
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-triggered click reaction in an Alzheimer's disease model: in situ bifunctional drug synthesis catalyzed by neurotoxic copper accumulated in amyloid-β plaques†
Zhi
Du
ab,
Dongqin
Yu
ac,
Xiubo
Du
d,
Peter
Scott
 e,
Jinsong
Ren
a and
Xiaogang
Qu
*a
e,
Jinsong
Ren
a and
Xiaogang
Qu
*a
aLaboratory of Chemical Biology and State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: xqu@ciac.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100039, China
cUniversity of Science and Technology of China, Hefei, Anhui 230029, China
dCollege of Life Sciences and Oceanography, Shenzhen Key Laboratory of Microbial Genetic Engineering, Shenzhen University, Shenzhen, 518060, China
eDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
First published on 27th September 2019
Abstract
Cu is one of the essential elements for life. Its dyshomeostasis has been demonstrated to be closely related to neurodegenerative disorders, such as Alzheimer's disease (AD), which is characterized by amyloid-β (Aβ) aggregation and Cu accumulation. It is a great challenge as to how to take advantage of neurotoxic Cu to fight disease and make it helpful. Herein, we report that the accumulated Cu in Aβ plaques can effectively catalyze an azide–alkyne bioorthogonal cycloaddition reaction for fluorophore activation and drug synthesis in living cells, a transgenic AD model of Caenorhabditis elegans CL2006, and brain slices of triple transgenic AD mice. More importantly, the in situ synthesized bifunctional drug 6 can disassemble Aβ–Cu aggregates by extracting Cu and photo-oxygenating Aβ synergistically, suppressing Aβ-mediated paralysis and diminishing the locomotion defects of the AD model CL2006 strain. Our results demonstrate that taking the accumulated Cu ions in the Aβ plaque for an in situ click reaction can achieve both a self-triggered and self-regulated drug synthesis for AD therapy. To the best of our knowledge, a click reaction catalyzed by local Cu in a physiological environment has not been reported. This work may open up a new avenue for in situ multifunctional drug synthesis by using endogenous neurotoxic metal ions for the treatment of neurodegenerative diseases.
Introduction
Cu is indispensable to life and plays an important role in energy generation, signal transduction, cellular respiration and antioxidant defense.1 The fluctuation of Cu is associated with a wide range of diseases, including genetic diseases like Menkes and Wilson's diseases; neurodegenerative diseases such as Alzheimer's, Parkinson's, Huntington's, and prion diseases; metabolic disorders and even cancer.1,2 For diseases related to Cu accumulation, the most common treatment strategy is the direct use of Cu-chelators. However, systemic administration of chelating agents may cause adverse side effects because of indiscriminate metal chelation.3 It is a promising strategy to harness toxic Cu under pathological conditions for defense against disease rather than just to chelate Cu.4–6Due to its high efficiency, fast kinetics, and mild reaction conditions, the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction has received special attention in many biochemical applications such as bioconjugation,7–11 biosensing,12–14 and drug synthesis.15,16 Many kinds of Cu sources are efficient for CuAAC bioorthogonal reactions. Besides typical Cu(I) complexes and the reduction of Cu(II) complexes,17 various Cu-containing nanoparticles (NPs) are increasingly used for CuAAC reactions, including Cu, Cu2O and CuO NPs,18,19 Cu-immobilized inorganic and polymer NPs,20–22 as well as Cu-doped semiconductor NPs.23,24
A major drawback of the CuAAC reaction in biological systems is Cu-triggered reactive oxygen species (ROS) from Fenton's reaction.25 An additional but usually neglected side effect is that the addition of exogenous Cu-catalysts might disturb Cu homeostasis in physiological milieu. In terms of intracellular chemistry, a click reaction using endogenous Cu as the catalyst holds a special attraction.
Alzheimer's disease (AD), affecting about fifty million people worldwide, is suggested to be strongly correlated to amyloid-β (Aβ) aggregation and metal dysregulation.26–28 The typical concentration of Cu in Aβ plaques reaches up to 0.4 mM, while the amount of Cu in a normal brain is 0.2–1.7 μM.29,30 Inspired by an aspartate-coordinated Cu catalyst that can accelerate the click reaction at low parts per million levels,31 herein we have found that Aβ–Cu aggregates can catalyze the CuAAC reaction in cells, an AD model of Caenorhabditis elegans (C. elegans) and the brain slices of triple transgenic AD (3 × Tg-AD) mice. To the best of our knowledge, a click reaction catalyzed by local Cu in the physiological environment has not been reported. Our work will promote the application of bioorthogonal chemistry in living systems, and may open up a new avenue for in situ multifunctional drug synthesis using endogenous neurotoxic metal ions for the treatment of neurodegenerative diseases.
Results and discussion
Aβ40–Cu aggregates were prepared by incubating Aβ40 (100 μM) with equimolar CuCl2 at 37 °C for 8 h and then characterized by atomic force microscopy (AFM, Fig. S1†). A dialysis experiment demonstrated that 97% of Cu was bound to Aβ40, analyzed by inductively coupled plasma mass spectrometry (ICP-MS, Table S1†). Aβ–Cu aggregates were used as Cu-accumulated Aβ plaque mimics to catalyze the click reaction, and conversion of non-fluorescent coumarin 1 into fluorescent 3 was chosen as the model reaction (Fig. 1a). The azido group of 1 is capable of quenching the fluorescence via internal charge transfer (ICT).32,33 Upon reaction with alkyne 2, the lone pair of the azide is delocalized to the triazole and the fluorescence is sharply enhanced.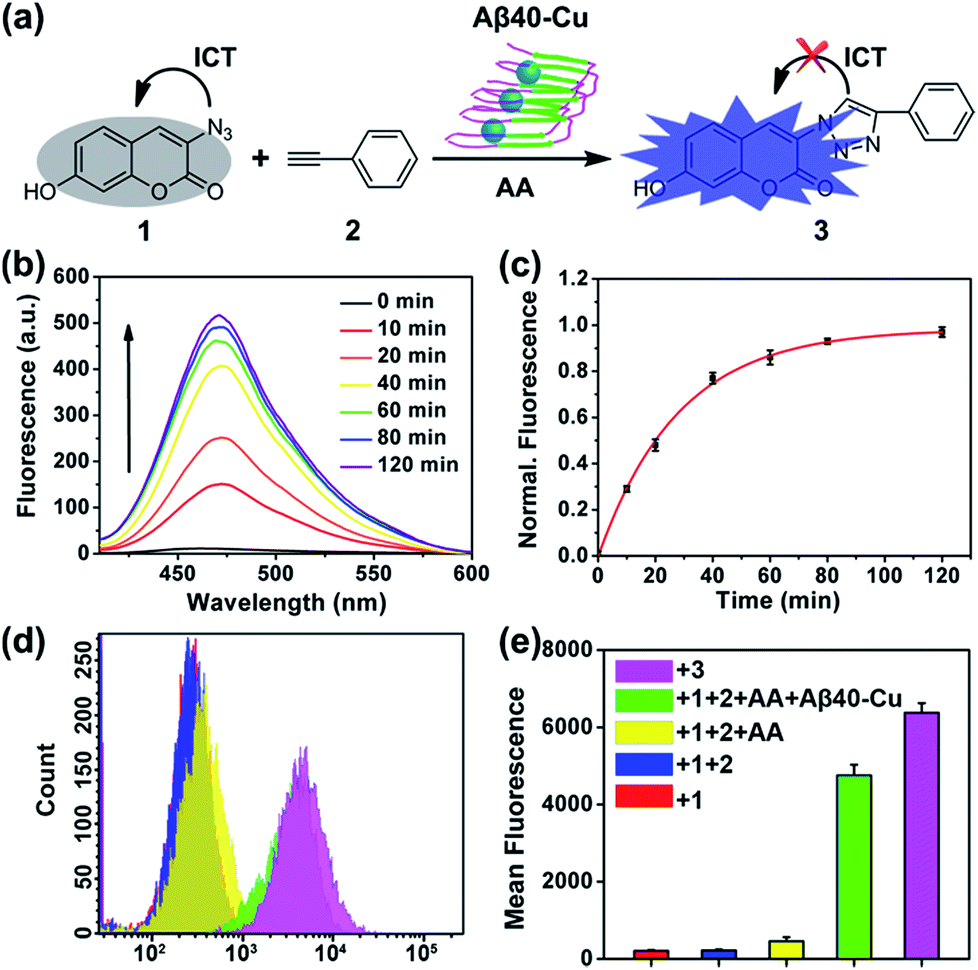 | ||
| Fig. 1 Aβ40–Cu-catalyzed fluorogenic click reaction. (a) Scheme of the click reaction to synthesize fluorescent probe 3. (b) Fluorescence spectra at different times of the reaction medium in HEPES buffer (pH 7.4). (c) Normalized fluorescence intensity at 475 nm versus the reaction time (λex = 405 nm). The fluorescence intensity of 3 (50 μM) was defined as 100%. (d) Flow cytometry detection of synthesized 3 in PC12 cells. (e) Mean fluorescence analysis of (d). PC12 cells incubated with 3 (50 μM) were used as the control. Each experiment was repeated three times. Error bars indicate ± standard deviation (s.d.). | ||
For catalysis of the click reaction in vitro, Aβ40–Cu aggregates (10 μM), ascorbic acid (AA, 50 μM) as a reducing agent, and compounds 1 (50 μM) and 2 (50 μM) were mixed at room temperature. As expected, Aβ40–Cu aggregates efficiently catalyzed the fluorogenic click reaction with a 51-fold fluorescence increase (Fig. 1b). Calculated from the fluorescence intensity, the conversion efficiency was about 96% (Fig. 1c). Moreover, the CuAAC reaction for different ratios of Aβ40 to Cu was explored. As shown in Fig. S2,† the conversion efficiency was almost the same for different ratios of Aβ40 to Cu. The result verified that the Aβ40–Cu species, rather than free Cu, is responsible for the CuAAC reaction. This conclusion was further supported by the data shown in Fig. S3.† Aβ42–Cu aggregates were also prepared and characterized by AFM (Fig. S4†). As shown in Fig. S5,† Aβ42–Cu aggregates can efficiently catalyze the CuAAC reaction.
Inspired by the above results, the performance of Aβ40–Cu for catalyzing the fluorogenic click reaction in living cells was explored. PC12 cells were treated with Aβ40–Cu aggregates (10 μM) for 12 h before compound 1 (50 μM), 2 (50 μM), and AA (50 μM) were incubated with the cells for a further 12 h. Flow cytometry (Fig. 1d and e) in conjunction with the fluorescence images (Fig. S6†) validated that Aβ40–Cu could activate the profluorophore. Mass spectrometry (MS, Fig. S7†) further verified the feasibility of Aβ40–Cu aggregates as a Cu catalyst for the CuAAC reaction in living cells. It has been reported that large Aβ aggregates are able to enter cells via macropinocytosis.34–36 Therefore, we conducted an immunofluorescent staining experiment to investigate whether Aβ40–Cu aggregates can enter cells. As shown in Fig. S8,† PC12 cells incubated with Aβ aggregates show noteworthy reactivity to Aβ fibril antibody mOC78, which means that Aβ–Cu aggregates can attach to the cytomembrane or enter cells.
We further assessed the feasibility of an Aβ–Cu-catalyzed click reaction in a transgenic AD model of C. elegans CL2006, which expresses Aβ1–42 in muscle cells. Firstly, CL2006 worms were cultured in nematode growth medium (NGM) containing 1 mM CuCl2 for 24 h to form Cu-accumulated Aβ plaques.37 The Cu-treated worms were then transferred to the NGM containing compound 1 (1 mM), 2 (1 mM), and AA (1 mM). As shown in Fig. S9 and S10,† a click reaction was promoted in a Cu-treated AD model of CL2006 worms after 12 h.
Organotypic brain slice cultures largely preserve brain architecture and possess major disease features, providing a practical platform for direct medication and real-time monitoring therapeutic effects.38,39 Therefore, this technology is widely used for studying neurodegenerative diseases. In addition, it has been observed that copper levels are elevated in senile plaques, the hippocampus, and cortex of AD model mice.40–42 Herein, we also investigated whether the click reaction can be catalyzed by Cu-accumulated plaques in the brain slices of triple transgenic AD (3 × Tg-AD) model mice.43 After incubation for 12 h, the fluorescence of the brain slices was much more intense for 3 × Tg-AD mice than for normal mice, which demonstrated that the local Cu in brain slices from 3 × Tg-AD mice can catalyze the CuAAC click reaction (Fig. S11 and S12†).
Repeated systemic administration is often hindered by absent target specificity and unpredictable toxicity in non-targeted tissue.44,45 Drugs may also suffer from rapid deactivation, poor pharmacokinetics, and low tissue absorption.46In situ drug synthesis within living cells or organisms through targeted bioorthogonal catalysis may overcome the above mentioned drawbacks because it can greatly improve drug concentration at desired sites and reduce off-target effects.47–52 We speculated that in situ drug synthesis through CuAAC reaction would probably mitigate the cytotoxicity caused by the Aβ–Cu aggregates. Recent studies have indicated that the aggregation properties and cytotoxicity of Aβ are closely related to the hydrophobicity of the protein.53,54 Photo-oxygenating Aβ is capable of disintegrating Aβ aggregates through increasing the protein polarity, which is an attractive strategy for AD therapy.55–58 However, in the presence of Cu, just oxygenating Aβ may not entirely block its fibrillation because Cu can still promote protein aggregation by coordinating to Aβ. Considering that Cu and Aβ are two critical pathogenic factors of AD,59,60 we envisioned that in situ synthesized bifunctional drug agent with Aβ-oxygenating and Cu-chelating properties could effectively disassemble Aβ–Cu aggregates in vivo.
To this end, compound 6 was rationally designed based on the following considerations: (1) thioflavin T (ThT) and its analogues can selectively photo-oxygenate amyloid aggregates.61,62 Without binding to β-sheet aggregates, photo-excited ThT undergoes twisted intramolecular charge transfer and transforms light energy to heat; (2) propargylglycine 5 has often been used for synthesizing transition metal chelators through a “click-to-chelate” approach.63,64 Therefore, the bifunctional compound 6 was constructed by integrating an Aβ-specific photosensitizer with a Cu prochelator (Fig. 2a).
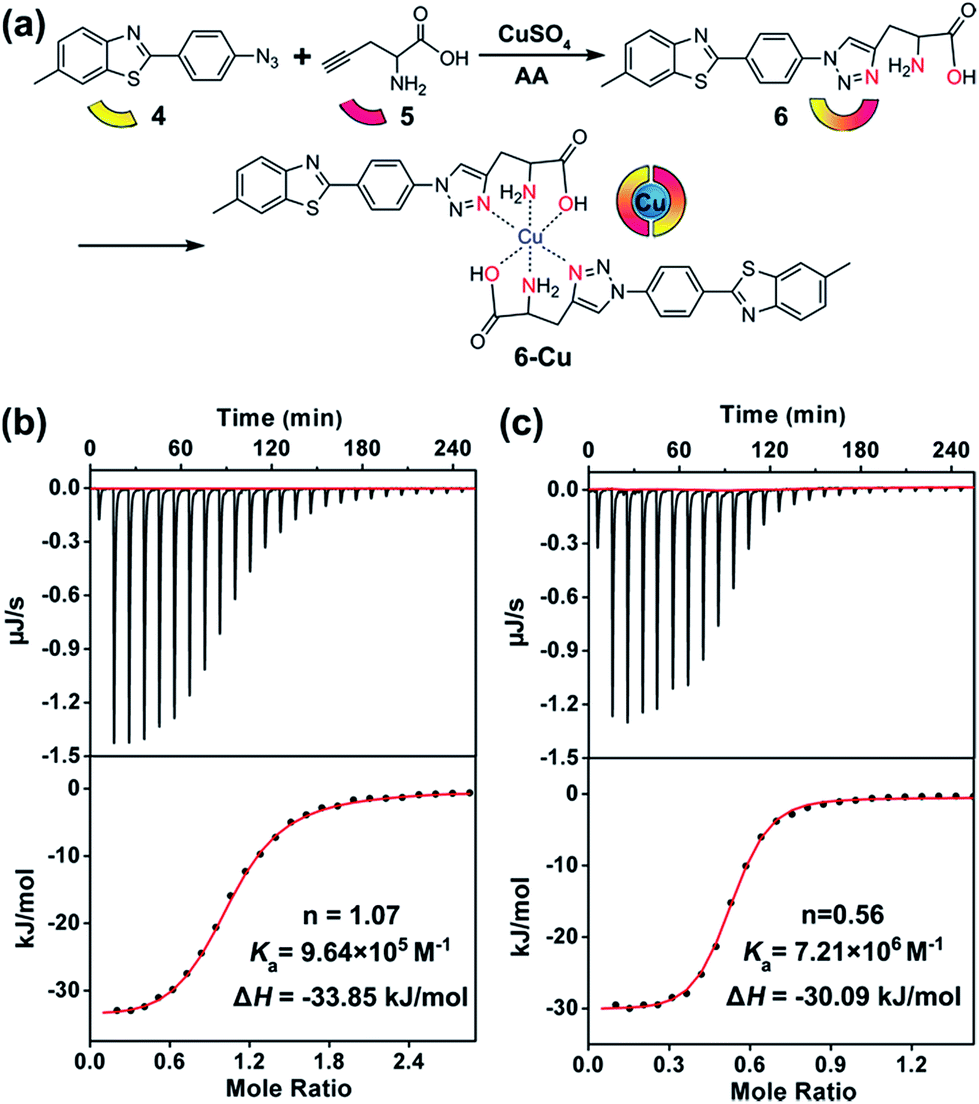 | ||
| Fig. 2 Cu-chelating properties of 6. (a) Scheme of the synthesis of the bifunctional compound 6. (b and c), ITC data of CuCl2 binding to Aβ40 and compound 6, respectively. The bottom panels show the fitting results. Details are described in the Experimental section. | ||
Spectral titration6 and isothermal titration calorimetry (ITC) experiments65–67 were carried out to measure the binding stoichiometry (n) and binding affinity of compound 6 with Cu. After adding CuCl2, the absorbance of compound 6 at 321 nm was slightly decreased (Fig. S13†). A breakpoint was observed at a ratio of 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, illustrating the binding stoichiometry of Cu and 6 was 1:2. For ITC assays, a weaker competitor glycine was added to prevent the precipitation of Cu. As shown in Fig. 2b and c, without considering the competing chelation of glycine,66 apparent binding constants (Ka) of Cu to Aβ40 and compound 6 were 9.64 × 105 M−1 and 7.21 × 106 M−1, respectively. The affinity of compound 6 to Cu was appropriate to disrupt Aβ–Cu interaction (Table S1†), without affecting the normal function of the metal enzymes.67 The binding affinity between compound 5 and Cu was too low to detect by ITC, showing that propargylglycine and glycine have equivalent chelating ability to Cu. Fig. S14† indicates that chelating Cu slightly decreases the fluorescence of compound 6.
:1, illustrating the binding stoichiometry of Cu and 6 was 1:2. For ITC assays, a weaker competitor glycine was added to prevent the precipitation of Cu. As shown in Fig. 2b and c, without considering the competing chelation of glycine,66 apparent binding constants (Ka) of Cu to Aβ40 and compound 6 were 9.64 × 105 M−1 and 7.21 × 106 M−1, respectively. The affinity of compound 6 to Cu was appropriate to disrupt Aβ–Cu interaction (Table S1†), without affecting the normal function of the metal enzymes.67 The binding affinity between compound 5 and Cu was too low to detect by ITC, showing that propargylglycine and glycine have equivalent chelating ability to Cu. Fig. S14† indicates that chelating Cu slightly decreases the fluorescence of compound 6.
We then assessed the ability of compound 6 to photo-oxygenate Aβ and inhibit its aggregation. 1,3-Diphenylisobenzofuran (DPBF)68 was used to detect singlet oxygen (1O2) generated by photo-excited compound 6. Glycerol, which can increase the solution viscosity, was used to restrict the bond rotation of the ThT analogue 6. In contrast to 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, compound 6 produced more 1O2 in a glycerol/HEPES mixed solution (Fig. S15†).55,61 Aβ40 (10 μM) and compound 6 (20 μM) were irradiated with UV light (1 W cm−2) for 1 h and then photo-oxygenated Aβ was detected by MS. Fig. S16† illustrates that both compounds 4 and 6 can effectively oxygenate Aβ40.
Aβ aggregation kinetics were measured with Nile Red (NR), a widely used dye for studying fibril formation.69 As shown in Fig. S17,† the effect of ThT analogues 4 and 6 on NR fluorescence is negligible. Fig. S18† demonstrates that 4 and 6 can inhibit Aβ40 aggregation under light illumination. 8-Anilinonaphthalene-1-sulfonate (ANS), the fluorescence of which is strongly enhanced in the presence of the hydrophobic protein, was also applied to monitor Aβ fibrillation (Fig. S19†). ANS fluorescence was greatly enhanced for native Aβ40 after incubation for 7 days, in contrast to the slight fluorescence change for photo-oxygenated Aβ40. This implies that photo-oxygenation decreased Aβ40 fibrillation and, possibly, toxicity, through increasing protein polarity.56
The inhibition effect of compound 6 on Cu-accelerated Aβ40 aggregation was then evaluated. After UV light (1 W cm−2) irradiation for 1 h, Aβ40 monomer (10 μM) and Cu (10 μM) were incubated with 2 equiv. of 4 or 6 for 24 h. Then, NR assays, circular dichroism (CD), and AFM were conducted to monitor Aβ40 aggregation. As the incubation period progressed, Cu induced Aβ40 to form β-sheet-rich fibrils, while compound 6 blocked this process (Fig. 3). However, due to the lack of a Cu-chelating moiety, compound 4 could not inhibit Cu-induced Aβ40 aggregation.
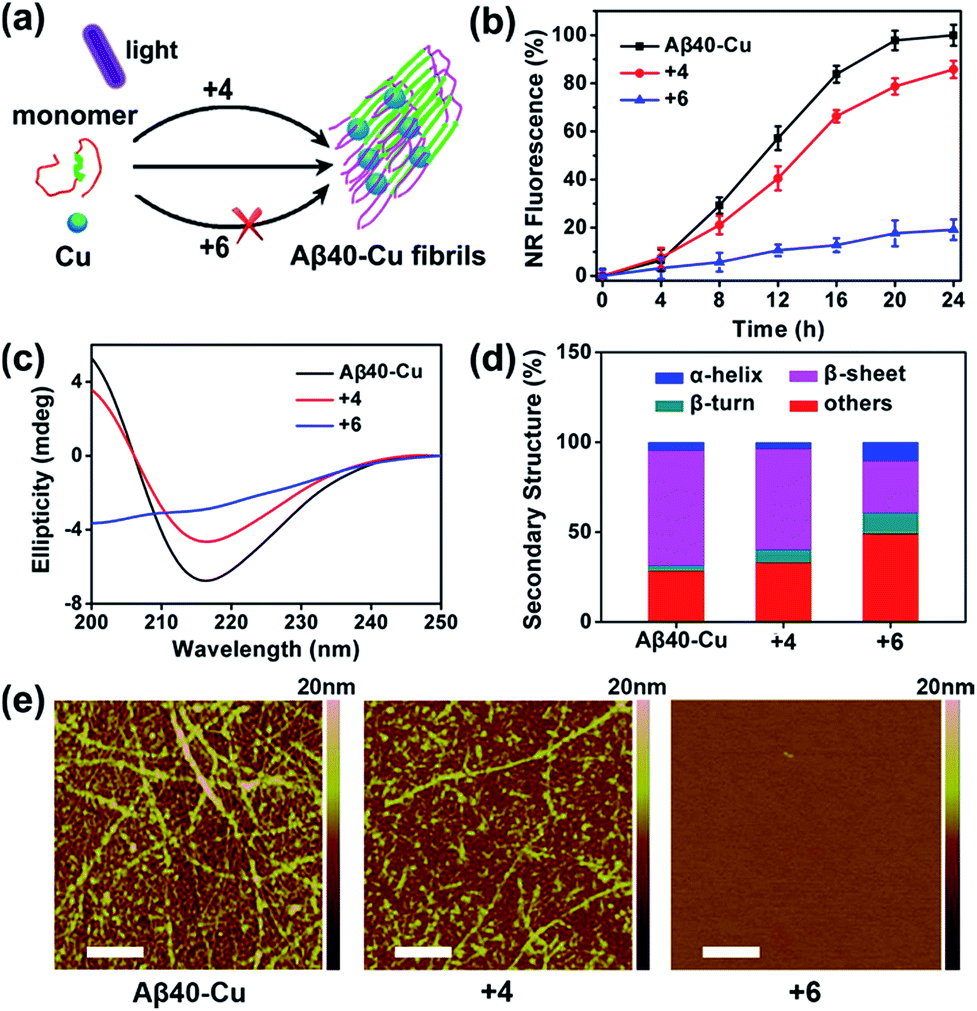 | ||
| Fig. 3 Inhibition of Cu-induced Aβ40 aggregation. (a) Scheme of the interruption of Cu-induced Aβ40 fibrillation with compound 6 under light illumination. (b) Fibrillation kinetics of Aβ–Cu alone or in the presence of compounds 4 and 6, monitored by NR. Each experiment was repeated three times. Error bars indicate ± s.d. (c) CD spectra of Aβ40–Cu alone or in the presence of compounds 4 and 6. (d) Secondary structure analysis of Aβ40–Cu alone or in the presence of compounds 4 and 6. (e) AFM images of Aβ40–Cu alone or in the presence of compounds 4 and 6. Scale bars: 500 nm. | ||
Furthermore, photo-activated compound 6 could also dissociate Aβ40–Cu aggregates. As shown in Fig. S20,† Aβ40–Cu aggregates further assembled into long fibrils after incubation for 24 h. However, NR fluorescence gradually decreased for Aβ40–Cu aggregates when incubated with compound 6, indicating that the Aβ40–Cu aggregates were dissociated over the course of time in the presence of compound 6. AFM and ANS assays further supported that compound 6 dissociates Aβ40–Cu aggregates (Fig. S20†). Compound 6 also inhibits and disassembles Cu-induced Aβ42 aggregates (Fig. S21 and S22†).
Given the above results, we assessed the neuroprotective effect of compound 6 on the cytotoxicity triggered by Aβ40–Cu aggregates. Immunofluorescence analysis showed the co-localization of compound 6 and anti-Aβ antibodies in PC12 cells (Fig. S23†). This implied that photo-activated compound 6 reduces non-specific oxygenation to lipid, DNA or other proteins in the cells.61 Methyl thiazolyl tetrazolium (MTT) assays revealed that 6 protects against Aβ–Cu-mediated cellular toxicity under light illumination (Fig. S24†).
We further evaluated the effectiveness of disassembling Aβ–Cu aggregates by 6 in brain slices of 3 × Tg-AD mice. Aβ plaques were labeled with NR to monitor the therapeutic effects. As shown in Fig. S25,† many Aβ aggregates could be observed in the hippocampus and cortex. For brain slices treated with compound 6 and UV light, the fluorescence of NR became weak after incubating for 12 h (Fig. S25, Table S2†), indicating that photo-activated 6 was able to reduce Cu–Aβ aggregates in brain slices. The immunofluorescence study further confirmed the effectiveness of clearing Aβ plaques using photo-excited compound 6 (Fig. S26†).39
We next investigated whether Aβ40–Cu aggregates could act as a catalyst for the in situ synthesis of the bifunctional drug 6 in cells. It has been proven that silica nanoparticles can penetrate the blood–brain barrier (BBB) by inducing tight junction loss and cytoskeleton arrangement.70,71 Moreover, certain types of silica-based nanoparticles have been approved by the Food and Drug Administration (FDA) for clinical use.72 Herein, H2O2-responsive mesoporous silica nanoparticles (MSN) were used as a controlled-release nanocarrier to deliver the prodrugs to desired sites.73,74 Firstly, phenylboronic acid (BA) was modified on MSN and then MSN–BA was loaded with the prodrugs (4, 5, and AA). Immunoglobulin G (IgG) acted as a capping agent and the formed boronates were sensitive to H2O2.75 The successful fabrication of MSN–IgG was validated by transmission electron microscopy (TEM, Fig. S27†), nitrogen adsorption–desorption isotherms (Fig. S28, Table S3†), Fourier transform infrared spectroscopy (FTIR, Fig. S29†), and thermogravimetric analysis (Fig. S30†). As shown in Fig. S31,† MSN–IgG efficiently released the loaded prodrug 4 when treated with 0.5 mM H2O2. It has been proved that H2O2 level is remarkedly elevated in AD because of Aβ–Cu76–78 and H2O2 levels in partial area of cells can be 1 mM.79 Table S4† shows that MSN can enter the cells.
To evaluate the therapeutic potential of in situ synthesized drug 6 in cells, PC12 cells were incubated with Aβ40–Cu aggregates and prodrug-loaded MSN–IgG. As shown in Fig. 4a, prodrug-loaded MSN–IgG was responsive to H2O2 induced by Aβ40–Cu aggregates,75 and the prodrugs (4, 5, and AA) were released in the cells. Then Aβ40–Cu aggregates catalyzed the click reaction and compound 6 was in situ synthesized, as verified by MS (Fig. S32†). Moreover, the yield of compound 6 increased upon an increase in the concentration of Aβ–Cu (Fig. S33†). In this regard, in situ drug synthesis without the use of the exogenous Cu-catalyst could be self-triggered and self-regulated by the level of Aβ–Cu aggregates in cells.
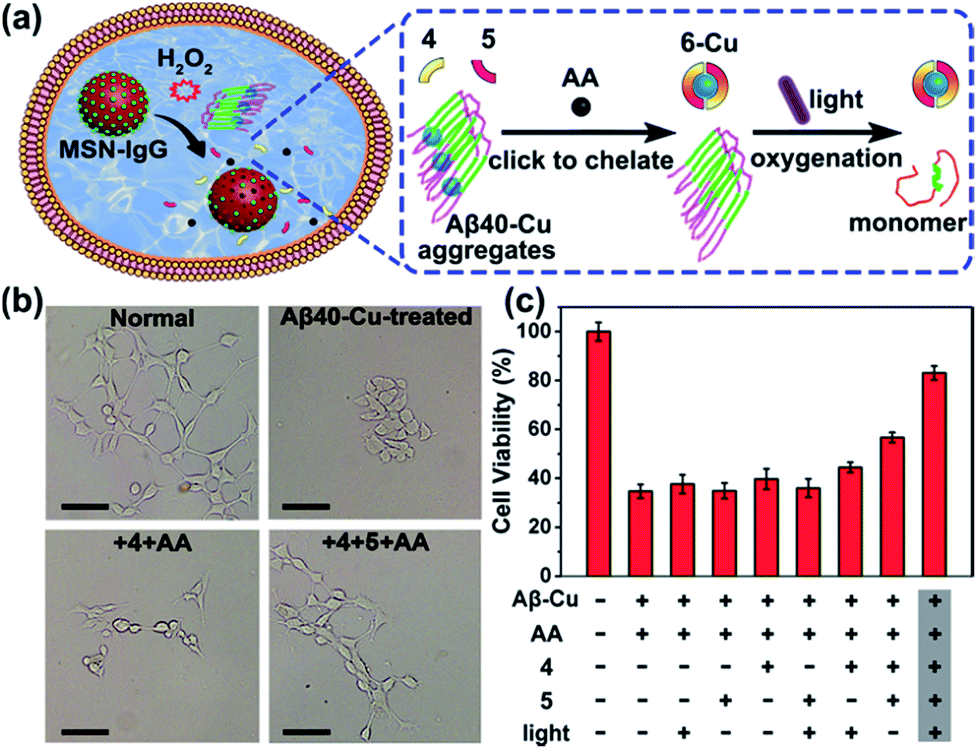 | ||
| Fig. 4 In situ synthesis of bifunctional compound 6 for mitigating Aβ40–Cu-induced cytotoxicity. (a) Overview of the in situ synthesis of compound 6 for disassembling Aβ40–Cu aggregates. (b) Microscope images of the normal PC12 cells and Aβ40–Cu-treated PC12 cells in the presence or absence of prodrug-loaded MSN. Scale bars: 20 μm. (c) MTT assays. Each experiment was repeated three times. Error bars indicate ± s.d. PC12 cells were incubated with Aβ40–Cu aggregates (10 μM) and prodrug-loaded MSN–IgG (0.1 mg mL−1) for 24 h and then irradiated with UV light (1 W cm−2) for 10 min in 20 min intervals 6 times. Cells were incubated for 24 h after the light irradiation and then MTT assays were carried out. | ||
As shown in Fig. 4b and c, Aβ40–Cu aggregates destroyed the cell morphology and caused cell death. In contrast, the in situ synthetic drug 6 restored PC12 cells to their normal morphologies and improved the cell viability from 37% to 82%. However, compound 4 did not display similar neuroprotective activity. Collectively, our results show that compound 6 mitigates the cytotoxicity induced by Aβ–Cu aggregates as a consequence of chelating Cu and photo-oxygenating Aβ.
The AD model C. elegans has been widely used to enhance the understanding of the molecular pathogenesis and identify promising therapeutic strategies.80,81 It has been reported that ThT rescues Aβ-mediated paralysis and increases the longevity of transgenic worms,82 and Cu-chelator clioquinol (CQ) ameliorates Aβ–Cu toxicity to C. elegans.83 As shown in Fig. S34,† photo-excited compound 6 can prolong the lifespan of C. elegans CL2006. We then explored whether the in situ synthesis of bifunctional compound 6 can suppress worm paralysis triggered by Aβ–Cu aggregates. CL2006 worms were cultured in NGM containing 1 mM CuCl2 for 24 h and then Cu-treated worms were transferred to NGM containing prodrug-loaded MSN–IgG (1 mg mL−1) for a further 24 h.
As shown in Fig. S35 and S36,† compound 6 was synthesized in Cu-treated CL2006 worms and 6 was able to co-localize with Aβ. Subsequently, it was confirmed that MSN can access worm tissue (Table S5†). As shown in Fig. 5, Aβ–Cu aggregates induce AD model CL2006 strain paralysis, reducing the lifespan, and behavioral defects,37,83 while the in situ synthesized drug 6 decreases Aβ deposits, rescues Aβ-associated paralysis and improves the motility of the CL2006 strain.
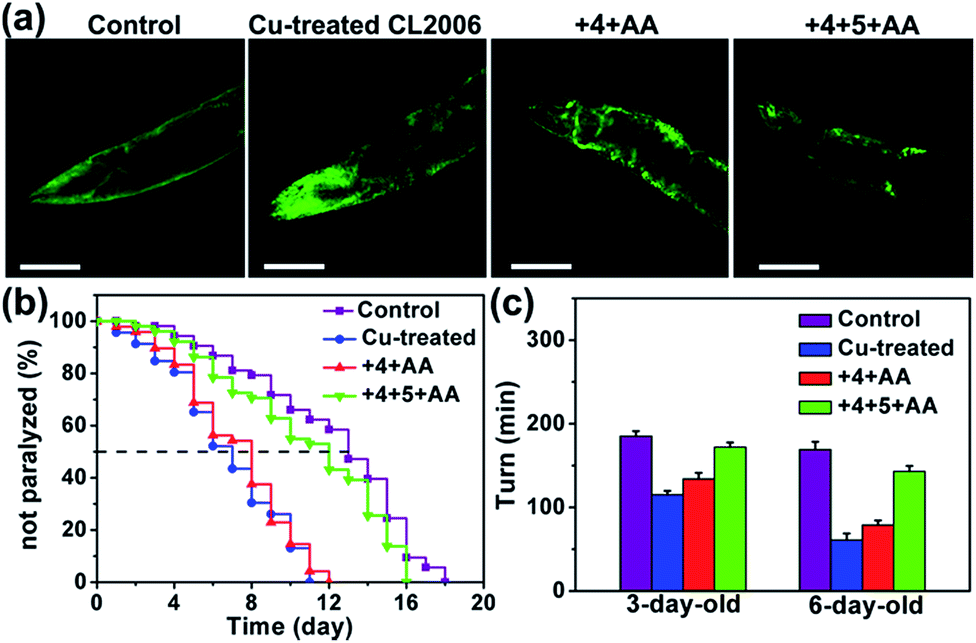 | ||
| Fig. 5 The in situ synthesis of compound 6 for suppressing the toxicity induced by Aβ–Cu fibrils in worms. (a) Representative ThT-staining images of N2 and Cu-treated CL2006 in the presence or absence of prodrug-loaded MSN on the 6th day. Scale bars: 40 μm. (b) Survival curves. (c) Quantification of the worm movement in M9 buffer (turns per minute). Each experiment was repeated three times. Error bars indicate ± s.d. Cu-treated worms were transferred to NGM containing prodrug-loaded MSN–IgG (1 mg mL−1) for 24 h before worms were irradiated with UV light (1 W cm−2) for 10 min in 20 min intervals 6 times. A wild-type N2 strain was used as the control. | ||
Conclusion
In summary, the CuAAC click reaction has been realized by endogenous Cu in vivo, instead of using exogenous Cu-catalysts. We have demonstrated that Aβ–Cu aggregates can catalyze a click reaction in cells, an AD model of C. elegans and brain slices of 3 × Tg-AD mice. The reaction is self-triggered and regulated by the level of Aβ–Cu aggregates. Harnessing the synergy of photo-oxidizing Aβ and chelating Cu, the in situ synthesized drug 6 can dissociate Aβ–Cu aggregates, reduce Aβ burden, suppress Aβ-mediated paralysis and diminish the locomotion defects of the AD model CL2006 strain. Our work provides new insights into in situ drug synthesis against AD using bioorthogonal chemistry.Experimental section
Synthesis of compound 4
A mixture of 2-(4-aminophenyl)-6-methyl-1,3-benzothiazole (1 mmol) and NaNO2 (1.2 mmol) in 5 M HCl (10 mL) was stirred at 4 °C for 30 min. Then, a NaN3 (4 mmol) solution was added dropwise and the mixture was stirred for 2 h at 4 °C. After adjusting the pH to 7, the resulting yellow precipitate was filtered off and washed with water, affording compound 4 (189 mg, 71%). 1H NMR (600 MHz, CDCl3), δ (ppm): 8.08–8.03 (m, 2H), 7.93 (d, J = 8.3 Hz, 1H), 7.68 (s, 1H), 7.33–7.28 (m, 1H), 7.14–7.09 (m, 2H), 2.50 (s, 3H). MS (ESI) m/z: [M + H]+ calcd for C14H10N4S, 267.3; found, 267.2.Synthesis of compound 6
Compound 4 (1 mmol), 5 (1.1 mmol), CuSO4 (0.1 mmol) and AA (0.2 mmol) were added to a solution of H2O and ethanol (1:1, 20 mL) and stirred in the dark at 37 °C for 24 h. Then, ethylenediaminetetraacetic acid disodium salt (EDTA, 0.3 mmol) was added and the mixture was stirred for 1 h to chelate Cu. The ethanol was then removed under reduced pressure and the pH of the solution was adjusted to 7. The resulting yellow precipitate was filtered off and washed with saturated salt water, affording compound 6 as a yellow solid (345 mg, 91%). 1H NMR (600 MHz, D2O/DCl), δ (ppm): 7.15 (s, 1H), 6.34 (d, J = 8.3 Hz, 2H), 6.19 (d, J = 8.3 Hz, 2H), 5.98 (d, J = 9.1 Hz, 2H), 5.60 (d, J = 8.5 Hz, 1H), 2.86 (t, J = 6.4 Hz, 1H), 1.91 (qd, J = 15.9 Hz, 2H), 0.46 (s, 3H). MS (ESI) m/z: [M − H]− calcd for C19H17N5O2S, 378.4; found, 378.4.
Peptide preparation
Aβ1–40 and Aβ1–42 were purchased from ChinaPeptides Co., Ltd. The peptides were dissolved in hexafluoroisopropanol (HFIP) and stored at −20 °C as the stock solution. Before use, the solvent HFIP was evaporated under a gentle stream of nitrogen and Aβ was re-dissolved in HEPES buffer (20 mM, pH 7.4). For preparing Aβ40–Cu aggregates, 100 μM Aβ40 was incubated with an equimolar amount of CuCl2 at 37 °C for 8 h and then diluted to the corresponding concentration before use. For preparing Aβ42–Cu aggregates, 100 μM of Aβ42 was incubated with an equimolar amount of CuCl2 at 37 °C for 4 h and then diluted to the corresponding concentration before use. The Aβ–Cu species were dialyzed to remove any free Cu before the click reaction.ITC measurements
ITC experiments were carried out on a NANO ITC System (TA Instruments Inc., USA) in HEPES buffer (20 mM, pH 7.4) with stirring at 300 rpm at 25 °C. Aβ or compound 6 was placed in the cell and the CuCl2 solution was loaded via a syringe. To prevent the precipitation of Cu2+, 4 equiv. of glycine were added to the CuCl2 solution. All solutions were degassed for 20 min before titration. The apparent binding constant (Ka), binding enthalpy (ΔH), and binding stoichiometry (n) were determined by fitting ITC data following information from the literature without considering the competing effects of glycine with Cu2+.66,67 When Cu2+–glycine was taken into consideration, the Ka values of Cu to Aβ and compound 6 were 1.63 × 109 M−1 and 1.04 × 1010 M−1, respectively.65 For binding between Aβ and Cu2+, [Aβ] = 20 μM, [Cu2+] = 200 μM. For binding between 6 and Cu2+, [6] = 40 μM, [Cu2+] = 200 μM.CD measurements
CD spectra were collected at 37 °C at a speed of 5 nm min−1 over the wavelength range from 200 to 250 nm. The secondary structure was analyzed using the BeStSel program (http://bestsel.elte.hu/).84C. elegans experiments
Transfected C. elegans strain CL2006 constitutively expresses human Aβ1–42 in muscle tissues and thus displays an age-related progressive defect of muscle-specific motility. CL2006 and wild-type (N2) strain were fed with E. coli (OP50) and cultured at 20 °C in NGM. For worm paralysis assay, N2 and CL2006 worms were cultured at 20 °C on new NGM plates (about 50 worms per plate) after egg synchronization. Worms were classified as paralyzed if they failed to respond to a touch-provoked movement. For ThT-staining experiments, worms were fixed with 4% paraformaldehyde for 24 h at 4 °C and rendered permeable in permeabilization buffer (1% Triton X-100, 5% β-mercaptoethanol) for a further 24 h at 37 °C. After washing with PBS, the worms were immersed in ThT solution (2 mM) for 5 min. After washing with PBS, the worms were mounted on slides and observed using a fluorescence microscope.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21431007, 21533008, 21820102009, 21871249, 91856205) and the Frontier Science Key Program of CAS (QYZDJ-SSW-SLH052) and 20190701028GH from Jilin Province.References
- J. A. Cotruvo Jr, A. T. Aron, K. M. Ramos-Torres and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4400–4414 RSC
.
- Y. Liu, Q. Su, M. Chen, Y. Dong, Y. Shi, W. Feng, Z. Y. Wu and F. Li, Adv. Mater., 2016, 28, 6625–6630 CrossRef CAS PubMed
- Q. Wang and K. J. Franz, Acc. Chem. Res., 2016, 49, 2468–2477 CrossRef CAS PubMed
- W. Wu, L. Yu, Q. Jiang, M. Huo, H. Lin, L. Wang, Y. Chen and J. Shi, J. Am. Chem. Soc., 2019, 141, 11531–11539 CrossRef CAS PubMed
- S. Bakthavatsalam, M. L. Sleeper, A. Dharani, D. J. George, T. Zhang and K. J. Franz, Angew. Chem., Int. Ed., 2018, 57, 12780–12784 CrossRef CAS PubMed
- J. Geng, M. Li, L. Wu, J. Ren and X. Qu, J. Med. Chem., 2012, 55, 9146–9155 CrossRef CAS PubMed
- J. F. Reuther, J. L. Dees, I. V. Kolesnichenko, E. T. Hernandez, D. V. Ukraintsev, R. Guduru, M. Whiteley and E. V. Anslyn, Nat. Chem., 2018, 10, 45–50 CrossRef CAS PubMed
- Y. Sun, S. Hong, R. Xie, R. Huang, R. Lei, B. Cheng, D. E. Sun, Y. Du, C. M. Nycholat, J. C. Paulson and X. Chen, J. Am. Chem. Soc., 2018, 140, 3592–3602 CrossRef CAS PubMed
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed
- Y. Zeng, J. Q. Ren, A. G. Shen and J. M. Hu, J. Am. Chem. Soc., 2018, 140, 10649–10652 CrossRef CAS PubMed
- G. Liu, G. Shi, H. Sheng, Y. Jiang, H. Liang and S. Liu, Angew. Chem., Int. Ed., 2017, 56, 8686–8691 CrossRef CAS PubMed
- W. Zheng, H. Li, W. Chen, J. Zhang, N. Wang, X. Guo and X. Jiang, Small, 2018, 14, e1703857 CrossRef PubMed
- D. S. Tyler, J. Vappiani, T. Caneque, E. Y. N. Lam, A. Ward, O. Gilan, Y. C. Chan, A. Hienzsch, A. Rutkowska, T. Werner, A. J. Wagner, D. Lugo, R. Gregory, C. Ramirez Molina, N. Garton, C. R. Wellaway, S. Jackson, L. MacPherson, M. Figueiredo, S. Stolzenburg, C. C. Bell, C. House, S. J. Dawson, E. D. Hawkins, G. Drewes, R. K. Prinjha, R. Rodriguez, P. Grandi and M. A. Dawson, Science, 2017, 356, 1397–1401 CrossRef CAS PubMed
- I. Glassford, C. N. Teijaro, S. S. Daher, A. Weil, M. C. Small, S. K. Redhu, D. J. Colussi, M. A. Jacobson, W. E. Childers, B. Buttaro, A. W. Nicholson, A. D. MacKerell Jr, B. S. Cooperman and R. B. Andrade, J. Am. Chem. Soc., 2016, 138, 3136–3144 CrossRef CAS PubMed
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed
- F. Alonso, Y. Moglie and G. Radivoy, Acc. Chem. Res., 2015, 48, 2516–2528 CrossRef CAS PubMed
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS PubMed
- J. Chen, J. Wang, Y. Bai, K. Li, E. S. Garcia, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2018, 140, 13695–13702 CrossRef CAS PubMed
- C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed
- B. Wang, J. Durantini, J. Nie, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2016, 138, 13127–13130 CrossRef CAS PubMed
- J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598–1601 CrossRef CAS PubMed
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS PubMed
- P. C. Ke, M. A. Sani, F. Ding, A. Kakinen, I. Javed, F. Separovic, T. P. Davis and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 6492–6531 RSC
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed
- A. Nakamura, N. Kaneko, V. L. Villemagne, T. Kato, J. Doecke, V. Dore, C. Fowler, Q. X. Li, R. Martins, C. Rowe, T. Tomita, K. Matsuzaki, K. Ishii, K. Ishii, Y. Arahata, S. Iwamoto, K. Ito, K. Tanaka, C. L. Masters and K. Yanagisawa, Nature, 2018, 554, 249–254 CrossRef CAS PubMed
- C. Hureau and P. Faller, Biochimie, 2009, 91, 1212–1217 CrossRef CAS PubMed
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603–4606 CrossRef CAS PubMed
- C. Le Droumaguet, C. Wang and Q. Wang, Chem. Soc. Rev., 2010, 39, 1233–1239 RSC
- R. Zeineddine and J. J. Yerbury, Frontiers in Physiology, 2015, 6, 277–284 CrossRef PubMed
- Y. Li, D. Cheng, R. Cheng, X. Zhu, T. Wan, J. Liu and R. Zhang, PLoS One, 2014, 9, e99939 CrossRef PubMed
- J. J. Yerbury, Prion, 2016, 10, 119–126 CrossRef CAS PubMed
- Y. Luo, J. Zhang, N. Liu, Y. Luo and B. Zhao, Sci. China: Life Sci., 2011, 54, 527–534 CrossRef CAS PubMed
- J. A. Varela, J. P. Dupuis, L. Etchepare, A. Espana, L. Cognet and L. Groc, Nat. Commun., 2016, 7, 10947–10956 CrossRef CAS PubMed
- H. Sun, J. Liu, S. Li, L. Zhou, J. Wang, L. Liu, F. Lv, Q. Gu, B. Hu, Y. Ma and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5988–5993 CrossRef CAS PubMed
- W. Liu, H. Dong, L. Zhang and Y. Tian, Angew. Chem., Int. Ed., 2017, 56, 16328–16332 CrossRef CAS PubMed
- Y. Luo, L. Zhang, W. Liu, Y. Yu and Y. Tian, Angew. Chem., Int. Ed., 2015, 54, 14053–14056 CrossRef CAS PubMed
- M. W. Bourassa, A. C. Leskovjan, R. V. Tappero, E. R. Farquhar, C. A. Colton, W. E. Van Nostrand and L. M. Miller, Biomed. Spectrosc. Imaging, 2013, 2, 129–139 CAS
- G. L. Song, C. Chen, Q. Y. Wu, Z. H. Zhang, R. Zheng, Y. Chen, S. Z. Jia and J. Z. Ni, Metallomics, 2018, 10, 1107–1115 RSC
- M. Hoop, A. S. Ribeiro, D. Rösch, P. Weinand, N. Mendes, F. Mushtaq, X.-Z. Chen, Y. Shen, C. F. Pujante, J. Puigmartí-Luis, J. Paredes, B. J. Nelson, A. P. Pêgo and S. Pané, Adv. Funct. Mater., 2018, 28, 1705920 CrossRef
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. Pan, Z. Yuan, L. E. Otterbein and B. Wang, Nat. Chem., 2018, 10, 787–794 CrossRef CAS PubMed
- Y. Li, Q. Zou, C. Yuan, S. Li, R. Xing and X. Yan, Angew. Chem., Int. Ed., 2018, 57, 17084–17088 CrossRef CAS PubMed
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2019, 58, 6987–6992 CrossRef CAS PubMed
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC
- A. M. Perez-Lopez, B. Rubio-Ruiz, V. Sebastian, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaria and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548–12552 CrossRef CAS PubMed
- T. Volker and E. Meggers, Curr. Opin. Chem. Biol., 2015, 25, 48–54 CrossRef PubMed
- A. Unciti-Broceta, E. M. Johansson, R. M. Yusop, R. M. Sanchez-Martin and M. Bradley, Nat. Protoc., 2012, 7, 1207–1218 CrossRef CAS PubMed
- X. Zhang, R. Huang, S. Gopalakrishnan, R. Cao-Milán and V. M. Rotello, Trends in Chemistry, 2019, 1, 90–98 CrossRef
- R. Krishnan, J. L. Goodman, S. Mukhopadhyay, C. D. Pacheco, E. A. Lemke, A. A. Deniz and S. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11172–11177 CrossRef CAS PubMed
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS PubMed
- J. Ni, A. Taniguchi, S. Ozawa, Y. Hori, Y. Kuninobu, T. Saito, T. C. Saido, T. Tomita, Y. Sohma and M. Kanai, Chem, 2018, 4, 807–820 CAS
- Z. Du, N. Gao, X. Wang, J. Ren and X. Qu, Small, 2018, 14, e1801852 CrossRef PubMed
- A. Aliyan, B. Kirby, C. Pennington and A. A. Marti, J. Am. Chem. Soc., 2016, 138, 8686–8689 CrossRef CAS PubMed
- B. I. Lee, S. Lee, Y. S. Suh, J. S. Lee, A. K. Kim, O. Y. Kwon, K. Yu and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 11472–11476 CrossRef CAS PubMed
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2018, 119, 1221–1322 CrossRef PubMed
- M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. Wee Yong, S. Tsutsui, P. K. Stys and T. Storr, Chem. Sci., 2017, 8, 5636–5643 RSC
- A. Taniguchi, Y. Shimizu, K. Oisaki, Y. Sohma and M. Kanai, Nat. Chem., 2016, 8, 974–982 CrossRef CAS PubMed
- D. Ozawa, H. Yagi, T. Ban, A. Kameda, T. Kawakami, H. Naiki and Y. Goto, J. Biol. Chem., 2009, 284, 1009–1017 CrossRef CAS PubMed
- J. Notni and H. J. Wester, Chem.–Eur. J., 2016, 22, 11500–11508 CrossRef CAS PubMed
- N. Zabarska, A. Stumper and S. Rau, Dalton Trans., 2016, 45, 2338–2351 RSC
- L. Q. Hatcher, L. Hong, W. D. Bush, T. Carducci and J. D. Simon, J. Phys. Chem. B, 2008, 112, 8160–8164 CrossRef CAS PubMed
- X. Hu, Q. Zhang, W. Wang, Z. Yuan, X. Zhu, B. Chen and X. Chen, ACS Chem. Neurosci., 2016, 7, 1255–1263 CrossRef CAS PubMed
- H. Zhang, C. Zhang, X. Y. Dong, J. Zheng and Y. Sun, J. Mol. Recognit., 2018, 31, e2697 CrossRef PubMed
- Z. Wang, Y. Zhang, E. Ju, Z. Liu, F. Cao, Z. Chen, J. Ren and X. Qu, Nat. Commun., 2018, 9, 3334–3347 CrossRef PubMed
- R. Mishra, D. Sjolander and P. Hammarstrom, Mol. BioSyst., 2011, 7, 1232–1240 RSC
- D. Ye, S. Anguissola, T. O'Neill and K. A. Dawson, Nanoscale, 2015, 7, 10050–10058 RSC
- X. Liu, B. Sui and J. Sun, Biomaterials, 2017, 121, 64–82 CrossRef CAS PubMed
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS PubMed
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 28, 5553–5585 CrossRef CAS PubMed
- J. Geng, M. Li, L. Wu, C. Chen and X. Qu, Adv. Healthcare Mater., 2012, 1, 332–336 CrossRef CAS PubMed
- P. Shi, M. Li, J. Ren and X. Qu, Adv. Funct. Mater., 2013, 23, 5412–5419 CrossRef CAS
- G. F. da Silva and L. J. Ming, Angew. Chem., Int. Ed., 2005, 44, 5501–5504 CrossRef CAS PubMed
- G. F. da Silva and L. J. Ming, Angew. Chem., Int. Ed., 2007, 46, 3337–3341 CrossRef CAS PubMed
- W. M. Tay, G. F. da Silva and L. J. Ming, Inorg. Chem., 2013, 52, 679–690 CrossRef CAS PubMed
- K. E. Broaders, S. Grandhe and J. M. Frechet, J. Am. Chem. Soc., 2011, 133, 756–758 CrossRef CAS PubMed
- T. M. Dawson, T. E. Golde and C. Lagier-Tourenne, Nat. Neurosci., 2018, 21, 1370–1379 CrossRef CAS PubMed
- Y. Guan, Z. Du, N. Gao, Y. Cao, X. Wang, P. Scott, H. Song, J. Ren and X. Qu, Sci. Adv., 2018, 4, eaao6718 CrossRef PubMed
- S. Alavez, M. C. Vantipalli, D. J. Zucker, I. M. Klang and G. J. Lithgow, Nature, 2011, 472, 226–229 CrossRef CAS PubMed
- K. E. Matlack, D. F. Tardiff, P. Narayan, S. Hamamichi, K. A. Caldwell, G. A. Caldwell and S. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4013–4018 CrossRef CAS PubMed
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04387j |
| This journal is © The Royal Society of Chemistry 2019 |