
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClosed-to-open-shell ground state photoswitching of thienyl-based acylhydrazones†
Martin Šetek a,
Valentino L. P. Guerraa,
Harry Robsona,
Anna O. Geleveryaa,
Ondřej Maxaa,
Anna Lamancováab,
Václav Eignera,
Dana Nachtigallovábc,
Ján Tarábekb and
Petr Kovaříček*a
a,
Valentino L. P. Guerraa,
Harry Robsona,
Anna O. Geleveryaa,
Ondřej Maxaa,
Anna Lamancováab,
Václav Eignera,
Dana Nachtigallovábc,
Ján Tarábekb and
Petr Kovaříček*a
aUniversity of Chemistry and Technology Prague, Technická 5, 166 28, Prague 6, Czechia. E-mail: petr.kovaricek@vscht.cz; Tel: +420 220 444 165
bInstitute of Organic Chemistry and Biochemistry, Flemingovo nám. 542/2, 160 00, Prague 6, Czechia
cIT4Innovations, VSB-Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava Poruba, Czech Republic
First published on 3rd April 2025
Abstract
Photoswitches convert a photon to molecular information through a reversible change in bonding or configuration. Applications of photoswitches are determined by their different properties in the two states, spanning from catalysis, to allosteric activation, to photopharmacology, up to materials and devices such as memories that can store optical information in chemical bonds. For the latter, operation in the solid state and different means to read/write/erase the information must be possible. These features, however, are challenging to achieve due to close molecular packing and intermolecular interactions. Here, we present acylhydrazones that photoswitch even in the solid state and adopt an open-shell triplet ground state electronic configuration in the photoswitched form, which is very long-lived. The optically stored information can thus be read magnetically and completely erased by applying an electrochemical potential. In addition, ab initio calculations identified the closed-to-open-shell pathway and explained the singlet–triplet crossing. Photoswitching of the closed/open-shell ground state electronic structure is a fundamentally new feature, which can bring new opportunities and applications. We demonstrate it in an example of the open-shell state of the photoswitch used as an initiator of radical benzylic bromination and radical debromination of an aromatic ring. However, the presented phenomenon is robust and insensitive to conditions, thus showing potential in a broad range from heterogeneous catalysis to spintronics.
Introduction
Photoswitching is an intramolecular transformation between two states triggered by light absorption.1–5 Based on the photochrome structure, photoswitches are grouped into prototypical families such as azobenzenes,5,6 stilbenes,7 spiropyrans,4 indigos,8 diarylethenes,9 fulgides,10 and (acyl)hydrazones.2,11 Photoswitching most commonly occurs via isomerization of a double bond or electrocyclisation.1 The character of this transformation translates into property differences between the two switched forms and hence into the application potential. Configurational switching has been used for catalytic site shielding,12 allosteric activation,13 photocomplexation14,15 and photopharmacology.16,17 Electrocyclizations change the conjugated backbone and finds applications in (organo)catalysis,18,19 smart materials,4,20,21 and optical memories.3,9,10 For memories, however, the read-out of the information should occur by non-optical means so that secondary light does not alter the stored information. Electrical22 or magnetic23 reading could be an option if the magnetic or electronic properties change significantly on photoswitching, i.e. if the two forms have different electronic configurations, such as closed- and open-shell. Moreover, a radical form of open-shell species is highly interesting for the initiation and catalysis of reactions.Acylhydrazones are known to perform reversible photoisomerisation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond between the more thermodynamically stable E-form and the Z-form.2,24–26 The reverse isomerization (Z → E backswitching) can occur both thermally (T-type photoswitches) or exclusively by excitation at different wavelengths (P-type), depending on the isomerization energy barrier and spectral separation of the two forms.
N bond between the more thermodynamically stable E-form and the Z-form.2,24–26 The reverse isomerization (Z → E backswitching) can occur both thermally (T-type photoswitches) or exclusively by excitation at different wavelengths (P-type), depending on the isomerization energy barrier and spectral separation of the two forms.
P-type switching is particularly interesting for optical information storage3,9,10 and catalysis because the properties associated with the Z-form do not revert at a significant rate at a given temperature.11,21,27 Further properties are required for application in practical devices: (1) the information must be written reliably, i.e. with the highest possible quantum yield;3 (2) the information reading must not alter the stored information, that is, the quantum yield of backswitching should be much lower or not occur during the reading process;28 and (3) the photoswitching properties must be achieved in the solid state.29–31 For catalysis, additional features such as stability under the reaction conditions and solubility are required. Achieving solid-state photoswitching is a paramount challenge due to efficient intermolecular quenching of excited states and the high steric hindrance imposed by molecular close packing.
Here, we present a series of acylhydrazones with the thiophene moiety in the azomethine part of the molecules to (1) form long-lived open-shell radicals in their photoswitched form, (2) erase the stored information by an electric impulse, and (3) operate in the solid state. This alone demonstrates the potential of the series of acylhydrazones for application in solid-state devices. We further show their application in catalysis.
Results and discussion
Acylhydrazones bearing the 2-thienyl group on the azomethine side have been reported2 to exhibit a large bathochromic shift of absorbance in the photoswitched form, so that both forms can be irradiated specifically without the contribution of the other. The large bathochromic shift is particularly intriguing because both E- and Z-forms are anticipated to be rather planar and thus no dramatic change in conjugation is expected. Therefore, the anticipated planarity of the Z-state is in dispute and hereafter we address this state as the ‘photoswitched form’ to discuss its geometry below. Synthesis of acylhydrazones is a trivial procedure (Fig. 1), providing the corresponding products in high yields. The starting compounds are available commercially at low cost or can be readily synthesized by conventional methods. The ease of procurement allows straightforward scale-up.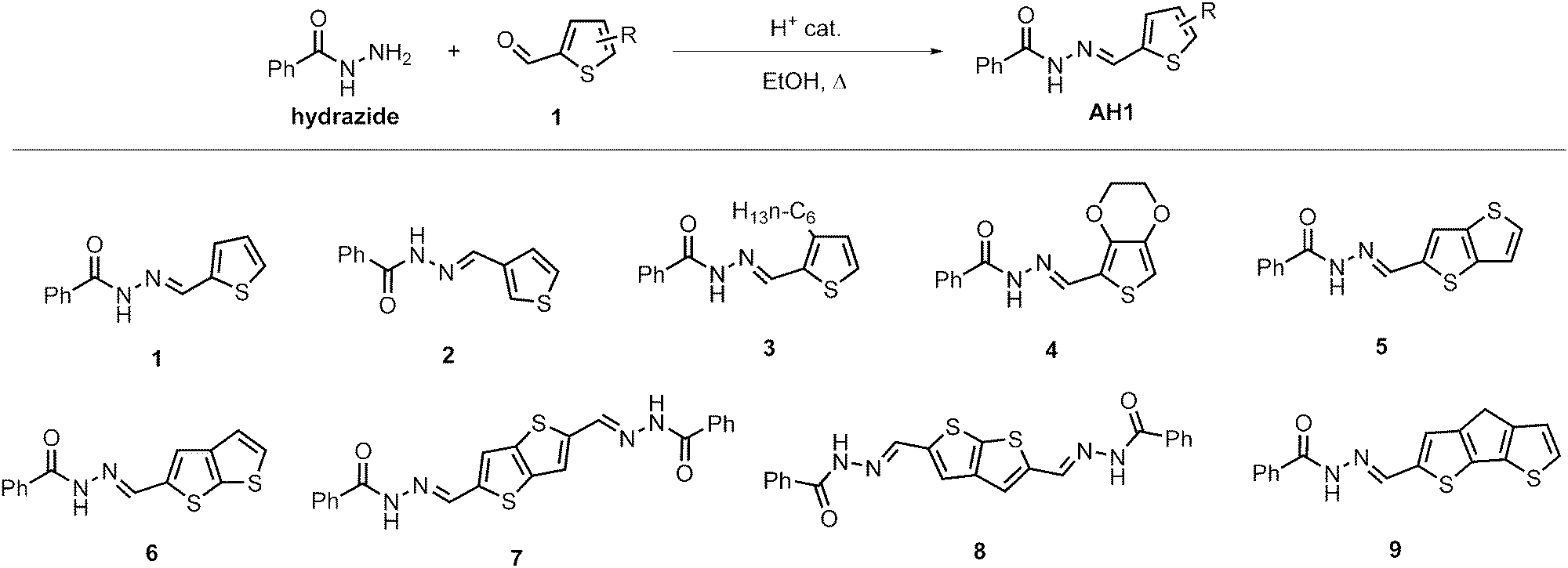 | ||
| Fig. 1 Scheme of acylhydrazone synthesis and the list of prepared photoswitches with their acronyms used in this study. | ||
The series of prepared derivatives (Fig. 1) showed similar photophysical properties, i.e. a large bathochromic shift of absorbance on photoswitching, negligible thermal back conversion in the solid state, and slow back-isomerisation by visible light for all 2-thienyl-based acylhydrazones in acetonitrile solution (AH1, AH3–9; see also the ESI†). The absorbance maxima in both E- and photoswitched forms shift bathochromically with extended conjugation from UV to the blue visible range (400–450 nm) in the case of thienothiophene derivatives (Fig. 2). Furthermore, excellent switching repeatability was observed in the case of AH1 and AH3–9 (see the Photoswitching section, ESI†).
 | ||
| Fig. 2 UV-vis spectra of AH5 recorded during irradiation at the 340 nm absorbance maximum. Similar results are observed for all 2-thienyl based acylhydrazones. Excellent separation of new absorbance features associated with the photoswitched form was confirmed in the whole series (see the ESI† for experimental traces of other derivatives). | ||
AH2 was an exception to the series. It did not show any new absorbance features in the UV-vis spectra upon irradiation and therefore the Z-form cannot be specifically addressed photochemically (Fig. S6, ESI†). AH7 and AH8 could also be predicted to be different due to potential two-stage photoswitching. However, UV-vis spectra recorded during irradiation show isosbestic points. This suggests that only one of the acylhydrazone moieties can be isomerized even at prolonged irradiation times as the UV-Vis spectra do not evolve after 5 minutes of irradiation (Fig. S25 and S29, ESI†). This is supported also by the 1H NMR spectrum, in which the loss of symmetry leads to the emergence of a new set of signals (Fig. S27, ESI†). The 1H NMR spectra remained the same even at a prolonged irradiation time (12 h). We have determined the quantum yield for the E → photoswitched-form isomerization of all AHs (e.g. the quantum yield of AH1 is 0.39; see Section S4, ESI†), which is in line with other reported acylhydrazone photoswitches.2
The photoswitching properties obtained in acetonitrile solutions motivated us to study our set of acylhydrazones in the solid state. Thin films prepared by drop-casting acylhydrazone AH1–AH9 solutions indeed showed reversible photoswitching (see the ESI† for solid-state photoswitching). The anticipated severe steric demands of switching in the solid state were shown to be less significant by the XRD single crystal structure (Section S12, ESI†). Crystal packing showed void volumes around the phenyl group of the hydrazide part. Importantly, the CN E → photoswitched-form transformation can occur through flipping of the carbonyl moiety, accompanied by rotation around the carbonyl–phenyl σ-bond without actual rotation of the benzene ring. In this scenario, most of the steric obstacles are obviated and the free space around the phenyl group provides the necessary degree of freedom to facilitate the flipping of the rather small N–NH–(CO)– group. However, despite our efforts, we did not achieve photoswitching of the single crystal nor were we able to obtain a monocrystal of the photoswitched form.
Due to scattering in solid films of the photoswitches causing significant spectral broadening, we have prepared ‘solid solutions’ of AH1–9 in a poly(methyl methacrylate) (PMMA) matrix and repeated photoswitching studies (see Section S6, ESI,† for photoswitching of AHs in the PMMA matrix). In all cases, the recorded spectra followed the same trends as those of acetonitrile solutions, i.e. a new bathochromic absorbance of photoswitched isomers, a long-lived photoswitched state, and slow back-isomerization by visible light. These experiments have two important implications. First, steric hindrance and intermolecular quenching of excited states in the crystal packing or polymer matrix do not prohibit switching of these acylhydrazones. Second, the bathochromically shifted absorbance feature of the photoswitched form is of the monomolecular origin because excimer formation is difficult to imagine in the PMMA matrix.
We used cyclic voltammetry (CV) and in situ UV-vis spectroelectrochemistry to clarify the particular behavior of 2-thienyl-based acylhydrazones. CV showed up to three irreversible oxidation waves in the range of 1.6 to 2.0 V for all acylhydrazones (Section S7, ESI†), and double acylhydrazones AH7 and AH8 also exhibited a reversible reduction peak at around −0.4 V. For spectroelectrochemistry, we irradiated the acylhydrazone solution in acetonitrile with light (wavelength adjusted to the most bathochromic absorbance maximum) to isomerize to the photoswitched form. After reaching the photostationary state (PSS), we performed cyclic voltammetry and monitored the UV-vis spectra. When a CV cycle of 0 V → 2.5 V → −2.0 V → 0 V was applied, a dramatic and rapid change observed in the UV-Vis spectrum indicated complete reversal of the photoswitched form to the E-form (Fig. 3, kinetics in the ESI†). This behavior is common to all 2-thienyl-based acylhydrazones in the studied series. Switching under cyclic voltammetry is interesting, as it shows that the optical information stored in the configuration of the molecule can be efficiently erased by applying an electrochemical stimulus. In essence, AHs act as “electroswitches”. We attribute the switching to the radical cation propagation in the medium, which has been described on azobenzenes.32
 | ||
| Fig. 3 UV-Vis spectra of AH5 photoisomerization (black to blue) and the spectrum after the CV cycle (red). The CV cycle led to a dramatic and instantaneous change in the UV-Vis spectrum, indicating a complete reversal to the E-form. The 1H NMR spectrum shows that only the photoswitched form of AH5 was transformed back to aldehyde after CV. | ||
The body of data obtained from spectroscopy and electrochemistry of the photoswitched form led us to suspect an open-shell electron configuration. We have employed electron paramagnetic resonance (EPR) studies in solution in the presence of a spin trap. To investigate this, AH5 was converted from E- to the photoswitched form in the presence of a spin trap (N-t-butyl-α-phenylnitrone, by irradiating with 340 nm light). In the resulting solution, an EPR spectrum gave a clear signature of a trapped radical species (Fig. 4). The solution without the trap agent did not show any detectable signal. Therefore, the magnetic properties of the compound can be “read” by an independent method (EPR) from the one that caused photoswitching.
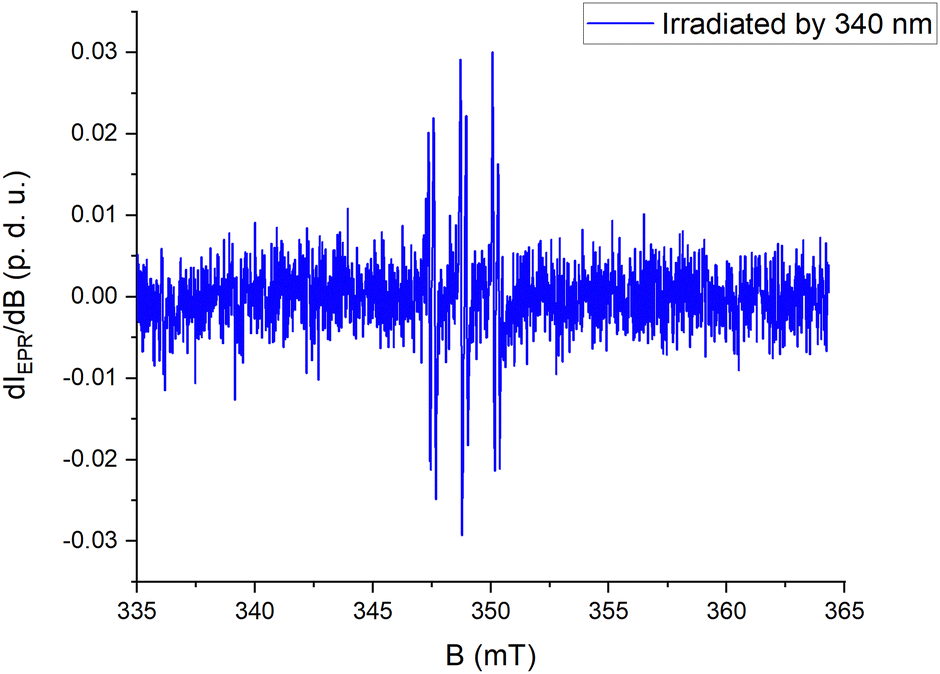 | ||
| Fig. 4 EPR spectrum (X-band) of the trapped radical species between irradiated AH5 and the spin trap agent in acetonitrile. Concentrations were set to c(AH5) = 10 mM and c(N-t-butyl-α-phenylnitrone) = 45 mM. The EPR spectrum was centered at giso = 2.0065. The hyperfine splitting structure comes from a (1 × 14N) = 1.35 mT and a (1 × 1H) = 0.22 mT. | ||
To shed light on the electronic structure and transitions, calculations of the ground and excited states were performed. All vertical excitation energies were calculated using the Complete Active Space Second Order Perturbation Theory (CASPT2) method, with the zero-order wavefunction obtained with state-averaged complete active space (SA-CASSCF) on AH1 and AH2. These two systems are the smallest in the investigated series and thus accessible at such a high level of calculations. Both AH1 and AH2 were investigated to compare the molecules that exhibit photoswitching associated with the bathochromic absorbance shift (AH1) with those that do not (AH2).
The AH1 ground state optimization indicated a mix of two ground state geometries close in energy, differing only in thiophene ring conformation (s-cis and s-trans, Fig. S75, ESI†). Calculation of vertical transitions to S1–3 produced results consistent with the experimentally measured UV-Vis spectra (Tables S1, S2 and Fig. S1, ESI;† summarized in Fig. 5). Relaxation of these excited states follows three pathways leading to local conformational minima (Table S3, ESI,† “radiative geometry 1”, “radiative geometry 2” and “switching geometry” in Fig. 5). The radiative geometries have a very similar torsion angle along the CN bond to the ground state structure (Tables S3 and S7, ESI†). Their relaxation by photon emission leads to a fluorescence spectrum that agrees very well with the experiment (Fig. 5).
 | ||
| Fig. 5 The proposed scheme for the photoswitching and radiative decay of AH1 upon excitation from the lowest energy ground state geometry to the S1–S3 states. Optimization of these states pointed at three geometries: “radiative geometry 1”, “radiative geometry 2” and “switching geometry” shown in the bottom center. In the “switching geometry”, SOC allowed S0 → T1 crossing, followed by further relaxation to T1 minima. Various energy differences are highlighted with their corresponding wavelengths and compared to experimental UV-vis spectra (top left) and fluorescence spectra (bottom left). T1 → T2 excitation represents a pathway for the back photoswitching. The molecular orbitals with occupancy close to 1 of the open-shell electronic structure in S1 and T1 are depicted in the bottom right. | ||
The “switching geometry” of AH1 has some unique properties. Along the S1 relaxation pathway to the “switching geometry”, the torsion angle along the CN bond changes from almost parallel to basically perpendicular (Table S3, ESI†). In this geometry, the S0 state most closely resembles the S1 state in the ground state geometry with both having open-shell character corresponding to an imine π → π* transition.
It is hence assigned that the ground state of AH1 undergoes vertical excitation to the S1 state in the “ground state geometry”, which relaxes to the S0 state in the “switching geometry” through a conical intersection. It is from here that conversion to the triplet state most likely occurs due to (1) the high S0 → T1 spin–orbit coupling (SOC) of 22 cm−1 calculated in this geometry (Table S4, ESI†), and (2) the lower energy of T1 than that of S0. This T1 state in the switching geometry relaxes further by 0.46 eV or 0.53 eV to two geometrically almost identical T1 minima. Calculation of T1 → T2 vertical excitations from these geometrical minima suggested peaks at 3.04 eV and 3.33 eV (408 and 372 nm, respectively), which is in excellent agreement with the observed bathochromically shifted absorbance of AH1 in the photoswitched state. Calculations also revealed a shallow conformational energy surface around these T1 geometries (Section S9.4, ESI†), which supports the observed long lifetime of these triplet species in both solid and solution states. Also, due to this conformational flexibility, describing the photoswitching of 2-thienyl-based acylhydrazones as E ↔ Z isomerization is an oversimplification, although generally used also for azobenzenes.33 Hence, we assign the AH1 photoswitching to conversion of the closed-shell E-isomer into the triplet ground state photoswitched form adopting a perpendicular orientation of the azomethine and hydrazide parts of the photoswitch. The process of conversion is summarized in Fig. 5.
Calculations of AH2 show important differences. Relaxation from the vertically excited S1 state also showed pathways to three possible geometries. However, the CN torsion angle does not approach perpendicularity in any of them. In contrast to AH1, for all S1 minima geometries identified for AH2 (Table S5, ESI†), the lowest electronic states reveal a closed-shell character. Furthermore, the S1 minima of AH2 is the most similar in geometry to that in AH1, which does not have the inversion of the closed shell state and the open shell (imine π → π*) state. Finally, although the calculated spin–orbit coupling elements at these geometries are relatively large, suggesting potential intersystem crossing, the energy ratios favor decay to the system with a closed-shell structure rather than a structure with radical character. Thus, in the case of AH2, the photoswitching occurs between closed-shell species and can be described as a plain E ↔ Z process. The observed decrease of absorbance during photoswitching is consistent with the calculated lower oscillator strength of the Z-isomer.
Localization of singly occupied orbitals in AH1 at both T1 minimum and S0 in the switching geometry shown in Fig. 5 indicated separation of radicals on the two constituents, thiophene ring and the hydrazide. These constituents are very close to each other in the molecule. Likely due to the proximity and delocalization of the two spin sites (radical centers), we could not detect any ΔMs = ±2 EPR (double quantum) signal characteristic of the triplet electronic configuration. This also explains why we were able to record clean NMR spectra of the photoswitched forms; although radicals are generally considered detrimental to NMR spectroscopy characterization, triplet states can provide sharp and easy-to-interpret spectra depending on the spin coupling. Furthermore, NMR experiments with an internal standard (t-BuOH) showed that all species originating from photoswitching, i.e. the E- and the photoswitched form, are observable in the NMR spectrum (Fig. S4, ESI†).
Therefore, we made an effort to separate the two radical centers and designed a new system – DAE-2AH1 (Fig. 6), which combines two AH1 motifs with a diarylethene (DAE) photoswitch (bis-thienyl in our case). DAE undergoes photochemical electrocyclization, which brings the two decoupled aromatic rings in the open state in conjugation in the cyclized form.9,20 We substituted the two unoccupied α-positions of the two thiophene rings with AH1 motifs by grafting aldehyde functions followed by condensation with benzhydrazide. Upon irradiation with 340 nm light, we observed a gradual development of a strong purple color characteristic of the closed form of DAE. Using NMR and UV-vis, we also confirmed that both AH1-sites were converted to the photoswitched state as well. In this photoswitched form, the two hydrazide parts of DAE-2AH1 are isolated by the torsion angle around the CN bond, but the two thiophene moieties are conjugated. This allows the thiophene singly occupied states to recombine and therefore the two spin sites on hydrazone moieties are separated from each other in space.
 | ||
| Fig. 6 (a) Catalysis of radical halogenation to the benzyl position by AH5 in a photoswitched form. (b) The new system featuring both an AH double bond and a DAE ring, which can be closed and opened upon irradiation and can catalyze radical dehalogenation in its photoswitched form. | ||
The triplet character of the photoswitched form of AH5 acts as a diradical, which inspired us to use it as an initiator in a model radical reaction – radical bromination of toluene in the benzylic position by N-bromosuccinimide (NBS) (Section S11, ESI†). We mixed AH5 either in the E-form or in the photoswitched form with toluene and NBS in acetonitrile and monitored the conversion of toluene to benzyl bromide by HPLC for three hours. The reaction was conducted in the dark to avoid secondary irradiation effects. Subsequently, the solvent was evaporated, and the product formation was confirmed by NMR. Reaction without any form of AH5 served as a negative control. While no benzyl bromide was detected in the negative control after three hours, the run with as-synthesized AH5 showed 11% conversion, and the run with AH5 pre-irradiated to the PSS reached 60% conversion over the same period. This clearly demonstrates that the triplet state lasts sufficiently long even under the conditions of radical bromination by NBS and acts as an efficient initiator of the reaction.
Based on the above-mentioned successful application of AH5, we decided to demonstrate also the power of DAE-2AH1 to drive radical reactions. Radical dehalogenation of 2-bromoanisole was chosen as a model reaction. We mixed DAE-2AH1 in the photoswitched form with 2-bromoanisole and tributylstannane in DMF and monitored the conversion of 2-bromoanisole to anisole by HPLC. The reaction was also conducted in the dark to avoid secondary irradiation effects. The presence of the product was confirmed by comparison with the standards by HPLC and also by 1H NMR. We also used 1H NMR to prove that the reaction without DAE-2AH1 does not yield the product.
Conclusions
We synthesized a series of thienyl-based acylhydrazones in high yields using the reaction of selected hydrazides and aldehydes (AH1–9). We observed a similar photoswitching of all 2-thienyl derivatives. Furthermore, we observed strikingly different photoswitching in the 3-thienyl derivative. Photoswitching is repeatable and the photoswitched form is stable according to the long half-life, which is above 6 hours. Solid-state photoswitching was observed in pure films and solid solutions in a PMMA matrix. The optically stored information is completely erased electrochemically at reasonable potentials. We attributed the specific spectral features of photoswitched isomers of 2-thienyl acylhydrazones to the open-shell triplet configuration by EPR and ab initio calculations. Further calculations provided excellent agreement of experimental spectra with calculated predictions for triplet states. Thorough theoretical investigation of excited states and their geometry optimization by multireference methods allowed us to describe the isomerization pathway. Finally, we demonstrated the application potential of the presented acylhydrazone systems in catalysis as radical initiators. The closed-to-open-shell reversible photoswitching is an unprecedented phenomenon allowing manipulation with spin multiplicity between two long-lived states. This opens up unique opportunities in many fields such as catalysis, information storage, spintronics, and materials science.Author contributions
P. K. designed the study. M. Š., A. G. and O. M. synthesized the compounds. M. Š., V. G., H. R. and O. M. performed spectroscopic and electrochemical experiments. M. Š. and V. G. determined the quantum yields. A. L. and D. N. designed and performed the ab initio calculation study. J. T. performed, analyzed and interpreted the EPR data. V. E. measured, analyzed and interpreted the XRD data. M. Š. performed the catalytic study. M. Š., H. R. and P. K. consolidated all data, discussed the results and cowrote the manuscript. All authors contributed to the manuscript editing.Data availability
All the data is available in the repository under DOI: 10.5281/zenodo.14681032.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Czech Science Foundation Grant No. 22-11299M ‘Reaction networks at phase interfaces for dynamic self-assembly’. The team was also supported by the Experientia Foundation 2021 start-up grant. The research leading to these results was supported by the Johannes Amos Comenius Programme, European Structural and Investment Funds, project ‘CHEMFELLS V’ (No. CZ.02.01.01/00/22_010/0003004). D. N. acknowledges the European Union under the REFRESH – Research Excellence for Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition.References
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- D. J. van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2013, 43, 148–184 RSC.
- G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- A. Kanaya, Y. Takashima and A. Harada, J. Org. Chem., 2011, 76, 492–499 CrossRef CAS PubMed.
- C.-Y. Dennis Huang and S. Hecht, Chem. – Eur. J., 2023, 29, e202300981 CrossRef PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1740 CrossRef CAS PubMed.
- G. Men and J.-M. Lehn, Chem. Sci., 2019, 10, 90–98 RSC.
- S. Yang, D. Larsen, M. Pellegrini, S. Meier, D. F. Mierke, S. R. Beeren and I. Aprahamian, Chem, 2021, 7, 2190–2200 CAS.
- D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef CAS PubMed.
- G. Vantomme and J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 3940–3943 CrossRef CAS PubMed.
- H. Fu, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2023, 145, 19554–19560 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- P. Kobauri, F. J. Dekker, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202300681 CrossRef CAS PubMed.
- R. Dorel and B. L. Feringa, Chem. Commun., 2019, 55, 6477–6486 RSC.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, Nat. Catal., 2018, 1, 516–522 CrossRef CAS.
- M. Kathan, P. Kovaříček, C. Jurissek, A. Senf, A. Dallmann, A. F. Thünemann and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 13882–13886 CrossRef CAS PubMed.
- M. Kathan, C. Jurissek, P. Kovaříček and S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2378–2382 CrossRef CAS.
- I. Hnid, D. Frath, F. Lafolet, X. Sun and J.-C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732–7736 CrossRef CAS PubMed.
- N. E. Beyrouti, F. Houard, M. Cordier, E. Trzop, S. Rigaut, B. L. Guennic, K. Bernot and L. Norel, Chem. Commun., 2023, 59, 5265–5268 RSC.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem. – Eur. J., 2011, 17, 248–258 CrossRef CAS PubMed.
- G. Vantomme, S. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- J. Holub, G. Vantomme and J.-M. Lehn, J. Am. Chem. Soc., 2016, 138, 11783–11791 CrossRef CAS PubMed.
- M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 128, 14446–14447 CrossRef CAS PubMed.
- R. Falkenburg, M. J. Notheis, G. Schnakenburg and L. K. S. von Krbek, Org. Biomol. Chem., 2023, 21, 4993–4998 RSC.
- N. Eleya, S. Ghosh, E. Lork and A. Staubitz, J. Mater. Chem. C, 2021, 9, 82–87 RSC.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem. – Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed.
- K. Munkerup, D. Romanov, T. Bohinski, A. B. Stephansen, R. J. Levis and T. I. Sølling, J. Phys. Chem. A, 2017, 121, 8642–8651 CrossRef CAS PubMed.
- A. Mostad, Chr Rømming, Chr Rømming, S. Hammarström, R. J. J. C. Lousberg and U. Weiss, Acta Chem. Scand., 1971, 25, 3561–3568 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2345919. For ESI and crystallographic data in CIF or other electronic format, see DOI: https://doi.org/10.1039/d5tc00826c |
| This journal is © The Royal Society of Chemistry 2025 |