
Solution-processed MoS2 nanotubes/reduced graphene oxide nanocomposite as an active electrocatalyst toward the hydrogen evolution reaction†
Long Song,
Mengjia Zhao,
Xiaoxue Li,
Zhipan Zhang* and
Liangti Qu
Key Laboratory of Cluster Science, Ministry of Education of China, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: zhipan@bit.edu.cn; Tel: +86-10-68918608
First published on 21st July 2016
Abstract
Advanced materials for electrocatalytic water splitting are essential to the area of renewable energy. In this work, we report a hierarchical 3D nanostructure of MoS2 nanotubes supported on a reduced graphene oxide network (MNTs@rGO) through a surfactant-assisted lyophilization process. The resulting MNTs@rGO exhibited good electrocatalytic activity in the hydrogen evolution reaction (HER) relative to other representative MoS2-based electrocatalysts, with an onset overpotential of 180 mV, a small Tafel slope of 69 mV dec−1 as well as a large cathodic current density (38.91 mA cm−2 at an overpotential of 300 mV). Linear sweep voltammetry (LSV) tests and electrochemical impedance spectroscopic (EIS) measurements reveal that the MNT building blocks with high exposure of surface atoms function as the active catalytic sites in the HER and the rGO support serves as the conductive footstone connecting all catalytic sites with fast electron transport. The EIS characterization also demonstrates that the electron transfer at the catalyst/electrolyte interface is the rate-limiting step in the catalyzed HER and assembling MoS2 nanosheets into nanotubes can significantly facilitate the HER. The current results are deemed to provide new insights into next-generation HER catalysts with high activity and low cost.
Introduction
Owing to its high specific energy (120–142 MJ kg−1) and environmentally-friendly combustion to form water, hydrogen has been considered as one of the most promising alternatives to complement traditional fossil fuels. Generally, a catalyst is used to facilitate the hydrogen evolution reaction (HER) from electrochemically splitting water.1–4 To date, while platinum-based materials including polycrystalline platinum [Pt(pc)], carbon-supported platinum nanoparticles (Pt/C), PtFeCo trimetallic tristar nanostructures and Li+–Ni(OH)2–Pt have shown high catalytic exchange current densities and small Tafel slopes,5–7 their high costs and low abundance limit their large-scale industrial applications.8,9 Consequently, developing noble-metal-free HER electrocatalysts with high activity is of great theoretical value and practical importance. To date, various catalysts have been extensively studied including non-noble-metal alloys (NiFeMo, CoMo),10 transition metals sulfides (MoS2 single-layer polytypes)/nitrides (W2N nanowire arrays)/phosphides (Fe0.5Co0.5P) as well as carbides (Mo2C nanowires).11–14 Among all these materials, two-dimensional (2D) layered transition metal dichalcogenides (TMDs) have exhibited remarkable catalytic activities thanks to their unique crystal structures, wide range of chemical compositions and distinctive material properties. TMDs feature strong intralayer covalent bonding and weak interlayer van der Waals force and demonstrate different electrochemical activities between the edge sites with uncoordinated atoms and the fully coordinated terrace sites, making them a versatile and inexpensive platform to study the catalytic process of HER.15,16In particular, recent results have shown that molybdenum disulfide (MoS2) can be a promising cost-effective alternative to Pt for the generation of hydrogen from water. For instance, Ji et al. successfully exfoliated two-dimensional MoS2 nanosheets from commercial MoS2 powders through a direct dispersion and ultrasonication method.17 The resulting nanosheets were directly deposited onto glassy carbon electrode substrate via a drop-casting method and exhibited an excellent electrocatalytic activity toward the HER (1.26 and 6.36 mA cm−2 at an overpotential (η) of 150 and 200 mV respectively). Xie et al. put forward a scalable pathway to realize controllable defect engineering in MoS2 ultrathin nanosheets by using a high concentration of precursors and different amounts of thiourea to promote partial cracking of the catalytically inert basal planes and the exposure of additional active edge sites.18 The defect-rich MoS2 ultrathin nanosheets exhibited excellent HER activity, with a small onset overpotential of 120 mV, a large cathodic current density (13 mA cm−2 at η = 200 mV) as well as a small Tafel slope of 50 mV dec−1. Moreover, this material showed excellent electrochemical stability comparable to that of the commercial Pt/C catalyst. Unfortunately, freshly prepared MoS2 nanosheets tend to aggregate after exfoliation, leading to a loss of active sites and/or interfacial catalytic surface area. Additionally, the high resistivity of MoS2 induces a large internal voltage loss during electron transport to the collection electrode, resulting in a concomitant high overpotential during the HER. A series of strategies have been employed to effectively alleviate the aggregation problem and promote the HER. Chung et al. controllably synthesized 2D MoS2 small flakes exposed to the edge-terminated sites into hierarchical nanostructures via a facile hydrothermal reaction, revealing the linear relationship between exchange current density and the number of sulfur edges existed and demonstrating that the active sites for MoS2 nanosheets were sulfur edges.19 By controlling the MoS2 morphology with the formation of nano-assembled spheres with the assembly of small-size fragments of MoS2, the resulting material has high electrocatalytic HER activity (a very low onset potential of 54 mV and a Tafel slope of 100 mV dec−1) and high thermodynamic stability (similar activity after 3000 cycles). In another study, Lu et al. designed a MoS2 film with a ‘superaerophobic’ surface composed of vertically aligned nanoplates and significantly improved the removal rate of small gas bubbles during the HER.20 Apart from these strategies, anchoring active MoS2 nanostructures onto a 3D conductive architecture with a high active surface area and interconnected pore channels can be another viable choice to retain the initial morphology of MoS2 building blocks. Graphene, an ideal two-dimensional single atomic carbon sheet with an extremely high specific surface area of about 2600 m2 g−1 in theory and high electrical conductivity of 106 S cm−1, has demonstrated several critical advantages in acting as the support for catalysts to boost the active surface area and facilitate charge transfer/mass transport during the catalysis. Consequently, the integration of MoS2 nano-blocks onto interconnecting 3D carbon networks can be a powerful method to obtain highly active catalysts for the HER. Recently, we have prepared a composite catalyst composed of iron and nickel layered double hydroxides (FeNi-LDHs) grown on 3D graphene networks via a one-step hydrothermal process and the catalyst demonstrated higher electrocatalytic activity for the oxygen evolving reaction than the conventional planar electrode.21 Aiming to profit from the high electrocatalytical performance of MoS2 nanotubes (MNTs) and the desirable interfacial properties of a porous graphene network, we herein synthesize a hierarchical 3D nanostructure of MoS2 nanotubes supported on reduced graphene oxide network (MNTs@rGO) through a surfactant-assisted lyophilization process. The unique MNTs@rGO system apparently possesses many advantages over the conventional MNTs or simple mixtures of MNTs and rGO catalysts in the HER. On one hand, the MNTs building blocks benefit from the high exposure of surface atoms that function as the active catalytic sites in the HER and the intimate contact between MNTs and graphene sheets ensures a facile electron flow at the MNTs/rGO interface, thus eliminating the bottleneck of electron transport through different layers within the highly resistive MoS2. On the other hand, the rGO support serves as the conductive footstone for the MNTs and creates an interconnected 3D framework connecting all catalytic sites with fast electron transport and sufficient microporous/macroporous channels that promote the diffusion of gas molecules and electrolytes in the HER. As a result, the MNTs@rGO shows a high catalytic current density (38.91 mA cm−2 at an overpotential of 300 mV) and excellent stability, showing its potential as an active catalyst in the HER. Meanwhile, electrochemical impedance spectroscopic (EIS) measurements reveal that the electron transfer at the MNTs@rGO/electrolyte interface is the rate-limiting step in the catalyzed HER, rendering new insights to develop next-generation HER catalysts with a high activity and low cost.
Experimental
Materials synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 10 μL P123 aqueous solution (10 mg mL−1) was added under vigorous stirring. Lyophilization was applied to prevent the aggregation of graphene sheets and MoS2 nanotubes during the drying process. After that, the as-prepared MNTs@GO was put in a tube furnace and annealed at 200 °C for 2 hours under Ar/H2 atmosphere to reduce GO to rGO and enhance the electrical conductivity. As the mass ration of MoS2 nanotubes to rGO may strongly affect the electrocatalytic performance of the MNTs@rGO samples towards the HER reactions, control samples with different MNTs to GO mass ratios of 0.5:1, 1:1 and 5:1 were prepared in the same manner by varying the amount of MoS2 nanotubes added into the GO solution. For comparison, commercial bulk MoS2 anchored on rGO (BM@rGO) was also prepared under the same conditions by using commercial available bulk MoS2 powder (BM) as the starting material.
:1 and 10 μL P123 aqueous solution (10 mg mL−1) was added under vigorous stirring. Lyophilization was applied to prevent the aggregation of graphene sheets and MoS2 nanotubes during the drying process. After that, the as-prepared MNTs@GO was put in a tube furnace and annealed at 200 °C for 2 hours under Ar/H2 atmosphere to reduce GO to rGO and enhance the electrical conductivity. As the mass ration of MoS2 nanotubes to rGO may strongly affect the electrocatalytic performance of the MNTs@rGO samples towards the HER reactions, control samples with different MNTs to GO mass ratios of 0.5:1, 1:1 and 5:1 were prepared in the same manner by varying the amount of MoS2 nanotubes added into the GO solution. For comparison, commercial bulk MoS2 anchored on rGO (BM@rGO) was also prepared under the same conditions by using commercial available bulk MoS2 powder (BM) as the starting material.Characterizations
The morphologies and elemental mappings of the samples were characterized by field-emission scanning electron microscopy (FE-SEM, JSM-7001F) and transmission electron microscopy (TEM, JEM-2100). X-ray photoelectron spectroscopy (XPS) data were obtained with an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Al Kα radiation. Nitrogen adsorption–desorption isotherms were measured on a Quantachrome Autosorb-IQ gas adsorption analyzer at 77 K. Pore size distribution and specific surface area were obtained through the non-local density functional theory (NLDFT) and the Brunauer–Emmett–Teller (BET) methods from the adsorption branch of the isotherm at a relative pressure range of p/p0 = 0–1.0. All the electrochemical measurements were performed in a three-electrode system at a computer controlled potentiostat equipped with a frequency response analyzer (Autolab, Model 204).Electrochemical measurements
The electrocatalytic activity of pure MNTs and BM were also prepared and evaluated for comparison. In a typical procedure, 16 mg sample was ultrasonically dispersed in 3.6 mL ethanol–isopropanol solution with volume ratio of 3:1 to form a homogeneous ink, into which 0.4 mL Nafion solution (5 wt%) was added. Then, 5 μL of the ink was loaded onto a glassy carbon electrode with 5 mm diameter (loading of ca. 0.102 mg cm−2). Linear sweep voltammetry (LSV) was carried out in 0.5 M H2SO4 aqueous solution (degassed with N2). A platinum foil and Ag/AgCl (in 3 M KCl solution) were used as the counter electrode and reference electrode, respectively. All of the polarization curves were corrected for the IR losses and all of the potentials were calibrated to a reversible hydrogen electrode (RHE). For the impedance measurement, the frequency range was 0.1–10 kHz and the magnitude of the modulation signal was 10 mV. The obtained spectra were fitted with Z-View software (v2.8b, Scribner Associates Inc.) in terms of appropriate equivalent circuits.
Results and discussion
The morphology of the as-prepared MNTs was first characterized by the scanning electron microscopy (SEM). As shown in Fig. 1a, the MNTs feature a tubular morphology with uniform diameters of 380–400 nm and lengths of 1.3–1.5 μm. The inner hollow structure and rough surface can be seen clearly in the low-magnification transmission electron microscopy (TEM) images (Fig. 1b). The high-resolution TEM image (Fig. 1c) shows the lattice spacing of 0.62 nm, corresponding to the interlayer distance of (002) crystal plane of MoS2. The transparent nature of the edge reveals that the number of stacked layers is less than ten, suggesting the MNTs are built up by few-layered MoS2 nanoplates. The thin feature of the MNTs was further confirmed by the X-ray diffraction (XRD) analysis (Fig. 1d). The weak peak of (002) diffraction at 14.39° indicates a disordered packing of few-layer MoS2 and broad features between 30 and 60° imply the amorphous nature of the as-prepared MNTs.24 The surface states of the MNTs were further probed by the X-ray photoelectron spectroscopy (XPS) analysis in Fig. 1e and f. The high-resolution XPS spectrum exhibits Mo 3d5/2 and Mo 3d3/2 peaks with binding energy of 229.10 eV and 232.25 eV, respectively, corresponding to the Mo4+ species in MoS2.22,25,26 The presence of a small feature at about 235.58 eV is attributed to the Mo6+ 3d5/2 peak of MoO3, which presumably arises from the incomplete decomposition of ammonium molybdate during the hydrothermal treatment. The peaks located at 161.90 eV and 163 eV are characteristic of the S2− 2p3/2 and S2− 2p1/2 components of MoS2 (Fig. 1f), respectively.22,27,28 Overall, the calculated sulfur to molybdenum (S/Mo) atomic ratio is about 1.93, which is in agreement with the stoichiometric ratio of MoS2. The specific surface area and pore structure were analyzed through the Brunauer–Emmett–Teller (BET) method. As illustrated in Fig. 1g, the N2 adsorption/desorption loop of MNTs shows a typical type-IV isotherm for mesoporous materials, rendering a high specific surface area of 112 m2 g−1. As compared to the BM sample that is composed of heavily stacked 2D MoS2 layers (Fig. S1†), the specific surface area of MNTs is more than seven times larger than that of the BM. Meanwhile, in contrast to the absence of micropores in the BM (Fig. 1h), a significant presence of micropores and mesopores ranging from 1 to 4 nm in the MNTs is confirmed by the pore size distribution derived from the non-local density function theory (NLDFT) and attributed to mesopores channels in the tubular structure resulting from the disordered assembly of MoS2 few-layered nanoplates. Consequently, compared to the heavily stacked BM, such a porous structure will lead to a large catalyst/electrolyte interface with more exposed electrocatalytic active sites, potentially leading to an improved catalytic performance in the HER.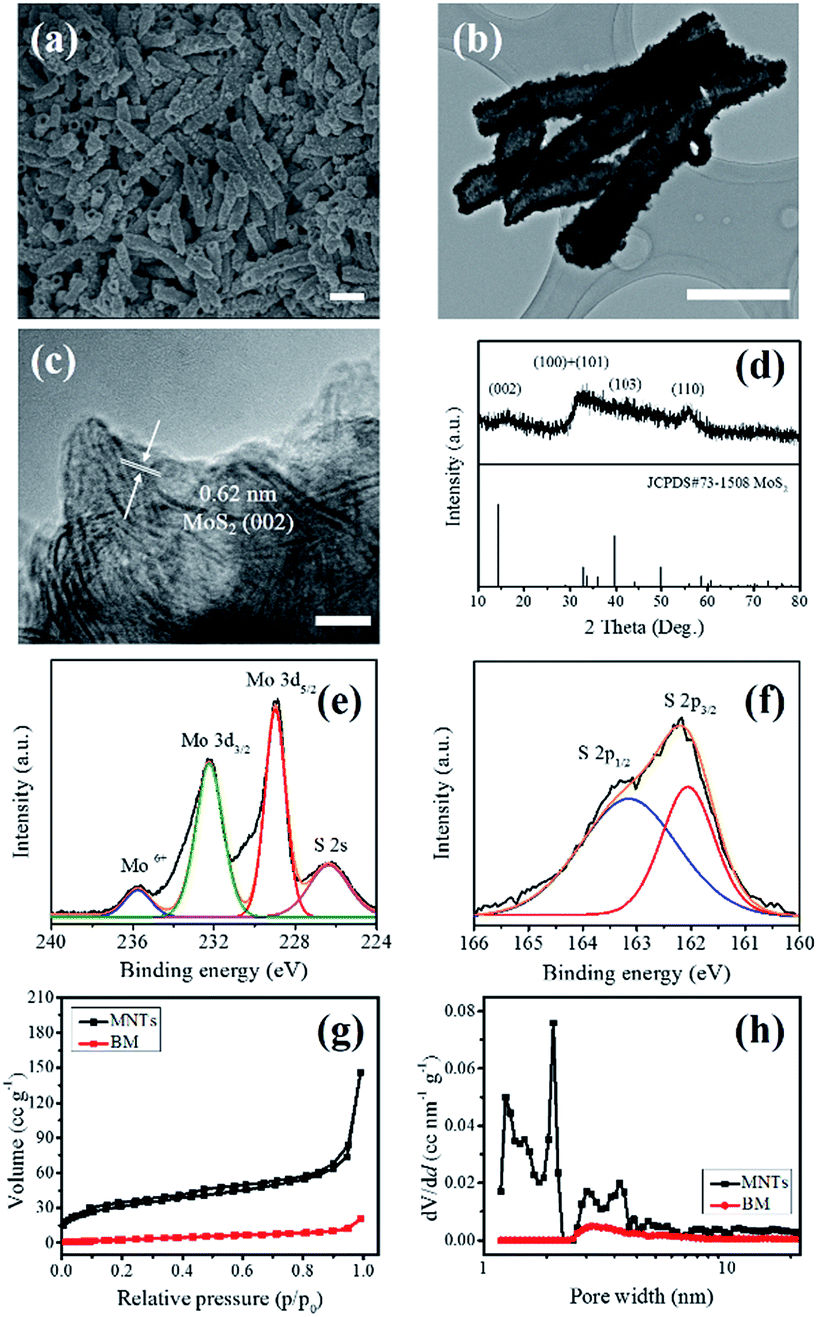 | ||
| Fig. 1 (a) SEM image of the as-prepared molybdenum disulfide nanotubes (MNTs). (b) TEM image of the typical MNTs. (c) Magnified TEM image of the typical MNTs. The scale bars in (a), (b) and (c) are 1 μm, 1 μm and 5 nm, respectively. (d) XRD pattern of the typical MNTs. High-resolution XPS spectra of (e) Mo6+, Mo 3d and S 2s peaks, and (f) S 2p peaks. (g) N2 adsorption/desorption isotherms, and (h) corresponding pore size distribution of the as-prepared MNTs and commercial bulk molybdenum disulfide (BM), respectively. | ||
The MNTs were anchored on the reduced GO to form the hybrid catalyst of MNTs@rGO by lyophilization. The morphology of the sample was characterized by SEM and TEM. As shown in Fig. 2a, the MNTs are randomly attached to the rGO sheets and the nanocomposite features a loose 3D structure similar to that of a typical 3D porous graphene network.29 The translucent nature of the rGO sheets is clearly visible in the TEM image (Fig. 2b), suggesting that the rGO network was built up by few-layered graphene sheets, and the characteristic (002) interplane spacing of MoS2 can be still resolved under higher magnifications in the TEM image (Fig. 2c). The elemental mappings in Fig. 2d–g show the presence of C element across the rGO network as well as the coexistence and homogeneous distribution of Mo and S elements in the MNTs. Additionally, the XPS survey spectrum of MNTs@rGO in Fig. 2h confirms the presence of C 1s, O 1s, Mo 3d and S 2p without any impurities. The corresponding peak positions of Mo4+ and S2− in the high-resolution XPS spectrum (Fig. 2i and j) remain unchanged as compared with that of the MNTs. The binding energy peaks located at 229.10 and 232.25 eV correspond to the Mo4+ 3d5/2 and Mo4+ 3d3/2 components of MoS2, respectively. The peaks at 162 and 163 eV can be indexed to the S2− 2p3/2 and S2− 2p1/2 components of MoS2, respectively. Besides, the XRD pattern of MNTs@rGO is shown in Fig. S2† and compared with those of rGO, BM, BM@rGO and MNTs as well. All these characterizations indicate that the MNTs have been retained intact and successfully integrated onto the rGO network.
 | ||
| Fig. 2 (a) SEM image of the as-prepared MNTs@rGO nanocomposite. (b) TEM image and (c) magnified view of the typical MNTs@rGO. The scale bars in (a), (b) and (c) are 1 μm, 500 nm and 5 nm, respectively. (d) SEM image showing the MNT supported on rGO sheets and (e–g) corresponding C-, Mo- and S-elemental mappings. Scale bars are 1 μm for (d–g). (h) XPS spectrum of the MNTs@rGO nanocomposite and high-resolution XPS spectrum showing (i) Mo6+, Mo 3d and S 2s peaks, and (j) S 2p peaks. | ||
The electrocatalytic activity of the MNTs@rGO in the HER was investigated by depositing it on a glassy carbon electrode (GCE) and measuring the cathodic current in 0.5 M H2SO4 aqueous solution. To study the effect of MNTs to rGO ratio on the catalytic performance of MTNs@rGO, the mass ratio of MNTs to rGO was purposely tuned from 0.5:1, 1:1, 2.5:1 to 5:1. The evolution of MNTs@rGO morphology with different ratios of MTNs to rGO is shown in Fig. 3. With the increasing amount of MNTs in the composite, the presence of the MNTs became more conspicuous on the rGO network, where large aggregates of MNTs apparently formed when the mass ratio of MNTs to rGO reached 5:1 (Fig. 3d). As pure rGO provides negligible catalytic current in the HER, the electrocatalytic current density (Jcat) improves with the increasing amount of electrocatalytically active MNTs contents until a mass ratio of 2.5:1 (Fig. 3e). Unfortunately, further increase of the MNTs in the composite caused a significant decrease in the catalytic activity, possibly due to the fact that excessive stackings of the MNTs eliminate effective catalytic sites as well as interrupt the connection between the highly resistive MNTs and the underlying conductive rGO network, retarding the electron transport from MNTs to the electrode and thus leading to a deteriorated catalytic performance. Therefore, for simplicity, only the MNTs@rGO sample prepared with MNTs to rGO mass ratio of 2.5:1 was examined below and the catalyst mass loading was additionally optimized to be 0.102 mg cm−2 (Fig. S3†).
 | ||
| Fig. 3 SEM images of MNTs@rGO with mass ratio (MNTs to rGO) of (a) 0.5:1, (b) 1:1, (c) 2.5:1 and (d) 5:1. All the scale bars are 1 μm. (e) The polarization curves of MNTs@rGO with different mass ratio (MNTs to rGO). | ||
Fig. 4a compares the polarization curves of different samples of BM, MNTs, BM@rGO and MNTs@rGO. Due to the low conductivity and lack of catalytic surface area, BM demonstrated very poor catalytic activity with a current density of less than 1 mA cm−2 at a large η of 400 mV. In comparison, the Jcat of the MNTs is significantly larger than that of the BM partly owing to their higher specific surface area. Meanwhile, loading BM or MNTs on the rGO network also boosted the Jcat and it is apparent that MNTs@rGO exhibited the highest Jcat. The catalytic current is observed to initiate at an onset potential of about −180 mV versus reversible hydrogen electrode (RHE), and beyond this potential, Jcat rises rapidly to about 38.91 mA cm−2 at η = 300 mV. It's worth noting that both MNTs@rGO and BM@rGO show better HER activities than their counterparts without the rGO network, verifying that the electrical coupling between the catalytically active sites and the underlying rGO sheets in an interconnected network renders rapid electron transport from the less-conducting catalyst to the electrode.30 To investigate the kinetics of the HER catalyzed by these samples, the individual Tafel plots were measured and plotted in Fig. 4b. The Tafel slope of the MNTs@rGO is 69 mV dec−1, suggesting the release of molecular hydrogen is the rate-limiting step.31 In contrast, higher slopes are found in the MNTs (80 mV dec−1), BM@rGO (83 mV dec−1) and BM (121 mV dec−1), respectively, demonstrating a retarded increase of the HER rate with the rising overpotential due to the absence of high catalytic surface area and/or fast electron transport channels. For the MNTs@rGO, an overpotential of 250 mV is needed to achieve a benchmark Jcat of 10 mA cm−2, outperforming most graphene and/or MoS2-based HER catalysts in the literature (Fig. 4c and Table S1†), including rGO paper supported MoS2 nanoflowers (285 mV),32 nanoporous MoS2 (270 mV)33 and MoS2 composite consist of nanosheets and quantum dots (260 mV).34 Durability is another key factor in evaluating the performance of non-precious-metal electrocatalysts in the HER. Herein, the accelerated degradation measurement was adopted to investigate the stability of the MNTs@rGO. Cyclic voltammetric (CV) sweeps were continuously measured between 0.1 V and −0.60 V (versus RHE) at a scan rate of 50 mV s−1 for 1000 cycles, and as shown in Fig. 4d, the polarization curves of the MNTs@rGO almost overlap at cycle 1 and cycle 1000, highlighting the excellent stability of the composite catalyst. The Jcat exhibits only slight degradations after a long-term electrolysis for more than 1000 cycles, which may be caused by the consumption of H+ in the electrolyte and/or the accumulation of H2 bubbles on the surface of the electrode.35 The effective electrochemical surface areas of the solid–liquid interfaces for MNTs@rGO and BM@rGO were calculated by measuring the corresponding electrochemical double layer capacitance (Cdl) using a CV method (Fig. S4†).36 A potential range of 0.15–0.25 V (versus RHE) was selected due to the absence of any faradaic current in this region for both catalysts.37 The MNTs@rGO exhibits an enhanced Cdl of 8 mF cm−2 that is about 3.3 times higher than that of the BM@rGO (2.3 mF cm−2) (Fig. 4e) and more importantly, this improvement in electrochemical surface area is lower than the 20-fold increase in Jcat (for example, 38.91 mA cm−2 for MNTs@rGO versus 1.83 mA cm−2 for BM@rGO at η = 300 mV), suggesting that the MNTs@rGO possesses a better intrinsic catalytic activity than the BM@rGO. The calculated turnover frequency (TOF) for each active site of the MNTs@rGO reaches 0.221 s−1 at η = 300 mV (Fig. 4f), which is about 30 times higher than that of BM@rGO (0.007 s−1 at η = 300 mV).38
 | ||
| Fig. 4 (a) The polarization curves in the HER and (b) the corresponding Tafel plots of catalysts BM, MNTs, BM@rGO and MNTs@rGO measured in 0.5 M H2SO4 with a scan rate of 5 mV s−1. (c) Comparison of overpotential values for different HER catalysts to reach a benchmark current density of 10 mA cm−2. (d) Durability test for the MNTs@rGO catalyst showing negligible performance loss after 1000 CV cycles from 0.1 to −0.6 V at a scan rate of 50 mV s−1. (e) Plots showing the extraction of the double layer capacitance (Cdl) for the MNTs@rGO and BM@rGO. (f) The plot of TOFs with respect to S atoms at different overpotentials. | ||
Finally, electrochemical impedance spectroscopy (EIS) was employed to scrutinize the kinetics of the HER reaction catalyzed by the MNTs@rGO and BM@rGO. Fig. 5a plots the typical Nyquist plots of the EIS spectra for both catalysts recorded at the same overpotential of 250 mV, with an enlarged view of the high frequency region shown in Fig. 5b. The semicircle at higher frequencies represents a time constant that is largely potential-independent and is related to the contact between the GCE and the catalyst, whereas the semicircle at lower frequencies is potential-dependent and denotes the charge transfer process at the interface of the catalyst and the electrolyte. In addition, a 45° line feature can be observed between these two semicircles in the Nyquist plot and is believed to be associated with the slow electron transport in the catalyst layer.39 As can be seen clearly in Fig. 5a and b, the MNTs@rGO features a much lower impedance compared to the BM@rGO, indicating a significantly promoted electron transfer between the catalyst and the proton in the electrolyte. To decouple the influence of catalyst conductivity and intrinsic catalytic activity, a series of EIS spectra were recorded by changing the applied bias and the corresponding equivalent circuit based on the transmission line model is shown in the inset of Fig. 5a.31,40 Here, Rs is the serial resistance of the whole electrolytic cell system, RE and CPEE are the charge transfer resistance and the constant phase element for the capacitance at the electrode/catalyst interface, respectively. DX is the extended element representing the electronic transport in the solid phase of the catalyst, the ionic transport in the electrolyte and the electrochemical recombination. By appropriately fitting the EIS spectra with the equivalent circuit, the charge transfer resistance (Rct) at the catalyst/electrolyte interface, the electron transport resistance (Rm) across the catalyst layer and the electrochemical capacitance (Cint) at the catalyst/electrolyte interface could be obtained and detailed values of fitted parameters are listed in Tables S2 and S3.† Fig. 5c plots the Rct values for the MNTs@rGO and BM@rGO at different overpotentials and both catalysts feature a decreasing Rct with the increasing overpotential, as would be predicted by the Butler–Volmer kinetics. More importantly, Rct of the MNTs@rGO is at least two orders of magnitudes smaller than that of the BM@rGO at any bias in the tested overpotential region, representing a remarkably more facile charge transfer from the catalyst to the proton in the electrolyte and thus well consistent with its noticeably higher catalytic activity in the HER. Additionally, while Rm is less dependent on the overpotential (Fig. 5d), the Rm value of the MNTs@rGO is also smaller than that of the BM@rGO, implying a faster electron percolation across the catalyst layer. Meanwhile, the MNTs@rGO possesses a considerably larger Cint than the BM@rGO (Fig. 5e), well corresponding to a larger electrochemical surface at the catalyst/electrolyte interface found in the CV measurement (Fig. 4e). The associated characteristic time for electron transfer across the catalyst/electrolyte interface in the HER and electron transport process can be approximately quantified by the time constants of individual RC time constants, e.g. τct = (Rct × Cint)1/2 and τm = (Rm × Cint)1/2, where smaller τct and τm represent a larger rate of proton reduction on the electrocatalyst and a faster electron transport inside the catalyst layer. Fig. 5f summarizes the evolution of τct and τm for the MNTs@rGO and BM@rGO at different overpotentials. Overall, τct values for both samples are more heavily dependent on the overpotentials than τm, in accordance with the observed difference between Rct and Rm. τct becomes considerably smaller as the overpotential rises and in the meantime, the rate of HER is believed to significantly increase. It is more important to note that over the measured overpotential region, τm is always smaller than τct for both samples, suggesting that the catalytic activity is limited by the electron transfer at the catalyst/electrolyte interface rather than the electron transport within the catalyst layer. At any given overpotential, a far smaller τct is found in the MNTs@rGO as compared to the BM@rGO, proving the much better intrinsic catalytic activity for the MNTs@rGO in the HER. In addition, despite of the smaller Rm found in the MNTs@rGO, a larger τm is surprisingly observed when compared to the BM@rGO at the same overpotential, possibly due to the increased pathlength and/or slower electron transport rate of the MNTs induced by its amorphous nature. However, it should be emphasized that this phenomenon seems to have no adverse effects on the HER catalyzed by the MNTs@rGO in the tested overpotential range, as τm is still considerably smaller than the corresponding τct at high overpotentials. As predicted, the τct of the BM@rGO is orders of magnitudes larger than τct over all the overpotentials. Since most of the electrons tend to transfer through the outer layer of the BM, the coordinatively unsaturated S atoms inside the bulk material will contribute to the electron transport but are not available in the process of absorbing protons and desorbing hydrogen molecules in the HER, leading to the poor electrocatalytic activity. In contrast, the MNTs feature a much higher percentage of surface S atoms that can serve as the electrocatalytically active sites and thus possess a significantly smaller τct (i.e. higher catalytic performance) over the BM.
 | ||
| Fig. 5 (a) Full-scale Nyquist plots for the MNTs@rGO and BM@rGO at an overpotential of 250 mV. The inset shows the equivalent circuit of the catalytic system. (b) A magnified view of Nyquist plots in (a). (c) to (f) are plots of the extracted parameters of Rct, Rm, Cint, τct and τm against the overpotentials for the MNTs@rGO and BM@rGO, respectively. | ||
Conclusions
In summary, rationally assembled hierarchical architecture of MoS2 nanotubes on 3D graphene network has been achieved through a simple surfactant-assisted lyophilization process. The polarization curve and Tafel measurement confirm that the composite catalyst is highly active towards hydrogen generation from water with an excellent stability over 1000 consecutive CV cycles. The EIS characterization reveals that rather than the electron transport across the catalyst layer, the electron transfer at the catalyst/electrolyte interface is the rate-limiting step in the catalyzed HER and assembling MoS2 nanosheets into nanotubes significantly facilitates the HER. The current results unambiguously demonstrate that the MNTs@rGO is an ideal noble-metal-free electrocatalyst and increasing the surface atom ratio to promote the electron transfer across the catalyst/electrolyte interface is more effective than improvement in electron transport to boost the catalytic activity, rendering new insights into designing highly-efficient and low-cost catalysts for the HER.Acknowledgements
We are grateful for the financial support from the 973 programs (2011CB013000 and 2011CBA00701) of China, the State Key Laboratory of Solidification Processing in NWPU (No. SKLSP201601) and the Technology Foundation for Selected Overseas Chinese Scholar, MOHRSS of China (No. 3190036821401).Notes and references
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553 CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529 CrossRef CAS.
- N. Du, C. Wang, X. Wang, Y. Lin, J. Jiang and Y. Xiong, Adv. Mater., 2016, 28, 2077 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256 CrossRef CAS PubMed.
- Z. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168 CrossRef CAS PubMed.
- F. Li, L. Zhang, J. Li, X. Lin, X. Li, Y. Fang, J. Huang, W. Li, M. Tian, J. Jin and R. Li, J. Power Sources, 2015, 292, 15 CrossRef CAS.
- A. W. Jeremiasse, J. Bergsma, J. M. Kleijn, M. Saakes, C. J. N. Buisman, M. C. Stuart and H. V. M. Hamelers, Int. J. Hydrogen Energy, 2011, 36, 10482 CrossRef CAS.
- X. Fan, Y. Yang, P. Xiao and W. Lao, J. Mater. Chem. A, 2014, 2, 20545 CAS.
- V. Chakrapani, J. Thangala and M. K. Sunkara, Int. J. Hydrogen Energy, 2009, 34, 9050 CrossRef CAS.
- J. Kibsqaard, C. Tsai, K. Chan, J. D. Benck, J. K. Nrskov, F. A. Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 Search PubMed.
- L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387 CAS.
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2664 RSC.
- H. Wang, Q. Zhang, H. Yao, Z. Liang, H. W. Lee, P. C. Hsu, G. Zheng and Y. Cui, Nano Lett., 2014, 14, 7138 CrossRef CAS PubMed.
- S. Ji, Z. Yang, C. Zhang, Z. Liu, W. W. Tjiu, I. Y. Phang, Z. Zhang, J. Pan and T. Liu, Electrochim. Acta, 2013, 109, 269 CrossRef CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. David Lou and Y. Xie, Adv. Mater., 2013, 25, 5807 CrossRef CAS PubMed.
- D. Y. Chung, S. K. Park, Y. H. Chung, S. H. Yu, D. H. Lim, N. Jung, H. C. Ham, H. Y. Park, Y. Piao, S. J. Yoo and Y. E. Sung, Nanoscale, 2014, 6, 2131 RSC.
- Z. Lu, W. Zhu, X. Yu, H. Zhang, Y. Li, X. Sun, X. Wang, H. Wang, J. Luo, X. Lei and L. Jiang, Adv. Mater., 2014, 26, 2683 CrossRef CAS PubMed.
- Y. Li, M. Zhao, Y. Zhao, L. Song and Z. Zhang, Part. Part. Syst. Charact., 2016, 33, 158 CrossRef CAS.
- P. P. Wang, H. Sun, Y. Ji, W. Li and X. Wang, Adv. Mater., 2014, 26, 964 CrossRef CAS PubMed.
- C. G. Hu, H. H. Cheng, Y. Zhao, Y. Hu, Y. Liu, L. M. Dai and L. T. Qu, Adv. Mater., 2012, 24, 5493 CrossRef CAS PubMed.
- S. Appel, A. Volman, L. Houben, Y. Gelbstein and M. B. Sadan, J. Mater. Sci., 2014, 49, 7353 CrossRef CAS.
- H. Yu, C. Ma, B. Ge, Y. Chen, Z. Xu, C. Zhu, C. Li, Q. Ouyang, P. Gao, J. Li, C. Sun, L. Qi, Y. Wang and F. Li, Chem.–Eur. J., 2013, 19, 5818 CrossRef CAS PubMed.
- J. Xiao, X. Wang, X. Q. Yang, S. Xun, G. Liu, P. K. Koech, J. Liu and J. P. Lemmon, Adv. Funct. Mater., 2011, 21, 2840 CrossRef CAS.
- V. O. Koroteev, L. G. Bulusheva, I. P. Asanov, E. V. Shlyaknova, D. V. Vyalikh and A. V. Okotrub, J. Phys. Chem. C, 2011, 43, 21199 Search PubMed.
- G. Eda, H. Yamaguchi, D. Viory, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111 CrossRef CAS PubMed.
- Y. Zhao, C. Hu, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881 CrossRef CAS PubMed.
- C. B. Ma, X. Qi, B. Chen, S. Bao, Z. Yin, X. J. Wu, Z. Luo, J. Wei, H. L. Zhang and H. Zhang, Nanoscale, 2014, 6, 5624 RSC.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- S. Xu, D. Li and P. Wu, Adv. Funct. Mater., 2015, 25, 1127 CrossRef CAS.
- Y. Zhao, F. Zhao, X. Wang, C. Xu, Z. Zhang, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2014, 53, 13934 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 Search PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274 CrossRef CAS PubMed.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262 RSC.
- H. Vrubel, T. Moehl, M. Grätzel and X. Hu, Chem. Commun., 2013, 49, 8985 RSC.
- K. C. Pham, Y. H. Chang, D. S. McPhail, C. Mattevi, A. T. S. Wee and D. H. C. Chua, ACS Appl. Mater. Interfaces, 2016, 8, 5961 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11147e |
| This journal is © The Royal Society of Chemistry 2016 |