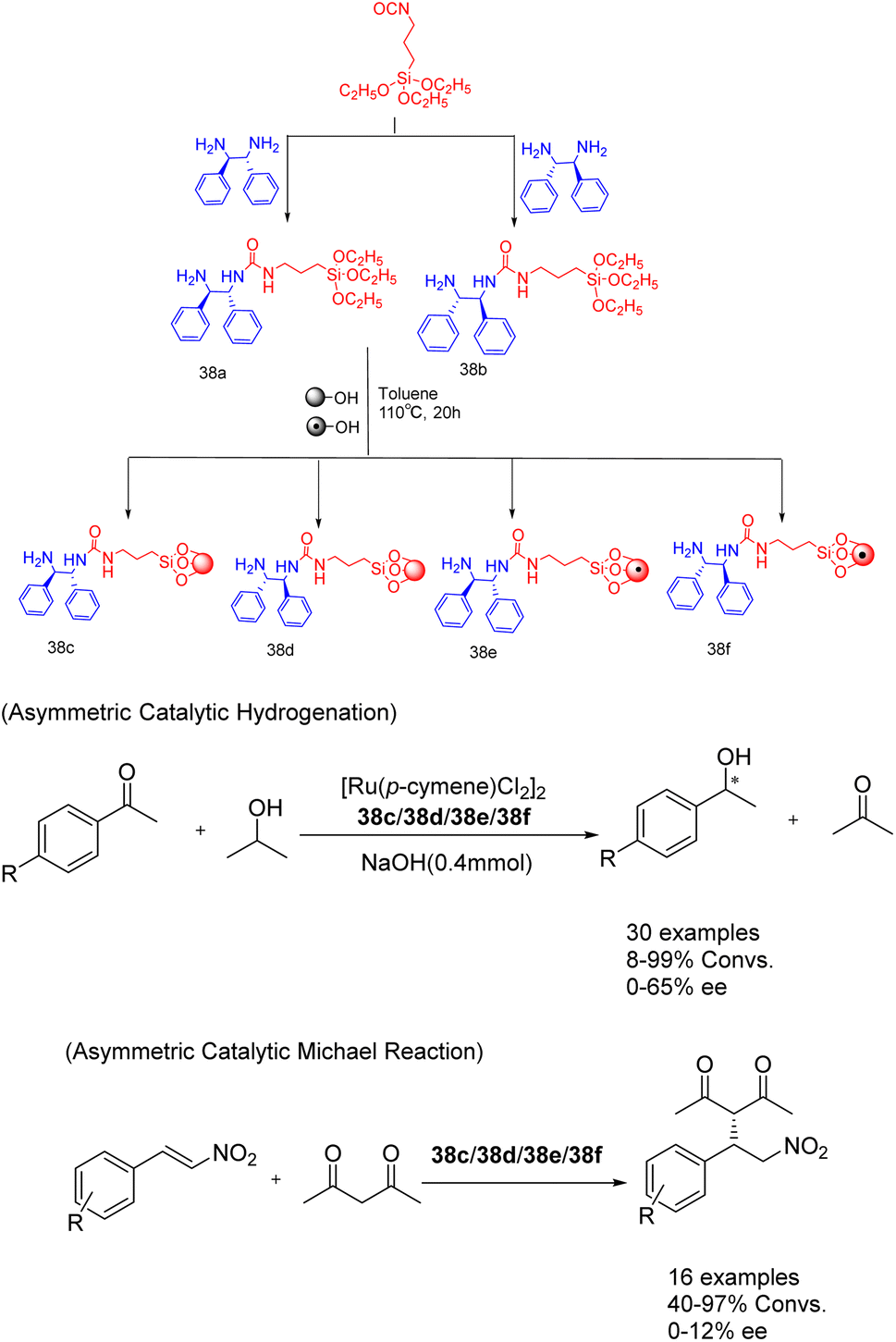 | ||
| Scheme 38 Synthesis and application of chiral ureamine bifunctional catalysts supported by silica in asymmetric reactions. | ||
(S)-Acetochlor is one of the most important herbicides. Its production scale is > 10000 tons per year, which is currently achieved by asymmetric hydrogenation of imines using a homogeneous Xyliphos-Ir catalyst, which is reported to show 2 × 106 tons (6 × 105 h−1 TOF). In 2023, Kobayashia et al. reported that a highly active and durable asymmetric hydrogenation heterogeneous catalyst was successfully developed by immobilizing Xyliphos ligand on core/shell SiO2/PS for the continuous flow synthesis of key intermediates of (S)-propachlor.128 When in the absence of solvent, the carrier showed a high turnover frequency and a long turnover time (more than 120 hours). The system can continuously produce chiral pesticide intermediates for more than 120 hours. The total yield can reach 1.45 × 105 and 1.12 × 103 h−1, and the space-time yield is about 1 kg L−1 d−1. After the long-term operation, the output will not decline further (Scheme 39).
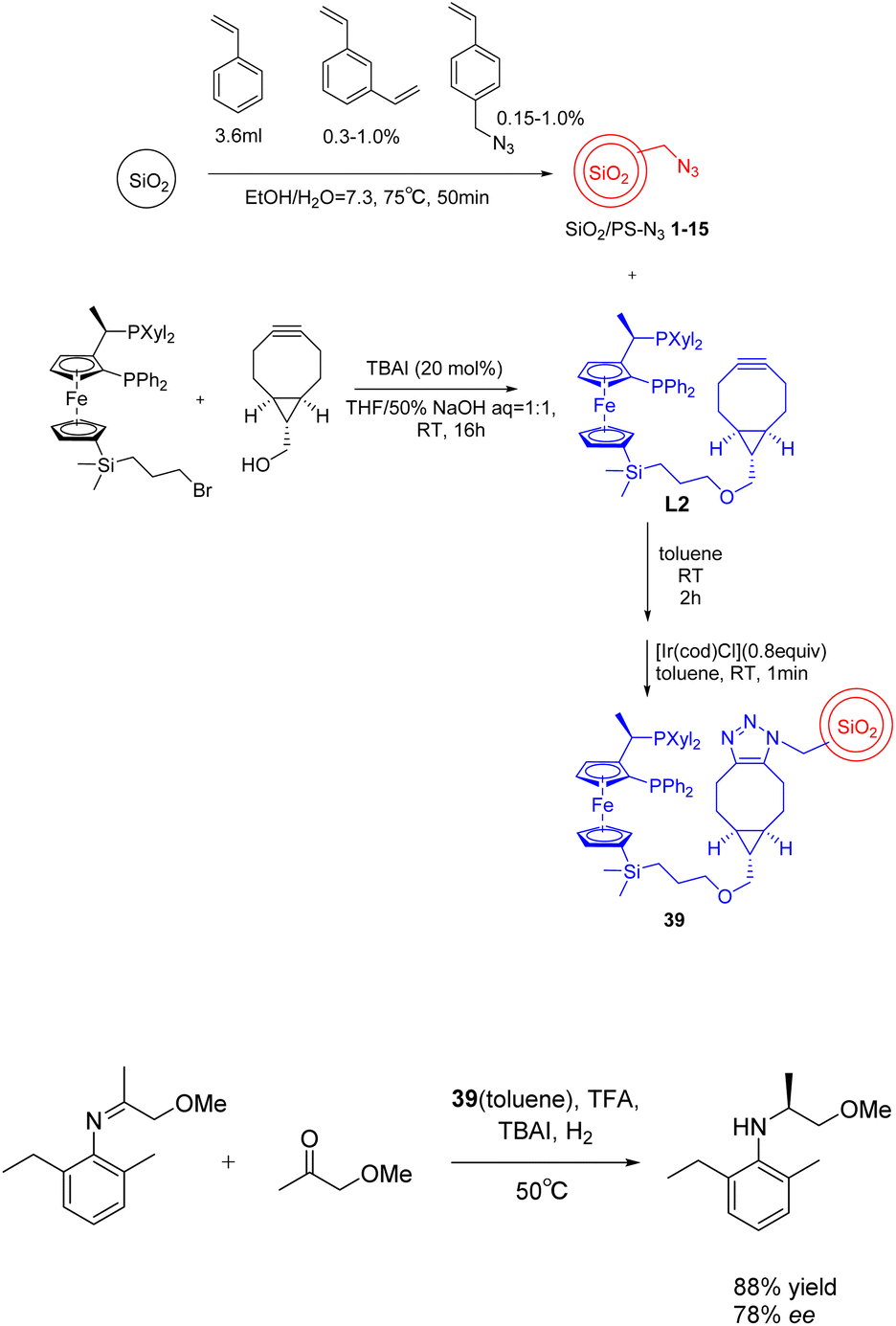 | ||
| Scheme 39 Synthesis and application of SiO2/PS-Xyliphos-Ir catalyst in asymmetric hydrogenation reaction. | ||
The catalyst supported by silica exhibits a significant specific surface area and pore size, thereby enhancing the reaction surface area and resulting in superior catalytic performance. It demonstrates excellent resistance to the reaction environment and catalyst surface at elevated temperatures and pressures, thereby minimizing deactivation during prolonged usage. Moreover, the catalyst can be easily regenerated and calcined, enabling its reuse and cost savings. The preparation process is uncomplicated and cost-effective. However, in certain catalytic reactions operating at elevated temperatures, the stability of the catalyst may be compromised as a result of alterations in its structure. Typically, the catalyst tends to generate silicates in conjunction with active constituents. The mechanical robustness is relatively low, the bond between the metallic constituent and the support material is not particularly strong, and the silica itself is prone to sintering. Furthermore, due to its inert nature, the silica encounters challenges in effectively collaborating with the active component to facilitate catalytic processes.
In addition, whether it is a soluble catalyst or an insoluble catalyst, the recycling idea is to recycle the catalyst, and the recycling of by-products is not mentioned. In 2016, Moberg reported the application of cyclic reaction networks in asymmetric catalysis, converting secondary enantiomers into starting reactants to generate target products. The formation of products and the regeneration of starting reactants are carried out through different mechanisms and pathways to improve the yield and enantioselectivity of target products.129 In the latest research, it has been found that the catalytic performance of supported catalysts is gradually better than that of unsupported catalysts.130
4 Conclusions
Recyclable catalysts have always been a research hotspot. This paper reviews the research progress of recyclable catalysts in the field of asymmetric catalysis since 2017. The recyclable catalyst is divided into two parts: soluble catalyst and insoluble catalyst. It can be seen that both soluble catalysts and insoluble catalysts have been widely studied. At the same time, they have been applied to various reactions, and have achieved good catalytic effects and maintained certain catalytic activity after repeated use. Recyclable catalysts not only save resources and reduce production costs; moreover, it simplifies the process flow, improves production efficiency, adheres to the development principles of green sustainability, and develops efficient and environmentally friendly organic synthesis methods. This is an important contribution in this field.This review objectively evaluates the strengths and weaknesses of these related catalytic systems. The advantages of soluble catalysts are as follows: (1) there is no problem of intermolecular mass transfer resistance, which improves the catalytic efficiency; (2) the catalyst can be recycled to improve the utilization rate. At the same time, the soluble catalyst also has certain disadvantages: (1) the influence of the carrier on the microenvironment; (2) the recovery method is to precipitate the soluble catalyst by solvent precipitation method, and then filter and recover it. In the process of catalyst recovery, there will inevitably be incomplete catalyst precipitation, and the amount of catalyst will be lost. For insoluble catalysts, there are two cases. The first case is the insoluble catalyst adsorbed or encapsulated by the carrier. The advantages are as follows: (1) the carrier is generally inert material, which increases the stability of the catalyst; (2) the catalyst can be recycled several times, which improves the utilization rate of the catalyst. However, its disadvantages are as follows: (1) the loading method of adsorption or encapsulation is easy to be desorbed or unwrapped due to the weak force, which leads to the leaching of the catalyst; (2) the problem of mass transfer resistance between molecules reduces the catalytic efficiency. The second case is the insoluble catalyst supported on the carrier by covalent bond. The advantages are as follows: (1) the covalent bond is strong, which increases the number of catalyst recycling and improves the utilization rate; (2) with a larger conjugated structure, the catalyst tends to be more stable. However, it also has some disadvantages that cannot be ignored: (1) there is a problem of mass transfer resistance between molecules, which reduces the catalytic efficiency; (2) sometimes there is a limited specific surface area, narrow pore size, reducing the catalytic efficiency.
Recyclable catalysts will continue to be developed in the future. However, there are still some pressing problems: (1) the synthesis of some catalysts inevitably uses toxic solvents of type I or II, which is particularly important for the pharmaceutical industry, because the residual solvents will affect the biological testing of potential candidate drugs. Therefore, it is necessary to detect the residues of toxic solvents in the final product (for example, DMF residues should not exceed 1500 ppm according to ICH guidelines). (2) The catalytic effect of supported catalysts is sometimes lower than that of unsupported catalysts. In order to solve the above problems, optimizing the reaction conditions, developing more catalysts and catalytic systems with better performance, reducing catalyst leakage, and developing more environmentally friendly process routes will be the future development direction in this field.
Author contributions
Writing-review and editing, G. P. H., W. Q. R. and S. Y. Z.; visualization, Z. Y. Z. and W. H.; supervision. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the Qin Chuangyuan's team building project (grant no. 2022KXJ-101), two chain integration project (grant no. S2022-YF-LLRH-12-0002), and the National Natural Science Foundation of China (grant no. 21272272) for the support.References
- C. Mingfeng, M. Gao, M. Suástegui, Y. Mei and Z. Shao, Metab. Eng., 2020, 58, 94–132 CrossRef PubMed.
- R. Raghuvanshi, A. Grayson, I. Schena, O. Amanze, K. Suwintono and R. Quinn, Metabolites, 2019, 9(8), 165 CrossRef CAS PubMed.
- X. Hang, X. Su, M. Guo, R. An, Y. Mou, Z. Hou and C. Guo, Eur. J. Med. Chem., 2020, 198, 112360 CrossRef PubMed.
- T. Xu, L. Wang, H. Wu, D. Jiang, N. Qi and Z. Gou, J. Cleaner Prod., 2023, 387, 135754 CrossRef CAS.
- S. Youyi, W. Congxu, G. Guizheng, F. Qiang, X. Zhiyuan, L. Dan and L. Yaqing, Appl. Surf. Sci., 2019, 469, 553–563 CrossRef.
- Z. Y. Yao, Y. G. Wen, X. Rui, Y. Li, L. Bo and W. G. Peng, Angew. Chem., Int. Ed., 2020, 59, 23291–23298 CrossRef PubMed.
- D. Entwistle, M. E. Kopach, M. R. Hickey, J. D. Hayler, F. Roschangar, P. J. Dunn, F. Gallou, S. Hughes, F. J. Weiberth, S. G. Koenig, A. Steven, G. Moine, M. C. Bryan and P. G. Richardson, Green Chem., 2018, 20, 5082–5103 RSC.
- R. E. Carles and P. M. A, Chem. Rec., 2019, 19(9), 1872–1890 CrossRef PubMed.
- H. Lin, H. Qu and W. Chen, et al., J. Catal., 2019, 372, 19–32 CrossRef CAS.
- G. ED, A. AS, H. AS, B. KC, S. JF, B. SR, B. SF and C. Matteo, Adv. Mater., 2021, 33(44), e2104533 CrossRef PubMed.
- S. E. Seo and C. J. Hawker, Macromolecules, 2020, 53, 3257–3261 CrossRef CAS.
- C. Avelino, H. Garcia and A. Leyva, J. Catal., 2006, 240(2), 87–99 CrossRef.
- F. Rania and Y. Selim, J. Polym. Environ., 2023, 31(6), 2285–2293 CrossRef.
- A. Ayushi, S. Avtar and C. H. Kumar, Curr. Org. Chem., 2023, 27(2), 130–152 CrossRef.
- Z. Xu, J. Li, X. Wang, T. Wang, D. Li and Z. Ao, Mater. Today Chem., 2023, 29, 101403 CrossRef CAS.
- H. Talukdar, G. Saikia, A. Das, S. Y. Sultana and N. S. Islam, J. Polym. Environ., 2023, 121, 249–263 CAS.
- F. Li, J. L. Kan, B. J. Yao and Y. B. Dong, Angew. Chem., Int. Ed., 2022, 61(25), e202115044 CrossRef CAS PubMed.
- K. Zhao, X. Wang, D. He, H. Wang, Bo. Qian and F. Shi, Catal. Sci. Technol., 2022, 12(16), 4962–4982 RSC.
- D. Cartagenova, S. Bachmann, K. Püntener, M. Scalone, M. A. Newton, F. A. P. Esteves, T. Rohrbach, P. P. Zimmermann, J. A. V Bokhoven and M. Ranocchiari, Catal. Sci. Technol., 2022, 12, 954–961 RSC.
- R. Tao, X. Ma, X. Wei, Y. Jin, L. Qiu and W. Zhang, J. Mater. Chem. A, 2020, 8(34), 17360–17391 RSC.
- B. Altava, M. I. Burguete, E. G. Verdugo and S. V. Luis, Chem. Soc. Rev., 2018, 47(8), 2722–2771 RSC.
- N. Kann, Molecules, 2010, 15(9), 6306–6331 CrossRef CAS PubMed.
- G. Szőllősi, Catal. Sci. Technol., 2018, 8, 389–422 RSC.
- T. Taniike, R. Baba, M. Terano, P. Chammingkwan and A. Thakur, J. Catal., 2018, 357, 69–79 CrossRef.
- C. Zhang, Y. Fan, S. Wang, S. Liu, X. Chu and E. Tang, Curr. Org. Synth., 2023, 21(1), 47–60 CrossRef PubMed.
- D. R. Bhatt, K. C. Maheria J and K. Parikh, RSC Adv., 2015, 5(16), 12139–12143 RSC.
- W. Hou, Q. Wang, Z. Guo, J. Li, Y. Zhou and J. Wang, Catal. Sci. Technol., 2017, 7(4), 1006–1016 RSC.
- B. Taleb, R. Jahjah, D. Cornu, M. Bechelany, M. A. Ajami, G. Kataya, A. Hijazi and M. H. E. Dakdouki, Molecules, 2023, 28(22), 7541 CrossRef CAS PubMed.
- F. Chen, M. Y. Jin, D. Z. Wang, C. Xu, J. Wang and X. Xing, ACS Catal., 2022, 12(22), 14429–14435 CrossRef CAS.
- B. R. Javle and A. K. Kinage, ChemistrySelect, 2018, 3, 2623–2625 CrossRef CAS.
- G. Kang, S. Lin, A. Shiwakoti and B. Ni, Catal. Commun., 2014, 57, 111–114 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115(13), 6621–6686 CrossRef CAS PubMed.
- J. Li, X. Li, Y. Ma, J. Wu, F. Wang, J. Xiang, J. Zhu, Q. Wang and J. Deng, RSC Adv., 2013, 3, 1825–1834 RSC.
- Z. Xu, X. Yu, X. Sang and D. Wang, Green Chem., 2018, 20(11), 2571–2577 RSC.
- X. Liu, C. Chen, Y. Xiu, A. Chen, L. Guo, R. Zhang, J. Chen and Z. Hou, Catal. Commun., 2015, 67, 90–94 CrossRef CAS.
- F. Wang, S. Zhang, C. Xie and X. Jin, RSC Adv., 2023, 13(40), 27865–27872 RSC.
- G. D. Y. Deepa, P. Chaudhary, M. J. Aalam, D. R. Meena and S. Singh, Chirality, 2019, 32(1), 64–72 CrossRef PubMed.
- M. J. Aalam, P. Deepa, D. R. Chaudhary, G. D. Meena and S. Singh, Chirality, 2021, 34(1), 134–146 CrossRef PubMed.
- D. Mohd, J. Aalam, P. Kumar and S. Singh, Tetrahedron Lett., 2023, 116, 154343 CrossRef.
- Z. Radai, N. Z. Kiss and G. Keglevich, Curr. Org. Chem., 2018, 22(6), 533–556 CrossRef CAS.
- F. R. Bisogno, R. Fernández, J. M. Lassaletta and G. D. Gonzalo, Molecules, 2021, 26(2), 355 CrossRef CAS PubMed.
- X. L. Zhang, S. R. Sheng, M. H. Wei and X. L. Liu, Phosphorus Sulfur Silicon Relat, Elem, 2017, 192(5), 513–517 CAS.
- S. Turkyilmaz and C. S. Wilcox, Tetrahedron Lett., 2017, 58(21), 2031–2033 CrossRef CAS PubMed.
- B. Zhang and L. Shi, Catal. Lett., 2019, 149(10), 2836–2843 CrossRef CAS.
- H. Zheng, L. Cai, M. Pan, M. Uyanik, K. Ishihara and X.-S. Xue, J. Am. Chem. Soc., 2023, 145(13), 7301–7312 CrossRef CAS PubMed.
- Y. Tang, Q. Wang, L. Wu, K. Liu, W. Wang, Y. Shen, Y. Xue and S. Dai, React. Funct. Polym., 2020, 150, 104544 CrossRef CAS.
- Y. Xia, Y. Ning, M. Liu and F. E. Chen, J. Catal., 2023, 417, 35–40 CrossRef CAS.
- N. O. Torbus, B. Mendrek, A. Kowalczuk, W. Wałach, B. Trzebicka and A. U. Wesołek, Polymers, 2021, 13(17), 2892 CrossRef PubMed.
- L. Zhou, P. Ji, X. Wang, D. Qi, T. Chen and J. Inorg, Organomet. Polym. Mater., 2023, 1–9 Search PubMed.
- X. Li, J. Dong, G. Ma and N. Ma, J. Catal., 2022, 411, 84–92 CrossRef CAS.
- Z. Jipeng, L. Runhui and S. Gonghua, Chem. J. Chin. Univ., 2023, 44(4), 28–36 Search PubMed.
- N. Zhang, Z. Sun and C. Wu, ACS Catal., 2022, 12(8), 4777–4783 CrossRef CAS.
- V. G. Desyatkin, M. V. Anokhin, V. O. Rodionov and I. P. Beletskaya, Russ. J. Org. Chem., 2017, 52(12), 1717–1727 CrossRef.
- Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Soc. Rev., 2022, 51(7), 2444–2490 RSC.
- Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Acc. Chem. Res., 2021, 54(13), 2894–2904 CrossRef CAS PubMed.
- A. Salamatmanesh, M. K. Miraki, E. Yazdani and A. Heydari, Catal. Lett., 2018, 148(10), 3257–3268 CrossRef CAS.
- B. Han, L. Zhao, Y. Song, Z. Zhao, D. Yang, R. Liu and G. Liu, Catal. Sci. Technol., 2018, 8, 2920–2927 RSC.
- A. M. Rabie, M. Shaban, M. R. Abukhadra, R. Hosny, S. A. Ahmed and N. A. Negm, J. Mol. Liq., 2019, 279, 224–231 CrossRef CAS.
- R. A Peralta, M. T Huxley, J. Albalad, C. J. Sumby and C. J. Doonan, Inorg. Chem., 2021, 60(16), 11775–11783 CrossRef PubMed.
- I. Y. Jeon, H. J. Noh and J. B. Baek, Molecules, 2018, 23(3), 657 CrossRef PubMed.
- S. A. Chhanda and S. Itsuno, J. Catal., 2019, 377, 543–549 CrossRef CAS.
- S. A. Chhanda and S. Itsuno, Tetrahedron, 2020, 76(24), 131247 CrossRef CAS.
- A. Nasery, G. Imanzadeh, M. Zamanlu, Z. Soltanzadeh and T. Öztürk, Tetrahedron, 2023, 147, 133656 CrossRef CAS.
- H. L. Nguyen, C. Gropp and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142(6), 2771–2776 CrossRef CAS PubMed.
- J.-B. Xiong, D.-D. Ban and Y.-J. Zhou, et al., Sci. Rep., 2022, 12(1), 15876 CrossRef CAS PubMed.
- F. E. R. González, V. Niebla, M. V. V. Tundidor, L. H. Tagle, R. M. Trasanco, D. Coll, P. A. Ortiz, N. Escalona, E. Pérez, I. A. Jessop, C. A. Terraza and A. T. Camba, React. Funct. Polym., 2021, 167, 104998 CrossRef.
- N. Rajeswari, E. R. Nagarajan and D. Kumar, J. Macromol. Sci., Part A: Pure Appl.Chem., 2004, 41(2), 189–209 CrossRef.
- A. Nodzewska, A. Wadolowska and M. Watkinson, Coord. Chem. Rev., 2019, 382, 181–216 CrossRef CAS.
- B. Wang, R. B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142(34), 14399–14416 CrossRef CAS PubMed.
- Z. Li, Y. Zhi, P. Shao, H. Xia, G. Li, X. Feng, X. Chen, Z. Shi and X. Liu, Appl. Catal., B, 2019, 245, 334–342 CrossRef CAS.
- B. Ma, Y. Xu, F. Hu, G. Zhang, X. Zheng, Z. Wang, H. Qiao, D. Yang and L. Mi, Macromol. Chem. Phys., 2022, 223(6), 2100462 CrossRef CAS.
- C. Li, W. Wang, L. Yan and Y. Ding, Front. Chem. Sci. Eng., 2017, 12(1), 113–123 CrossRef.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, ACS Sustain. Chem. Eng., 2019, 7(5), 5065–5071 CrossRef CAS.
- J. C. Wang, X. Kan, J. Y. Shang, H. Qiao and Y. B. Dong, J. Am. Chem. Soc., 2020, 142(40), 16915–16920 CrossRef CAS PubMed.
- Y. Chen, Y. Zhang and J. Huo, J. Solid State Chem., 2022, 310, 123047 CrossRef CAS.
- J. Hu, J. Zhang, Z. Lin, L. Xie, S. Liao and X. Chen, Chem. Mater., 2022, 34(11), 5249–5257 CrossRef CAS.
- Y. Zhang, T. Mao, L. Hao, T. Sun, T. Wang, P. Cheng, Y. Chen, Z. Wang and Z. Zhang, Macromol. Rapid Commun., 2023, 44(11), e2200722 CrossRef PubMed.
- S. Morales, F. G. Guijarro, J. L. G. Ruano and M. B. Cid, J. Am. Chem. Soc., 2014, 136(3), 1082–1089 CrossRef CAS PubMed.
- F. Li, J. L. Kan, B. J. Yao and Y. B. Dong, Angew. Chem., Int. Ed., 2022, 61(25), e202115044 CrossRef CAS PubMed.
- X. Wang, M. Liu, Y. Liu, S. Shang, C. Du, J. Hong, W. Gao, C. Hua, H. Xu, Z. You, J. Chen and Y. Liu, J. Am. Chem. Soc., 2023, 145(49), 26900–26907 CrossRef CAS PubMed.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2017, 139(24), 8277–8285 CrossRef CAS PubMed.
- J. He, B. Luo, H. Zhang, Z. Li, N. Zhu, F. Lan and Y. Wu, J. Colloid Interface Sci., 2022, 606(Pt 2), 1333–1339 CAS.
- X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139(25), 8693–8697 CrossRef CAS PubMed.
- C. T. Ding, J. Q. Yuan, M. Y. Xie, Q. Y. Liu, Z. G. Yao, S. Y. Zhang, R. N. Zhang, H. Wu and Z. Y. Jiang, Chin. J. Polym. Sci., 2023, 42, 141–158 CrossRef.
- R. Liu, C. Wei, X. Liu, Y. Qu, L. Yan, H. Zhao, Y. Wu, H. Cao, J. Li and H. Wang, J. Mater. Sci.: Mater. Electron., 2023, 34(3), 159 CrossRef CAS.
- R. Gao, G. Zhang, F. Lu, L. Chen and Y. Li, Front. Chem., 2021, 9, 687183 CrossRef CAS PubMed.
- W. Zhang, H. Zuo, Z. Cheng, Y. Shi, Z. Guo, N. Meng, A. Thomas and Y. Liao, Adv. Mater., 2022, 34(18), 2104952 CrossRef CAS PubMed.
- L. Xu, J. Cui, S. Gao, J. Wang, J. Liu, H. Jia, Z. Zhang, F. Miao and Y. Zang, Microporous Mesoporous Mater., 2022, 341, 112075 CrossRef CAS.
- J. Huang and S. R. Turner, Polym. Rev., 2018, 58(1), 1–41 CrossRef CAS.
- Z. Jia, K. Wang, B. Tan and Y. Gu, ACS Catal., 2017, 7, 3693–3702 CrossRef CAS.
- J. P Joseph, C. Miglani, A. Singh, D. Gupta and A. Pal, Soft Matter, 2020, 16(10), 2506–2515 RSC.
- Y. Luo, Y. Mei, Y. Xu and K. Huang, Nanomaterials, 2023, 13(18), 2514 CrossRef CAS PubMed.
- C. Liu, W. Xu, D. Xiang, Q. Luo, S. Zeng, L. Zheng, Y. Tan, Y. Ouyang and H. Lin, Catal. Lett., 2020, 150(9), 2558–2565 CrossRef CAS.
- R. Porta, M. Benaglia, R. Annunziata, A. Puglisi and G. Celentano, Adv. Synth. Catal., 2017, 359(14), 2375–2382 CrossRef CAS.
- C. Liu, L. Zheng, D. Xiang, S. Liu, W. Xu, Q. Luo, Y. Shu, Y. Ouyang and H. Lin, RSC Adv., 2020, 10(29), 17123–17128 RSC.
- T. Wang, W. Wang, Y. Lyu, K. Xiong, C. Li, H. Zhang, Z. Zhan, Z. Jiang and Y. Ding, Chin. J. Catal., 2017, 38(4), 691–698 CrossRef CAS.
- Z. Ren, L. Fang, M. Cui, T. Lv, H. Wu, Z. Feng, Z. Feng, C. Meng and L. Ren, Ind. Eng. Chem. Res., 2023, 62(26), 10057–10068 CrossRef CAS.
- X. Chen, H. Zhu, W. Wang, H. Du, T. Wang, L. Yan, X. Hu and Y. Ding, ChemSusChem, 2016, 9(17), 2451–2459 CrossRef CAS PubMed.
- S. J. D. Smith, R. J. Mulder, C. M. Doherty, M. R. Hill, X. Mulet, H. Mahdavi, C. D. Wood and P. H. M. Feron, Sci. China Mater., 2022, 65(7), 1937–1942 CrossRef CAS.
- Y. Zhang, Z. Zhang, S. Ma, J. Jia, H. Xia and X. Liu, J. Mater. Chem. A, 2021, 9(45), 25369–25373 RSC.
- X. Y. Huang, Q. Zheng, L. M. Zou, Q. Gu, T. Tu and S. L. You, ACS Catal., 2022, 12(8), 4545–4553 CrossRef CAS.
- A. J. Emerson, A. Chahine, S. R. Batten and D. R. Turner, Coord. Chem. Rev., 2018, 365, 1–22 CrossRef CAS.
- Y. Wang, L. Liu, Y. Wang, K. Sang, C. Zhang and T. Satoh, Polym. Chem., 2023, 14(10), 1124–1134 RSC.
- O. Cohen, K. V. Novikova, H. Toledo and M. S. Silverstein, ACS Appl. Polym. Mater., 2023, 5(8), 5824–5833 CrossRef CAS.
- W. Jiang, H. Yue, P. Shuttleworth, P. Xie, S. Li and J. Guo, Polymers, 2019, 11(3), 486 CrossRef PubMed.
- M. Valle, L. Martín, A. Maestro, J. M. Andrés and R. Pedrosa, Polymers, 2018, 11(1), 13 CrossRef PubMed.
- N. R. Bennedsen, S. Kramer and S. Kegnæs, Catal. Sci. Technol., 2020, 10(22), 7697–7705 RSC.
- B. Shrimant, U. K. Kharul and P. P. Wadgaonkar, React. Funct. Polym., 2018, 133, 153–160 CrossRef CAS.
- A. R Antonangelo, N. Hawkins, E. Tocci, C. Muzzi, A. Fuoco and M. Carta, J. Am. Chem. Soc., 2022, 144(34), 15581–15594 CrossRef PubMed.
- S. Fajal, S. Dutta and S. K Ghosh, Mater. Horiz., 2023, 10(10), 4083–4138 RSC.
- H. Zhang, X. Han and Y. Zhao, J. Electroanal. Chem., 2017, 799, 84–91 CrossRef CAS.
- D. Khalili, S. Lavian and M. Moayyed, Tetrahedron Lett., 2020, 61(6), 151470 CrossRef CAS.
- S. Ye, F. Luo, T. Xu, P. Zhang, H. Shi, S. Qin, J. Wu, C. He, X. Ouyang, Q. Zhang, J. Liu and X. Sun, Nano Energy, 2020, 68, 104301 CrossRef CAS.
- J. U. Lee, W. Lee, S. S. Yoon, J. Kim and J. H. Byun, Appl. Surf. Sci., 2014, 315, 73–80 CrossRef CAS.
- K. Berijani, A. Farokhi, H. H. Monfared and C. Janiak, Tetrahedron, 2018, 74(18), 2202–2210 CrossRef CAS.
- X. Wang, J. Guo, F. Qie and Y. Yan, J. Mater. Sci., 2019, 54(9), 6908–6916 CrossRef CAS.
- M. A. E. Sater, M. Mellah, D. Dragoe, E. Kolodziej, N. Jaber and E. Schulz, Chem.–Euro. J., 2021, 27(36), 9454–9460 CrossRef PubMed.
- S. M. Badr, M. Azlouk, E. Zor, H. Bingol and M. Durmaz, Mol. Catal., 2022, 526, 112383 CrossRef CAS.
- M. Azlouk, M. Durmaz, E. Zor and H. Bingol, Mater. Chem. Phys., 2020, 239, 122298 CrossRef CAS.
- M. Shukla, K. C. Barick, H. G. Salunke and S. Chandra, Mol. Catal., 2021, 502, 111367 CrossRef CAS.
- A. Ciogli, D. Capitani, N. D. Iorio, S. Crotti, G. Bencivenni, M. P. Donzello and C. Villani, Eur. J. Org Chem., 2019, 2019(10), 2020–2028 CrossRef CAS.
- P. Lakhani and C. K. Modi, Mol. Catal., 2022, 525, 112359 CrossRef CAS.
- L. Wu, Y. Li, J. Meng, R. Jin, J. Lin and G. Liu, ChemPlusChem, 2018, 83(9), 861–867 CrossRef CAS PubMed.
- J. Long, G. Liu, T. Cheng, H. Yao, Q. Qian, J. Zhuang, F. Gao and H. Li, J. Catal., 2013, 298, 41–50 CrossRef CAS.
- R. Porta, M. Benaglia, R. Annunziata, A. Puglisi and G. Celentano, Adv. Synth. Catal., 2017, 359(14), 2375–2382 CrossRef CAS.
- Z. An, Y. Dai, Y. Jiang and J. He, Asian J. Org. Chem., 2019, 8(8), 1539–1547 CrossRef CAS.
- Y. Gök, I. T. Aykut and H. Z. Gök, Appl. Organomet. Chem., 2020, 34(12), e6015 CrossRef.
- T. Kuremoto, T. Yasukawa and S. Kobayashi, Adv. Synth. Catal., 2024, 366(4), 757–761 CrossRef CAS.
- C. Moberg, Acc. Chem. Res., 2016, 49(12), 2736–2745 CrossRef CAS PubMed.
- Y. Zhu, J. Zhong and F. Xiong, J. Catal., 2024, 430, 115366 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |