
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of crystallinity and local disorder on the luminescence properties of solvothermally synthesized LuPO4:Pr3+ nanocrystals†
Sanu Bifal
Maji
 *a,
Alexander
Vanetsev
a,
Vitali
Nagirnyi
a,
Kirill
Chernenko
b,
Eduard
Feldbach
a,
Jekaterina
Kozlova
a,
Hugo
Mändar
a,
Ivo
Romet
a,
Mihkel
Rähn
a and
Marco
Kirm
a
*a,
Alexander
Vanetsev
a,
Vitali
Nagirnyi
a,
Kirill
Chernenko
b,
Eduard
Feldbach
a,
Jekaterina
Kozlova
a,
Hugo
Mändar
a,
Ivo
Romet
a,
Mihkel
Rähn
a and
Marco
Kirm
a
aInstitute of Physics, University of Tartu, W. Ostwald Str. 1, 50411 Tartu, Estonia. E-mail: sanu.bifal.maji@ut.ee
bMAX IV Laboratory, Lund University, P.O. Box 118, SE-22100, Lund, Sweden
First published on 9th April 2025
Abstract
In this study, we present the solvothermal synthesis of LuPO4:Pr3+ (1%) nanocrystals in a dimethyl sulfoxide–water solvent system. The as-prepared nanocrystals exhibit a relatively high degree of crystallinity, along with significant short-range disorder, particularly in the anionic sublattice, as revealed by XRD and Raman spectroscopy data. Low temperature luminescence spectroscopy under VUV excitation showed that this disorder hinders the energy transfer from the host lattice to the Pr3+ ions, resulting in the absence of the 4f15d1→4f2 emission under high energy excitation. Thermal annealing of as-prepared nanocrystals significantly reduces the degree of local disorders enhancing UV-C luminescence in the 4.4–5.5 eV range by improving energy transfer processes from the LuPO4 host to Pr3+ ions. Time-integrated luminescence spectra reveal up to a five-fold increase in UV-C emission intensity after annealing. UV-C emissions due to Pr3+ ions in LuPO4 are efficiently excited in the deep intrinsic absorption region at 45 eV, which simulates the energy conversion process for radiotherapy applications. Time-resolved studies identify two types of Pr3+ ions. One group consists of strongly perturbed ions near the nanoparticle surface, influenced by structural imperfections, while the other includes weakly perturbed ions located in the bulk. These results highlight the critical role of local structural imperfections almost undetectable by XRD in energy transfer processes involving the host lattice.
1. Introduction
Recently, research efforts to improve the efficiency of radiotherapy by using rare-earth luminescent materials have gained increased interest. Radiotherapy is one of the most widely used techniques to treat various tumors, such as skin, prostate, lung, and cervix carcinomas.1 In radiotherapy, ionizing radiations such as X-rays, electrons, and particle beams transfer energy into cells, generating reactive oxygen species (ROS) (e.g., singlet oxygen).2,3 When the ROS concentration overwhelms the body's antioxidant defense mechanisms, oxidative stress leads to cellular damage.4 However, these treatment techniques are limited by two factors: the maximum radiation dose that can be delivered to tumorous cells without significantly damaging adjacent normal cells, and the presence of oxygen and other compounds suitable for ROS generation.5 A promising approach to overcome these limitations is the use of UV-C photon-emitting materials to induce additional cellular damage.1,5 UV-C radiation can directly be absorbed and can damage cellular DNA or indirectly increase ROS generation.3 For instance, Miwa et al. demonstrated UV-C radiation-induced destruction of human pancreatic carcinoma cells, showing a 70% cell death rate under a 25 J m−2 UV-C dose.6 This highlights the potential of UV-C photon-emitting materials for improving radiotherapy efficiency.Several studies have explored rare-earth luminescent materials for their ability to emit high-energy photons and their suitability in radiotherapy. For example, Vistovskyy et al. investigated Pr3+ and Eu3+-doped phosphates, demonstrating their efficient UV photon emission and the critical role of crystallinity and particle size in determining luminescence performance.7 Building on these findings, LuPO4:Pr3+, known for its promising luminescence properties, was chosen as the focus of the present work. The ability of such materials to function as UV-C photon emitters under high-energy excitation makes them suitable candidates for enhancing radiotherapy outcomes.8 To achieve effective delivery and performance, nanoparticles of high-density compounds like LuPO4:Pr3+ must be dispersible and stable in colloidal solutions. This allows their transportation to the targeted sites using nanocarrier molecules (e.g., polymeric nanoparticles, micelles, liposomes, etc.) modified with ligands that specifically recognize tumor cell receptors.9,10 The overall size of the system (including the nanoparticles and carrier molecules) should remain as small as possible, typically within the range of 10 to 200 nm.11 Moreover, smaller carrier molecules result in higher cell uptake compared to larger, micro-sized carriers, allowing for better access to cellular and intracellular targets.12 A larger surface area-to-volume ratio in smaller particles also leads to faster and more efficient release of the active agents at the targeted site.13
Achieving optimal luminescence performance for nanoparticles in such nanocarriers depends heavily on their crystallinity.14 Local disorder, especially in the form of structural defects and short-range and long-range disordering at the nanoscale, can have significant effects on the luminescence properties.15 In many cases, as-prepared nanoparticles exhibit high levels of short-range disorder, particularly in the anionic sublattice.14,16 This local disorder can lead to the distortion of the unit cell, impairing energy transfer processes and reducing luminescence efficiency. Surface defects and imperfections, such as dangling bonds or absorbed species, can also serve as quenching centers, further hindering the occurrence of radiative transitions.
Annealing at high temperatures is a well-known method to reduce local disorders and improve the crystallinity of materials.7,17 This process not only aids in recrystallization but also reduces the structural imperfections, facilitating better energy transfer from the host lattice to the Pr3+ ions. By minimizing the influence of these local disorders, the overall luminescence efficiency of the nanoparticles can be significantly enhanced, making them more suitable for practical applications in radiotherapy. In this work, we explore a novel solvothermal synthesis method to produce LuPO4:Pr3+ nanocrystals in a dimethyl sulfoxide (DMSO) water solvent system. A doping concentration of 1% Pr3+ was selected based on previous studies, which demonstrated that lower doping levels (e.g., 0.1%) result in inefficient energy transfer, whereas higher concentrations (e.g., 3%) lead to concentration quenching.18 This ensures an optimal balance between strong luminescence and minimal quenching losses. We examine how annealing affects their structure, size, and luminescence properties, paying particular attention to the effects of local disorder on luminescence performance. Specifically, we compare the as-prepared nanocrystals, which exhibit significant local disorders, with annealed nanocrystals that show enhanced crystallinity and improved luminescence.
2. Materials and Methods
LuPO4:Pr3+ (1%) nanocrystals were synthesized by the solvothermal method in DMSO–water media. The reagents used for this synthesis were Lu(NO3)3·6H2O (Sigma-Aldrich, 99.9% purity), Pr(NO3)3·6H2O (Sigma-Aldrich, 99.99% purity), (NH4)2HPO4 (Sigma-Aldrich, >99% purity), DMSO (Honeywell Riedel-de-Haën, 99.7% purity) and Milli-Q water.In our typical synthesis process, 9.9 mmol solution of Lu(NO3)3 and 0.1 mmol (1%) solution of Pr(NO3)3 were prepared in 20 mL and 5 mL of DMSO–water mixed solvent, respectively. The solvent used for the synthesis was a mixture of DMSO and Milli-Q water in different ratios, namely, for the sample L1 – 30% DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70% water, L2 – 50% DMSO:50% water, respectively. Both solutions were mixed together using a magnetic stirrer for 15 minutes. Simultaneously 10 mmol solution of (NH4)2HPO4 was prepared in 25 mL of DMSO–water solvent. The (NH4)2HPO4 solution was added to the above solution of rare earth nitrates in a dropwise manner under vigorous stirring for 30 minutes. The resultant mixture was transferred to the 100 mL Teflon autoclave, treated at 200 °C for 3 hours, and then cooled down to room temperature. The sample was then centrifuged at 10000 rpm (Heraeus Multifuge centrifuge) for 10 minutes, and the transparent supernatant was discarded. The precipitate was washed several times with double distilled water and ethanol and then dried at 100 °C for 2 hours. The powders obtained with two different DMSO ratios (L1 and L2) will be further referred to as “as-prepared L1” and “as-prepared L2”. Half of the synthesized powders were then annealed at 1000 °C in an argon atmosphere for 2 hours and will be further referred to as “annealed L1” and “annealed L2”, respectively. An argon gas atmosphere was used during annealing to prevent the oxidation of Pr3+ to Pr4+. As-prepared and annealed powders were then studied using various analytical and spectroscopic techniques described below.
:70% water, L2 – 50% DMSO:50% water, respectively. Both solutions were mixed together using a magnetic stirrer for 15 minutes. Simultaneously 10 mmol solution of (NH4)2HPO4 was prepared in 25 mL of DMSO–water solvent. The (NH4)2HPO4 solution was added to the above solution of rare earth nitrates in a dropwise manner under vigorous stirring for 30 minutes. The resultant mixture was transferred to the 100 mL Teflon autoclave, treated at 200 °C for 3 hours, and then cooled down to room temperature. The sample was then centrifuged at 10000 rpm (Heraeus Multifuge centrifuge) for 10 minutes, and the transparent supernatant was discarded. The precipitate was washed several times with double distilled water and ethanol and then dried at 100 °C for 2 hours. The powders obtained with two different DMSO ratios (L1 and L2) will be further referred to as “as-prepared L1” and “as-prepared L2”. Half of the synthesized powders were then annealed at 1000 °C in an argon atmosphere for 2 hours and will be further referred to as “annealed L1” and “annealed L2”, respectively. An argon gas atmosphere was used during annealing to prevent the oxidation of Pr3+ to Pr4+. As-prepared and annealed powders were then studied using various analytical and spectroscopic techniques described below.
The phase composition of samples was characterized by X-ray diffraction (XRD). Diffraction patterns were measured using Bragg–Brentano optics on an X-ray diffractometer SmartLabTM (Rigaku, Japan) working at source power of 8.1 kW (Cu Kα radiation, λ = 0.1540598 nm). The X-ray true size of crystallites was calculated by Voigt analysis of peak shapes assuming a cylindrical shape model.19 The used instrumental broadening of reflections was based on measurements of the standard reference material SRM-660 (LaB6). Cell parameters were calculated using the whole powder X-ray diffraction pattern fitting algorithm.20 Data from databases PDF-2 (version 2023, from International Center of Diffraction Data, USA) and ICSD (version 2021, from FIZ Karlsruhe, Germany) were used for crystalline phase identification and calculation of diffraction patterns, respectively. For studying the crystallinity and vibrational spectra of the LuPO4:Pr3+ nanocrystals, Raman spectroscopy was applied. A micro-Raman setup (Renishaw in Via) equipped with a continuous multi-line argon laser (Stellar-Pro) operating at 514 nm and an optical DM Microscope (Leica Microsystems) were used for the measurements of Raman scattering spectra. The detailed description of the experimental setup has been presented elsewhere.21 A FEI Titan Themis 200 Cs-corrected (scanning) transmission electron microscope (STEM) operated at 200 kV in the STEM mode was used to study the morphology of as-prepared and annealed LuPO4:Pr3+ nanocrystals. High-angle annular dark-field (HAADF) and bright-field (BF) images were simultaneously captured from the same locations on the sample. For the STEM analysis, a particle suspension in 2-propanol was drop cast onto a carbon film-covered 300-mesh copper TEM grid.22,23
Time-integrated (TI) luminescence spectroscopy of as-prepared and annealed LuPO4:Pr3+ nanocrystals in the temperature range of 6–300 K was carried out using the FinEstBeAMS beamline located at the 1.5 GeV storage ring of the MAX IV synchrotron radiation laboratory. The low-temperature measurement (6.8 K) was chosen to minimize thermal quenching, enabling a more detailed analysis of intrinsic energy transfer mechanisms and spectral characteristics, while the room-temperature measurement (300 K) provides insight into the material's performance under practical conditions, particularly for potential applications in scintillation and biomedical applications. The detailed description of the beamline design, its performance and photoluminescence setup used can be found in ref. 24–26. The multi-bunch operation mode of a 1.5 GeV storage ring was applied in recording the TI luminescence spectra using an Andor CCD camera (Newton DU970P-BFV) in the spectral range of 200–700 nm and a Hamamatsu photon counting head H8259–01 sensitive in a spectral range of 200–900 nm mounted in two different exit ports of an Andor Shamrock (SR-303i) 0.3 m spectrometer. The typical spectral resolutions in measuring the emission spectra were 8.2 and 3.1 nm using the photon counting head and CCD detector, respectively. The time-integrated excitation spectra were obtained by recording the emission spectra for each exciting photon energy using the CCD camera. During data analysis, the desired excitation spectrum was generated allowing for detailed analysis by integrating intensity over a specific wavelength (energy) range of the selected emission band. Moreover, all spectra are corrected for the sensitivity of the detection channel.
The time resolved luminescence studies were performed only at 6–7 K using an ultra-fast Hamamatsu R3809U-50 MCP-PMT detector in the single-bunch operation mode of the 1.5 GeV storage ring at MAX IV. The time resolution of the detection system, i.e. an instrumental response function (IRF), was determined to be 174 ps. The details of the time-resolved luminescence method implemented at FinEstBeAMS and the technical information of equipment in use can be found in ref. 27 and 28. The photon flux of the beamline was recorded with an AXUV-100G photodiode for normalizing excitation spectra to the same amount of incident photons. In all experiments the slit width of the primary monochromator was set to 150 μm, which corresponded to the spectral resolution better than 4 meV at 10 eV (i.e., a resolving power of ∼2500) of the exciting photon beam.25
3. Results and discussion
3.1. Phase composition and morphology of LuPO4:Pr3+ (1%) powders
The XRD phase analysis of solvothermally synthesized powders showed that all observed reflections (Fig. 1) for both as-synthesized and annealed samples of L1 and L2 can be indexed to the tetragonal phase of LuPO4 (PDF-2 file number 84-337 and ICSD collection code 162336, space group I41/amd). This demonstrates that the solvothermal synthesis method can successfully produce the tetragonal phase of LuPO4. Furthermore, the solvothermal synthesis method offers an advantage over water-based techniques, as it facilitates the formation of hydrated rare-earth phosphates, which typically crystallize in a hexagonal crystal structure due to the incorporation of water molecules.29–31 The cell parameters of the as-prepared LuPO4:Pr3+ samples (Table 1) were considerably larger compared to the ones of undoped LuPO4 (a = 0.67827, c = 0.59467).32 This can be explained by a larger ionic radius of Pr3+ (127 pm) compared to that of Lu3+ (98 pm) for the VIII coordination.33 The estimation of the crystallite shape and size was performed using a cylindrical model, which accounts for the anisotropic growth of the crystallites. This anisotropy, evident from the XRD analysis, reflects differences in the crystallite size along specific crystallographic directions. The Voigt analysis of XRD peak shapes showed that the mean diameters of the as-prepared LuPO4:Pr3+(1%) samples (L1 and L2) were approximately 9 and 10 nm, respectively, with the size along the [001] direction being slightly larger than that along the [100] and [010] directions (Table 1). | ||
| Fig. 1 XRD patterns of as-prepared (lower curve) and annealed (upper curve) LuPO4:Pr3+ (1%) powder synthesized in L1 (30% DMSO–70% water) solvent and L2 (50% DMSO–50% water) solvent. The calculated diffraction pattern including the indices of reflections of tetragonal LuPO4 is shown at the topmost part of the figure. | ||
| Type of the sample | a, nm | c, nm | 〈2r〉100, nm | 〈h〉001, nm |
|---|---|---|---|---|
| L1 (30% DMSO–70% water) | 0.6824(2) | 0.5970(2) | 9(1) | 19(2) |
| Annealed L1 (1000 °C) | 0.6800(2) | 0.5965(2) | 19(1) | 25(2) |
| L2 (50% DMSO–50% water) | 0.6825(2) | 0.5972(2) | 10(1) | 14(2) |
| Annealed L2 (1000 °C) | 0.6800(2) | 0.5965(2) | 23(1) | 27(2) |
Annealing of LuPO4:Pr3+samples (annealed L1 and L2) increased the size of crystallites up to 17 and 21 nm, respectively, and decreased the degree of anisotropy (see Tables 1 and 3). After annealing, a decrease in cell parameters was observed, but the final values were still significantly larger in comparison to the undoped LuPO4. This decrease of cell parameters may be related to a lower disorder rate in the annealed samples compared to that in as-prepared samples. It is well-known that local disorder can lead to the dilation of the unit cell34 and synthesis at relatively low temperature could result in the formation of a disordered phase. On the other hand, there is no impurity phase contribution observed in the XRD patterns, as well as no sign of a significant amount of an amorphous phase. Thus, we can assume that the samples possess long-range ordering, while short-range disorder in the first and/or second coordination sphere may be present.
In order to study structural peculiarities, we compared the results of XRD studies with those of Raman spectroscopy, which is specifically sensitive to the short-range ordering.35 The unit cell of tetragonal LuPO4 consists of Lu3+ at the D2d lattice site, which is 8-fold coordinated by oxygen atoms (Fig. 2), forming a dodecahedron with triangular planes and providing a single site for the isovalent Pr3+ dopant ion.36 According to the literature data, there are twelve Raman active vibrational modes in the LuPO4 structure.37 All these vibrational modes, except the one at 321 cm−1, are well-observed in both annealed LuPO4:Pr3+ samples (see Fig. 3 and Table 2), however, these are absent in the as-prepared LuPO4:Pr3+ samples, which confirms the hypothesis of short-range disordering. The nature of this disorder requires additional structural studies, but we tentatively suggest that it is related to the displacement (most likely rotation) of the PO4 groups in the lattice. Another possibility is the protonation of the PO4 group, which has been observed in the case of YPO4 microwave-hydrothermal synthesis and has been shown to cause the displacement of oxygen atoms in the lattice and lowering the symmetry of the PO4 group.38 Given the light weight of oxygen, such displacement does not strongly affect the XRD pattern (apart from a slight increase of the cell parameters), but might have a significant effect on Raman modes, as all of them are related to the vibrations involving oxygen atoms. Our hypothesis is that random shifts of oxygen atoms in the lattice lead to full disappearance of Raman bands similar to an amorphous phase, though the XRD pattern shows long ranged ordering in the material. The XRD measurement is more sensitive to the contribution of heavy Lu atoms, resulting in well-defined XRD patterns.
 | ||
| Fig. 2 The crystal structure model of the tetragonal unit cell of LuPO4 in polyhedral presentation: green atoms – Lu, red atoms – P; oxygen atoms are located at the vertices of polyhedra, but not presented for the sake of the image clarity. The figure has been prepared using VESTA software. | ||
 | ||
| Fig. 3 Raman spectra of the as-prepared and annealed LuPO4:Pr3+ nanocrystals recorded at 300 K. The band at 520 cm−1 in the spectra of as-prepared samples is related to the silicon substrate. | ||
| S. No | Raman modes | Experimental results of this work (cm−1) | Experimental data from ref. 37 (cm−1) |
|---|---|---|---|
| 1. | Eg(1) | 133.2 | 133 |
| 2. | B1g(1) | 139.4 | 139 |
| 3. | Eg(2) | 186.4 | 187 |
| 4. | Eg(3) | 306.2 | 306 |
| 5. | B2g | 330.4 | 331 |
| 6. | A1g(1) | 488.5 | 490 |
| 7. | Eg(4) | 582.7 | 582 |
| 8. | B1g(3) | 665.4 | 665 |
| 9. | A1g(2) | 1010.9 | 1010 |
| 10. | Eg(5) | 1031.9 | 1031 |
| 11. | B1g(4) | 1069.4 | 1069 |
The STEM images of as-prepared LuPO4:Pr3+ samples demonstrate elongated nanoparticles of irregular shape (see Fig. 4(1(a) and 2(a)). Such shape is in good correlation with the results of XRD pattern analysis showing the anisotropic growth along the [001] direction. Although the irregular appearance suggests imperfect formation of defined facets, this may be partly due to the limited resolution of the STEM images (Fig. 4(1(b), (c) and 2(b), (c)), as some facets can be discerned. The growth of nanoparticles proceeds along the most energetically favorable crystallographic direction, [001], which is typical for tetragonal LuPO4 crystals. The observed shapes suggest that primary nuclei grew preferentially along the [001] direction and stacked along this axis to form the nanoparticles visible in the STEM images. Quantitative size distribution analysis of the STEM images of over 400 nanoparticles for both as-prepared LuPO4:Pr3+ samples is shown in Fig. S1 in the ESI† data. Due to the anisotropic nature of the formed nanoparticles, it was impossible to calculate a single valid mean size from the STEM images. Thus, the mean width and mean length of the synthesized nanoparticles, as well as their degree of anisotropy, were estimated. Additionally, we had calculated the mean quadratic value of the mean width and mean length in order to compare this single value with the average crystallite size obtained from the XRD patterns (Table 3). The mean quadratic values for the as-prepared L1 and L2 samples are 12 ± 2 nm and 16 ± 1 nm, respectively. The degree of anisotropy between the mean length and the mean width of as-prepared nanoparticles is 2.50 and 3.56 for L1 and L2, respectively.
 | ||
| Fig. 4 The first row [1(a), 1(d), 2(d) and 2(a)] STEM images are of different LuPO4:Pr3+ (1%) nanocrystal families displaying uniformity of the particles. The middle row images [1(b), 1(e), 2(b) and 2(e)] are high resolution TEM images for smaller LuPO4:Pr3+ (1%) nanocrystal agglomerates. The bottom row [1(c), 1(f), 2(c) and 2(f)] shows high-resolution STEM images taken at a 5 nm resolution, with visible lattice fringes indicating crystallinity. | ||
| S. No. | Sample name | STEM size (400 particles) | Degree of anisotropy using STEM (length/width) | X-ray mean true diameter of cylindrical shape crystallites (nm) 〈2r〉 | Degree of anisotropy using XRD | ||
|---|---|---|---|---|---|---|---|
| Mean width (W) (nm) | Mean length (L) (nm) | Mean quadratic value (nm)
|
|||||
| 1. | As-prepared L1 | 6 ± 1.0 | 16 ± 2.5 | 12 ± 2 | 2.50 | 9 | 1.85 |
| 2. | Annealed L1 | 17 ± 1.6 | 21 ± 2.3 | 19 ± 2 | 1.27 | 19 | 1.38 |
| 3. | As-prepared L2 | 6 ± 0.8 | 22 ± 2.0 | 16 ± 1 | 3.56 | 10 | 1.42 |
| 4. | Annealed L2 | 22 ± 2.2 | 27 ± 2.6 | 25 ± 3 | 1.23 | 23 | 1.17 |
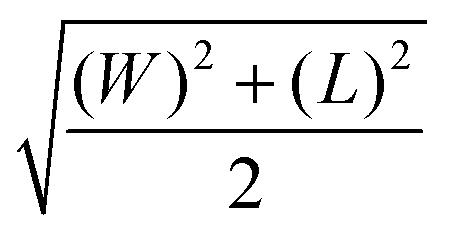
There is a good correlation between the mean quadratic values of particle dimensions calculated from STEM images and the average crystallite size obtained from XRD analysis (Table 3). This allows us to suggest that the degree of crystallinity is already rather high in as-prepared LuPO4:Pr3+ nanoparticles. However, this contradicts the results of Raman spectroscopy, which shows no bands in the recorded spectra of such nanoparticles. Thus, as mentioned above, it is possible that the long-range order in as-prepared nanoparticles, deduced from the XRD analysis, is accompanied by the local disorder, most likely in the anionic sublattice, revealed in the Raman shift spectra. After annealing, the nanoparticles become relatively isotropic (the degree of anisotropy is 1.27–1.23) and their mean particle sizes and crystallinity increase. Also, the annealed nanoparticles become well-shaped with clearly defined facets (see Fig. 4(1(e) and 2(e)). In this process, nanoparticles tend to form densely sintered agglomerates with thin low-angle boundaries. This improvement is due to the recrystallization and Ostwald ripening during annealing. The mean quadratic values between the mean width and the mean length of the annealed nanoparticles, calculated using the same procedure as for the as-prepared nanoparticles (as shown in Fig. S1, ESI†), are 19 ± 2 nm and 25 ± 3 nm, respectively (Table 3). These values are quite comparable to the sizes calculated from the XRD patterns, showing the increase in crystallinity and possibly also a decrease in anisotropy.
The evolution of nanoparticle morphology during annealing at high temperature in an inert atmosphere is qualitatively different from solvothermal growth. During the solvothermal synthesis, particles grow intensively along one direction with unrestricted mass flow from the solution. However, the low temperature limits recrystallization and the formation of well-defined facets, resulting in nanoparticles with numerous structural imperfections. In contrast, thermal annealing in an inert atmosphere promotes more isotropic and slower growth due to limited mass flow between particles, while the significantly higher processing temperature (1000 °C) drives an intensive recrystallization process. This process reduces the concentration of structural imperfections and enhances the overall crystallinity of the calcined nanoparticles. Another important aspect after annealing is the formation of dense agglomerates of nanoparticles with thin low-angle borders between them. For many processes such agglomerates can act as a single bulk material, as grain boundaries are thin and easily penetrable. However, in the applications where well-dispersed nanoparticles are required, as in biological or optoelectronic systems, the agglomeration can be a limiting factor. Potential strategies to mitigate this effect include the surface modification, the calcination in matrices, or the use of the surfactants during post-processing.39,40 While this study focuses on structural and luminescence properties, future work could explore these methods to improve particle dispersion for the practical applications demanding the use of stable colloids of nanoparticles.
3.2. Photoluminescence study of the as-prepared and annealed LuPO4:Pr3+ (1%) nanocrystals under synchrotron radiation excitation
The luminescence properties of LuPO4:Pr3+ nanocrystals are largely influenced by the interaction between the electronic states of the host material and Pr3+ impurity ions. This interaction results in various relaxation processes that govern the energy transfer from the host to rare earth ions. Using the P. Dorenbos methodology, which applies to all rare earth elements, one can predict the energy level scheme of Pr3+ in LuPO4, showing the positions of the 4f and 5d energy levels in the electronic band structure of the host41 (see Fig. 5). As shown in Fig. 5, a strong crystal field of LuPO4 positions the 5d energy bands slightly below the 4f 1S0 energy level, enabling dipole and parity-allowed interconfigurational 4f15d1→4f2 radiative transitions, which are responsible for the broad-band Pr3+ emission observed usually in the UV-C spectral region. In the case of close positions of the 5d term and 1S0 level, narrow emission lines due to the 4f2→4f2 transitions can also be observed both through the direct intra-center excitation and through the host excitation via energy transfer processes populating the respective levels.42 Furthermore, intrinsic emissions, such as broad-band self-trapped exciton emission peaked near 3 eV, can also be present in LuPO4 under the host excitation in the excitonic and fundamental absorption region.43,44 | ||
| Fig. 5 Schematic representation of the fundamental electronic structure of LuPO4:Pr3+, based on P. Dorenbos’ methodology.17,41 The host near-edge energy bands, impurity levels and the Pr3+ 4f15d1→4f2 transitions (blue arrows) are illustrated. | ||
As an example of overview emission spectra recorded for the synthesized LuPO4:Pr3+ samples, a spectrum recorded under the excitation of the annealed L1 powder with 45 eV photons at 6.5 K is shown in Fig. 6. Indeed, strong emission bands from 5.5 to 4.4 eV are observed in the UV region that occur due to the parity allowed inter-configurational 4f15d1→3H4, 3H5, 3H6 and 3F2,3,4 (4f2) radiative transitions in Pr3+ ions adhering to the spin selection rule (ΔS = 0) because the initial excited 4f15d1 state is triplet. The corresponding emission maxima in our synthesized LuPO4:Pr3+ (1%) nanocrystals are located at 5.31 eV (233 nm), 5.06 eV (245 nm), 4.71 eV (263 nm), and 4.56 eV (271 nm), which are in good agreement with the already reported emission bands in LuPO4:Pr3+ materials.17,18 The non-elementary emission bands near 4.5 eV (271 nm) formed from various overlapping sub-bands spectrally better resolved at low temperatures (see Fig. 5 and 6) are due to the transitions to the 3F2,3,4 multiplet. Moreover, few relatively weak narrow bands that can be distinguished in the visible spectral region are connected with the 4f2→4f2 transitions of Pr3+ ions. Remarkably, no sign of self-trapped exciton emission could be detected in the spectra. These circumstances allowed us to focus on a detailed study of the UV 4f15d1→4f2 emission of synthesized nanoparticles, highly promising for radiotherapy applications. Excitation by high-energy 45 eV photons results in intense UV-C emissions and these conditions simulate the energy conversion process needed for improvement of radiotherapy efficacy.
 | ||
| Fig. 6 TI emission spectra of Pr3+ doped LuPO4 nanoparticles excited with 45 eV photons at 6.5 K. | ||
 | ||
| Fig. 7 TI PL excitation spectra of the as-prepared and annealed LuPO4:Pr3+ (1%) nanocrystals at 6.8 K recorded for the 4f15d1→3H5 luminescence peaked at 5.06 eV (the signal is integrated for the emission energy range from 4.9 to 5.1 eV). | ||
During the post-measurement analysis, the excitation spectra of the 4f15d1→3H5 transition were obtained by integrating the emission recorded using the CCD detector in the energy range from 4.9 eV to 5.1 eV. The features of the excitation spectra in the range from 5.4 eV to 7.5 eV correspond to the intra-center 4f2→4f15d1 excitation of Pr3+ ions and are in agreement with the data already published.17,47 There are two distinct excitation bands, peaking at 5.41 eV and 6.54 eV, corresponding to the transitions of the first ([4f5d1]1) and second ([4f5d1]2) crystal-field components as labeled in ref. 48. In the higher energy region, the features in the excitation spectra are less pronounced, but low intensity peaks near 7 and 7.8 eV can be identified as corresponding to the excitation into the higher d-states [4f5d1].3,4 These peaks are less pronounced or almost absent for the as-prepared samples; however, in general the features of intra-center excitation spectra of the as-prepared and annealed samples are quite similar. For the annealed samples, in full agreement with the literature data,17,49 the intrinsic absorption starts at 8.5 eV with the exciton creation region, due to electronic excitation formation within the tetrahedral PO43− units. The exciton peak energy of 8.8 eV for our samples is in good agreement with the value earlier obtained at 10 K for LuPO4:Pr3+ powders.49 The band gap value can be estimated from the minimum in the excitation spectra shown in Fig. 7. It is equal to 9.2 eV at 7 K, being in good correspondence with the value 9.3 eV obtained for the LuPO4:Nd3+ (1%) powder.43 The process of the excitation of Pr3+ ions in the intrinsic absorption region will be discussed in detail in Section 3.2.3. Moreover, the excitation spectra of both as-prepared LuPO4:Pr3+ (1%) L1 and L2 samples are completely featureless above 7.5 eV and demonstrate negligible intensity in this region. This is an indication of the absence of energy transfer from the 5d states lying higher than the [4f5d1]2 component and also from the intrinsic absorption region. To verify this finding, we performed a series of measurements of emission spectra of the as-prepared L1 and L2 samples excited with different photon energies and compared them with the annealed samples as discussed in detail in below sections.
 | ||
| Fig. 8 Effect of annealing on 4f15d1→4f2 luminescence intensities of Pr3+ ions under intra-center excitation at 6.54 eV of the as-prepared and annealed LuPO4:Pr3+ (1%) nanocrystals at 6.8 K. The intensities of the as-prepared and annealed L1, L2 samples are shown on the left and right intensity axes, respectively. | ||
Fig. 9 and Fig. S2 (ESI†) depict emission spectra recorded under intra-center interconfigurational 4f2→4f15d1 excitation of Pr3+ ions at 6.54 eV, at the excitation in the host excitonic region at 8.54 eV, as well as in the region of interband transitions at 9.4 and 45 eV. The PL spectra of the annealed LuPO4:Pr3+ (1%) nanocrystals show efficient Pr3+ luminescence due to the 4f15d1→3H4, 3H5, 3H6, and 3F2,3,4 (4f2) transitions under excitation in all above-listed energy ranges. However, the Pr3+ luminescence intensity is remarkably lower in the as-prepared LuPO4:Pr3+ (1%) nanocrystals, being particularly very weak under excitation in the excitonic region and practically absent under excitation in the fundamental absorption region. These observations are in accordance with the excitation spectra recorded for the as-prepared and annealed LuPO4:Pr3+ (1%) nanocrystals (Fig. 8). The nature of such observations most likely lies in the high degree of local disorder in the as-prepared samples, which is also demonstrated by the Raman spectra (see Fig. 3). The correlation between the Raman and PL excitation spectra is an interesting fact, especially considering that the XRD patterns that are traditionally used to characterize the degree of crystallinity or disorder of the crystal lattice do not show qualitative differences between as-prepared and annealed samples. Our preliminary hypothesis suggests that Raman spectroscopy is much more sensitive to a short-range disorder, mainly related to the rotation or shift of the PO4 groups from their regular positions. Another possibility is the lowering of the symmetry of the PO4 group due to protonation,38 which can also significantly affect Raman bands. In both scenarios, the annealing procedure would remedy this short-range disorder, enhancing the rearrangement within the crystal lattice and promoting recrystallization. Consequently, the Raman spectra (Fig. 3) and PL excitation spectra (Fig. 7) of the annealed samples exhibit all the expected characteristics of the crystalline LuPO4 material due to the short-range ordering of the crystal lattice and the healing of structural imperfections by annealing. The as-prepared samples are supposed to have a highly disordered lattice, which causes the dispersion of phonon modes of multiple defect sites and their strong broadening, smoothing the Raman spectrum. A direct comparison of phonon energies before and after annealing is not possible, as the as-prepared samples exhibited no Raman-active vibrational modes, while all expected Raman lines appeared after annealing (see Fig. 3 and Table 2). The appearance of Raman lines indicates the improved structural ordering, which also improves energy transfer efficiency and eliminates luminescence quenching centers, as supported by the increased luminescence intensity and lifetime (see Section 3.2.4)
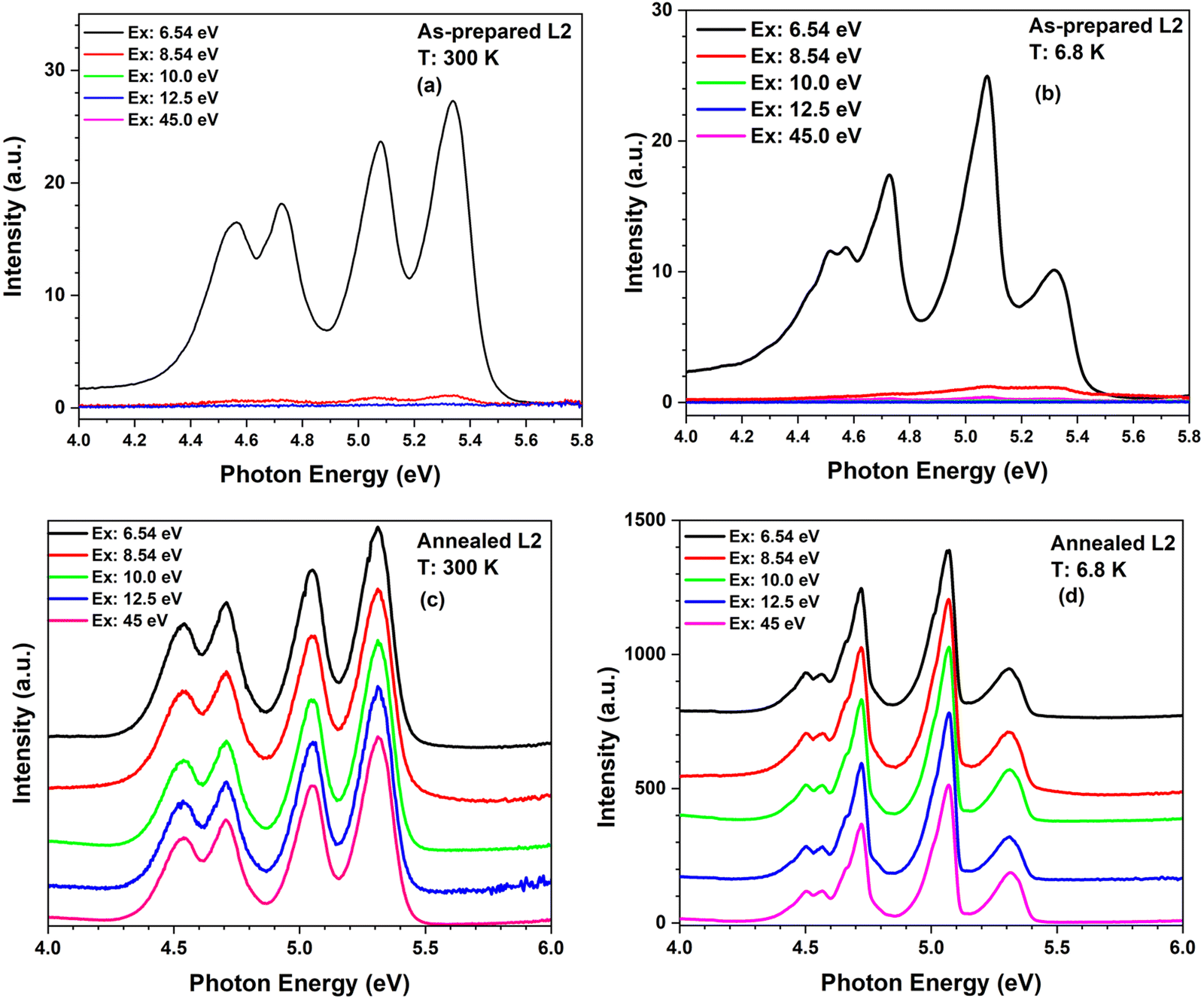 | ||
| Fig. 9 Pr3+ 4f15d1→4f2 luminescence spectra of the as-prepared (a) and (b) and annealed (c) and (d) LuPO4:Pr3+ (1%) nanocrystals (L2 sample) excited by photons with different energies at 300 and 6.8 K. The luminescence spectra of the annealed sample (c) and (d) are presented as a stacked graph, to better highlight the enhanced luminescence intensity and more distinct spectral features observed under different excitation energies (6.54 to 45 eV). Similar graphs for the L1 sample are shown in Fig. S2 of the ESI† data. | ||
In the case of the nanosized LuPO4:Pr3+, the luminescence intensity is strongly influenced by the particle size, crystallinity, structural imperfections (including short-range disorder) and the thermalization length of electrons within the host material, which is typically on order of a few tens of nanometers.17,50 The structural and morphology studies (XRD, STEM images and Raman spectra shown in Section 3.1) proved that the size of nanocrystals is below the thermalization length of mobile electrons typical of scintillation materials17,50 in as-prepared LuPO4:Pr3+ (1%) characterized by the presence of short-range disorder and structural imperfections. These factors increase the probability of non-radiative losses of mobile charge carriers, finally accounting for the reduced luminescence intensity of Pr3+ ions in these as-prepared nanocrystals excited in the host absorption region (see Sections 3.2.1 and 3.2.3). Nonetheless, the Pr3+ ion emission is still revealed in the as-prepared LuPO4:Pr3+ (1%) nanocrystals excited through the intra-center absorption of the Pr3+ ions in the two pronounced lower lying 4f–5d bands peaked at 5.4 and 6.6 eV (Fig. 6). In contrast, the annealing of the LuPO4:Pr3+(1%) nanocrystals reduces significantly the concentration of structural imperfections and increases short-range order, facilitating the appearance of bright Pr3+ 4f15d1→4f2 luminescence when excited in excitonic and fundamental absorption regions (Fig. 8), which positions the annealed LuPO4:Pr3+(1%) nanocrystals as promising candidates for medical and scintillating applications.
 | ||
| Fig. 10 Effect of annealing on the 4f15d1→3H4, 3H5, 3H6 luminescence decay of the Pr3+ ions for (a) L1 and (b) L2 nanocrystals, recorded under the intra-center excitation of the Pr3+ ion by the 6.54 eV photons at 7 K. Experimental data (green and blue lines) are fitted by the bi-exponential decay function, which is shown by red lines. The decay curves are normalized at their maxima for better comparison. | ||
| S. No. | Sample names | Radiative transitions (Pr3+ 4f15d1 → 3H4,5,6) | |
|---|---|---|---|
| τ 1 | τ 2 | ||
| 1. | As-prepared L1 | 1.6 ± 0.3 ns (97.3%) | 7.5 ± 0.5 ns (2.7%) |
| 2. | Annealed L1 | 3.4 ± 0.4 ns (61.6%) | 10.6 ± 0.6 ns (38.4%) |
| 3. | As-prepared L2 | 3.2 ± 0.2 ns (79.5%) | 14.4 ± 0.4 ns (20.5%) |
| 4. | Annealed L2 | 5.0 ± 0.3 ns (52.6%) | 16.1 ± 0.5 ns (47.4%) |
3.3. Time-resolved photoluminescence spectroscopy of the as-prepared LuPO4:Pr3+ (1%) nanocrystals under synchrotron radiation excitation
Building on the insights gained from the decay kinetics, we studied the time-resolved excitation spectra of as-prepared samples to further explore the relaxation dynamics of the Pr3+ 4f15d1→4f2 luminescence. These spectra provide critical information about the fast and slow relaxation processes associated with strongly and weakly perturbed Pr3+ ions. By leveraging synchrotron radiation excitation, it becomes possible to probe relaxation dynamics within the nanosecond time range as demonstrated in Pr3+ doped BaLuF5 nanoparticles.52 To investigate these relaxation processes in LuPO4:Pr3+ (1%) nanocrystals, the time-resolved PL excitation spectra of the 4f15d1→3H5 luminescence (5.06 eV) were recorded at 7 K using synchrotron radiation at FinEstBeAMS. The detailed methods for measuring, plotting, and analyzing the TR excitation spectra are provided in ref. 52. The excitation spectra are shown in two time-windows, short and long, marked as STW (δt = 0 and Δt = 4 ns) and LTW (δt = 4 and Δt = 20 ns), respectively, which are selected based on decay kinetics studies (see Fig. 9) to distinguish the faster relaxation processes, dominated by strongly perturbed Pr3+ ions, from the slower processes, characteristic of weakly perturbed Pr3+ ions. The time δt = 0 ns is defined by the arrival of the VUV excitation pulse, and the luminescence signal is integrated over Δt = 4 ns in the STW. In the LTW, the integration over Δt = 20 ns starts at δt = 4 ns after the excitation pulse (Fig. 11). | ||
| Fig. 11 Time resolved PL excitation spectra of the as-prepared LuPO4:Pr3+ (1%) L1 (a) and L2 (b) nanocrystals recorded at 6.8 K for the 4f15d1→3H5 luminescence at 5.06 eV in the STW (δt = 0 and Δt = 4 ns) and LTW (δt = 4 and Δt = 20 ns). The spectra are normalized to the highest intensity of the excitation band for better comparison. | ||
The excitation spectra recorded in the two-time windows reveal clearly visible differences. In the STW, the 4f2→4f15d1 excitation bands exhibit a redshift of approximately 0.038 eV for L1 and 0.03 eV for L2 compared to the 4f2→4f15d1 excitation bands in the LTW, which remain at relatively higher energies. This redshift in the STW is attributed to strongly perturbed Pr3+ ions probably situated near the nanoparticle surface, where structural imperfections and short-range disorder dominate. Conversely, the LTW spectra reflect weakly perturbed Pr3+ ions located in the bulk of the nanoparticles, where the influence of surface effects is significantly reduced. In the excitonic excitation region, the STW spectra show a blueshift of approximately 0.19 eV for both L1 and L2 compared to the LTW spectra. This opposite trend highlights the contrasting behavior of excitonic states in the strongly disordered crystal region with respect to that of the 4f15d1 states, further emphasizing the role of structural and morphological variations in energy transfer processes.
Thus, in conclusion, the time-resolved PL excitation spectra provide a comprehensive understanding of the relaxation dynamics in LuPO4:Pr3+ nanocrystals. The observed spectral shifts and trends confirm the strong influence of structural imperfections and crystallinity on the Pr3+ luminescence, with annealing treatments offering a promising approach to mitigate these imperfections. By reducing the disorder rate and enhancing energy transfer efficiency, annealing significantly improves the luminescence efficiency of LuPO4:Pr3+ nanocrystals, making them highly suitable for advanced luminescence applications.
Conclusions
This study highlights the pivotal role of local structural disorder in influencing the energy transfer processes and luminescence properties of LuPO4:Pr3+ nanocrystals. The high degree of short-range disorder in the as-prepared nanocrystals, undetected by XRD, was found to impair energy transfer between the host lattice and Pr3+ ions, suppressing luminescence excitation through the host-to-ion transfer. A significant outcome of this work is the observation of bright Pr3+ 4f15d1→4f2 emissions in annealed nanocrystals, positioning these materials as promising candidates for medical and optical applications. It is important to note that excitation by high-energy 45 eV photons in the deep fundamental absorption of LuPO4 results in UV-C emissions, demonstrating that analogous energy conversion is expected in radiotherapy treatment. Through decay kinetics and time-resolved excitation spectroscopy, this study further identified the presence of two distinct Pr3+ ion environments, which are strongly perturbed Pr3+ ions located at the nanoparticle surface and weakly perturbed Pr3+ ions situated in the bulk of the nanoparticles. Our research also showed that Raman spectroscopy proved to be a practical and informative tool for characterizing nanomaterials synthesized at low temperatures, clarifying the contradiction in the results provided by XRD and luminescence spectroscopy for as-prepared and annealed nanocrystals. Raman spectroscopy clearly showed that thermal annealing of our nanocrystals drastically improved the local ordering in the crystal lattice, resulting in significantly improved UV-C luminescence intensity. These findings highlight the importance of post-synthesis treatment in optimizing the performance of nanocrystals for practical applications, particularly in the biomedical field and other high-performance applications.Data availability
All data supporting the findings of this study are included within the article and its ESI,† ensuring transparency and reproducibility. Additional raw datasets generated during this study are available from the corresponding author upon reasonable request. On behalf of all co-authors, we affirm our commitment to facilitating the accessibility and integrity of the data presented in this work, contributing to the advancement of scientific knowledge.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the MAX IV Lab staff for their valuable contribution to our research. We are also grateful to Dr Tavo Romann and Dr Valter Kiisk for their assistance with the Raman experiments. The financial support from the GSFMT Graduate School, Estonian Research Council (grant PRG629) and Center of Excellence (TK 210) is gratefully acknowledged. We acknowledge the MAX IV Laboratory for the beamtime on the FinEstBeAMS beamline under Proposal 20211020 and 20221357. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research Council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. The activities of Estonian researchers at the FinEstBeAMS beamline of the MAX IV Lab were partially funded within the MAX-TEENUS project (grant no. 2014-2020.4.01.20-0278 by the ERDF funding in Estonia granted to the University of Tartu) and by the Estonian Research Council project TT20.References
- R. Baskar, K. A. Lee, R. Yeo and K.-W. Yeoh, Int. J. Med. Sci., 2012, 9, 193–199 CrossRef PubMed.
- T. L. de Jager, A. E. Cockrell and S. S. Du Plessis, in Ultraviolet Light in Human Health, Diseases and Environment, ed. S. I. Ahmad, Springer International Publishing, Cham, 2017, vol. 996, pp. 15–23 Search PubMed.
- M. R. Squillante, T. Jüstel, R. R. Anderson, C. Brecher, D. Chartier, J. F. Christian, N. Cicchetti, S. Espinoza, D. R. McAdams, M. Müller, B. Tornifoglio, Y. Wang and M. Purschke, Opt. Mater., 2018, 80, 197–202 CrossRef CAS PubMed.
- B. Poljšak and R. Dahmane, Dermatol. Res. Pract., 2012, 1–4 Search PubMed.
- I. Villa, C. Villa, R. Crapanzano, V. Secchi, M. Tawfilas, E. Trombetta, L. Porretti, A. Brambilla, M. Campione, Y. Torrente, A. Vedda and A. Monguzzi, ACS Appl. Mater. Interfaces, 2021, 13, 12997–13008 CrossRef CAS.
- S. Miwa, S. Yano, Y. Hiroshima, Y. Tome, F. Uehara, S. Mii, E. V. Efimova, H. Kimura, K. Hayashi, H. Tsuchiya and R. M. Hoffman, J. Cell. Biochem., 2013, 114, 2493–2499 CrossRef CAS.
- V. Vistovskyy, T. Malyi, A. Vas’kiv, M. Chylii, N. Mitina, A. Zaichenko, A. Gektin and A. Voloshinovskii, J. Lumin., 2016, 179, 527–532 CrossRef CAS.
- M. Müller, S. Espinoza, T. Jüstel, K. D. Held, R. R. Anderson and M. Purschke, Radiat. Res., 2019, 193, 82 Search PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS.
- G. Tiwari, R. Tiwari, S. Bannerjee, L. Bhati, S. Pandey, P. Pandey and B. Sriwastawa, Int. J. Pharm. Invest., 2012, 2, 2 CrossRef PubMed.
- B. Yu, H. C. Tai, W. Xue, L. J. Lee and R. J. Lee, Mol. Membr. Biol., 2010, 27, 286–298 CrossRef CAS.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- H. M. Redhead, S. S. Davis and L. Illum, J. Controlled Release, 2001, 70, 353–363 CrossRef CAS PubMed.
- H. Li, X. Pu, J. Yin, X. Wang, S. Yao, H. M. Noh and J. H. Jeong, J. Am. Ceram. Soc., 2016, 99, 954–961 CrossRef CAS.
- Y. Asscher, G. Dal Sasso, L. Nodari, I. Angelini, T. Boffa Ballaran and G. Artioli, Phys. Chem. Chem. Phys., 2017, 19, 21783–21790 RSC.
- A. O. Shilov, R. V. Kamalov, M. S. Karabanalov, A. V. Chukin, A. S. Vokhmintsev, G. B. Mikhalevsky, D. A. Zamyatin, A. M. A. Henaish and I. A. Weinstein, Nanomaterials, 2023, 13, 3109 CrossRef CAS PubMed.
- J. Kappelhoff, J.-N. Keil, M. Kirm, V. N. Makhov, K. Chernenko, S. Möller and T. Jüstel, Chem. Phys., 2022, 562, 111646 CrossRef CAS.
- S. Espinoza, M. Volhard, H. Kätker, H. Jenneboer, A. Uckelmann, M. Haase, M. Müller, M. Purschke and T. Jüstel, Part. Part. Syst. Charact., 2018, 35, 1800282 CrossRef.
- H. Mändar, J. Felsche, V. Mikli and T. Vajakas, J. Appl. Crystallogr., 1999, 32, 345–350 CrossRef.
- H. Mändar, T. Vajakas, J. Telsche and R. E. Dinnebier, J. Appl. Crystallogr., 1996, 29, 304 CrossRef.
- A. Niilisk, T. Kahro, V. Kiisk, M. Rähn, H. Alles, J. Aarik and V. Sammelselg, Open Phys., 2015, 13, 34–40 CAS.
- M. Visnapuu, M. Rosenberg, E. Truska, E. Nõmmiste, A. Šutka, A. Kahru, M. Rähn, H. Vija, K. Orupõld, V. Kisand and A. Ivask, Colloids Surf., B, 2018, 169, 222–232 CrossRef CAS.
- M. Rosenberg, M. Visnapuu, K. Saal, D. Danilian, R. Pärna, A. Ivask and V. Kisand, Nanomaterials, 2021, 11, 3384 Search PubMed.
- R. Pärna, R. Sankari, E. Kukk, E. Nõmmiste, M. Valden, M. Lastusaari, K. Kooser, K. Kokko, M. Hirsimäki, S. Urpelainen, P. Turunen, A. Kivimäki, V. Pankratov, L. Reisberg, F. Hennies, H. Tarawneh, R. Nyholm and M. Huttula, Nucl. Instrum. Methods Phys. Res., Sect. A, 2017, 859, 83–89 Search PubMed.
- K. Chernenko, A. Kivimäki, R. Pärna, W. Wang, R. Sankari, M. Leandersson, H. Tarawneh, V. Pankratov, M. Kook, E. Kukk, L. Reisberg, S. Urpelainen, T. Käämbre, F. Siewert, G. Gwalt, A. Sokolov, S. Lemke, S. Alimov, J. Knedel, O. Kutz, T. Seliger, M. Valden, M. Hirsimäki, M. Kirm and M. Huttula, J. Synchrotron Radiat., 2021, 28, 1620–1630 CrossRef CAS PubMed.
- V. Pankratov, R. Pärna, M. Kirm, V. Nagirnyi, E. Nõmmiste, S. Omelkov, S. Vielhauer, K. Chernenko, L. Reisberg, P. Turunen, A. Kivimäki, E. Kukk, M. Valden and M. Huttula, Radiat. Meas., 2019, 121, 91–98 Search PubMed.
- J. Saaring, A. Vanetsev, K. Chernenko, E. Feldbach, I. Kudryavtseva, H. Mändar, R. Pärna, V. Nagirnyi, S. Omelkov, I. Romet, O. Rebane and M. Kirm, J. Alloys Compd., 2021, 883, 160916 CrossRef CAS.
- S. I. Omelkov, K. Chernenko, J. C. Ekström, A. Jurgilaitis, A. Khadiev, A. Kivimäki, A. Kotlov, D. Kroon, J. Larsson, V. Nagirnyi, D. V. Novikov, V.-T. Pham, R. Pärna, I. Romet, J. Saaring, I. Schostak, E. Tiirinen, A. Tõnisoo and M. Kirm, J. Phys.: Conf. Ser., 2022, 2380, 012135 CrossRef CAS.
- P. Li, Y. Liu, Y. Guo, X. Shi, G. Zhu and H. Zuo, Ceram. Int., 2015, 41, 6620–6630 CrossRef CAS.
- X. Zhang, M. Zhang, Y. Zhu, P. Wang, F. Xue, J. Gu, H. Bi and Y. Qian, Mater. Res. Bull., 2010, 45, 1324–1329 CrossRef CAS.
- Y. Zhang and H. Guan, J. Cryst. Grow., 2003, 256, 156–161 CrossRef CAS.
- Y. Ni, J. M. Hughes and A. N. Mariano, Am. Mineral., 1995, 80, 21–26 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A:Found. Adv., 1976, 32, 751–767 CrossRef.
- A. Debelle, A. Boulle, A. Chartier, F. Gao and W. J. Weber, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 90, 174112 CrossRef.
- S. Loridant, Catal. Today, 2021, 373, 98–111 CrossRef CAS.
- W. O. Milligan, D. F. Mullica, G. W. Beall and L. A. Boatner, Inorg. Chim. Acta, 1982, 60, 39–43 CrossRef CAS.
- A. Sanson, M. Giarola, M. Bettinelli, A. Speghini and G. Mariotto, J. Raman Spectrosc., 2013, 44, 1411–1415 CrossRef CAS.
- G. Gouadec and P. Colomban, Prog. Cryst. Growth Charact. Mater., 2007, 53, 1–56 CrossRef CAS.
- R. Javed, M. Zia, S. Naz, S. O. Aisida, N. U. Ain and Q. Ao, J. Nanobiotechnol., 2020, 18, 172 CrossRef PubMed.
- R. P. Bagwe, L. R. Hilliard and W. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS.
- P. Dorenbos, Phys. Rev. B:Condens. Matter Mater. Phys., 2012, 85, 165107 CrossRef.
- A. M. Srivastava, J. Lumin., 2016, 169, 445–449 CrossRef CAS.
- V. N. Makhov, N. Y. Kirikova, M. Kirm, J. C. Krupa, P. Liblik, A. Lushchik, C. Lushchik, E. Negodin and G. Zimmerer, Nucl. Instrum. Methods Phys. Res., Sect. A, 2002, 486, 437–442 CrossRef CAS.
- Y. Takebuchi, M. Koshimizu, K. Ichiba, T. Kato, D. Nakauchi, N. Kawaguchi and T. Yanagida, Materials, 2023, 16, 4502 CrossRef CAS.
- W. T. Carnall, G. L. Goodman, K. Rajnak and R. S. Rana, J. Chem. Phys., 1989, 90, 3443–3457 CrossRef CAS.
- I. Romet, É. Tichy-Rács, K. Lengyel, E. Feldbach, L. Kovács, G. Corradi, K. Chernenko, M. Kirm, D. Spassky and V. Nagirnyi, J. Lumin., 2024, 265, 120216 CrossRef CAS.
- A. M. Srivastava, M. Jennings and J. Collins, Opt. Mater., 2012, 34, 1347–1352 CrossRef CAS.
- A. M. Srivastava, A. A. Setlur, H. A. Comanzo, W. W. Beers, U. Happek and P. Schmidt, Opt. Mater., 2011, 33, 292–298 CrossRef CAS.
- V. V. Mikhailin, D. A. Spassky, V. N. Kolobanov, A. A. Meotishvili, D. G. Permenov and B. I. Zadneprovski, Radiat. Meas., 2010, 45, 307–310 CrossRef CAS.
- Z. Wang, Y. Xie, L. W. Campbell, F. Gao and S. Kerisit, J. Appl. Phys., 2012, 112, 014906 CrossRef.
- T. S. Malyy, V. V. Vistovskyy, Z. A. Khapko, A. S. Pushak, N. E. Mitina, A. S. Zaichenko, A. V. Gektin and A. S. Voloshinovskii, J. Appl. Phys., 2013, 113, 224305 CrossRef.
- S. B. Maji, A. Vanetsev, H. Mändar, V. Nagirnyi, K. Chernenko and M. Kirm, Opt. Mater., 2024, 154, 115781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00391a |
| This journal is © The Royal Society of Chemistry 2025 |