
Cu/Mg/Al/Zr non-noble metal catalysts for o-phenylphenol synthesis†
Yongping Zeng*a,
Tianchi Zhanga,
Yueyang Xub,
Peili Hua,
Ting Yea,
Zhehua Jiaa and
Shengui Juc
aCollege of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, China. E-mail: ypzeng@yzu.edu.cn
bGuodian Science and Technology Research Institute, Nanjing, 210031, China
cThe State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing, 210009, China
First published on 12th January 2016
Abstract
Cu/Mg/Al/Zr hydrotalcite-like precursors with Zr4+:(Al3+ + Zr4+) atomic ratios between 0 and 1 were prepared by co-precipitation methods. The precursors were analyzed using X-ray diffraction (XRD), scanning electron microscopy (SEM), thermogravimetric (TG) analysis and Fourier transform infrared spectroscopy (FT-IR). The results confirmed that well-defined layered double hydroxides (LDH) can be synthesized when the added Zr content is less than 0.25 in terms of Zr4+/(Al3+ + Zr4+) atomic ratio. The catalysts of Cu/Zn/Al/Zr mixed oxides can be obtained via thermal decomposition of hydroxide precursors, and can be used in dehydrogenation of 2-(1-cyclohexenyl) cyclohexanone (CHCH) to ortho-phenylphenol (OPP). Copper particles inside the catalyst act as active sites for dehydrogenation. Transmission electron microscopy (TEM), XRD, N2O chemisorption and N2 adsorption–desorption were performed to investigate the effect of Zr content on determining the copper particle size. Based on the catalytic performance test, it was concluded that the conversion of CHCH depends on the copper particle size of these catalysts.
1. Introduction
Many investigations have been made on the dehydrogenation of alicyclic compounds. For instance, the only important industrial synthesis of o-phenylphenol (OPP) is still the cyclohexanone method, in which 2-(1-cyclohexenyl)cyclohexanone (CHCH) is obtained from cyclohexanone via aldol condensation and dehydration. At present, the catalysts used for OPP dehydrogenation generally use noble metals as the active ingredients such as Pt.1,2 Only a few studies of the non-noble metal catalysts in the OPP synthesis process have been reported in the literature.3 Thus, the synthesis of non-precious metal dehydrogenation catalysts is very important to minimize both capital and operative expenses. Cu-based catalysts are applied in various industrial hydrogenation processes, and most of the catalysts when reduced with care can afford a high dispersion of copper, showing good activity. However, the catalysts still suffered from thermal sintering and poisoning due to poor dispersion. It is shown that the Cu-containing catalysts with high dispersion show good catalytic selectivity and activity in some reactions such as hydrogenation.4–6 However, Cu-containing catalysts are not widely applied for dehydrogenation reactions of organic compounds.Hydrotalcite (HT)-like compounds are a new type of interesting materials which have been applied as ion exchangers, catalyst supports, catalysts, and composites.7–14 Their empirical formula is [MII1−xMIIIx(OH)2]x+[Ax/nn−]x−·mH2O, where MII is a divalent cation such as Cu2+, Zn2+, or Mg2+, MIII is a trivalent cation such as Fe3+, Al3+, or Ga3+. An− is an anion with a negative charge such as CO32− and NO3−, and m is the molar amount of water in the interlayers.15,16 The as-synthesized metallic oxides exhibit highly homogeneously dispersed M3+ and M2+ at the atomic level, high specific surface area, high sintering stability, and strongly basic properties.17–19 Thermal treatments leads to dehydroxylation, dehydration, and destruction of interlayer anions, and form the basic metallic oxides with high surface area, high dispersion of the active centers and better resistance to sintering.20 The homogeneous distributions of metallic oxides in precursors can form the highly dispersed and much stable active metallic centers on catalyst surface after calcination and reduction.21 Thus, well dispersed Cu-based catalysts with high stability, large surface area, and moderate or strong basic properties can be synthesized via the thermal decomposition of the hydrotalcite-like precursors.
In addition, the dispersibility and size of copper particles might significantly affect the stability of the catalyst. Some literatures reported the relevant deactivation mechanism for commercial catalysts.22–24 The low catalytic stability is currently a limiting factor in applying Cu-containing catalysts in the hydrogenation process. How to solve this problem is still the key to applying the Cu-containing catalysts in the hydrogenation process. The introduction of Zr has been reported to improve the surface basicity and Cu dispersion, and hence to influence the activity of catalysts.25–28 Furthermore, the presence of Zr can provide high stability and resistance to redox cycles.29 However, very few studies reported the effect of addition of zirconium on tuning the particle size of Cu inside the catalyst. The detailed structure–activity relationship of the Zr–Cu material is hence lacking and is the subject of this work.
In this work, a series of Cu/Mg/Al/Zr catalysts obtained from Cu/Mg/Al/Zr layered double hydroxides were prepared with Zr4+/(Al3+ + Zr4+) atomic ratios between 0 and 1. Catalyst were characterized using X-ray diffraction (XRD), scanning electron microscope (SEM), N2 adsorption–desorption, transmission electron microscope (TEM), high-magnification transmission electron microscope (HRTEM), N2O chemisorption, thermogravimetric (TG), H2 temperature programmed reduction (H2-TPR), Fourier transform infrared spectroscopy (FT-IR) and CO2 temperature programmed desorption (CO2-TPD).
2. Experimental
2.1. Preparation of catalysts
The Cu/Mg/Al/Zr hydrotalcite-like samples were prepared by co-precipitation method. The Cu2+/Mg2+/(Al3+ + Zr4+) atomic ratio was kept at 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.5:2 in the starting solution, while the atomic ratio of Zr4+/Al3+ was changed from 0 to 1. Calculated amount of Cu(NO3)2·3H2O, Mg(NO3)2·6H2O, Zr(NO3)4·5H2O and Al(NO3)3·9H2O were added into deionized water to prepare a 200 mL mixed salt solution A, where the total concentration of (Cu2+ + Mg2+ + Al3+ + Zr4+) is 0.9 mol L−1. Another solution (called B) was an aqueous of Na2CO3 and NaOH, in which the concentration of Na2CO3 and NaOH are both 1 mol L−1. The solutions A and B were slowly added to a four-necked flask with vigorous stirring and heated to the reaction temperature of 60 °C with the pH maintained at 9.5. The resulting precipitant were aged for 6 hours and recovered with filtration, then washed with distilled water to remove the trace sodium. Drying was performed in the air at 90 °C for 12 hours prior to the calcination conducted in air at 450 °C for 4 hours. Then the obtained powders were ground and extruded to strip catalysts. The catalysts were reduced using H2 at 300 °C for 2 hours, followed by reacted at 330 °C. Synthesized Cu/Mg/Al/Zr catalysts were denoted as CMAZ-N, where N = 0, 1, 2, 3 and 4 denoting the Zr4+/(Al3+ + Zr4+) ratio of 0, 0.25, 0.5, 0.75 and 1, respectively.
:3.5:2 in the starting solution, while the atomic ratio of Zr4+/Al3+ was changed from 0 to 1. Calculated amount of Cu(NO3)2·3H2O, Mg(NO3)2·6H2O, Zr(NO3)4·5H2O and Al(NO3)3·9H2O were added into deionized water to prepare a 200 mL mixed salt solution A, where the total concentration of (Cu2+ + Mg2+ + Al3+ + Zr4+) is 0.9 mol L−1. Another solution (called B) was an aqueous of Na2CO3 and NaOH, in which the concentration of Na2CO3 and NaOH are both 1 mol L−1. The solutions A and B were slowly added to a four-necked flask with vigorous stirring and heated to the reaction temperature of 60 °C with the pH maintained at 9.5. The resulting precipitant were aged for 6 hours and recovered with filtration, then washed with distilled water to remove the trace sodium. Drying was performed in the air at 90 °C for 12 hours prior to the calcination conducted in air at 450 °C for 4 hours. Then the obtained powders were ground and extruded to strip catalysts. The catalysts were reduced using H2 at 300 °C for 2 hours, followed by reacted at 330 °C. Synthesized Cu/Mg/Al/Zr catalysts were denoted as CMAZ-N, where N = 0, 1, 2, 3 and 4 denoting the Zr4+/(Al3+ + Zr4+) ratio of 0, 0.25, 0.5, 0.75 and 1, respectively.
2.2. Characterization of samples
X-ray diffraction (XRD) patterns were measured by the X-ray diffraction instrument (Bruker AXS, D8 Advance) with the wavelength of the Cu Kα radiation source being 1.5406 Å. The intensity data were collected over the 2θ range of 5–70° with a step size of 0.05° and counting time of 1 s each point under the condition of 40 kV and 40 mA. SEM images of hydrotalcite-like precursors and catalysts were obtained by a field emission scanning electron microscopy (Hitachi, S-4800II). Thermal decomposition and stability for the catalyst precursors was studied by thermogravimetry analysis (TG-DTG, PerkinElmer Pyris 1) under the N2 atmosphere (30 mL min−1) with the heating rate of 10 °C min−1 from 50 °C to 750 °C. The infrared spectra of samples were recorded by a Fourier transform spectroscopy (FT-IR, Bruker, Tensor 27), and measurements were performed in the wavenumber range of 400–4000 cm−1. Transmission Electron Microscope (TEM) images of low-magnification were collected using a Tecnai 12 model machine for low-magnification observe samples. High-magnification Transmission Electron Microscope (HRTEM) was performed using Tecnai G2 F30 S-TWIN model machine for high-magnification to observe the samples. The specific surface areas of the samples were computed from N2 adsorption–desorption isotherms obtained at the temperature of liquid N2 on a BELSORP-MAX instrument. Firstly, the powders were outgassed at the temperature of 200 °C for 6 h to ensure a clean surface before the measurement of the adsorption isotherms. H2-TPR was performed on the instrument Finesorb-3010. The catalyst was firstly treated in Ar (30 mL min−1) with a 10 °C min−1 heating rate to 200 °C and kept it at 200 °C for 30 min. After the samples were cooled down to 100 °C, TPR were conducted in the mixture of H2/Ar (molar ratio 1:9) flowing at 30 mL min−1 at a 10 °C min−1 heating rate until 500 °C. Finally, the catalyst was cooled to room temperature in the presence of Ar. The exposed surface area of copper (SCu) and the dispersion of copper (DCu) were determined by adsorption of N2O and performed on Finesorb-3010 instrument, using the method similar to that reported by Xia et al.30,31 Catalysts were reduced (the procedure is described in TPR section) in H2/Ar (molar ratio 1:9) mixture with a flow rate of 30 mL min−1 at a heating rate of 10 °C min−1 until 450 °C and was kept at 330 °C for 30 min. The consumption amount of H2 in the TPR was recorded as X. Then the reactor bed was purged in Ar to 50 °C. The mixture of N2O/Ar (the molar ratio 1:4, 30 mL min−1) was changed to oxidize copper atoms of surface to Cu2O at the temperature of 50 °C for 1 hour. The reactor bed was then flushed with pure Ar to get rid of the oxidant. Another TPR procedure was conducted in the mixture of H2/Ar (molar ratio 1:9) with a flow rate of 30 mL min−1 at a 10 °C min−1 heating rate up to 330 °C and was kept at 330 °C for 30 min. The consumption of H2 in this TPR procedure was recorded as Y. The exposed surface area of copper and the dispersion of copper were computed by the following equations:| SCu = (2Y × NA)/(1.4 × 1019 × X × MCu) (m2 g−1) | (1) |
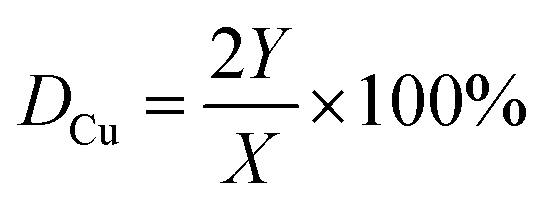 | (2) |
2.3. Catalytic reactions
The dehydrogenation reactions of CHCH were performed in a home-made set-up with a tubular quartz glass flow reactor (Φ 19 × 560 mm). The reactor temperature was measured by a thermocouple located in the middle of the catalyst bed. The dehydrogenation reactions were carried out at 330 °C and atmospheric pressure. All catalysts were pre-treated by reduction under H2 (flow rate of 20 mL min−1) at 300 °C for 2 hours. Adjusting the H2 flow rate at 10 mL min−1, the reagent was added into the reactor using a syringe pump. The reaction products were dissolved in alcohol and cooled to room temperature and analyzed using a gas chromatograph equipped with a flame ionization detector (FID) detector.3. Results and discussions
3.1. Textural and structural properties of the precursors
The XRD patterns of the precursors are shown in Fig. 1. It is shown that the structure crystallinity of the samples decreases with increase of Zr amount. The reflection peaks for CMAZ-0 and CMAZ-1 show a well-crystallized hydrotalcite structure (JCPDS 48-0601). The introduction of Zr in CMAZ-1 still exist the formation of hydrotalcite structure, which was also observed by Jeong and Gao et al.33,34 But, further increasing the amount of Zr, the characteristic peaks of hydrotalcite structure disappeared and the amorphous structure occurred instead. The amorphous structure occurred probably because of the introduction of the distortions in the hydrotalcite sheets as a result of the replacement of Al3+ by Zr4+.35 Another reason may be the formation of amorphous precipitates (i.e. Mg and Zr salts of hydroxyl carbonates and hydroxides).36 In these systems, hydrotalcite-like microcrystalline phases consisting of small poorly ordered layers are more likely to be present, leading to the loss of crystallinity for the samples. Therefore, we expect that Zr-containing hydrotalcite-like compounds are the main component in these precursors, especially for the composition of Zr4+/(Al3+ + Zr4+) less than 0.25. | ||
| Fig. 1 XRD patterns of the Cu/Mg/Al/Zr precursors with various Zr content. | ||
SEM images of all precursors are shown in Fig. 2. It can be observed that a well-developed layered and plate-like structure of hydrotalcite prepared by co-precipitation method without Zr added, could be easily identified in Fig. 2a.37,38 When the Zr4+/(Al3+ + Zr4+) ratio is up to 0.25 (i.e. CMAZ-1), the materials still exhibit a plate-like structure of hydrotalcite, though a destructed morphology is observed (Fig. 2b). Also, it can be found that the plate-like structures are dense-stacking and homogeneous. However, when the amount of Zr increases as shown in Fig. 2c–e, platelet particles tend to break into small pieces for another three samples35 These samples consist of irregular particles that existed in diverse and larger agglomerates with poor crystallinity, which is consistent with the XRD results. We can conclude that the Zr/Al ratio has an obvious influence on the crystal morphology.
 | ||
| Fig. 2 SEM images of uncalcined samples: (a) CMAZ-0, (b) CMAZ-1, (c) CMAZ-2, (d) CMAZ-3, and (e) CMAZ-4. | ||
FT-IR (ESI†) was used to identify anion-cation and bond types for the interlayers of the precursors, shown in Fig. S1.† The intense and broad band at about 3500 cm−1 is ascribed to the bond stretching mode of H2O molecules of interlayers and the hydroxyl groups on the layers (νOHstr). The broad bands show that the hydroxyl species are hydrogen bonded. A weak absorption band occurs at about 1600 cm−1 because of the deformation bending modes of H2O molecules. The bands between 1300 and 1550 cm−1 are due to bond stretching modes of the carbonate species. CMAZ-0 and CMAZ-1 exhibit medium vibration at about 420 cm−1 ascribed to O–Metal–O bonds in the sheets. This vibration band gradually disappeared, which can be ascribed to the increase of the Zr content.36 It also shows that introduction of Zr has a significant effect on the crystallinity of hydrotalcite layer structure.
To clarify the thermal decomposition process during the calcination, TG and differential thermal gravity (DTG) analysis techniques were performed (Fig. S2 in the ESI†). The results show that there are three major weight losses for all samples. The first peak of weight loss occurred at 100–200 °C, which can be ascribed to the removal of physically adsorbed H2O molecules in the interlayers. This suggests that the temperature around 150 °C was sufficient for the removal of physisorbed water. The second weight loss occurred at 250–400 °C, which may be ascribed to the decomposition of carbonate ions and laminates hydroxy dehydration. There are some differences for the third weight loss in these materials. The peak of weight loss peak for CMAZ-0 occurred at 600–650 °C. The weight loss peak for CMAZ-1 drops to 500–550 °C compared to CMAZ-0, which is ascribed to the introduction of Zr in the structure. This might reduce the electrostatic interaction between the anions and layers, that can result in lower thermal stability.39,40 For the three samples of CMAZ-2, -3 and -4, the third stage almost disappeared, which implies that hydrotalcite laminate structure disappeared with the increase of Zr content.
3.2. Textural and structural properties of the calcined samples
Fig. 3 shows the XRD patterns for the samples calcined after H2 reduction for 2 hours. It can be seen that there are no characteristic peaks of hydrotalcite-like compounds in the XRD patterns. This shows the lamellar framework collapsed and the structure is decomposed. Therefore, crystalline compounds containing Mg and Al were not detected. There are no characteristic peaks of Zr in XRD patterns of CMAZ-0, CMAZ-1 and CMAZ-2, which indicate that Zr was also in the amorphous state.26,41 Cu0 is the main crystal phase in the CMAZ-0, CMAZ-1 and CMAZ-2. But, with the increase of Zr content (CMAZ-3 and CMAZ-4), the monoclinic zirconia (m-ZrO2, 2θ = 31.5°) characteristic peak occurs.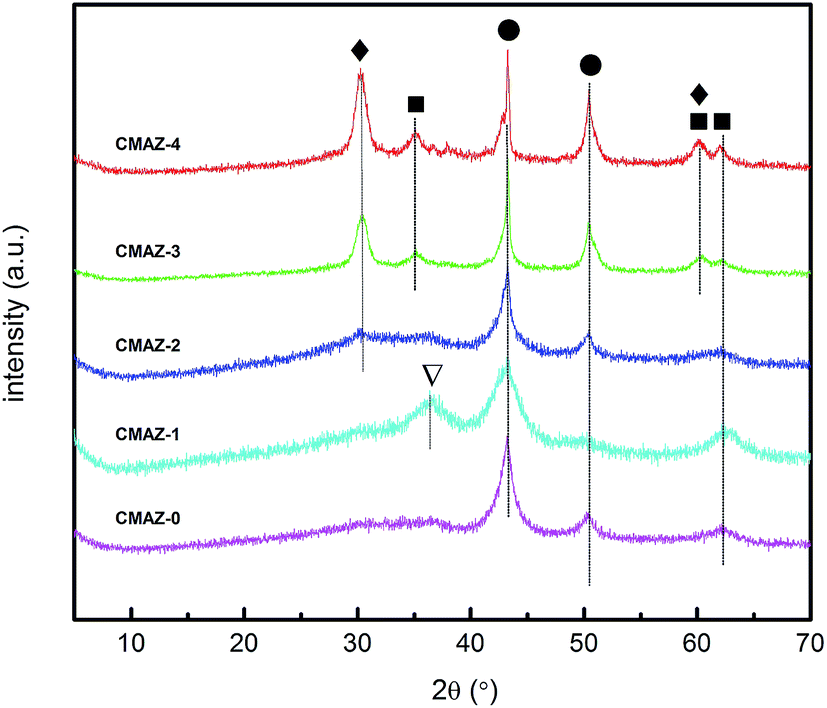 | ||
| Fig. 3 XRD patterns of the calcined Cu/Mg/Al/Zr catalysts after reduction at 300 °C. (●) Cu0; (■) CuO; (▽) Cu2O; (◆) m-ZrO2. | ||
To investigate the effect of Zr content on the size of copper particles, the full width at half maxima (FWHM) in the XRD patterns and the size of the copper particles for different samples are listed in Table 1. The FWHM of the Cu0 particles was calculated by Gaussian equation and the d(Cu0)XRD was calculated by Scherrer formula (eqn (3)).
 | (3) |
The results show that the average size of Cu0 crystalline was 3.97–13.23 nm after H2 reduction. As the Zr content changes, the size of cooper particles reaches the minimum value (3.97 nm) in these samples. Further increase of the Zr content results in the increase of the copper particles size. It is clear that the particle size of Cu is related to the amount of Zr added. Through adding the amount of Zr in the catalyst, the size of copper particles can be controlled. However, excessive introduction of Zr may cause the growth of copper particle.
Fig. 4 shows a representative set of TEM images for copper particles synthesized at two different compositions (CMAZ-1 and CMAZ-2) with the statistics of size distributions for copper particles in Fig. 4. The mean diameter and min/max diameter were calculated by counting 100 particles (CMAZ-1) and 50 particles (CMAZ-2) with a loupe from the TEM image of 195000 magnifications. From Fig. 3 we can observe that CMAZ-1 and CMAZ-2 only present characteristic peaks of copper. Thus, we can confirm that particles in these two samples are copper nanoparticles, which is confirmed by Zhang et al.42 A copper particle size on surface of CMAZ-1 and CMAZ-2 was obtained from the corresponding size histograms. For CMAZ-1, the average particle size is 3.85 nm, which is smaller than 7.79 nm for CMAZ-2. It can be observed that copper particles are well dispersed in the oxide matrix. Additionally, the dispersion of copper particles in CMAZ-1 is much better than that CMAZ-2. This means that the introduction of Zr can tune the size of the copper particles and have a significant impact on the dispersion of the copper particles in the catalyst.29 In order to fully understand the inner structure of Cu particles in CMAZ-1, the high-magnification TEM (HRTEM) are obtained shown in Fig. 5. Cu particles size in Fig. 5a is corresponding to the CMAZ-1 sample. Fig. 5b depicts the fringe spacing of 0.21 nm ensures the presence of Cu (111) plane. HRTEM elemental mapping images is shown in Fig. S3 (ESI†), each element is well dispersed in the catalyst, which illustrated the high dispersion of Cu/Mg/Al/Zr catalyst.
 | ||
| Fig. 4 TEM images of CMAZ-1 and CMAZ-2 catalysts after H2 reduction at 300 °C for 2 h. | ||
 | ||
| Fig. 5 (a) HRTEM images of CMAZ-1 catalysts after H2 reduction at 300 °C for 2 h. (b) HRTEM image of single particle from (a). | ||
The pore volume and surface area of the catalysts after reduction are shown in Table 2. The adsorption and desorption isotherms of samples are shown in Fig. S4 (ESI†). The maximum surface area is 134.07 m2 g−1 possessed by CMAZ-1. Although CMAZ-0 surface area compared with CMAZ-1 is almost the same (133.11 m2 g−1), but the pore volume is nearly twice lower than CMAZ-1, respectively 0.358 cm3 g−1 and 0.789 cm3 g−1. It is speculated that the introduction of Zr may result in smaller particle size, larger surface area and pore volume. Conversely, surface area and pore volume drop significantly in CMAZ-2, -3 and -4. This influence can be ascribed to distortion and destruction of hydrotalcite laminates. The distortion increases with the amount of Zr in the catalysts. The SBET of catalysts decreases because of the destruction and distortion of hydrotalcite. The adsorption and desorption isotherms of samples are shown in Fig. S4 (ESI†).
| Catalysts | Surface areaa (m2 g−1) | Pore volumea (cm3 g−1) | Cu surface areab (m2 g−1) | Cu dispersionb (%) |
|---|---|---|---|---|
| a After reduction at 300 °C.b Calculated from dissociative adsorption. | ||||
| CMAZ-0 | 133.11 | 0.358 | 81.85 | 12.08 |
| CMAZ-1 | 134.07 | 0.789 | 152.45 | 22.54 |
| CMAZ-2 | 97.046 | 0.340 | 15.75 | 2.32 |
| CMAZ-3 | 35.929 | 0.128 | 20.12 | 2.97 |
| CMAZ-4 | 66.655 | 0.409 | 41.40 | 6.11 |
The exposed catalyst surface area and dispersion of Cu were measured by N2O adsorption and are shown in Table 2. When Zr4+/(Al3+ + Zr4+) ratio is less than 0.25, the exposed surface area of Cu increased with the increase of Zr content. The addition of Zr also enhanced the Cu dispersion, which may be due to the effect of valence compensation and ion doping, i.e. the dissolved Zr in MgO crystal resulted in the formation of cation defects on the surface of Cu–MgO forming more active centers.43 This is consistent with the results observed by Velu et al.,44,45 who demonstrated that substitution of Zr could improve the dispersion of Cu. With further increase of the amount of Zr, Zr4+ may form the m-ZrO2 crystalline structure and cause the decrease of the proportion of Zr placed in the layered structures.46 Therefore, the existence of m-ZrO2 with little Zr4+ in the layered structures caused the lower DCu for the CMAZ-2, CMAZ-3 and CMAZ-4.
FT-IR spectra of calcined samples are shown in Fig. S5 (ESI†). Compared with the spectra of the precursors, the stretching vibration and absorption bands positions were retained meaning that the layered structure of hydrotalcite is not completely destroyed at the calcination temperature of 450 °C.
Fig. 6 shows the H2-TPR profiles for the calcined samples. As shown in Fig. 6, the TPR profiles for all samples with various amount of Zr show a broad band of H2 consumption at the range of 150–400 °C. To get more insights into TPR results, the band of H2 consumption were deconvoluted into two Gaussian forms denoted as α and β peaks, respectively. The peak positions and corresponding contributions are listed in the Table 3. The two peaks are attributed to the reduction of two different types of CuO phase. The β peak at high temperature is due to the reduction of dispersed CuO. The α peak is attributed to the reduction of highly dispersed CuO appearing at low temperature.47 As shown in Table 3, α peak proportion in CMAZ-0 only is 14.97%, the lowest Cu content of all the samples. The proportion suddenly increased to 85.32% in CMAZ-1 and reached the maximum. With the increasing amount of the Zr, this proportion decreases. The addition of Zr caused the broad α peak for the highly dispersed CuO phase. It is because Zr can strengthen the interactions of the Cu+/Cu2+, leading to the higher reduction temperature.35 Excessive Zr amount assists to form the m-ZrO2, as observed in the XRD profile (Fig. 3), thus resulting in the decrease of α peak proportion.
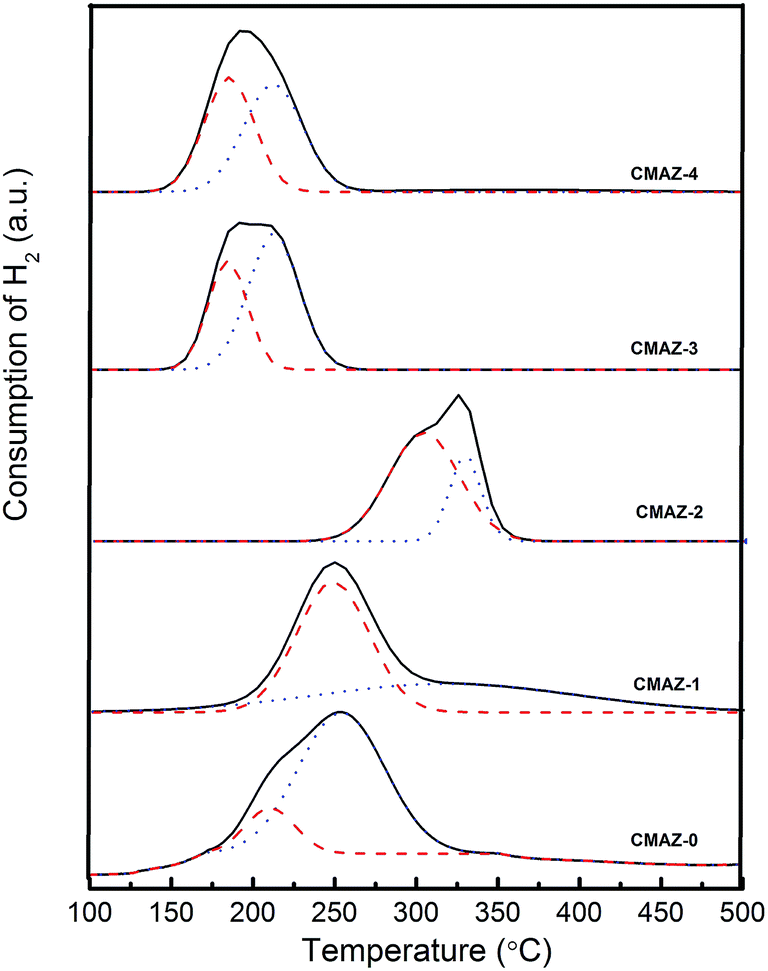 | ||
| Fig. 6 H2-TPR profiles of catalysts at 450 °C, black solid, red dash and blue dot lines correspond to the TPR, α and β peak, respectively. | ||
The surface basicity of the catalysts was detected with CO2-TPD and is shown in Fig. 7. The broad band of CO2 consumption was deconvoluted into three Gaussian forms denoted as weakly (α peak), moderately (β peak) and strongly (γ peak) basic sites. The weakly basic sites are ascribed to hydroxyl groups, moderately basic sites were related to Al–O, Zr–O or Mg–O pairs, and strongly basic sites can be related to oxygen atoms with low coordinations.35,47 The peak positions and corresponding contributions are also shown in Table 4. The strength of strongly basic sites is markedly enhanced for CMAZ-1 after the introduction of Zr. While, the strength of strongly basic sites decreased for subsequent CMAZ-2, CMAZ-3 and CMAZ-4. More specifically, in CMAZ-4, strongly basic sites disappeared. CMAZ-1 has the most strongly basic sites among these samples. The number of strongly basic sites increases because of the increase of Zr content in the layered structures. The higher electronic density of the layers in the samples with higher Zr content prohibits some oxygen atoms to coordinate with Zr atoms.48 On the other hand, the number of strongly basic sites decreases with further increase of Zr amount in the catalysts due to the transformation of amorphous zirconia (am-ZrO2) to monoclinic zirconia (m-ZrO2) (observed in Fig. 3). This transformation of structure may lead to less strongly basic sites.49
 | ||
| Fig. 7 CO2-TPD profiles of the catalysts with various Zr content, black solid, red dash, blue dot dash and dot lines correspond to the TPD, α, β and γ peak, respectively. | ||
| Sample no. | TPD peak position [temperature (°C)] and concentrationa (%) | ||
|---|---|---|---|
| Site α | Site β | Site γ | |
| a The proportion of different basic sites. | |||
| CMAZ-0 | 232 (6.01) | 446 (79.81) | 659 (14.18) |
| CMAZ-1 | 295 (23.43) | 439 (9.22) | 473 (67.35) |
| CMAZ-2 | 329 (44.44) | 447 (41.89) | 566 (13.67) |
| CMAZ-3 | 371 (72.73) | 385 (27.27) | — |
| CMAZ-4 | — | — | — |
3.3. Catalytic performance
Catalytic activity and selectivity of catalysts were tested in dehydrogenation of CHCH to OPP. Fig. 8 shows the selectivity and conversions as a function of the Zr4+/(Zr4+ + Al3+) atomic ratio. Clear differences can be observed between the nature of the catalysts and corresponding catalytic performance. There is a significant change in the activity observed with the increase of the amount of Zr on these samples. The average conversion of CHCH reaches around 98% for CMAZ-0 to CMAZ-3 and decreased to 67.5% for CMAZ-4. The selectivity to OPP is also shown in Fig. 8. There is a significant difference for these samples. The selectivity of OPP reached a maximum for Zr4+/(Zr4+ + Al3+) atomic ratio 0.25 (CMAZ-1). Further increase in the amount of Zr on the calcined samples leads to drastically toward 4.2% with Zr4+/(Zr4+ + Al3+) atomic ratio of 1. Cu/Mg/Al/Zr catalysts with a better dispersion was prepared with the introduction of an appropriate Zr amount. In addition, the sintering of catalysts can be effectively inhibited because the catalysts can form with a more suitable structural morphology, which was an important factor. The enhanced crystallinity enables more surface are of the catalyst support of the active sites for dehydrogenation. The dehydrogenation performance follows the similar trend as that of dispersion of Cu. For CMAZ-1 where Zr4+/(Zr4+ + Al3+) atomic ratio is low, the sample has a high pore volume as observed by BET measurement (Table 2). The catalysts exist in the form of the agglomeration of active sites at high Zr4+/(Zr4+ + Al3+) atomic ratio resulting in large-sized particles. From the CO2-TPD profiles in Fig. 7, the OPP selectivity is influenced by the distribution of basic sites on the surface of catalysts. The catalytic selectivity is also related to the amount of reducible copper, although the selectivity is not proportional to the atomic ratio of Zr4+/(Zr4+ + Al3+) shown in Fig. 8. The appropriate amount of Zr added in the sample is 0.25 for the atomic ratio of Zr4+/(Zr4+ + Al3+). H2-TPR and reaction results confirmed this observation. CMAZ-1 shows the highest proportion of low temperature peak (around 85.3%) in H2-TPR, and the better dispersed CuO in this sample. Guo and Wang et al. reported that catalytic activity of Cu-based dehydrogenation catalysts is related to well dispersed copper species.47,50 Therefore, the variation trends of catalytic activity of catalysts would be easy to understand in view of the similar trends of the dispersion of copper species, which was illustrated by XRD and TPR results. In addition, it is clear that the OPP selectivity is also affected by the Zr amount, which tuned the dispersibility of copper species. The exposed surface area of copper measured by chemisorption of N2O is an important parameter for Cu-containing catalysts. The catalytic activity depends on the exposed surface area of Cu. The exposed surface area of Cu increased obviously with the increase of Zr content. The maximum dispersion (22.5%) of Cu is obtained for CMAZ-1 and the selectivity of OPP also reached a maximum. The number of strongly basic sites decreases with further increase of Zr amount in the catalysts because of the transformation of amorphous zirconia (am-ZrO2) to monoclinic zirconia (m-ZrO2). This structural transformation may lead to less strongly basic sites. Zhou et al. studied on Cu/ZrO2 catalysts, which consisted of tetragonal zirconia with Cu content lower than 10 wt%.51 They referred to as the interaction between Cu species and the ZrO2 support which hindered the phase transformation to m-ZrO2 based on the vacancy model of oxygen.52 | ||
| Fig. 8 Effects of the amount of Zr on the conversion of OCC and the selectivity toward OPP at 330 °C after 2 h reaction. | ||
The goal of this investigation is to shed light on the effect of different Zr content on the catalytic properties in relation to byproducts. The dehydrogenation process consists of a sequence of hydrogenation or dehydrogenation reactions, in which the CHCH is successively transformed into o-cyclohexylcyclohexanone (OCC), o-cyclohexylphenol (OCHP) and finally OPP (Scheme 1).53 Besides, there are small amount of biphenyl (BP) and dibenzofuran (DBF) among byproducts. It is because that OPP can further dehydrogenated to DBF and dehydrated to BP. Catalytic selectivity for primary products of all catalysts is shown in Fig. 9. When OPP selectivity increases, the byproducts OCC and OCHP naturally decrease. Moreover, OCC is the main byproducts. According to the route shown in the Scheme, the OPP selectivity is significantly affected by the amount of Zr. Mean Cu0 crystalline sizes is the smallest in CMAZ-1 and OCC content is the lowest. This suggests that smaller copper particles size of catalyst results in the better copper dispersion. Thus, the conversion of CHCH to OPP is more effective. While mean Cu0 crystalline sizes increases, copper distribution becomes worse. The conversion of CHCH mainly occurs in the first step reaction and the main byproduct is OCC. In conclusion, copper particle size has a significant effect on the OPP catalytic activity and selectivity. The introduction of Zr can control the copper particle size. The small mean Cu0 crystalline size is in favor of the conversion of CHCH to OPP.
 | ||
| Scheme 1 Synthesis route of OPP from CHCH and its byproducts. | ||
 | ||
| Fig. 9 The catalytic selectivity for three primary products of all catalysts after 2 h reaction. | ||
Fig. 10 shows the catalytic selectivity to OPP with the reaction time. The corresponding conversions can be found in Table 1S (ESI†). During the reactions, CMAZ-1 always shows the highest catalytic performance and the selectivity is still above 65% after 7 hours reaction. With the increase of Zr content, CMAZ-4 almost lost its catalytic activity. The copper particles size and high dispersion of containing active copper centers on surface have a significant impact on the catalytic stability. Chraska et al. studied the effect of the particle size on the stability of the catalysts and observed that the stability for small particles can be illustrated by the lower surface energy of ZrO2.54 Purnama et al. observed a significant stabilizing effect of Cu centers in ZrO2.55 This may explain the stabilization of the Cu particles with a diameter of about 3 nm. It is clear that the CMAZ-1 catalyst prepared via co-precipitation method shows a stable catalytic performance during this reaction time.
 | ||
| Fig. 10 The stability for different Zr content catalysts at 330 °C. | ||
4. Conclusion
Cu–Mg–Al–Zr hydrotalcite-like compounds with different atomic ratio of Zr4+/(Al3+ + Zr4+) were successfully prepared by co-precipitation methods. The introduction of Zr can improve the catalytic properties of Cu–Mg–Al catalyst prepared via hydrotalcite-like precursors. The catalytic activity and selectivity for OPP synthesis are related to the size of Cu species and the amount of Zr in catalysts. The optimum content of Zr is about Zr4+/(Al3+ + Zr4+) = 0.25 (CMAZ-1), showing the highest catalytic stability and selectivity above 65% even after 7 hours of reaction. The existence of Cu nanoparticles is confirmed by analysis using XRD, TEM and HRTEM. Introducing Zr affects the size of Cu nanoparticles, smaller Cu nanoparticles always shows the better catalytic performance; and CO2-TPD results show that Zr can lower the content of Al on the catalyst, and generate the strongly basic sites. This may weaken the interaction between Al2O3 and CuO so to avoid the agglomeration of active sites. CMAZ-1 always shows the highest catalytic stability and the selectivity keep above 65% even after 7 hours reaction.Acknowledgements
We thank Dr Peyman Moghadam at Northwestern University and Prof. Gui Han at Yangzhou University for reading this manuscript and giving the valuable suggestions. This work was supported by the Natural Science Foundation of China (Grant No. 20806064 and 21106125), the Natural Science Foundation of Jiangsu Province, China (BK20131227). Generous allocations of test were provided by Testing Center of Yangzhou University and a Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- H. Goto and N. Shibamoto, Process for preparing of o-phenylphenol, US pat., 4088702, 1978.
- I. Juichi, Process for the production of o-phenylphenol, US pat., 4080390, 1978.
- W. Oskar and H. Schwarz, Process for preparing hydroxypiphenyl, US pat., 3932536, 1976.
- K. V. R. Chary, G. V. Sagar, D. Naresh, K. K. Seela and B. Sridhar, J. Phys. Chem. B, 2005, 109, 9437–9444 CrossRef CAS PubMed.
- J. Fei, Z. Hou, B. Zhu, H. Lou and X. Zheng, Appl. Catal., A, 2006, 304, 49–54 CrossRef CAS.
- D. Jin, B. Zhu, Z. Hou, J. Fei, H. Lou and X. Zheng, Fuel, 2007, 86, 2707–2713 CrossRef CAS.
- J. L. Shumaker, C. Crofcheck, S. A. Tackett, E. Santillan-Jimenez, T. Morgan, Y. Ji, M. Crocker and T. J. Toops, Appl. Catal., B, 2008, 82, 120–130 CrossRef CAS.
- C. Xu, Y. Gao, X. Liu, R. Xin and Z. Wang, RSC Adv., 2013, 3, 793–801 RSC.
- J. S. Valente, F. Tzompantzi, J. Prince, J. G. H. Cortez and R. Gomez, Appl. Catal., B, 2009, 90, 330–338 CrossRef CAS.
- M. Jablonska, A. E. Palomares and L. Chmielarz, Chem.–Eng. J., 2013, 231, 273–280 CrossRef CAS.
- A. Hanif, S. Dasgupta, S. Divekar, A. Arya, M. O. Garg and A. Nanoti, Chem.–Eng. J., 2014, 236, 91–99 CrossRef CAS.
- J. Agrell, H. Birgersson and M. Boutonnet, J. Power Sources, 2002, 106, 249–257 CrossRef CAS.
- Z. Yuan, Li. Wang, J. Wang, S. Xia, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 101, 431–440 CrossRef CAS.
- A. Serrano-Lotina, L. Rodriguez, G. Munoz and L. Daza, J. Power Sources, 2011, 196, 4404–4410 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- Z. Jiang, Z. P. Hao, J. J. Yu, H. X. Hou, C. Hu and J. X. Su, Catal. Today, 2005, 99, 3–4 Search PubMed.
- A. Alejandre, F. Medina, P. Salagre, X. Correig and J. E. Sueiras, Chem. Mater., 1999, 11, 939–948 CrossRef CAS.
- G. Busca, U. Costantino, F. Marmottini, T. Montanari, P. Patrono, F. Pinzari and G. Ramis, Appl. Catal., A, 2006, 310, 70–78 CrossRef CAS.
- L. H. Zhang, C. Zheng, F. Li, D. G. Evans and X. Duan, J. Mater. Sci., 2008, 43, 237–243 CrossRef CAS.
- J. Barrault, A. Derouault, G. Courtois, J. M. Maissant, J. C. Dupin, C. Guimon, H. Martinez and E. Dumitriu, Appl. Catal., A, 2004, 262, 43–51 CrossRef CAS.
- A. F. Lucrédio, G. Jerkiewickz and E. M. Assaf, Appl. Catal., A, 2007, 333, 90–95 CrossRef.
- D. Mott, J. Galkowski, L. Y. Wang, J. Luo and C. J. Zhong, Langmuir, 2007, 23, 5740–5745 CrossRef CAS PubMed.
- G. Prieto, J. Zecevic, H. Friedrich, K. P. de Jong and P. E. de Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef CAS PubMed.
- C. T. Campbell, S. C. Parker and D. E. Starr, Science, 2002, 298, 811–814 CrossRef CAS PubMed.
- A. Szizybalski, F. Girgsdies, A. Rabis, Y. Wang, M. Niederberger and T. Ressler, J. Catal., 2005, 233, 297–307 CrossRef CAS.
- Y. F. Li, X. F. Dong and W. M. Lin, Int. J. Hydrogen Energy, 2004, 29, 1617–1621 CrossRef CAS.
- J. H. Flores, D. P. B. Peixoto, L. G. Appel, R. R. de Avillez and M. I. P. da Silva, Catal. Today, 2011, 172, 218–225 CrossRef CAS.
- B. Lindstrom and L. J. Pettersson, Int. J. Hydrogen Energy, 2001, 26, 923–933 CrossRef CAS.
- J. Agrell, H. Birgersson, M. Boutonnet, I. M. Cabrera, R. Navarro and J. Fierro, J. Catal., 2003, 219, 389–403 CrossRef CAS.
- S. X. Xia, Z. L. Yuan, L. N. Wang, P. Chen and Z. Y. Hou, Appl. Catal., A, 2011, 403, 173–182 CrossRef CAS.
- S. X. Xia, R. F. Nie, X. Y. Lu, L. N. Wang, P. Chen and Z. Y. Hou, J. Catal., 2012, 296, 1–11 CrossRef CAS.
- S. X. Xia, L. P. Zheng, W. S. Ning, L. N. Wang, P. Chen and Z. Y. Hou, J. Mater. Chem. A, 2013, 1, 11548–11552 CAS.
- H. Jeong, K. I. Kim, T. H. Kim, C. H. Ko, H. C. Park and I. K. Song, J. Power Sources, 2006, 159, 1296–1299 CrossRef CAS.
- G. Chen, N. Xu, X. Li, Q. Liu, H. Yang and W. Li, RSC Adv., 2015, 5, 60128–60134 RSC.
- P. Gao, F. Li, H. J. Zhan, N. Zhao, F. K Xiao, W. Wei, L. S. Zhong, H. Wang and Y. H. Sun, J. Catal., 2013, 298, 51–60 CrossRef CAS.
- S. Velu, D. P. Sabde, N. Shah and S. Sivasanker, Chem. Mater., 1998, 10, 3451–3458 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS.
- S. H. Wang, Y. B. Wang, Y. M. Dai and J. M. Jehng, Appl. Catal., A, 2012, 439–440, 135–141 CrossRef CAS.
- L. H. Zhang, F. Li, D. G. Evans and X. Duan, Ind. Eng. Chem. Res., 2010, 49, 5959–5968 CrossRef CAS.
- X. X. Guo, F. Z. Zhang, D. G. Evans and X. Duan, Chem. Commun., 2010, 46, 5197–5210 RSC.
- H. Oguchi, T. Nishiguchi, T. Matsumoto, H. Kanai, K. Utani, Y. Matsumura and S. Imamura, Appl. Catal., A, 2005, 281, 69–73 CrossRef CAS.
- Q. Zhang, Y. Z. Zuo, M. H. Han, J. F. Wang, Y. Jin and F. Wei, Catal. Today, 2010, 150, 55–60 CrossRef CAS.
- X. An, J. L. Li, Y. Z. Zuo, Q. Zhang, D. Z. Wang and J. F. Wang, Catal. Lett., 2007, 118, 264–269 CrossRef CAS.
- S. Velu, K. Suzuki, M. Okazaki, M. P. Kapoor, T. Osaki and F. Ohashi, J. Catal., 2000, 194, 373–384 CrossRef CAS.
- S. Velu, K. Suzuki, M. P. Kapoor, F. Ohashi and T. Osaki, Appl. Catal., A, 2001, 213, 47–63 CrossRef CAS.
- D. Tichit, N. Das, B. Coq and R. Durand, Chem. Mater., 2002, 14, 1530–1538 CrossRef CAS.
- X. M. Guo, D. S. Mao, G. Z. Lu, S. Wang and G. S. Wu, J. Catal., 2010, 271, 178–185 CrossRef CAS.
- Z. Y. Ma, C. Yang, W. Wei, W. H. Li and Y. H. Sun, J. Mol. Catal. A: Chem., 2005, 227, 119–124 CrossRef CAS.
- Y. X. Liu, K. P. Sun, H. W. Ma, X. L. Xu and X. L. Wang, Catal. Commun., 2010, 11, 880–883 CrossRef CAS.
- L. C. Wang, Q. Liu, M. Chen, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Phys. Chem. C, 2007, 111, 16549–16557 CAS.
- R. X. Zhou, T. M. Yu, X. Y. Jiang, F. Chen and X. M. Zheng, Appl. Surf. Sci., 1999, 148, 263–270 CrossRef CAS.
- M. G. Sanchez and J. L. Gazquez, J. Catal., 1987, 104, 120–135 CrossRef CAS.
- K. Imafuku, J. Oda, K. Itoh and H. Matsumura, Bull. Chem. Soc. Jpn., 1974, 47, 1201–1202 CrossRef CAS.
- T. Chraska, A. H. King and C. C. Berndt, Mater. Sci. Eng., A, 2000, 286, 169–178 CrossRef.
- H. Purnama, F. Girgsdies, T. Ressler, J. H. Schattka, R. A. Caruso, R. Schomäcker and R. Schlögl, Catal. Lett., 2004, 94, 61–68 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, DG-DTG, N2 adsorption–desorption isotherms and elemental mapping images of catalysts. See DOI: 10.1039/c5ra25340c |
| This journal is © The Royal Society of Chemistry 2016 |