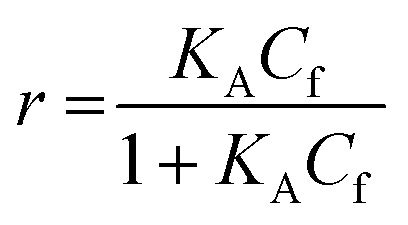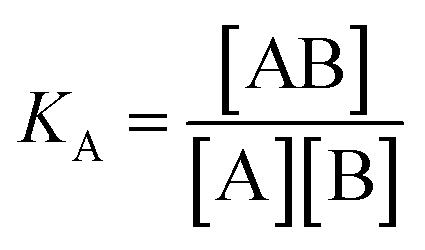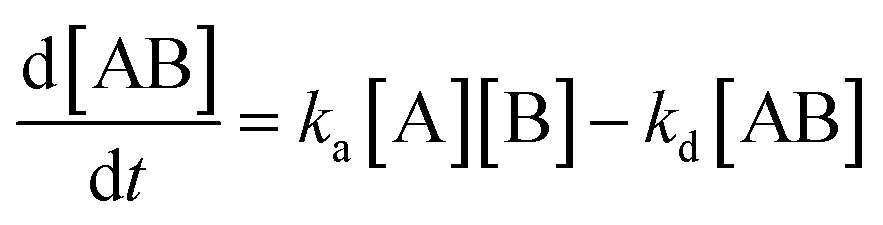DOI:
10.1039/C6RA05266E
(Paper)
RSC Adv., 2016,
6, 71652-71661
Molecular recognition of parallel quadruplex [d-(TTGGGGT)]4 by mitoxantrone: binding with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :4 stoichiometry leads to telomerase inhibition†
:4 stoichiometry leads to telomerase inhibition†
Received
28th February 2016
, Accepted 18th July 2016
First published on 18th July 2016
1. Introduction
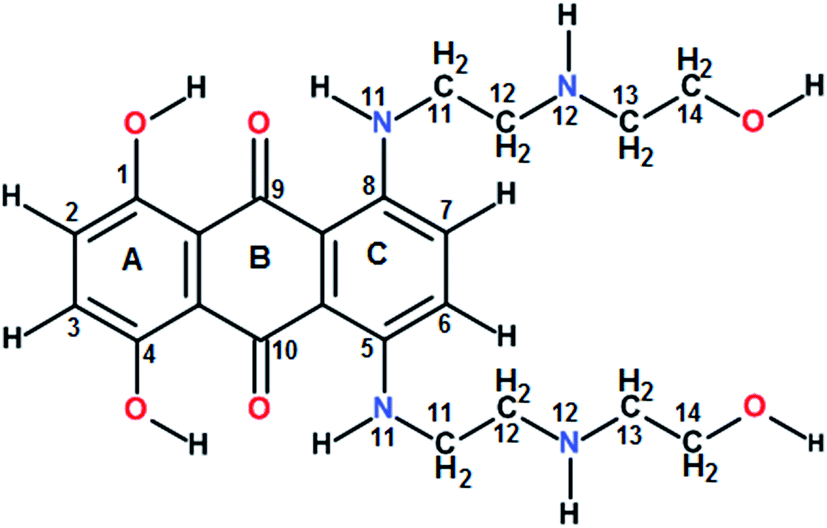
Guanine rich DNA forming G-quadruplex structures have been identified in human telomeres, gene promoters (c-myc, c-kit, K-ras and bcl-2), the immunoglobulin heavy chain switch region, ribosomal DNA, 5′-UTRs of RNA oncogenic sequences, etc. Several proteins bind, promote or non-catalytically disrupt their formation.1–6 Small molecules which bind to such scaffolds and stabilize them offer an effective method to inhibit telomerase activity and control gene expression.5 The mitoxantrone (Scheme 1), a semisynthetic antitumor antibiotic drug used for cancer chemotherapy, is known to act against topoisomerase-II. It has been found to bind to DNA quadruplex structures and inhibit the telomerase enzyme.7
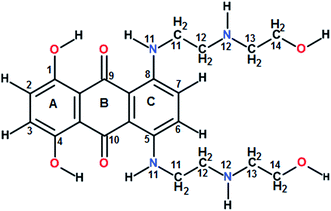 |
| | Scheme 1 Chemical structure of mitoxantrone. | |
The first report8 on inhibition of human telomerase activity by 2,6-diamidoanthraquinone with IC50 = 23 μM was attributed to increased stabilization of d-(TTAGGGT)4 by preliminary NMR experiments. Since then a series of anthracene and anthraquinone based compounds having 2 or 3 substituents at aromatic ring have been investigated9–12 for cytotoxicity, telomerase inhibition, improved binding affinity and increased stabilization with a view to ascertain role of substituent groups, position of substituent, length of intervening linkers/moieties, etc. It is imperative to link these findings with structural studies of drug–DNA quadruplexes for rational drug-designing. The structural studies are limited2–4 due to challenges in crystallization, complications in NMR spectra due to presence of multiple species, and difficulties in molecular modeling due to strong electrostatic field created by K+/Na+ ions near G-quartet. The X-ray crystallographic and NMR structures available in literature2 have to date shown preference for quadruplex component to have all strands in a parallel folding arrangement so as to perhaps maximize the planar surface available for ligand binding, particularly if end stacking or intercalation are the preferred modes of interaction. Out of two dozen structures available in literature, only about five have so far established binding of ligands at grooves of DNA.13–17 Few other studies have inferred binding at grooves by CD spectroscopy.18 Our laboratory has initiated a structure based approach to study efficacy of some of the natural products and their derivatives as G-quadruplex binding ligands. Since anthraquinones/amido-anthraquinones/anthracene derivatives are promising agents as G-quadruplex binders, as a first step, we looked at the structural aspects of binding of the well-known anthraquinone based compound mitoxantrone.
2. Materials and methods
The DNA sequence 7-mer [d-(TTGGGGT)], 22-mer d-[5′-GGGG(TTGGGG)3-3′], mitoxantrone and other chemicals like potassium di hydrogen phosphate (KH2PO4), dipotassium hydrogen phosphate (K2HPO4), potassium chloride (KCl), etc. used for buffer preparation were purchased from Sigma Chemical Co., USA Ltd. The DNA sequences for SPR experiments 5′ biotin labeled desalted DNA 7-mer d-TTGGGGT and 22-mer d-[5′-GGGG(TTGGGG)3-3′] was purchased from Sigma Chem. Co., USA. Mitoxantrone and DNA sequences were used without further purification. DNA sequence [d-(TTGGGGT)] was dissolved in 20 mM KBPES buffer (pH 7.0) containing 100 mM KCl, 10 mM K2HPO4 and 1 mM EDTA, which was heated at 90 °C for 5 minutes and was allowed to cool overnight at room temperature. The sample was stored at 4 °C and shaken gently at regular intervals to ensure its homogeneity. The concentration of DNA sequence and MTX were determined spectrophotometrically using the molar extinction co-efficient (ε) of 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 M−1 cm−1 for 7-mer DNA, 213400 M−1 cm−1 for 22-mer DNA at 256 nm (per strand) and 20900 M−1 cm−1 for MTX at 659 nm. The stock solution of MTX was prepared by dissolving 1 mg of MTX into 1 mL of phosphate buffer (pH 7.0). All the experiments were conducted at 298 K.
800 M−1 cm−1 for 7-mer DNA, 213400 M−1 cm−1 for 22-mer DNA at 256 nm (per strand) and 20900 M−1 cm−1 for MTX at 659 nm. The stock solution of MTX was prepared by dissolving 1 mg of MTX into 1 mL of phosphate buffer (pH 7.0). All the experiments were conducted at 298 K.
2.1 Telomerase repeat amplification protocol (TRAP) assay
Inhibition of telomerase enzyme by mitoxantrone was assessed by standard TRAP assay using TRAPeze XL Telomerase Detection Kit (Merck Millipore). MCF-7 breast line cancer cells are harvested and approximately 105 cells were pelleted and washed thrice with PBS. To extract telomerase, the cell pellet was resuspended in 200 μL of CHAPS Lysis buffer (0.5% CHAPS, 10 mM Tris–HCl, 1 mM MgCl2, 1 mM ethylene glycol tetra-acetic acid (EGTA), 5 mM β-mercapto ethanol, 0.1 mM benzamidine, 10% glycerol), and incubated in ice for 30 minutes. The lysate was centrifuged at 10000 rpm for 20 minutes at 4 °C, and the protein concentration of the supernatant was determined using Bradford's method of protein estimation. The supernatant was divided into aliquots and stored at −80 °C or used directly for TRAP assay. TRAP reaction mixture was prepared using 100 mM Tris–HCl (pH 8.3), 7.5 mM MgCl2, 315 mM KCl, 0.25% Tween 20, 5 mM EGTA, 0.5 mg mL−1 BSA, 60 μM of dATP, dGTP, dTTP, dCTP, and the oligonucleotides TS primer, RP Amplifluor primer, K2 Amplifluor primer and TSK2 template, 2 units of Taq polymerase, 500 ng μL−1 of cell extract and incubated at 30 °C for 30 minutes. The test samples were prepared by adding the desired concentration of MTX (0.01, 0.1, 1.0, and 10.0 μM) to the reaction mixture and a 4-step PCR reaction was carried out. Samples of the reaction mixture without ligand were also prepared with telomerase (telomerase positive), without telomerase (telomerase negative), with heat inactivated telomerase and without Taq polymerase (Taq negative). The assay products are examined directly by running on 15% non-denaturing PAGE and gel was stained with ethidium bromide and destained. 50 bp DNA ladder was used as a standard marker. The gel was illuminated in UV region and recorded using Bio-Rad Gel documentation system. Alternately, since TRAPeze XL Telomerase Detection Kit is an advanced Amplifluor primer containing kit, the PCR products were directly quantified using spectrofluorimeter. 20 μL of assay mixture was diluted to 600 μL with 10 mM Tris–HCl buffer pH 7.4 containing 0.15 M NaCl and 2 mM MgCl2. The fluorescence emission of sample was measured by exciting fluorescein at 495 nm and sulforhodamine at 600 nm, respectively. The relative fluorescence intensity was measured using the equation,
| ΔFL/ΔR = [FLo − FLneg]/[Ro − Rnotaq] |
where FLo is observed fluorescein emission in respective test samples, FLneg is fluorescein emission of telomerase negative control, Ro is observed sulforhodamine emission of respective test sample, Rnotaq is sulforhodamine emission of Taq negative control, ΔFL = FLo − FLneg is difference in fluorescein emission between respective test samples and telomerase negative control, ΔR = Ro − Rnotaq is difference in sulforhodamine emission between respective test samples and Taq negative control. Both negative controls have been obtained by using equivalent TRAP-PCR conditions as that of test samples.
2.2 Surface plasmon resonance (SPR) experiments
For SPR studies, the DNA samples were dissolved in HEPES buffer (0.01 M HEPES, 3 mM EDTA, 0.005% surfactant P20, 100 mM KCl) and heated at 90 °C for 5 minutes and was allowed to cool overnight at room temperature. The DNA samples at 50 nM concentration were applied to flow cells in streptavidin-derivatized sensor chips (BIACORE SA) by direct flow at 5 μL min−1 in a BIACORE 2000 Surface Plasmon Resonance (SPR) instrument, available at John F. Welch Technology Center, GE Healthcare Life sciences, Bengaluru, India. The flow cells 2 and 4 were used for DNA immobilization, whereas flow cells 1 and 3 were used as the respective blanks. The bound Response Unit (RU) for immobilization was 484.2 RUs and 486.7 RUs for 7-mer and 22-mer G-quadruplex DNA, respectively. Steady state binding experiments were performed with multiple injections of different concentrations of mitoxantrone in the range 15–360 μM (prepared by serial dilutions from a stock solution prepared in HEPES buffer containing 100 mM KCl) over the immobilized DNA surface at a flow rate of 30 μL min−1 at 25 °C. The flow was continued through the flow cells until a constant steady state response was obtained, where the rate of association and dissociation are equal. The ligand/analyte (MTX) solution was then replaced by buffer flow containing 1 M NaCl resulting in dissociation of the complex. Flow cell 1 and 3 was kept blank as control in the two runs carried out, to account for any signal generated to bulk solvent effects or any other effect not specific to MTX-DNA quadruplex interaction, such as probable aggregation. The reference response from the blank cells 1 and 3 were subtracted from the response in corresponding flow cells 2 and 4 containing quadruplex DNA, to give signal (RU, response units) that is directly proportional to the amount of ligand bound. The average of the data in the steady state region of each sensogram (RUavg) was determined by linear averaging on a selected time span and is plotted as a function of analyte concentration. A set of sensograms at different concentrations of MTX were thus obtained for binding of MTX to [d-(TTGGGGT)]4 and d-[5′-G4(T2G4)3-3′]. The predicted maximum response per bound compound in the steady-state region (RUmax) was determined from the DNA molecular weight, the amount of DNA on the flow cell, the compound molecular weight, and the refractive index gradient ratio of the compound and DNA for a 1:1 binding interaction as 1375 and 1174 in run 1 and 2, respectively. The observed RUavg value, at high concentrations of MTX are found to be slightly less than RUmax suggesting presence one binding site in these DNA sequences. The standard binding parameter r, moles of ligand bound/mole of DNA quadruplex is obtained as r = RUavg/RUmax. Cf is the concentration of the ligand/analyte in equilibrium with the complex and is fixed by the concentration of ligand in the flow cell solution. Equilibrium constant (KA) is obtained by fitting the results from the steady state region (measured RUavg or r versus Cf) with a single site model in the following equation and evaluating goodness of fit:| |
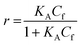 | (1) |
where KA is the macroscopic thermodynamic binding/affinity constants for one-site binding model since 1:1 binding model shows the best fit of data. The binding of MTX to quadruplex was also evaluated by following kinetics of association (rate constant ka) and dissociation (rate constant kd):| | |
[A]t=0 = 0, [B]t=0 = RUmax, [AB]t=0 = 0
| (3) |
| |
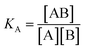 | (4) |
| |
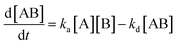 | (5) |
Global fitting of the association and dissociation curves was performed using software (BIAevaluation software 2.0.1, Biacore Inc.) to fit all sensograms to get ka and kd rate constants.
2.3 Titrations by absorbance measurements
Absorbance measurements were carried out by using CARY-100 Bio-spectrophotometer (Varian, USA) equipped with a thermostatic cell holder and quartz cuvette having optical path length of 1 cm. The samples were prepared in the absence and presence of various concentrations of DNA at MTX drug (D) to nucleotide quadruplex (N) ratio = 0.21–20.9 using a fixed concentration of 4.4 μM MTX. The samples were incubated for one hour at 298 K and spectra were recorded in the wavelength range 200–800 nm.
2.4 Job plot using fluorescence
In order to establish the binding stoichiometry of mitoxantrone with G-quadruplex sequence [d-(TTGGGGT)]4, continuous variation analysis procedure, Job plot, was used. The fluorescent intensity was measured by using Fluorolog-3 Spectrofluorimeter LS55 (make HORIBA JobinYvonSpex®) at a wavelength of λem = 678 nm using λex = 610 nm at a constant temperature 298 K for a mixture of MTX and DNA quadruplex, while the sum of their concentration is kept constant at 3 μM.
2.5 DNA thermal melting (Tm) studies
Tm experiments were conducted with a Cary Bio UV-Vis spectrophotometer (Varian Inc) interfaced to a microcomputer. A thermistor fixed into a reference cuvette was used to monitor the temperature. The G-quadruplex DNA was added to 1 mL of buffer (potassium phosphate buffer containing 100 mM KCl) in a 1 cm path length quartz cells and the concentration was determined by measuring the absorbance at 260 nm. Rate of melting was adjusted to 1 °C min−1 and data taken for the range of 25–95 °C. MTX was added progressively from a stock solution to a fixed concentration (20 μM) of quadruplex to get D/N ratio of 1.0, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, and 5.0. Tm was measured as rate of change of absorbance (A) with temperature (T), that is, dA/dT for each complex so obtained.
2.6 Nuclear magnetic resonance (NMR) experiments
A 2.13 mM solution of quadruplex was prepared by dissolving it in a mixture of 90% H2O and 10% D2O containing 20 mM KBPES buffer (pH = 7.0), 100 mM KCl, 10 mM K2HPO4 and 1 mM EDTA. Samples of mitoxantrone were prepared by dissolving it in 90% H2O and 10% D2O containing 20 mM potassium phosphate buffer (pH = 7.2) and 100 mM KCl. Drug (D):DNA quadruplex (N) complexes at different mole equivalent ratios, D/N = 0.25–4.0, were prepared by titrating appropriate concentrations of DNA and drug. Trimethylsilyl propionic acid (TSP) was used as the internal reference for proton NMR spectra. 31P resonances were referenced to 85% ortho-phosphoric acid. All NMR data were collected on a Bruker Avance AMX-500 MHz FT-NMR spectrometer located at Nuclear Magnetic Resonance Facility, Indian Institute of Technology Roorkee. The data obtained were processed with TOPSPIN version 1.3 software. 1D proton NMR spectra were recorded at different temperatures in the range 278–318 K with number of data points = 64 K, number of scans = 64–128, pulse width = 11.49 μs, FID resolution = 0.12 Hz per point, spectral width = 20 ppm and a delay time = 1.5 s. Receiver gain was optimized in each instance to obtain the best signal to noise ratio. The removal of water signal was accomplished using Watergate suppression. In temperature variable experiments, constant temperature is maintained in the range 278–363 K using Bruker variable temperature (BVT) control accessory. Homo-nuclear 1H–1H 2D phase sensitive COSY of uncomplexed and complexed DNA were carried out at D/N = 1.0 and 2.0 at 298 K, while NOESY experiments were carried out at D/N = 1.0 and 2.0 using mixing time τm = 100, 200, 250 ms at 298, 308 and 318 K as well as at D/N = 4.0 at 298 K using τm = 100, 250 ms. Typical parameters for 2D experiments were: 2048 data points along t2 dimension; 512 free induction decays in t1 dimension; pulse width = 9.5–12 μs; spectral width = 10000 Hz; no. of scans = 64–128; digital resolution = 2.30–4.60 Hz per point and relaxation delay = 2.0 s. 1H–13C HSQC was carried out for D/N = 2.0 using echo-anti echo pulse sequence with 1024 data points along t2 dimension; 256 free induction decays in t1 dimension; pulse width = 14.89 μs; spectral width = 10000 Hz × 25000 Hz; no. of scans = 48–56; digital resolution = 19.5 Hz per point in t2 and 125 Hz per point in t1 dimension; and relaxation delay = 2.0 s and SSB = 2.
All 31P NMR experiments were carried out using Broad Band Observed (BBO) probe. One dimensional 31P NMR spectra was taken at D/N ratios of 0.2, 0.5 and 1.0 at 278, 283 and 298 K using number of data points = 64 K, number of scans = 64, spectral width = 8 ppm, digital resolution = 0.03 Hz per point. 31P–1H shift Hetero-nuclear Multiple Bonded Correlation (HMBC) spectroscopy experiment was carried out at D/N = 2.0 at 298 K. The long range evolution delay of 65 ms was used.
2.7 Circular dichroism (CD)
The measurements were carried out on a PC controlled Applied Photophysics (Model Chirascan, UK) spectropolarimeter equipped with a programmable temperature controlled cell holder using a rectangular quartz cell of 0.1 cm path length. The instrument was purged continuously with clean nitrogen using high purity nitrogen generator (make SAS F-DGS, Every, France model HPNG10/1) prior to igniting the xenon arc lamp. All spectra were acquired in the wave length range 200–700 nm using 1 nm slit width at 1 nm interval and were averaged over 3 scans. Titrations were performed by progressively adding mitoxantrone to a fixed concentration of DNA quadruplex (8.2 μM) at D/N = 0.13–6.0. The titrations were repeated by adding DNA quadruplex in steps to a fixed concentration, 30 μM of mitoxantrone, at D/N = 0.6–7.0.
3. Results and discussion
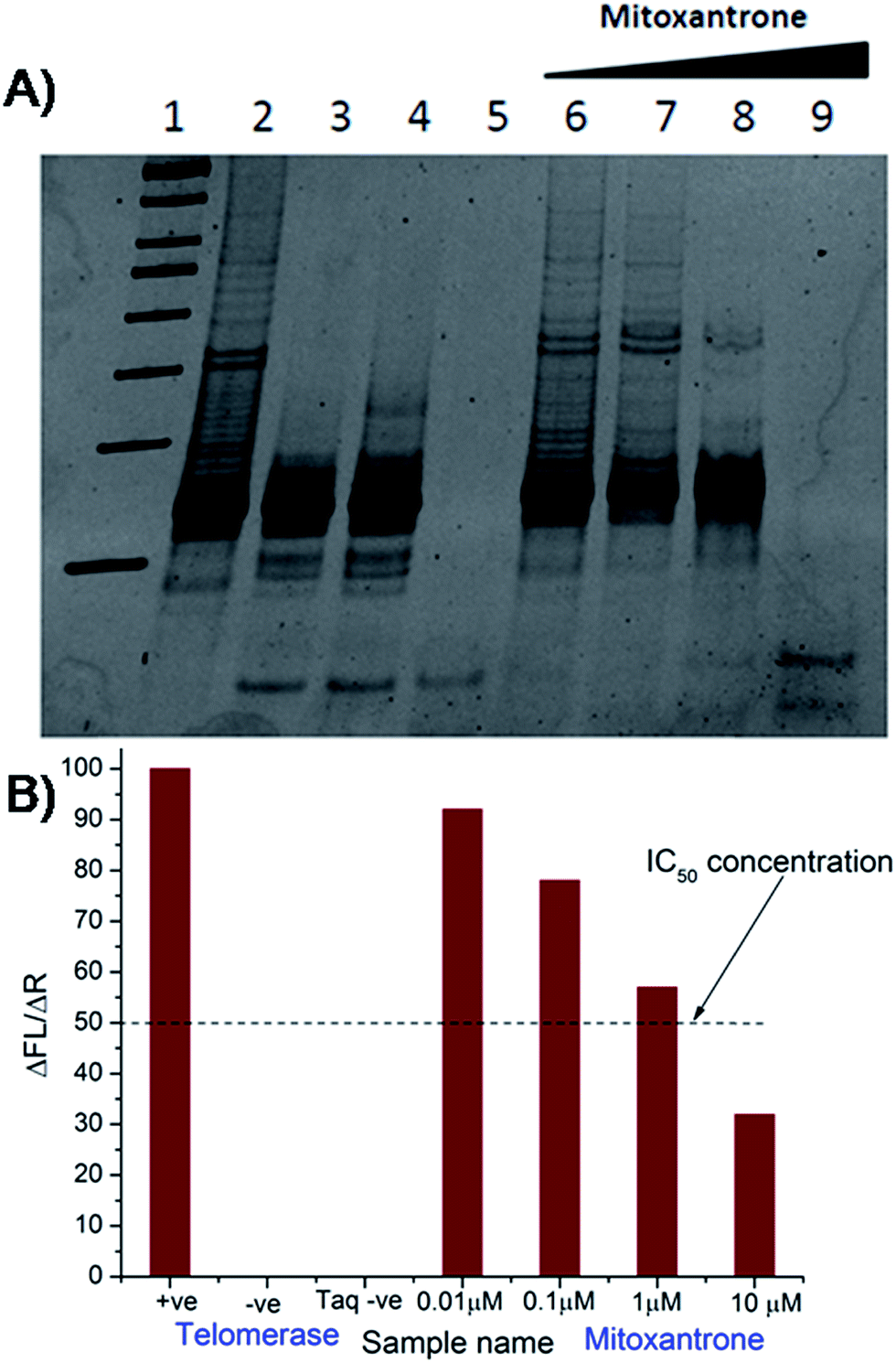
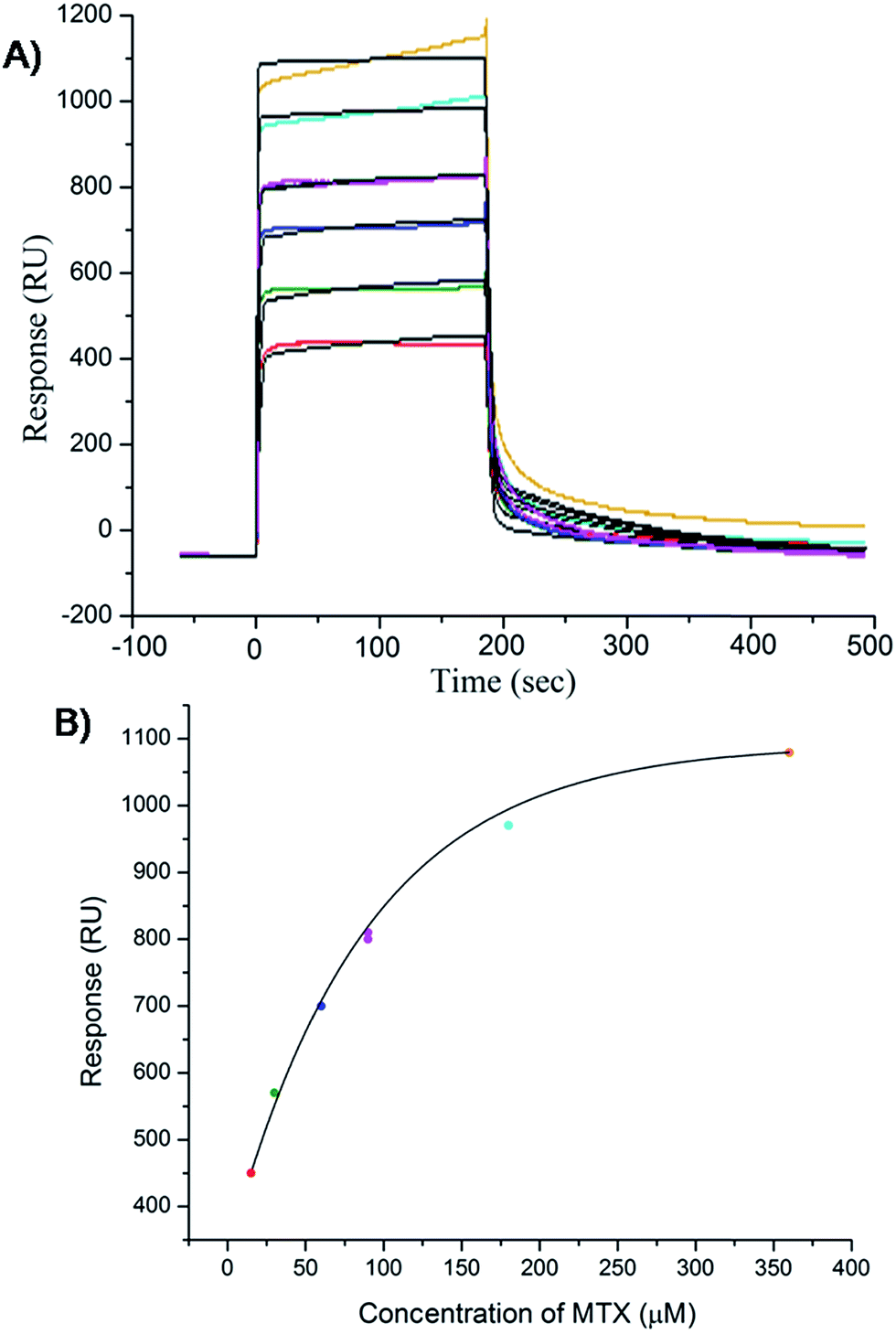
The results of inhibition of telomerase enzyme by different concentrations of mitoxantrone using TRAP assay are shown in Fig. 1A and B. The relative fluorescence intensity of the fluorescein and sulforhodamine amplifluor tag, used in primers, yields an estimate of IC50 = 2 μM [Fig. 1B]. To examine the binding characteristics and mode of interaction of mitoxantrone with G-quadruplex structures, we chose the model sequence [d-(TTGGGGT)] found in telomere of Tetrahymena thermophila for our study. We have used the uni-molecular quadruplex formed by the 22-mer d-[5′-G4(T2G4)3-3′] as well as the tetra-molecular parallel quadruplex [d-(TTGGGGT)]4 formed by 7-mer d-(TTGGGGT) for the determination of association constant by SPR, stoichiometry by Job's method of continuous variation and for titrations by absorption spectroscopy. The surface plasmon resonance sensograms showing kinetics of association and dissociation of interaction of mitoxantrone with 7-mer sequence at 298 K and the binding isotherm for determining affinity constant from steady state region are shown in Fig. 2A and B. The affinity constant KA ∼ 1.08 × 104 M−1 obtained from steady state analysis agrees reasonably well with that obtained from kinetic analysis, that is, 3.25 × 104 M−1. The results for the 7-mer and 22-mer sequences of quadruplex DNA [ESI Fig. 1A, B and Table 1†] were found to be comparable. It may be noted that the increase in steady state response with concentration (Fig. 2B) suggests that a specific interaction of ligand with G-quadruplex sequence does take place and the association constant may refer to the dominant mode of interaction in the concentration range 15–360 μM. Thus having established absence of non-specific binding, we proceeded ahead to ascertain independently the stoichiometry and structural aspects of complex formation by CD and NMR experiments.
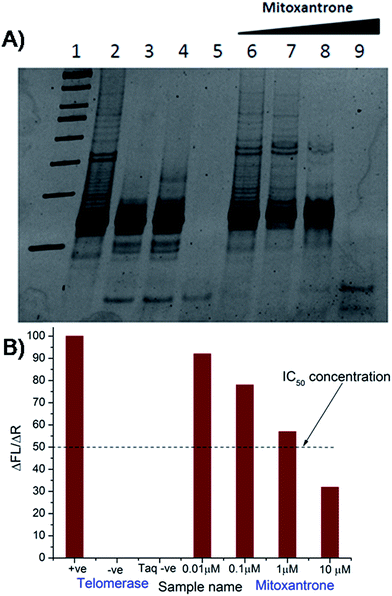 |
| | Fig. 1 Inhibition of human telomerase by mitoxantrone using TRAP assay. (A) 15% polyacrylamide gel showing effect of increasing concentration of mitoxantrone on telomerase inhibition – Lane 1: 50 bp DNA ladder, Lane 2: telomerase positive control, Lane 3: telomerase negative control, Lane 4: heat inactivated telomerase, Lane 5: Taq polymerase negative control, Lanes 6–9: mitoxantrone concentration, 0.01, 0.1, 1.0, and 10.0 μM. (B) Observed ratio of difference in fluorescein emission between respective test samples and telomerase negative control (ΔFL = FLo − FLneg) to difference in sulforhodamine emission between respective test samples and Taq negative control (ΔR = Ro − Rnotaq) in various experiments giving telomerase inhibitory concentration IC50 = 2 μM. | |
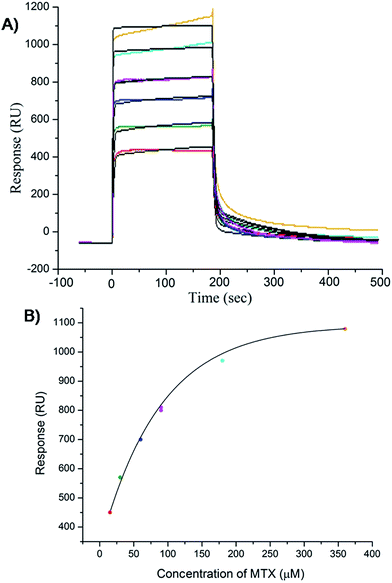 |
| | Fig. 2 Results of surface plasmon resonance for binding of mitoxantrone to [d-(TTGGGGT)]4 using HEPES buffer with 100 mM KCl. (A) Sensograms obtained for unbound mitoxantrone concentration of 15, 30, 60, 90, 180 and 360 μM, from bottom to top curves. (B) Binding plots showing Response Unit (RU) versus concentration of mitoxantrone from steady state data. | |
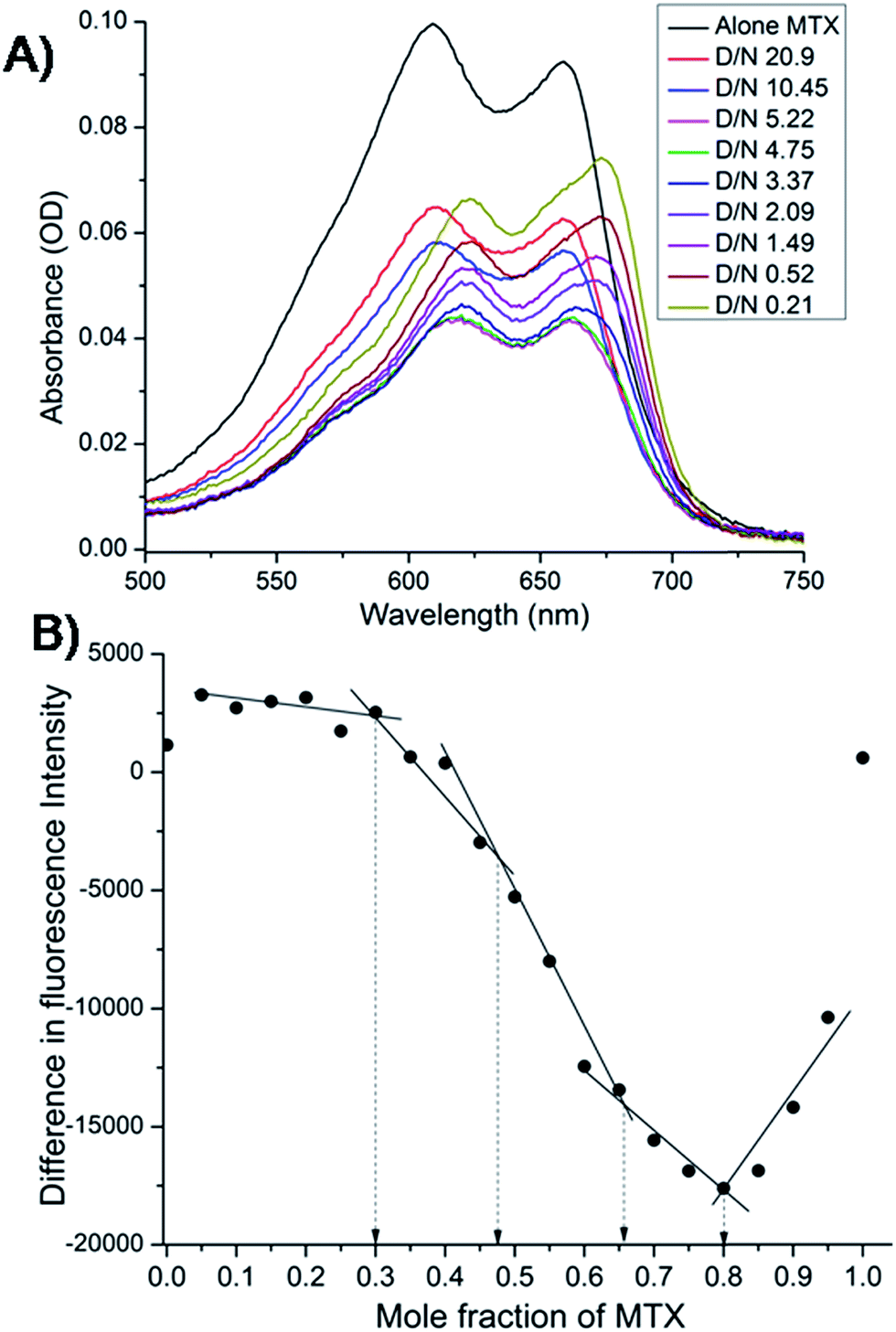
The mitoxantrone shows four absorption bands at 242, 276, 608 and 658 nm. Since DNA absorbs efficiently in the UV region, we monitored titrations by measuring the changes in the absorption in the visible region (λmax = 608 and 658 nm). Varying concentrations of tetramolecular [d-(TTGGGGT)]4 quadruplex were added to a fixed (4.4 μM) concentration of mitoxantrone in the mole equivalents of mitoxantrone (D) to quadruplex DNA (N) concentration ratio as 0.21–20.9. We did not observe a clear isobestic point [Fig. 3A], which points towards presence of multiple conformations in the complex. The next step was to determine binding stoichiometry independently through fluorescence using continuous variation (Job plot) analysis procedure. The fluorescent intensity was measured at a wavelength of λem = 678 nm using excitation wavelength λex = 610 nm at 298 K for a mixture of mitoxantrone and DNA quadruplex, while the sum of their concentrations was kept constant. The difference in emission intensity of mitoxantrone, ΔF = F–F0, in the presence (F) and absence (F0) of DNA plotted as a function of mole fraction of mitoxantrone shows several slope changes between approximately linear regions [Fig. 3B]. The data indicates stoichiometric ratios of 0.5:1, 2:1 and 4:1 in the mitoxantrone-[d-(TTGGGGT)]4 complex corresponding to mole fraction of mitoxantrone = 0.34, 0.66 and 0.80, respectively while that at 0.48 cannot be said with certainty. This confirms existence of multiple stoichiometric complexes. Existence of multiple binding sites have earlier been shown on binding of TMPyP4 and TPrPyP4 to parallel G-quadruplex.19
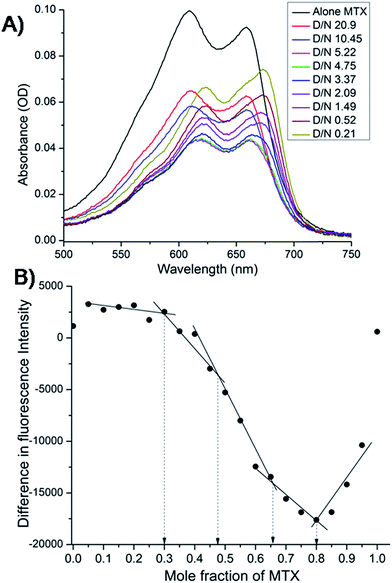 |
| | Fig. 3 (A) Selected traces from the titration of 4.4 μM mitoxantrone with increasing concentration of [d-(TTGGGGT)]4 between D/N ratios of 0.21–20.9 in 20 mM phosphate buffer with 100 mM KCl at 298 K using absorbance measurements in visible region. (B) Job plot for the binding of mitoxantrone to [d-(TTGGGGT)]4 in 20 mM phosphate buffer with 100 mM KCl at 298 K using fluorescence keeping the total concentration of ligand and DNA as 3 μM. | |
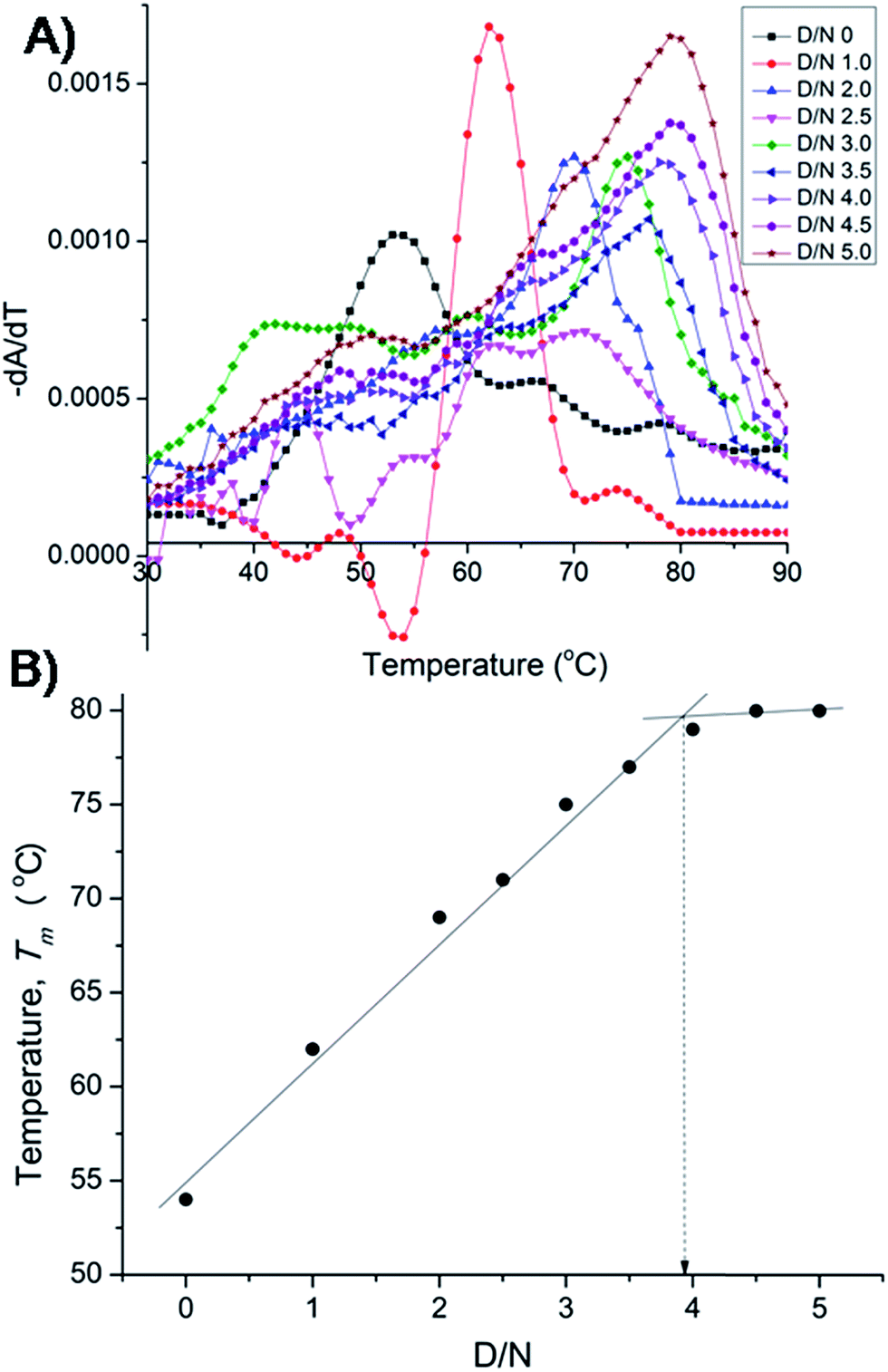
The results on binding equilibrium, stoichiometry and absorption spectroscopy were found to be similar for both 22-mer and 7-mer sequences and we therefore chose 7-mer for further studies since its structure has already been characterized by NMR.20 We measured change in melting temperature (ΔTm) in the folded → unfolded quadruplex at different D/N ratios using absorbance measurements at 260 nm. The results of thermal melting are shown in Fig. 4A and B. It is clearly seen that as the binding sites on the nucleic acid become saturated, the effect of increasing drug to a fixed concentration of nucleic acid has a diminishing effect on Tm.21,22 The Tm value levels off to a plateau [Fig. 4B] and the ratio D/N = 4.0 that corresponds to the break point refers to the maximum number of mitoxantrone molecules binding to DNA, that is, the stoichiometry of the complex is 4:1 at high D/N ratios. This is consistent with the results obtained from Job plot [Fig. 3B]. This result is somewhat intriguing since several of anthraquinone and anthracene analogs have only been found to have a drug to DNA stoichiometry of either 1.0 or 2.0.9,11
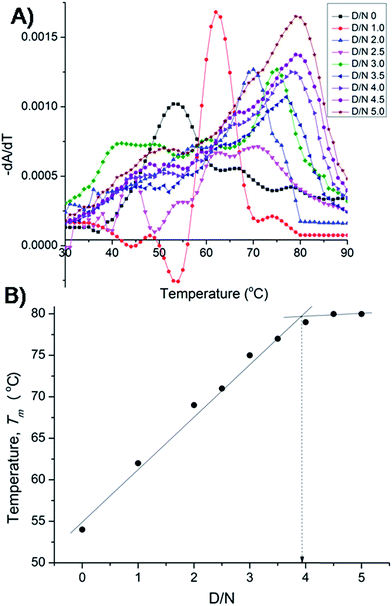 |
| | Fig. 4 (A) Thermal melting profiles showing derivative (dA/dT) as a function of temperature of uncomplexed DNA (10 μM) and that complexed at various D/N ratios in 20 mM phosphate buffer with 100 mM KCl. (B) Plot of observed Tm vs. D/N ratio. | |
Besides for the first time we have established n = 4 by two independent methods, that is, direct Job plot using fluorescence and melting profiles using absorbance (ΔTm) experiments. It is noteworthy that at D/N = 1.0 at which Tm = 62 °C, a high temperature phase melting transition occurs at a temperature >74 °C, presumably corresponding to 2:1 stoichiometry of the complex. The result is significant and shows that on adding one mole equivalent of mitoxantrone to DNA, stoichiometry of 2:1 is achieved. A distinct possibility exists that the drug binds as a dimer and we investigated the same further by NMR experiments. It is noteworthy that binding of mitoxantrone stabilizes DNA quadruplex (ΔTm) significantly by 26 °C. This value is reasonably high in view of the fact that groove binder peimine,16 neomycin–anthraquinone conjugate12 and intercalator actinomycin23 stabilize DNA quadruplex by 13.5, 23.7, and 19 °C, respectively. Some of the substituted acridines in which mode of binding has not established are however known to stabilize DNA quadruplex by 27–39 °C.10
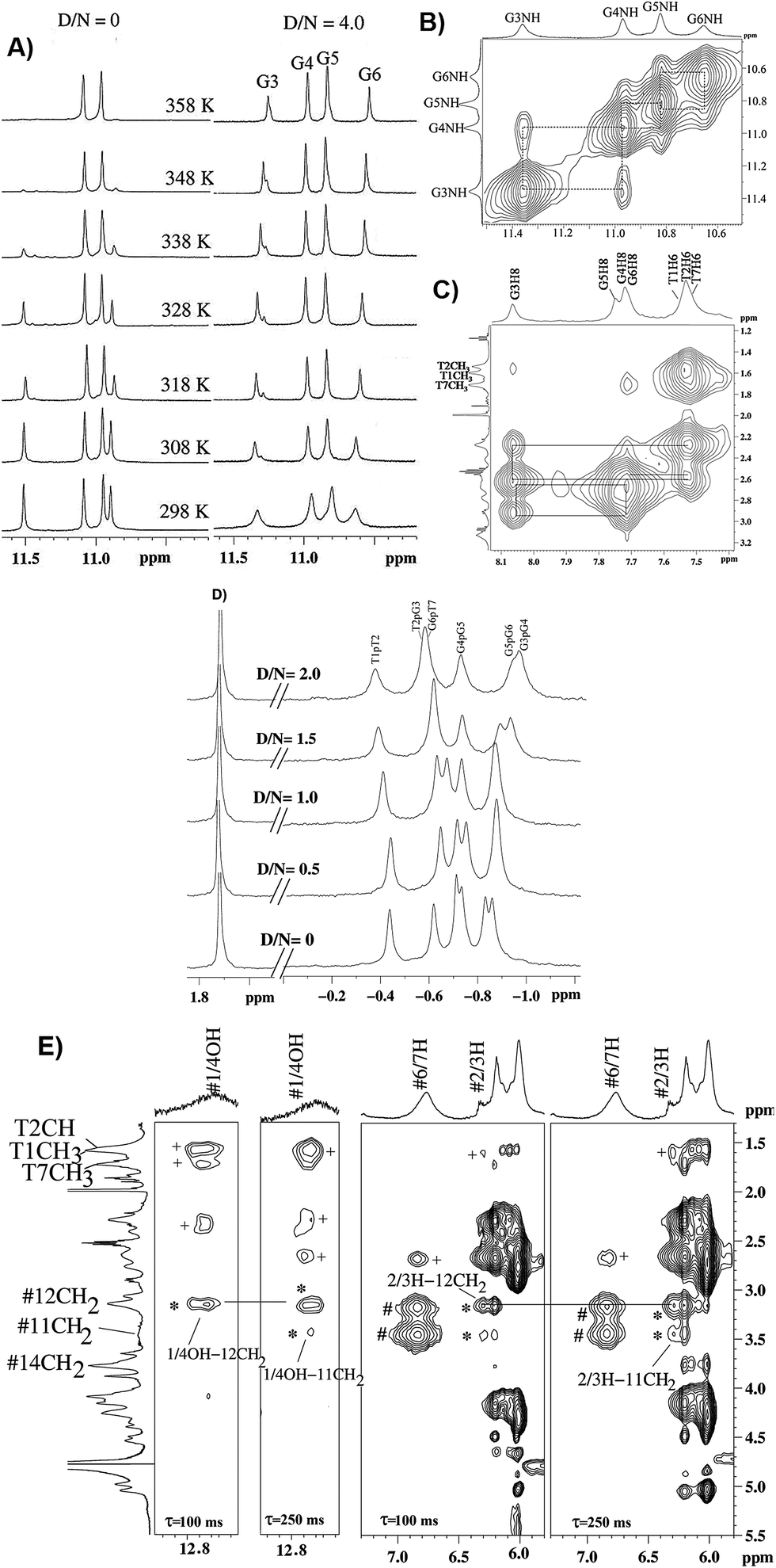
The titrations of mitoxantrone with 7-mer DNA quadruplex sequence were carried out following proton and phosphorous-31 NMR spectra at D/N = 0–4 by progressive addition of drug to 2.13 mM solution of quadruplex. The chemical shift variations in DNA protons saturated at D/N ratio of ∼4.0. On the other hand, the signals corresponding to ligand protons grew in intensity but did not show any significant shift in chemical shift values with increasing D/N ratio. The thermal denaturation behavior of G-quadruplex in the absence and presence of mitoxantrone, monitored through disappearance of GNH signals indicated a drug induced stabilization by more than 20 K [Fig. 5A]. The four GNH protons shifted upfield by 0.15–0.26 ppm with G6NH showing maximum upfield shift [Fig. 5A]. This cannot be attributed to intercalation mode of interaction since the NOESY spectra of G-quadruplex was not interrupted in presence of intermediate (D/N = 2.0) or saturating concentrations of mitoxantrone (D/N = 4.0). Several NOEs between base H8/H6-base H8/H6; adjacent GNH; GNH-preceding base H8; base H8/H6–H1′; and base H8/H6–H2′, H2′′ were easily observable [Fig. 5B and C].
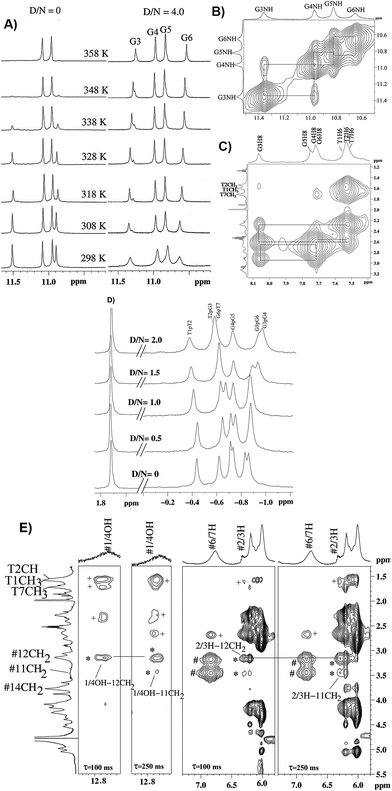 |
| | Fig. 5 (A) 1H spectra of imino region of uncomplexed 2.1 mM [d-(TTGGGGT)]4 and that of its complex with mitoxantrone in 20 mM phosphate buffer with 100 mM KCl at D/N = 4.0 at different temperatures. (B) Selected region of NOESY spectra at D/N = 4.0 showing NH–NH connectivity between adjacent G-quartet steps. (C) Selected region of NOESY spectra showing base H8/H6-sugar H2′, 2′′ sequential NOE connectivities at D/N = 4.0. (D) 31P spectra of uncomplexed DNA and its complex with mitoxantrone at D/N ratios of 0, 0.5, 1.0, 1.5 and 2.0. (E) Selected region of NOESY spectra of mitoxantrone complexed to [d-(TTGGGGT)]4 at D/N = 4.0 showing drug–drug intra molecular (#), drug–drug inter molecular (*) and drug–DNA inter molecular (+) NOE contacts. | |
On progressive addition of mitoxantrone to 7-mer sequence, the 31P signals showed continuous shift up to D/N = 2.0 [Fig. 5D]. The drift in resonances is sequence dependent, being −0.05, 0.02, −0.09, −0.01, −0.15, and 0.13 ppm for TpT2, T2pG3, G3pG4, G4pG5, G5pT6, and G6pT7, respectively. Apparently G5pG6 and G6pT7 are involved in interaction. It is known that 31P chemical shifts vary in response to local, sequence specific and induced environmental distortions in the duplex geometry.24 31P chemical shift has also been correlated with degree of duplex DNA unwinding resulting from increase in the length of sugar phosphate backbone to accommodate intercalation of drug chromophore. The observed downfield shift of G6pT7 resonance ∼0.13 ppm, is much lesser than that expected due to intercalation, that is, Δδ = 1.6–2.6 ppm,24 and therefore completely rules out opening of base pairs to allow intercalation mode of binding. Mitoxantrone thus appears to bind to DNA only externally. The small observed upfield shift in G3pG4 and G5pG6 and resonances may be attributed to their involvement in electrostatic interactions, perhaps with positive charge at 12NH of ligand.
The alternate mode of interaction, that is mitoxantrone chromophore stacking at two ends of G-quadruplex DNA, also does not occur since that would cause large noticeable upfield shifts in T1H6, T7H6, T1CH3 and T7CH3 protons, which are absent. In fact T1, T7 protons show minor shifts in the range −0.06 ppm (upfield) to +0.10 ppm (downfield). End stacking of mitoxantrone chromophore with DNA bases or intercalation within DNA base pairs would also be expected to cause uniformly large upfield shifts in its ring protons, particularly 2/3H, 1/4OH, 6/7H and 11NH protons. We observed upfield shift of 0.41–0.46 ppm in 2/3H and 1/4OH protons and practically negligible shift in 6/7H and 11NH protons. Thus NMR experiments completely rule out end stacking or intercalation as a mode of interaction. We observed NOE cross peaks of 2/3H protons with 11NH, 6/7H, 11CH2, 12CH2, 14CH2 as well as that of 1/4OH with 11CH2 and 12CH2 protons [Fig. 5E]. These are possible only if two mitoxantrone molecules stack over each other with head to tail orientation, that is, as a dimeric pair in anti-parallel orientation. The observed NOEs thus correspond to intermolecular short contacts between two MTX molecules within the dimeric mitoxantrone bound to DNA quadruplex.25 The fact that 2/3H and 1/4OH protons show significantly large shifts on binding, suggests that a dimer of mitoxantrone binds with ring A pointing close to the binding site on DNA. Several intermolecular drug to DNA cross peaks, particularly between 2/3H,1/4OH and alkyl side chain protons of mitoxantrone and T1H6, T2H6, T2CH3, G6H8, T7H6, T7CH3 and their corresponding sugar protons have been observed [Fig. 5E]. These observations clearly establish that two dimers of mitoxantrone bind at opposite sites at the grooves of DNA, close to T1pT2 and G6pT7. We are in the process of evaluating the conformation of the complex by restrained molecular dynamics simulations using experimental NOE restraints.
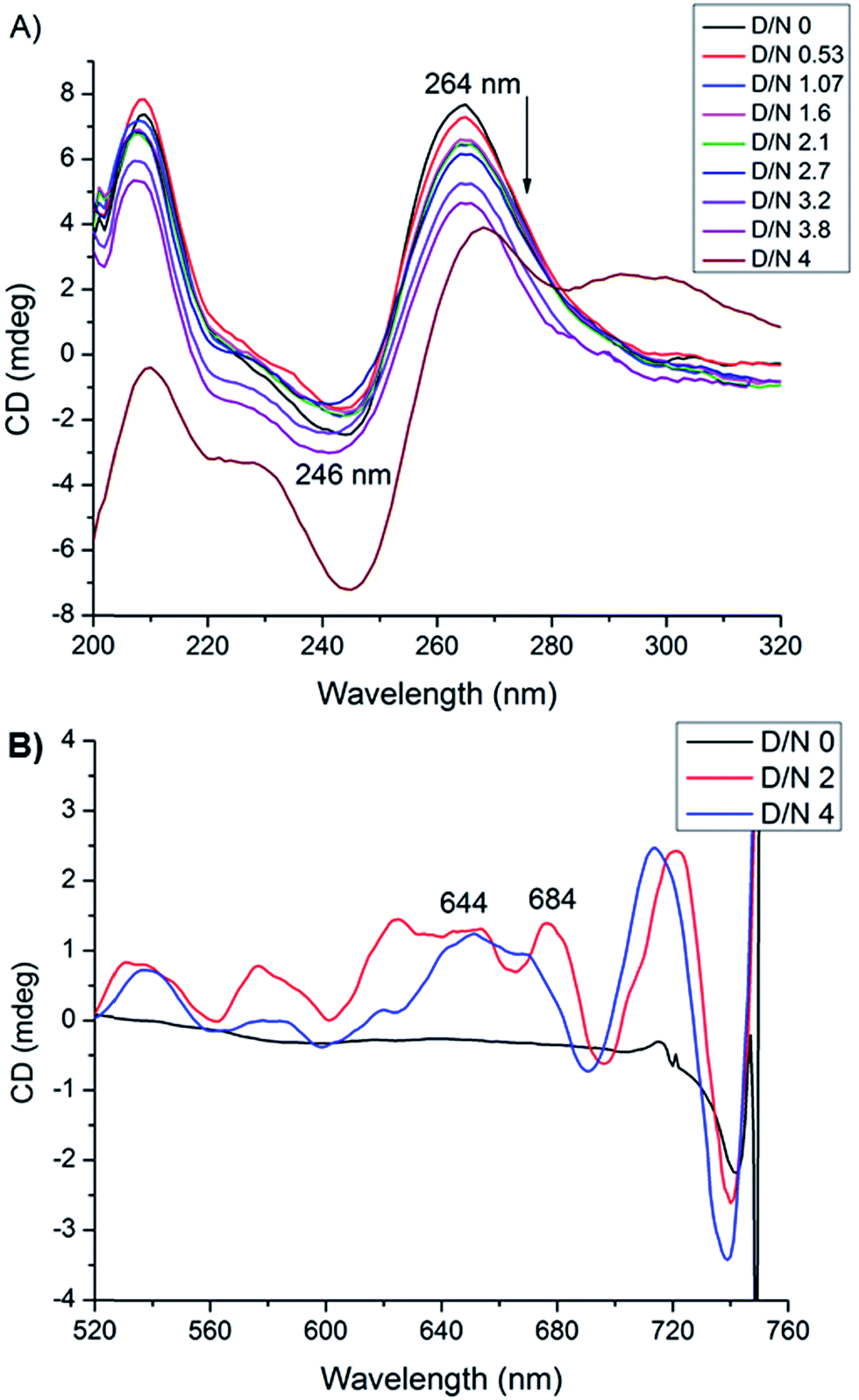
Circular dichroism spectroscopy gives signature peak of parallel stranded G-quadruplex at 264 nm and is a useful method of monitoring conformational changes due to binding.11,18 The titrations of DNA with mitoxantrone showed a change in 246 and 264 nm CD peaks up to 3.4 mdeg at D/N = 3.8 [Fig. 6A], which is consistent with groove binding mode as end stacking is expected to give practically no change in CD signals.18,26 We observed strong Induced CD (ICD) signals at 600–680 nm, the region of absorbance of mitoxantrone [Fig. 6B and ESI S2†], which is indicative of the change in the chirality of the proximal chemical environment of the ligand. A strong positive ICD is evident at ∼640 nm. In addition two exciton bands are evident at D/N = 2.0 and 4.0 with crossover points at 619 and 664 nm, which are the corresponding absorption maximum of mitoxantrone in the complex [ESI Fig. S2†]. The two exciton bands are opposite in sense, that is, within the exciton band, the +ve band appears at higher and lower wavelength with respect to corresponding cross over wavelength at 619 and 664 nm, respectively. Exciton splitting in the induced spectra indicates that mitoxantrone binds to the quadruplex grooves as one or more stacked species18,27,28 corroborating thereby our NMR results which show presence of dimer of mitoxantrone in the bound complex [Fig. 5E]. Further, we found that shape of the induced CD spectra, arising from contributions from 645 nm positive band and the 2 exciton bands, varied with D/N ratio. The ICD at 645 nm is prominent at low D/N ratios (D/N = 0.8) while the exciton bands due to stacked dimer formation dominate at higher D/N ratios. This confirms presence of multiple complexes, the relative mole fraction of which varies with D/N ratio. This observation is supported by our detailed NMR studies (accepted in Biochimie DOI: 10.1016/j.biochi.2016.07.005). Since the absence of any significant downfield shift in 31P resonances rules out intercalation as a mode of interaction at low D/N = 0.25–0.75, the positive ICD band at D/N = 0.8 at 645 nm pertains to external binding of MTX at grooves, predominantly as monomer. At D/N = 1.0, we observed only few cross peaks due to dimer formation of MTX in 2D NOESY spectra. However at D/N = 2.0 and 4.0, all cross peaks expected due to head to tail alignment of two MTX molecules in the form of stacked dimer, were observed which gives rise dominant bisignate ICD bands at high D/N ratios. The results also corroborate the general understanding that presence of induced exciton bands can be used as a method to distinguish between different binding modes.18 Also it supports the findings that groove binders weakly associate with telomere DNA sequences18 with KA ∼ 105 M−1. The end stacking generally results in much higher association constant, for example KA ∼ 3 × 107 M−1 on binding of BRACO-19 to 22-mer human telomere repeat.18 It has been suggested that groove binding to DNA quadruplex is likely to be seen only at high concentrations (∼few mM) in experiments such as NMR.18 The present study shows that this is not necessary as CD experiments have been conducted at relatively lower (∼8 μM) concentration of DNA quadruplex.
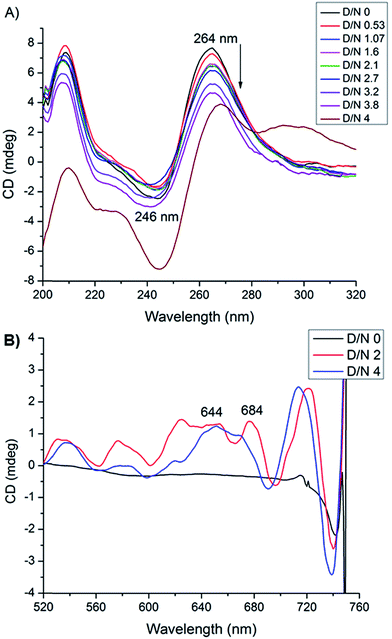 |
| | Fig. 6 (A) CD spectra of uncomplexed 8.2 μM [d-(TTGGGGT)]4 in 20 mM phosphate buffer with 100 mM KCl at 298 K and that complexed with mitoxantrone at different D/N ratios. (B) Induced CD spectra of 30 μM mitoxantrone on binding to [d-(TTGGGGT)]4 at different D/N ratios. | |
Out of about two dozen X-ray crystallographic/NMR structures of ligand-quadruplex DNA available in literature, only five structures have so far been shown to bind at grooves. These include crescent shaped distamycin13 and its derivative,14 a cyanine dye 2,2′-diethyl-9-methyl-selenacarbocyanine bromide,15 peimine/peiminine16 and bis(phenylethynyl) amide derivatives;17 of which the first three ligands bind as dimers. Several analogues involving two adjacent furan rings/other ligands have earlier been shown to be groove binders to duplex29,30 and quadruplex DNA18,29–34 on the basis of CD spectroscopy. Of all these ligands only peimine/peiminine are conjugated ring compounds. In this context, it may be concluded that it is not necessary for groove binders to be crescent shaped or having a shape which is fairly complementary to that of the groove. Recently it has been shown that non planar amino sugars conjugated to planar anthraquinone have potential as groove binders.12 Rigorous quantum mechanical calculations on binding of ligands to G-quadruplex DNA32,33 as well as Induced Fit simulations34 have shown that a variety of compounds can be accommodated at grooves resulting in positive or negative induced CD bands depending upon their relative orientation with grooves. Since the grooves of quadruplex DNA structures have different groove geometries than duplex DNA, as well as different patterns of donor–acceptor sites, compounds that bind only in the grooves should be able to achieve excellent structure-specific recognition affinity and specificity.
It is indeed intriguing to observe that mitoxantrone having conjugated planar rings could stack close to DNA quadruplex grooves as a dimer. Earlier it has been shown that mitoxantrone binds as monomer in pre-mRNA splicing regulatory element25 stabilizing the stem loop. The grooves in stem-looped RNA segment25 and DNA quadruplex are quite different and perhaps afford different mechanism for mitoxantrone to bind, that is, as monomer or dimer, respectively in the two cases. Even in a duplex DNA, it has been shown that methyl green can bind as dimer, with its x-axis containing two phenyl rings at an angle of 48 degrees to helix axis, depending upon salt and ligand concentration.35 Stacked species are of particular interest for recognition of quadruplexes since studies with duplex DNAs show that compounds that bind as stacked dimers have increased binding affinity and selectivity over similar compounds that bind as monomers.18 Similar stacking in the grooves of quadruplex DNA structures would appear to be a favorable way to selectively recognize quadruplexes with optimum interactions, perhaps employing an induced fit component, between the stacked heterocycles and the G bases of the quadruplex tetrads.18 It may be emphasized that dimer formation eliminates the possibility of complete intercalation.29,35
4. Conclusions
Thus we conclude that mitoxantrone stacks close to DNA quadruplex groove as dimer and the side chains involving 12NH and methylene groups at position 11–14 bind to DNA in threading mechanism due to the flexibility of side chains. A dual or multiple recognition modes may be used by ligands. Nevertheless the present results have shown that quadruplex groove-binding by mitoxantrone dimer enhances thermal stability and induces telomerase inhibition, which is a promising approach for the design of highly selective quadruplex-targeting agents. This group of compounds may serve as the starting point for the design of a new class of highly selective groove-binding molecules. Structure-specific recognition of quadruplex ends as well as grooves offers a very attractive strategy for the discovery and design of highly selective quadruplex DNA binding ligands with attractive biological properties.
Abbreviations
| TRAP | Telomerase repeat amplification protocol |
| SPR | Surface plasmon resonance |
| UTR | Untranslated regions of mRNA |
| IC50 | 50% enzyme inhibitory concentration |
| Tm | Melting temperature |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| Taq polymerase | Thermus aquaticus polymerase |
Acknowledgements
This work was supported by grant from Department of Science and Technology (DST), Govt. of India, grant number SB/SO/BB-081/2009. The fellowship to T. P. Pradeep from University Grants Commission (UGC) and S. Tripathi from Ministry of Human Resource and Development (MHRD), Govt. of India is thankfully acknowledged.
References
- D. J. Patel, A. T. Phan and V. Kuryavyi, Nucleic Acids Res., 2007, 35, 7429–7455 CrossRef CAS PubMed.
- S. Neidle, Curr. Opin. Struct. Biol., 2009, 19, 239–250 CrossRef CAS PubMed.
- S. Neidle, FEBS J., 2010, 277, 1118–1125 CrossRef CAS PubMed.
- S. M. Haider, S. Neidle and G. N. Parkinson, Biochimie, 2011, 93, 1239–1251 CrossRef CAS PubMed.
- C. Shan, J.-H. Tan, T.-M. Ou and Z.-S. Huang, Sci. China: Chem., 2013, 56, 1351–1363 CrossRef CAS.
- V. Sekaran, J. Soares and M. B. Jarstfer, J. Med. Chem., 2014, 57, 521–538 CrossRef CAS PubMed.
- H.-S. Huang, I.-B. Chen, K.-F. Huang, W.-C. Lu, F.-Y. Shieh, Y.-Y. Huang, F.-C. Huang and J.-J. Lin, Chem. Pharm. Bull., 2007, 55, 284–292 CrossRef CAS PubMed.
- D. Sun, B. Thompson, B. E. Cathers, M. Salazar, S. M. Kerwin, J. O. Trent, T. C. Jenkins, S. Neidle and L. H. Hurley, J. Med. Chem., 1997, 40, 2113–2116 CrossRef CAS PubMed.
- P. J. Perry, A. P. Reszka, A. A. Wood, M. A. Read, S. M. Gowan, H. S. Dosanjh, J. O. Trent, T. C. Jenkins, L. R. Kelland and S. Neidle, J. Med. Chem., 1998, 41, 4873–4884 CrossRef CAS.
- M. J. Moore, C. M. Schultes, J. Cuesta, F. Cuenca, M. Gunaratnam, F. A. Tanious, W. D. Wilson and S. Neidle, J. Med. Chem., 2006, 49, 582–599 CrossRef CAS.
- G. Zagotto, A. Ricci, E. Vasquez, A. Sandoli, S. Benedetti, M. Palumbo and C. Sissi, Bioconjugate Chem., 2011, 22, 2126–2135 CrossRef CAS PubMed.
- N. Ranjan, E. Davis, L. Xue and D. P. Arya, Chem. Commun., 2013, 49, 5796–5798 RSC.
- L. Martino, A. Virno, B. Pagano, A. Virgilio, S. Di Micco, A. Galeone, C. Giancola, G. Bifulco, L. Mayol and A. Randazzo, J. Am. Chem. Soc., 2007, 129, 16048–16056 CrossRef CAS PubMed.
- S. Cosconati, L. Marinelli, R. Trotta, A. Virno, S. De Tito, R. Romagnoli, B. Pagano, V. Limongelli, C. Giancola and P. G. Baraldi, J. Am. Chem. Soc., 2010, 132, 6425–6433 CrossRef CAS.
- W. Gai, Q. Yang, J. Xiang, W. Jiang, Q. Li, H. Sun, A. Guan, Q. Shang, H. Zhang and Y. Tang, Nucleic Acids Res., 2013, 41, 2709–2722 CrossRef CAS.
- Q. Li, J. Xiang, X. Li, L. Chen, X. Xu, Y. Tang, Q. Zhou, L. Li, H. Zhang and H. Sun, Biochimie, 2009, 91, 811–819 CrossRef CAS PubMed.
- J. Dash, P. S. Shirude, S.-T. D. Hsu and S. Balasubramanian, J. Am. Chem. Soc., 2008, 130, 15950–15956 CrossRef CAS PubMed.
- E. W. White, F. Tanious, M. A. Ismail, A. P. Reszka, S. Neidle, D. W. Boykin and W.
D. Wilson, Biophys. Chem., 2007, 126, 140–153 CrossRef CAS PubMed.
- M. W. Freyer, R. Buscaglia, K. Kaplan, D. Cashman, L. H. Hurley and E. A. Lewis, Biophys. J., 2007, 92, 2007–2015 CrossRef CAS PubMed.
- Y. Wang and D. J. Patel, J. Mol. Biol., 1993, 234, 1171–1183 CrossRef CAS PubMed.
- W. D. Wilson, F. A. Tanious, M. Fernandez-Saiz and C. T. Rigl, Drug–DNA interaction protocols, Meth. Mol. Biol., Springer, 1997, vol. 90, pp. 219–240 Search PubMed.
- L. Wang, C. Bailly, A. Kumar, D. Ding, M. Bajic, D. W. Boykin and W. D. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12–16 CrossRef CAS.
- J. S. Hudson, S. C. Brooks and D. E. Graves, Biochemistry, 2009, 48, 4440–4447 CrossRef CAS PubMed.
- D. G. Gorenstein, Methods Enzymol., 1992, 211, 254–286 CAS.
- S. Zheng, Y. Chen, C. P. Donahue, M. S. Wolfe and G. Varani, ACS Chem. Biol., 2009, 16, 557–566 CrossRef CAS.
- G. N. Roviello, C. Vicidomini, S. Di Gaetano, D. Capasso, D. Musumeci and V. Roviello, RSC Adv., 2016, 6, 14140–14148 RSC.
- E. Tuite and B. Norden, J. Am. Chem. Soc., 1994, 116, 7548–7556 CrossRef CAS.
- L. Wang, C. Carrasco, A. Kumar, C. E. Stephens, C. Bailly, D. W. Boykin and W. D. Wilson, Biochemistry, 2001, 40, 2511–2521 CrossRef CAS PubMed.
- E. W. White, Selective Recognition of Quadruplex DNA by Small Molecules, Ph.D Thesis, Department of Chemistry Georgia State University, USA, 2006.
- R. Nanjunda, C. Musetti, A. Kumar, M. A. Ismail, A. A. Farahat, S. Wang, C. Sissi, M. Palumbo, D. W. Boykin and W. D. Wilson, Curr. Pharm. Des., 2012, 18, 1934 CrossRef CAS PubMed.
- A. Paul, B. Maji, S. K. Misra, A. K. Jain, K. Muniyappa and S. Bhattacharya, J. Med. Chem., 2012, 55, 7460–7471 CrossRef CAS PubMed.
- I. Manet, F. Manoli, B. Zambelli, G. Andreano, A. Masi, L. Cellai, S. Ottani, G. Marconi and S. Monti, Photochem. Photobiol. Sci., 2011, 10, 1326–1337 CAS.
- J. Rubio-Magnieto, F. Di Meo, M. Lo, C. Delcourt, S. Clément, P. Norman, S. Richeter, M. Linares and M. Surin, Org. Biomol. Chem., 2015, 13, 2453–2463 CAS.
- C. Percivalle, C. Sissi, M. L. Greco, C. Musetti, A. Mariani, A. Artese, G. Costa, M. L. Perrore, S. Alcaro and M. Freccero, Org. Biomol. Chem., 2014, 12, 3744–3754 CAS.
- B. Nordén and F. Tjerneld, Chem. Phys. Lett., 1977, 50, 508–512 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05266e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.