
Biological activity of dendrimer–methylglyoxal complexes for improved therapeutic efficacy against malignant cells†
Srabanti Ghosh*a,
Prabal Chakrabortyb,
Adrita Chakrabartic,
Manosij Ghoshd,
Amit Mandale,
Partha Sahab,
Anita Mukherjeed,
Somobrata Acharyaa and
Manju Raycf
aCentre for Advanced Materials, Indian Association for the Cultivation of Science, Kolkata-700032, India. E-mail: ghosh.srabanti@gmail.com
bCrystallography and Molecular Biology Division, Saha Institute of Nuclear Physics, Kolkata-700064, India
cBiological Chemistry, Indian Association for the Cultivation of Science, Kolkata-700032, India
dCentre of Advanced Study, Department of Botany, University of Calcutta, Kolkata-700019, India
ePolymer Science Unit, Indian Association for the Cultivation of Science, Kolkata-700032, India
fDivision of Molecular Medicine, Bose Institute, Kolkata-700 054, India
First published on 5th January 2016
Abstract
The clinical application of methylglyoxal (MG, a normal human metabolite) for cancer therapy is limited by its facile enzymatic degradation. The present investigation was aimed at exploring the potential anticancer therapy of methylglyoxal loaded polyamidoamine (PAMAM) dendrimers with different terminal groups (PAMAM/MG). Uniform PAMAM-NH2/MG with an average particle size of 55 ± 5 nm and high encapsulation efficiencies (EE) of 82 ± 2% have been characterized by transmission electron microscopy (TEM) and spectroscopic techniques. Compared to the free MG, a slow release of MG from the dendrimer complex was ∼85 ± 2% after 24 hours, suggesting the potential of the dendrimers as a sustained drug delivery system. PAMAM-NH2/MG possesses biocompatibility with no hemolytic activity and is highly effective in growth inhibition of mice carcinoma and sarcoma cells. PAMAM-NH2/MG selectively reduced cell viability of Hela cells with an IC50 value of 0.4 ± 0.15 μg mL−1 while more than 90% of normal fibroblast cells have been found to be viable at a similar dose. Interestingly, even a lower dose of MG (∼250 times) in PAMAM-NH2/MG can effectively target Hela cells in comparison to free MG. TEM images demonstrated the ultra structural changes of Hela cells after being treated with PAMAM-NH2/MG and also confirmed cellular uptake. DNA damage as measured by comet assay was found to be dose dependent for mice carcinoma and sarcoma cells but no such genotoxic response was observed in human lymphocytes, after treatment with PAMAM-NH2/MG. Thus, dendrimer encapsulated MG might be an effective strategy to target the cancer cells and further improvements of the surface functionality of dendrimers can be used as a valuable tool for the development of novel therapeutics in nano-oncology.
Introduction
Nanomedicines have revolutionized cancer diagnosis and therapy via significantly improving the therapeutic efficacy as well as reducing the side effects of clinically established drugs.1,2 A range of therapeutic nanocarriers such as nanoparticles, liposomes, carbon nanotube, graphene oxide nanosheets and macromolecules etc. have contributed as novel nanoscale approaches through the encapsulation of drug molecules within the nanocarriers to facilitate targeted delivery.3–8 The important technological advantages of nanocarriers are the high stability, elevated carrier capacity, ease of incorporation of both hydrophilic and hydrophobic materials and feasibility of variable routes of oral administration and inhalation.9–11 To date, there are only a few clinically approved nanocarriers which can selectively bind cancer cells for the targeted delivery of antitumor therapeutics. In this regard, polymer conjugates with superior permeability and retention effect are ideal for drug delivery and can enhance the absorption of the drugs into tumor cells and the use of biocompatible as well as biodegradable polymers offers a promising way for enhancing the efficacy of existing drugs.12,13 With the significant advancement in surface engineering, the polyamidoamine (PAMAM) dendrimer based multivalent nanoplatform has become progressively redefined in terms of the modulation of surface chemistry, control of drug release kinetics and integration of multiple functionalities on single nanoplatforms.14–18 Polyamidoamine dendrimers are highly branched macromolecules possessing both the solvent filled interior core (nanoscale container) as well as a homogeneous, well-defined exterior surface functionality (nano-scaffold) which offer a powerful multifunctional platform for drug delivery, diagnostic imaging and clinical immunoassays etc.19–23 In contrast to traditional polymers, PAMAM possess exceptional structural properties such as monodispersity, high density peripheral functional groups, globular structure and multivalency which make them an efficient nanodevice for controlled and targeted delivery of therapeutic compounds. Moreover, ability to control their size (e.g., 1 nm to 135 nm) through the variation of generations and to implement surface and intramolecular functionalities designed to carry or trap desired drug molecules through covalent, hydrophobic, ionic, or hydrogen-bonding interactions are useful for drug delivery. With large numbers of surface groups (for example, a G-5 polyamidoamine (PAMAM) dendrimer with 128 surface amines), dendrimers have the capacity to take place in multivalent interactions. This unique structural feature offers an extraordinary combination of guest–host and interfacial surfaces which are amenable to a wide range of chemical modifications. Dendrimer conjugate plays a crucial part in determining optimum drug delivery to disease sites by protecting active drug efficacy while influencing sustained release with target specificity.24–26 We have chosen methylglyoxal (MG, a normal metabolite, Scheme 1) as a biocompatible small molecules which specifically kills cancer cells.27,28 This therapeutic molecule functions primarily by inhibiting the glycolysis and mitochondrial respiration exclusively of a wide variety of malignant cells without much adverse effect upon normal cells.29 MG, a highly reactive dicarbonyl compound toward the amino groups of proteins and nucleic acids and excessive accumulation of MG may induce modifications of mitochondrial proteins leading to dysfunction of the mitochondria and inhibits the respiration of both Ehrlich ascites carcinoma (EAC) cells and leukemic leukocytes without affecting the respiration of normal leukocytes. Moreover, previous detailed pharmacokinetic and toxicological study of MG from our group confirmed the safety and toxicity issues of MG for successful development of anticancer drug.27–29 Despite of superior anticancer property of MG, a major barrier to successful cancer treatment is its gradual enzymatic degradation.30–32 This has prompted interest in developing drug delivery system via reactive encapsulation in order to protect and enhance the efficacy of MG which might circumvent the problem of facile degradation. Specifically, conjugation of MG with dendrimer for cancer treatment has not reported so far. However, recently with an objective to enhance the efficacy of MG, polymer conjugated nanoformulations of MG have been developed with superior antimicrobial activity against multidrug resistant human pathogens.33,34 Here, we report a straightforward approach for the synthesis of PAMAM dendrimer based MG complexes with superior inhibition ability towards malignant cell growth through slow release of MG. Various spectroscopic measurements including UV-vis and FTIR were used to confirm the structure of MG loaded dendrimer whereas DLS and TEM measurements were performed to determine the morphology and the size of PAMAM/MG complexes. The cytotoxicity and the inhibitory effect of MG loaded dendrimer towards the malignant cells such as Ehrlich ascites carcinoma (EAC), sarcoma-180 and Hela were evaluated in vitro. These PAMAM/MG complexes are highly effective in malignant cell growth inhibition both for carcinoma and sarcoma in comparison to free MG. Thus, encapsulation of MG within the dendrimer matrix might be an effective strategy to selectively target the cancer cells.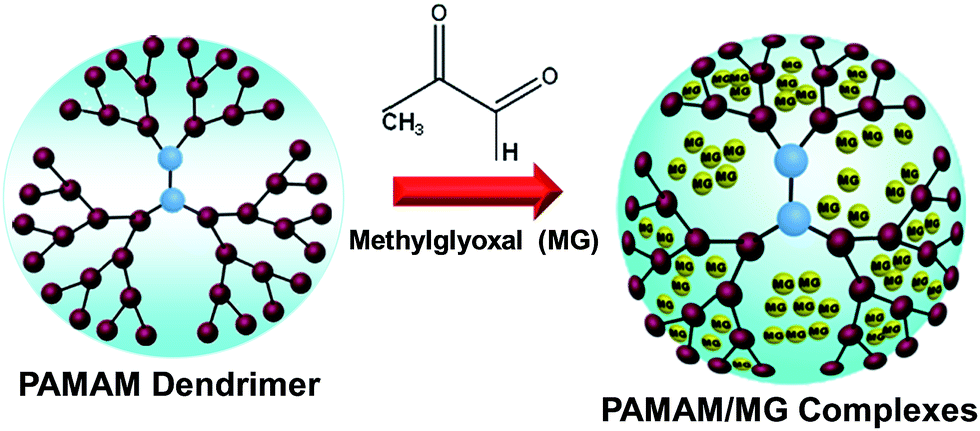 | ||
| Scheme 1 Formation of PAMMA/MG complexes. The MG molecules are physically encapsulated in the interior of PAMAM dendrimer matrix. | ||
Experimental
Materials
The methylglyoxal 40% solution, polyamidoamine dendrimer generation 5.0 was purchased from Sigma Aldrich, USA. A series dendrimers with different terminal groups namely amino (–NH2), hydroxyl (–OH) and carboxyl (–COOH) has been also purchased from Sigma Aldrich, USA. Sephadex G-50, trypan blue, cell culture media, 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT) and fetal bovine serum were purchased from Sigma Aldrich Co. (St Louis, MO, USA). Mouse myoblast (C2C12), Hela cells and human fibroblast cells (WI38) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Filter paper (0.2 μm) was procured from Merck Millipore (Billerica, MA, USA). All the other chemicals and biochemical were of analytical grade and purchased from Merck, Germany. Ultrapure Millipore water (18.2 MΩ) was used as solvent.Synthesis of PAMAM/MG complexes
Methylglyoxal was loaded in the PAMAM dendrimer matrix using an equilibrium dialysis method. The PAMAM dendrimer solution was dried under vacuum to remove methanol and dissolved in 1 mL deionized water. Then 50 μL MG solution (40%) was mixed with PAMAM-dendrimer (10−6 M) and stirred overnight slowly in dark. Then the solution was transferred to a dialysis bag (MWCO 8000) and dialyzed twice against deionized water under strict sink conditions for 10 min to remove free MG. For in vitro release studies, 5 mL of PAMAM/MG complex was taken in a dialysis bag separately and dialyzed under sink conditions. Then, 1 mL of aliquots was collected after every 1 h interval up to 48 h and drug release was estimated indirectly using spectrophotometrically and double-distilled water as blank. The encapsulation concentration of MG was determined by spectroscopic analysis using an ultraviolet-visible spectrophotometer (Cary Varian 50 scan UV-Vis optical spectrometer equipped with Cary Win UV software). The sample solution was treated with 5 M perchloric acid solution so that MG was released from the PAMAM. It was then derivatized with 1,2-diaminobenzene to produce 2-methylquinoxaline and estimated spectrophotometrically by measuring the absorbance at 336 nm. The concentrations of perchloric acid and 1,2-diaminobenzene in the reaction mixture were 0.5 M and 1.8 mM, respectively. Similarly, five known concentrations of MG and their corresponding absorbencies were recorded at 336 nm. A linear standard curve was obtained plotting known MG concentration versus absorbance. This was used to calculate the amount of MG form complex to dendrimer. Initially the optimization of MG loading by varying the different ratios of dendrimer to methylglyoxal (MG), for example, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, 1:100, and 1:200 and checked the loading concentration of MG by spectroscopic analysis. For, MG:polymer ratio of 1:100, the expected loading of MG is well matched with the calculated MG. For MG:polymer ratio of 1:200 release MG relatively faster than 1:100, which may be due to free available MG in the near vicinity of dendrimer molecule for high loading. In this regard, MG:polymer ratio of 1:100 has been considered as a suitable for the successful lading of MG in the dendrimer matrix.
:50, 1:100, and 1:200 and checked the loading concentration of MG by spectroscopic analysis. For, MG:polymer ratio of 1:100, the expected loading of MG is well matched with the calculated MG. For MG:polymer ratio of 1:200 release MG relatively faster than 1:100, which may be due to free available MG in the near vicinity of dendrimer molecule for high loading. In this regard, MG:polymer ratio of 1:100 has been considered as a suitable for the successful lading of MG in the dendrimer matrix.
Characterization
Transmission electron microscopy (TEM) was carried out on JEOL JEM-2010 with acceleration voltage of 200 kV. A drop of as-prepared PAMAM/MG solution was placed on a carbon-coated copper grid and dried before putting it on to the TEM sample chamber. The average size and morphology of PAMAM/MG was determined from TEM image. The FTIR spectra were recorded with Perkin Elmer, Spectrum GX equipment with a resolution of 2 cm−1 and scan range of 1000–4000 cm−1. Size distribution and zeta potential of PAMAM/MG complexes were determined by dynamic light scattering spectrophotometer (Model DLS-nanoZS, Zetasizer, Nanoseries, Malvern Instruments). The zeta potential was calculated from the electrophoretic mobility using the Smoluchowski equation with the help of commercial software. The results are expressed as mean values of three samples.Hemolytic activity
Toxicity to human red blood cells (RBCs) was assessed by a hemoglobin release assay.35 The absorbance at 450 nm was recorded using UV-Vis spectrophotometer. The detailed methodology has been described in ESI.† The percent of hemolysis was calculated as follows: hemolysis% = [(sample absorbance − negative control)/(positive control − negative control)] × 100%.Development of sarcoma in mice
Animal experiments were carried out in accordance with the guidelines of institutional ethics committee (IEC). Appropriate precautions were taken to minimize pain or discomfort to animals.Sarcoma tissue was developed in left hind leg of mice by intramuscular injection of 3-methyl-cholanthrene.
Ehrlich ascites carcinoma (EAC) cells
EAC cells were maintained in the intraperitoneal cavity of mice.36 Briefly, cells were harvested for 8–10 days post-inoculation in female Swiss albino mice before being collected from the peritoneal cavity in 3 mL of normal saline (0.9% NaCl) and centrifuged (2000 × g) for 5 minutes at 4 °C. Similarly sarcoma cells also collected and centrifuged (2000 × g) for 5 minutes at 4 °C. The resulting pellets were washed again in normal saline before they were re-suspended in the requisite amount of PBS at pH 7.4. Cells (4 × 103) were treated with various concentrations of MG, PAMAM dendrimer, and PAMAM/MG in a final reaction volume of 1 mL and incubated at 37 °C for 1 hour. A control tube with only EAC and sarcoma cells were maintained in a similar condition.Trypan blue exclusion assay
To measure the viable cell percentage, trypan blue dye was employed to stain the cells which do not have intact membrane.37 The viable cells exclude the dyes and are not stained, which is a standard method to detect cell death. EAC and sarcoma-180 cells lines are non adherent i.e. unable to make layer, so limited viability time period (within one hour) in suspension. First, EAC and sarcoma-180 cells were washed with PBS buffer pH 7.4 and then cells were incubated for 1 h at 37 °C in presence of varying concentration of PAMAM/MG complexes. Finally, 10 μL of the trypan blue dye was added to each cellular suspension. The viable cell was counted through a hemacytometer, and the viability values were compared with the sample of the negative control.Cell culture
Mouse myoblast, C2C12, Hela cells and human fibroblast cells (WI38) were maintained in DMEM growth medium (Dulbecco's modified Eagle's medium, Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS), 100 mg L−1 streptomycin and 100 IU mL−1 penicillin. The primary culture of the fetal mouse fibroblast was manipulated according to the regular processes. Cells were grown in 25 mL cell culture flask and incubated at 37 °C in a humidified atmosphere of 5% CO2 to approximately 70–80% confluence. Cells of 4–7 generations were used in this experiment.Cytotoxicity assays (MTT assay)
Human fibroblast cell line and Hela cells were used to determine the cytotoxicity of the as prepared PAMAM/MG complex. The number of surviving cells was expressed as percent viability = (the absorbance of the sample (treated cells) − background/the absorbance of the control (untreated cells) − background) × 100. Cellular toxicity of extracted contents from scaffolds was rated as follows: severe (<30%), moderate (30–60%), slight (60–90%), or non-cytotoxic (>90%) of MTT activity, compared to the control cells cultured in extract-free medium.Superoxide and nitrite production in macrophages
Macrophages isolated from the peritoneal cavity of mice with 10 mL RPMI 1640, the cells were washed twice and re-suspended in RPMI 1640 containing 10% heat-inactivated fetal bovine serum (FBS), 100 IU mL−1 penicillin and 100 μg mL−1 streptomycin. Briefly, after treated with PAMAM/MG, 105 cells were incubated with Nitro Blue Tetrazolium (NBT, 1 mg mL−1) for one hour at 37 °C in a humidified atmosphere of 5% CO2. After formation of a blue precipitate, cells were dissolved in an equal volume of 2 M KOH and dimethyl sulphoxide (DMSO). A blank was prepared by mixing of equal volume of 2 M KOH and DMSO. Readings were taken at 630 nm against blank. The concentrations of reduced NBT were determined by comparison to a standard curve constructed by using different concentrations of NBT directly. The nitric oxide production was evaluated by quantifying the nitrite accumulation using the Griess method as described in the literature.38 Briefly, each supernatant was mixed with the same volume of Greiss reagent (1% sulfanilamide in 5% phosphoric acid and 0.1% naphthylethylenediamine dihydrochloride in water) and absorbance of the mixture was determined at 550 nm. Nitrite concentrations in the medium were finally determined by using standard solutions of sodium nitrite (0–100 μM).DNA damage using comet assay in EAC and sarcoma-180 cells
EAC and sarcoma-180 cells were incubated for 3 h at 37 °C in RPMI-1640 media with different concentrations of PAMAM-NH2/MG. Following treatment, DNA damage was evaluated using alkaline comet assay according to the method of Singh et al.39 Images were scored using an image analysis system (Komet version 5.5, Kinetic Imaging Ltd; Andor Technology, Nottingham, UK) attached to a fluorescence microscope (Leica, Wetzlar, Germany) equipped with appropriate filters (N2.1). DNA percentage in comet tail was used to measure DNA damage.Genotoxicity of PAMAM/MG in human lymphocytes
Lymphocytes were isolated from fresh HPB according to the method of Boyum using Histopaque.40 Dose selection for genotoxicity study was based on initial screening using trypan blue dye exclusion method. The cut-off point as suggested by Henderson et al., was 70%. Following the initial screening, final treatment concentrations (0, 0.2, 0.4 and 0.8 μg mL−1) were selected for further experiments.41,42 The lymphocytes were processed for the detection of DNA damage as assessed by the alkaline comet assay after treated with PAMAM/MG.Microscopy imaging
The morphological changes of C2C12 myoblast mice cells, human fibroblast cells (WI38) and Hela cells treated with PAMAM/MG were visualized with a BX51WI fluorescence microscope (Olympus) equipped with 460–490 nm excitation filter setting and DP71 digital camera and DP-BSW software for image acquisition.Cell morphology observation
The morphological changes of Hela cells were investigated by Transmission Electron Microscopy (TEM) after treated with 0.8–8 μg mL−1 of PAMAM/MG. Aliquots of 20 μL of treated and untreated cell suspensions were deposited on glass cover slips. After being air dried for 1 h, the cover slips were fixed with a primary fixative solution containing 2.5% glutaraldehyde and 1% osmium tetroxide. Subsequently, the cells were dehydrated with sequential treatment with 30, 50, 70, 80, 90, and 100% ethanol for 15 min. Ultrathin sections (70 nm) were cut using Reichert Jung Ultracut. The sections were stained with 1% lead citrate and 0.5% uranyl acetate. Digital images of the treated and untreated cells were acquired using a JEOL field emission scanning electron microscope (JSM-6700F) operating at an accelerating voltage of 100 kV.Statistical analysis
Statistical analysis was performed using Origin 6 software. Each experiment was performed 3 to 5 times and results are expressed as mean ± SD. Student's t-test was performed for significance analysis and p < 0.05 was considered significant. Cellular viability data show representative data of at least three independent experiments.Results and discussion
Highly dispersed dendrimer/MG complex (PAMAM-NH2/MG) has been synthesized via physical loading within the amine terminated PAMAM dendrimer polymer matrix using an equilibrium dialysis method (Scheme 1). After dialysis, the content of MG has been determined through spectroscopic technique. The percent entrapment of MG in PAMAM/MG using amine terminated dendrimer has been found to be ∼82% ± 0.09 whereas that for carboxyl terminated dendrimer being 25% ± 0.2. The loading efficiency depends on surface functional groups of the dendrimer molecules and concentration of MG. The average sizes of the PAMAM-NH2/MG have been estimated to be ∼50 ± 5 nm from transmission electron microscopy (TEM) images using NH2 terminated dendrimer (Fig. 1a and b). The PAMAM-NH2/MG exhibit a spherical morphology without aggregation or adhesion by using TEM image (Fig. 1b). | ||
| Fig. 1 Structural characterization of PAMAM/MG using NH2 terminated dendrimer. (a) Transmission electron microscope (TEM) image of PAMAM-NH2/MG. (b) TEM image of PAMAM-NH2/MG at different magnification. (c) Size distribution histogram of PAMAM-NH2/MG. (d) Absorption spectra of pure amine terminated PAMAM dendrimer, methylglyoxal and PAMAM/MG. (e) FTIR spectra of PAMAM dendrimer and PAMAM-NH2/MG. (f) Zeta potential distribution of PAMAM-NH2/MG. | ||
At a fixed molar ratio, 1:100 [MG]:[polymer], PAMAM-NH2/MG was obtained with an average hydrodynamic diameter of 55 ± 5 nm with a relatively narrow size distribution, as determined by dynamic light scattering (DLS) measurements (Fig. 1c). Particle size and distribution are two important aspects in determining whether the as-prepared complexes are suitable for drug delivery. It has been proposed that the average sizes of nanoformulated drugs should be less than 500 nm that could cross the membranes of epithelial cells via endocytosis.43 Hence, PAMAM-NH2/MG complexes having size in the range 55 nm can be ideal candidates for drug delivery where size is concerned. A significant difference can be noticed in the average diameter of MG loaded complexes (Fig. S1a†) using carboxyl (PAMAM-COOH/MG) and hydroxyl (PAMAM-OH/MG) in the range of 60–110 nm with a mean diameter of 78 nm and 85 nm respectively (Table S1†). This suggests the loading of drug can influence the interaction between MG and surface functional group of the PAMAM dendrimer. Strong electrostatic interaction between MG and NH2 groups controls the growth and stabilization of the MG within the dendrimer matrix. It can be concluded that NH2-terminated dendrimer provides better stabilization than COOH terminated dendrimer.
Fig. 1d presents UV-Vis spectra of aqueous solutions of pure MG, PAMAM dendrimer and PAMAM-NH2/MG synthesized. The PAMAM dendrimer solution exhibits a distinct absorption band with λmax ranging between 280 and 285 nm which may arises from the intact dendrimer structure and specifically from its interior tertiary amine groups.44 On the other hand, aqueous MG solutions demonstrated an absorbance peak at 282 nm.45 On comparison of the three spectra, the most significant point is that the band 282 nm in dendrimer gets shifted to ∼307 nm in complexes and also becomes broadened. Solutions containing MG and PAMAM dendrimer became visibly light brown colored after mixing and became progressively darker in color with time. This suggests complex formation between PAMAM and MG. Furthermore, the formation of PAMAM-NH2/MG is corroborated by the observed results of FTIR measurement. Fig. 1e illustrates the FTIR spectra of pure PAMAM dendrimer and PAMAM-NH2/MG. On comparison of the two spectra the most significant point found is that the band at 3288 cm−1 in dendrimer gets shifted to 3437 cm−1 in complexes and also becomes wider. This particular band can be assigned for stretching mode of surface amine group (NH2) of the dendrimer and the observed shift is due to the coordination of NH2 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of MG.46,47 It can also be seen that the –NHCO– bands at 1637 cm−1 corresponding to CO vibration of dendrimer remained same but band at 1556 cm−1 due to N–H bending vibration completely disappeared during conjugation. Moreover, a new peak at 1725 cm−1 has appeared in the PAMAM-NH2/MG due to another carbonyl group from MG molecule (two carbonyl groups).48 This suggests that MG molecules are attached to the dendrimer through the surface amino (NH2) group as well as the –NHCO– moieties lying in the interior of dendrimer structure. Zeta potential or surface charge density is a critical parameter for effective interactions of drug molecules with cell membrane as well as the stability of PAMAM-NH2/MG complexes in aqueous solution.49 Due to the strong influence of pH on PAMAM dendrimer solution, the pH range of 5.5–6.5 has been used for the formulation of PAMAM/MG which was favored by the strong interaction of the oppositely charged solution.50 The surface charge of the PAMAM/MG complexes was measured by zeta potential, which depends on the terminal groups of the dendrimer as shown in Table S1.† Amine terminated and hydroxyl terminated dendrimer/MG complexes were positively charged, whereas carboxyl terminated dendrimer/MG complexes were negatively charged indicating that functional groups of dendrimer molecule is not significantly influenced after the formation of the complex with MG (Fig. S2†). Hence, the surface charge of the PAMAM/MG complexes depends on the availability of surface functional groups of the polymers. This further indicates that the dendrimer molecule imparts the surface charge tunability to the as-prepared PAMAM/MG complex.
O of MG.46,47 It can also be seen that the –NHCO– bands at 1637 cm−1 corresponding to CO vibration of dendrimer remained same but band at 1556 cm−1 due to N–H bending vibration completely disappeared during conjugation. Moreover, a new peak at 1725 cm−1 has appeared in the PAMAM-NH2/MG due to another carbonyl group from MG molecule (two carbonyl groups).48 This suggests that MG molecules are attached to the dendrimer through the surface amino (NH2) group as well as the –NHCO– moieties lying in the interior of dendrimer structure. Zeta potential or surface charge density is a critical parameter for effective interactions of drug molecules with cell membrane as well as the stability of PAMAM-NH2/MG complexes in aqueous solution.49 Due to the strong influence of pH on PAMAM dendrimer solution, the pH range of 5.5–6.5 has been used for the formulation of PAMAM/MG which was favored by the strong interaction of the oppositely charged solution.50 The surface charge of the PAMAM/MG complexes was measured by zeta potential, which depends on the terminal groups of the dendrimer as shown in Table S1.† Amine terminated and hydroxyl terminated dendrimer/MG complexes were positively charged, whereas carboxyl terminated dendrimer/MG complexes were negatively charged indicating that functional groups of dendrimer molecule is not significantly influenced after the formation of the complex with MG (Fig. S2†). Hence, the surface charge of the PAMAM/MG complexes depends on the availability of surface functional groups of the polymers. This further indicates that the dendrimer molecule imparts the surface charge tunability to the as-prepared PAMAM/MG complex.
In general, primary amines of dendrimer (RNH2) undergo nucleophilic addition with aldehydes or ketones of MG to give carbinolamines as intermediate which then dehydrate to give imines. When an aldehyde or ketone is treated with a primary amine, nitrogen substitutes for the oxygen of the carbonyl group to form a CN double bond (an imine). Additionally, secondary amines, R2NH, react with aldehydes or ketones to also give carbinolamines as intermediate which can only eliminate to give a CC since there is no N–H band then dehydrate to give enamines. MG loading to other dendrimers terminated with carboxylic acid or alcohol might be lower due to lack of primary amines. Other possibility, in presence of large number of OH functional groups at the surface of dendrimer molecule possibly forms acetal reacting with MG. In general, an aldehyde can react with an alcohol to form a hemiacetal and the equilibrium is shifted towards the acetal by using an excess of the alcohol or hydroxyl groups. It is also expected that the high surface charge would help to stabilize the complexes in aqueous solution through electrostatic repelling forces and also prevent aggregation. Hence, the preparation of MG loaded PAMAM dendrimer is based on the strong interaction between oppositely charged solution of amino group of dendrimer and carbonyl group of MG solution. The positively charged PAMAM-NH2/MG can be useful for the preparation of drug delivery systems in the cancer therapy. This is obvious that the release of the MG is significantly affected by the presence less interaction between COOH groups of dendrimer molecules and carbonyl groups of the MG. However, the release of MG from amine terminated dendrimer experienced an initial burst release which followed by a constant and continuous releases (Fig. 2).
 | ||
| Fig. 2 In vitro release studies of free MG, PAMAM-COOH/MG and PAMAM-NH2/MG (n = 3). Values represent mean ± SD. | ||
Fig. 2 illustrated approximately ∼75% ± 3% of the MG released in the first 12 h and after 24 h, the totally released MG reached ∼85% suggests the potential of using amine terminated PAMAM dendrimer for sustained drug release. The in vitro release profile of MG from the polymer complexes was investigated by using dialysis method. The release behavior of MG has been compared for both PAMAM/MG using amine and carboxyl terminated PAMAM dendrimer along with free MG. As shown in Fig. 2, nearly 90% ± 2% of pure MG molecules released out of the dialysis bag within 2 h. Similarly, for carboxyl terminated PAMAM dendrimer, ∼78 ± 2% of the MG released in the first 2 h, which was then followed by a constant release for the subsequent 48 h. The release of MG from PAMAM-OH/MG followed similar trend like carboxyl terminated PAMAM dendrimer (data not shown).
PAMAM/MG demonstrates high biocompatibility towards human red blood cells over a broad range of concentrations. Interestingly, with increasing the concentration of PAMAM/MG causes a hemolytic activity of 2.5% and 4.8% hemolysis using OH and NH2 terminated dendrimer and slight increase in the hemolytic activity of 5.2% using COOH terminated dendrimer under the similar conditions (Fig. 3a).
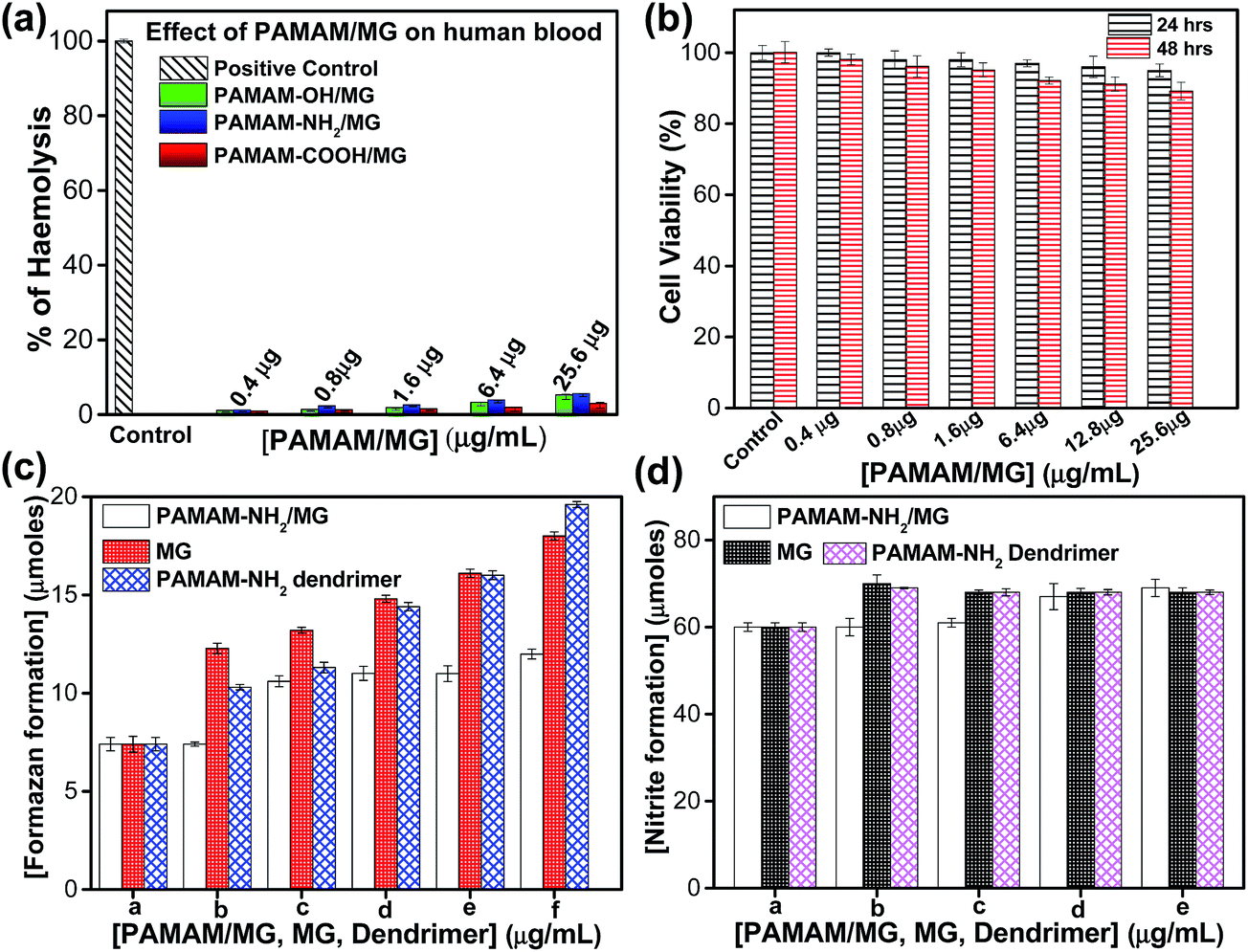 | ||
| Fig. 3 Biocompatibility test of PAMAM/MG complexes. (a) Hemolysis assay for PAMAM/MG complexes using Triton-X as a positive control using human blood cells. (b) Cell viability of normal mice myoblast cells (C2C12) after 24 (black line bar) and 48 (red line bar) hours treatment with different concentrations of PAMAM-NH2/MG as calculated from the MTT assay. (c) Superoxide and (d) nitrite generation in peritoneal macrophages of normal mice after treated with MG, PAMAM-NH2 dendrimer and PAMAM-NH2/MG. The values are represented as mean SD of three individual experiments. | ||
According to several studies, in vitro hemolysis varies from 5 to 25% which is suitable for in vivo applications.51 This indicates the as-prepared PAMAM-NH2/MG is suitable for drug delivery. As methylglyoxal is a normal human metabolite, did not show cytotoxicity towards human red blood cells over a broad range of concentrations. Similarly, none of the dendrimer with different surface functional groups employed in the present study released haemoglobin and hence, these were not cytotoxic to human erythrocytes which is consistent with earlier report.45 Since PAMAM-NH2/MG has higher encapsulation efficiency of MG with sustained release behavior, further study has been followed up on efficiency and cytotoxicity of PAMAM-NH2/MG against normal and malignant cells. Cytotoxic effects of PAMAM-NH2/MG against normal mice myoblast cells (C2C12) was examined by MTT cell proliferation assay (MTT, 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide). Importantly, PAMAM-NH2/MG was practically non-toxic for C2C12 cells upto 25.6 μg mL−1, since cell viability was higher than 90% (Fig. 3b) after 24 and 48 hours incubation periods. This suggests that the PAMAM-NH2/MG complexes are biocompatible to the normal mice C2C12 cells.
Study of the interaction between macrophages and nanoformulated drug is essential to design effective therapeutic strategies as drug molecules are primarily removed from circulation by the macrophages of the mononuclear phagocyte system during in vivo application.52 Superoxide and nitric oxide production during the interaction between MG, PAMAM dendrimer, PAMAM-NH2/MG with murine macrophages were evaluated using the nitroblue tetrazolium (NBT) reduction assay and the Griess method respectively. Macrophages produce oxygen and nitrogen-reactive metabolites as part of the host defense function or to interaction with nanoparticles.53,54 Nitric oxide (NO) is a key marker of activation of inflammation and ROS generations as well as oxidative stress measurement have been developed to mechanistically evaluate cytotoxic responses to nanomaterials. Fig. 3c illustrated that PAMAM-NH2/MG did not induce any ROS in macrophages. The effect of PAMAM-NH2/MG on the nitric oxide production was analyzed in cultured macrophages (Fig. 3d). Cells were incubated for 48 hours with PAMAM-NH2/MG alone and the production of nitric oxide was not detected. The results show that the PAMAM-NH2/MG are not cytotoxic and do not stimulate the production of nitric oxide by macrophages in the range of concentrations studied. However, PAMAM dendrimer produce superoxides under similar reaction media. It has been well documented that PAMAM dendrimer generate hydrogen peroxide with increasing dosages.55 On the other hand, MG can also activate macrophages via superoxide and nitrite production through the MAPK/NF-κB signaling pathway.34 But, in the present experimental concentration, MG did not induce significant oxygen and nitrogen-reactive species. Interestingly, after formation of complex, dendrimer are modified with MG and did not show toxicity to macrophages. Hence, the presence of the PAMAM-NH2/MG appears not to promote any inflammatory action or elicit a reactive response when in contact with macrophages.
The cytotoxicity of PAMAM-NH2/MG has been tested against EAC and sarcoma-180 cell lines by trypan blue exclusion assay. A concentration-dependent reduction of both EAC and sarcoma-180 cell viability with IC50 values of 0.25–0.3 μg mL−1 for PAMAM-NH2/MG was observed (Fig. 4a). Interestingly, PAMAM-NH2/MG which was found to be lethal for both the EAC and sarcoma cells but did not show significant cytotoxic effect in normal cells upto 25.6 μg mL−1 (Fig. 3b). Furthermore, growth inhibition kinetics has been studied by following both sarcoma and EAC cell death after treated with PAMAM-NH2/MG at different time intervals (Fig. 4b and c). In order to prove that inhibition arises from stable and effective PAMAM-NH2/MG, the effect of various terminal groups containing PAMAM/MG complexes and control experiments with individual precursors (MG and PAMAM dendrimer) have been carried out under the similar experimental conditions. However, both PAMAM/COOH and PAMAM/OH did not show significant toxicity against malignant cells as well as normal cells and more than 80% cells were found to be viable after treated under similar condition as shown in Fig. 4d. None of these precursors show any inhibition for EAC cells in the similar concentration range (Fig. S3†). Earlier report suggested that biocidal activity of MG was demonstrated at concentration ≥ 1000 μM mL−1 much higher concentration in comparison to PAMAM-NH2/MG.34 Thus, the biological activity of MG could be augmented by encapsulating within the polymer in a more controlled fashion.
 | ||
| Fig. 4 (a) Cellular viability of EAC and sarcoma-180 cells line after treated with different concentration of PAMAM-NH2/MG complexes by trypan blue exclusion assay. Cells were treated with PAMAM-NH2/MG at concentrations of 0.04, 0.2, 0.4, 0.8, 1.6 and 2.4 μg mL−1 for 1 hour. PAMAM-NH2/MG complexes reduced cell viability of both EAC and sarcoma-180 cells with IC50 values of 0.25–0.3 μg mL−1. Data are the mean ± SD for three independent experiments. (b) and (c) are the kinetic of growth inhibition of sarcoma-180 and EAC cells respectively. (d) Representative cellular viability of EAC cells after treated with different concentration of PAMAM-COOH/MG and PAMAM-OH/MG complexes. | ||
Moreover, we examined the cellular morphology in a monolayer culture after treating with different concentrations of PAMAM-NH2/MG (Fig. 5). Microscopic observations showed no distinct morphological changes for normal myoblast mice cells treated with 2.4 μg mL−1 PAMAM-NH2/MG indicating normal myoblast cells remain healthy after treating with PAMAM-NH2/MG up to this dose after 12, 24 and even 48 hours (Fig. 5b–d) with respect to untreated cells (Fig. 5a).
 | ||
| Fig. 5 PAMAM-NH2/MG complex induced morphological change of mouse myoblast EAC and sarcoma-180 cells by phase contrast microscopy. Phase contrast image of normal mouse myoblast cell (a) without PAMAM-NH2/MG and incubated with 2.4 μg mL−1 of PAMAM-NH2/MG for (b) 12 hours (c) 24 hours and (d) 48 hours respectively. Phase contrast images of EAC cells (e) in absence of PAMAM-NH2/MG and treated with PAMAM-NH2/MG (2.4 μg mL−1) for (f) 15 minutes (g) 30 minutes and (h) 60 minutes respectively. Phase contrast images of sarcoma cells (i) in absence of PAMAM-NH2/MG and treated with PAMAM-NH2/MG (2.4 μg mL−1) for (j) 15 minutes (k) 30 minutes and (l) 60 minutes respectively. Images were acquired at 10× magnification. | ||
In contrast, a dramatic change in cellular shapes of both malignant EAC cells and sarcoma-180 cells are observed after 15 min, 30 min and 60 min of treatment with 2.4 μg mL−1 PAMAM-NH2/MG as shown in Fig. 5f–h and Fig. 5j–l, respectively. The phase contrast image shows, after treatment with PAMAM-NH2/MG, both EAC and sarcoma-180 cells showed swelling, aggregation and irregularities in the plasma membrane. The control experiments with the individual precursors, MG and PAMAM dendrimer did not induce morphological changes for both normal and malignant cells at this experimental concentration as shown in Fig. S4.† This suggests the effective as well as selective malignant growth inhibition by PAMAM-NH2/MG is associated with the slow release of MG from the dendrimer.
PAMAM-NH2/MG induced DNA damage of EAC and sarcoma-180 cells was studied using comet assay. Fig. 6a–f illustrates the DNA-damaging effect on the nuclei of EAC and sarcoma cells upon treatment with the PAMAM-NH2/MG (0, 0.4 and 0.8 μg mL−1) for 2 hours. After treatment, nuclei from EAC and sarcoma-180 cells were isolated immediately.
 | ||
| Fig. 6 DNA damage assay of sarcoma-180 cells, EAC cells and human lymphocytes after treated with PAMAM-NH2/MG complexes. (a)–(c) Morphological change of DNA of sarcoma cells after treated with PAMAM-NH2/MG complexes (0, 0.4, and 0.8 μg mL−1), (d–f) morphological change of DNA of EAC cells after treated with PAMAM-NH2/MG complexes (0, 0.4, and 0.8 μg mL−1), morphological change of DNA of human lymphocytes in absence (g) and after treated with 0.8 μg mL−1 of PAMAM-NH2/MG complexes (h). (i) Comet assay data (% tail DNA) of sarcoma-180 cells, EAC cells and human lymphocytes after treated with PAMAM-NH2/MG complexes (0, 0.08, 0.4, and 0.8 μg mL−1). *p < 0.05. | ||
Comet parameter (% tail DNA) clearly indicated a dose dependent increase in DNA migration both in case of EAC (Fig. 6b, c and i) and sarcoma (Fig. 6e, f and i) cells as compared to control cells (Fig. 6a and d), which indicates the extent of DNA damage. On the other hand, human lymphocytes treatment with PAMAM-NH2/MG did not reveal any dose dependent response and tail DNA percent remained same (Fig. 6g–i). A comet-like tail suggests presence of a damaged DNA strand that migrates when electrophoreses was done. The length of the tail increases with the extent of DNA damage. An increase in DNA damage with increase in PAMAM-NH2/MG concentration was observed in cancer cells, whereas the normal human lymphocytes showed no further increase in DNA damage.
We have also tested the cytotoxicity of PAMAM-NH2/MG against human normal fibroblast cells (WI38) (red bar) and Hela cells (green bar). As shown in Fig. 7a, approximately 88 ± 1.8% of viability for human normal fibroblast cells (WI38) was measured after incubation with 2.4 μg mL−1 of PAMAM-NH2/MG for 24 h. In contrast, a significant reduction in the Hela cells viability was observed when the cells were exposed to the same concentration of the PAMAM-NH2/MG for the same incubation period. The viability of the Hela cells was decreased by 50 ± 2% and 90 ± 3% at 0.4 μg mL−1 and 2.4 μg mL−1 concentrations of PAMAM-NH2/MG, respectively. These results demonstrated a concentration dependent response of PAMAM-NH2/MG. The free MG and PAMAM dendrimer were also tested as the control groups. The viability of the WI38 cells and Hela cells were 94.0 ± 2% and 91 ± 1% respectively after treatment with bare MG at 0.8 μg mL−1 concentrations (Fig. 7b). Hence, PAMAM-NH2/MG was found to have significantly higher cellular toxicity than the free MG for Hela cells and exhibited considerable anticancer effects. Only dendrimer treatment showed nearly no cytotoxicity; ∼90 ± 1% of the WI38 cells were viable and also not cytotoxic to Hela cell (∼85 ± 1%) at the experimental concentration. Therefore, due to the complex formation with the rigid dendrimer molecules, it is expected to improve the cellular uptake and controlled slow release of the MG and consequently, PAMAM-NH2/MG could be an ideal anticancer medicine for the targeted therapy. Moreover, phase contrast microscopic observations showed no distinct morphological changes in the WI38 cells treated with up to 2.4 μg mL−1 PAMAM-NH2/MG as shown in Fig. 7c–f and the trypan blue exclusion method also indicated more than 95% cell viability (data not shown) indicating healthy cells which is also consistent with MTT assay result. However, PAMAM-NH2/MG showed significant morphological changes of Hela cells as evident from microscopic observations where the cells have a tendency to become rounded and form aggregate (Fig. 7g–j). This suggests that immobilization of MG in the dendrimer matrix can exert anticancer activity but has no cytotoxic effect on normal mammalian cells. Further, we have investigated the effect of PAMAM-NH2/MG on DNA of cells through 4,6-diamidino-2-phenylindole (DAPI) staining. The fluorescent microscopic studies after DAPI staining of untreated Hela cells does not show any fragmentation (Fig. 7k) and the corresponding DIC image also shows well-shaped healthy cells (Fig. 7l). However, PAMAM-NH2/MG treated Hela cells clearly exhibit nuclear fragmentation as highlighted with white arrow (Fig. 7m) and the DIC image corroborates the cellular damage (Fig. 7n). This suggests the potential selective cytotoxicity of PAMAM-NH2/MG towards cancer cells via DNA degradation.
 | ||
| Fig. 7 Effect of PAMAM-NH2/MG, control MG and PAMAM dendrimer on normal human fibroblast cells (WI38) and Hela cells. (a) Cell viability of normal fibroblast cells (WI38) (red bar) and Hela cells (green bar) after 24 hours treatment with different concentrations of PAMAM-NH2/MG as calculated from the MTT assay. (b) Cell viability of normal fibroblast cells (WI38) (red bar) and Hela cells (green bar) after 24 hours treatment with different concentrations of control MG and PAMAM dendrimer. The values are represented as mean SD of three individual experiments. Bright field images illustrating the overall cellular morphology of normal human fibroblast cells (WI38) without PAMAM-NH2/MG (c), after 24 h incubation in presence of (d) 0.4, (e) 0.8, (f) 2.4 μg mL−1 of NMG. Bright field images illustrating the cellular morphology of Hela cells without PAMAM-NH2/MG (g), after 24 h incubation in presence of (h) 0.4, (i) 0.8, (j) 2.4 μg mL−1 of PAMAM-NH2/MG. The bar represents a scale of 50 μm. Data are the mean ± SD for three independent experiments. (k) Bright field microscopic images of untreated Hela cells. (l) Fluorescence microscopic images of DAPI stained untreated Hela cells and (m) bright field microscopic images of Hela cells treated with 2.4 μg mL−1 of PAMAM-NH2/MG, (n) fluorescence microscopic images of DAPI stained Hela cells treated with 2.4 μg mL−1 of PAMAM-NH2/MG. Inset: a higher magnification DAPI stained Hela cells treated with 2.4 μg mL−1 of PAMAM-NH2/MG shows the fragmented nuclei. White arrows indicate fragmented nuclei. The bar represents a scale of 10 μm. | ||
Representative TEM images (Fig. 8) demonstrate the structural changes of the Hela cells after treated with PAMAM-NH2/MG for different incubation periods. Fig. 8a shows the untreated Hela cells with well-defined membranes. In contrast, morphological alterations of cells after exposure with 2.4 μg mL−1 of PAMAM-NH2/MG are visible at 6 hours in TEM images (Fig. 8b). After 24 hours, cells have lost their cellular integrity and large amount of cellular debris around the cell, indicating irreversible cell damage or cell death (Fig. 8c) compared to the untreated cells (Fig. 8a). Furthermore, the cellular uptake of PAMAM-NH2/MG has been followed in TEM images of 70 nm ultrathin sections of Hela cells (Fig. 8d–f). The PAMAM-NH2/MG were located at the cytoplasm (Fig. 8d and e) and also existed as aggregates wrapped with an endocytic vesicle within the cytoplasm near the cell membrane as shown in inset of Fig. 8e. This suggests a typical endocytosis procedure has occurred and the typical structure of the endosome then disappears which is consistent with literature.56 Importantly, Fig. 8f shows the PAMAM-NH2/MG were located inside the mitochondrion. It is clear that from these observations, PAMAM-NH2/MG complexes can be internalized into cells and MG can be released into the cytoplasm, resulting in cancer cell death.
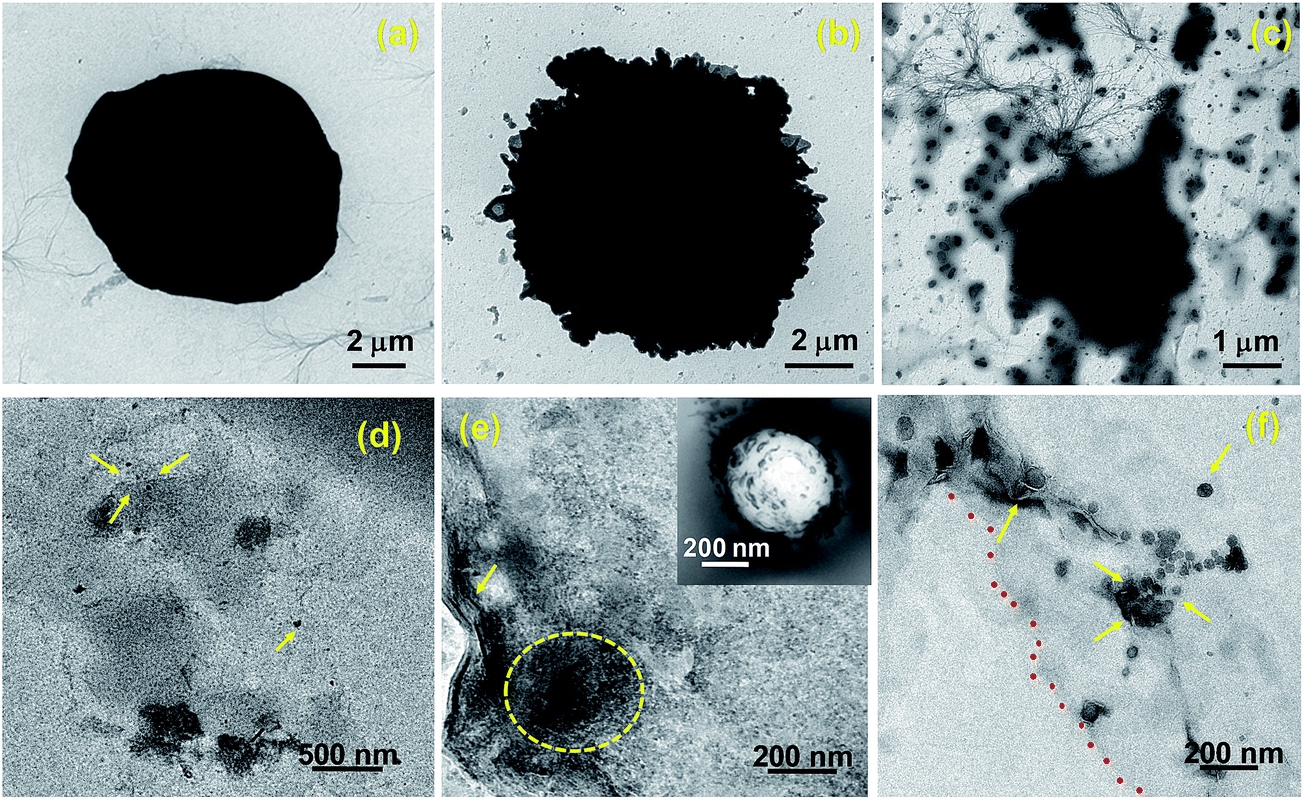 | ||
| Fig. 8 TEM images of Hela cells. (a) Untreated cells showed no abnormalities (b) whereas cells treated with PAMAM-NH2/MG for 6 hours showed membrane damage. (c) After 24 hours, cells have lost their cellular integrity with severe cell damage. (d), (e) Ultrathin sections of cells illustrate presence of complexes in cytoplasm (marked with yellow arrows and dotted yellow circle). Inset: shows large endosomes near the cell membrane with many PAMAM-NH2/MG inside. (f) Electron micrograph shows the presence of PAMAM-NH2/MG in mitochondria (marked with yellow arrows) and on the nuclear membrane (marked with red dots). | ||
Conclusion
Here, the bioactive end-groups of the dendrimer have been conjugated to biological active methylglyoxal yielding PAMAM/MG hybrid materials with high specificity and sensitivity, which could be of immense utility for drug delivery. The study concludes amine terminated PAMAM dendrimer to be superior carrier for MG based cancer therapy compared to carboxyl and hydroxyl based dendrimer. In order to shed light on the improvement of MG efficacy in PAMAM/MG, drug entrapment efficiency, in vitro release profile, toxicology, DNA damage assay and studies to understand the fundamental difference between normal and malignant cells studied have been conducted. The present work provides significant evidence for the selection of suitable “dendrimer terminal groups” and enhance the efficacy of MG to kill malignant cells. PAMAM/MG complexes are highly effective in growth inhibition of mice carcinoma and sarcoma-180 cells and remains biocompatible in normal mice myoblast cells. A similar trend was observed for human cervical cancer cells, where PAMAM-NH2/MG can selectively inhibit cancer cells without affecting normal human fibroblast cells. These observations suggest that the tumor cells are much more sensitive to PAMAM-NH2/MG than normal cells. Hence, bioactive agents with a high degree of target specificity and clinical therapeutic activity, represent an exciting direction for drug development. Further studies on immune response, in vivo anti-tumor activity as well as toxicity studies are under process to achieve further understanding of efficiency of PAMAM-NH2/MG.Acknowledgements
S. G. and M. R. gratefully acknowledge Council of Scientific and Industrial Research (CSIR), Government of India and Department of Science and Technology (DST) Nanomission, Government of India for financial support. The authors gratefully acknowledge Prof. D. D. Sarma, Indian Institute of Science, Bangalore for his valuable suggestions.Notes and references
- G. M. Whitesides, Nat. Biotechnol., 2003, 21, 1161–1165 CrossRef CAS PubMed.
- S. Ghosh, M. Ray, M. Das, A. Chakrabarti, A. H. Khan, D. D. Sarma and S. Acharya, Phys. Chem. Chem. Phys., 2014, 16, 5276–5283 RSC.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388–3432 CrossRef CAS PubMed.
- S. Thamphiwatana, W. Gao, D. Pornpattananangkul, Q. Zhang, V. Fu, J. Li, M. Obonyo and L. Zhang, J. Mater. Chem. B, 2014, 2, 8201–8207 RSC.
- Z. Liu, W. B. Cai, L. N. He, N. Nakayama, K. Chen, X. M. Sun, X. Y. Chen and H. J. Dai, Nat. Nanotechnol., 2007, 2, 47–52 CrossRef CAS PubMed.
- P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef CAS.
- A. Sharma, A. Khatchadourian, K. Khanna, R. Sharma, A. Kakkar and D. Maysinger, Biomaterials, 2011, 32, 1419–1429 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nature, 2007, 2, 751–760 CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- C. Shi, D. Guo, K. Xiao, X. Wang, L. Wang and J. Luo, Nat. Commun., 2015, 6, 7449–7462 CrossRef PubMed.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958–962 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS PubMed.
- Y. Cheng, L. Zhao, Y. Li and T. Xu, Chem. Soc. Rev., 2011, 40, 2673–2703 RSC.
- P. Kesharwani, R. K. Tekade and N. K. Jain, Nanomedicine, 2014, 9, 2291–2308 CrossRef CAS PubMed.
- R. K. Tekade, M. Tekade, M. Kumar and A. S. Chauhan, Pharm. Res., 2015, 32, 910–928 CrossRef CAS PubMed.
- C. C. Lee, J. A. MacKay, J. M. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- R. Iezzi, B. R. Guru, I. V. Glybina, M. K. Mishra, A. Kennedy and R. M. Kannan, Biomaterials, 2012, 33, 979–988 CrossRef CAS PubMed.
- D. A. Tomalia, A. Nayor and W. I. Goddard, Angew. Chem., Int. Ed. Engl., 1990, 29, 138–175 CrossRef.
- E. R. Gillies and J. M. J. Réchet, Drug Discovery Today, 2005, 10, 35–50 CrossRef CAS PubMed.
- H. Kobayashi and M. W. Brechbiel, Adv. Drug Delivery Rev., 2005, 57, 2271–2286 CrossRef CAS PubMed.
- J. Zhu and X. Shi, J. Mater. Chem. B, 2013, 1, 4199–4211 RSC.
- V. Jain and P. V. Bharatam, Nanoscale, 2014, 6, 2476–2501 RSC.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171–185 CrossRef CAS PubMed.
- Y. Lu, D. L. Slomberg, A. Shah and M. H. Schoenfisch, Biomacromolecules, 2012, 13, 3343–3354 CrossRef PubMed.
- Y. Lu, D. L. Slomberg, A. Shah and M. H. Schoenfisch, Biomacromolecules, 2013, 14, 3589–3598 CrossRef CAS PubMed.
- L. G. Együd and A. Szent-Györgyi, Science, 1968, 160, 1140 Search PubMed.
- D. Talukdar, S. Ray, M. Ray and S. Das, Drug Metab. Drug Interact., 2008, 23, 175–210 CAS.
- S. Biswas, M. Ray, S. Misra, D. P. Dutta and S. Ray, Biochem. J., 1997, 323, 343–348 CrossRef CAS PubMed.
- M. Ghosh, D. Talukdar, S. Ghosh, N. Bhattacharyya, M. Ray and S. Ray, Toxicol. Appl. Pharmacol., 2006, 212, 45–58 CrossRef CAS PubMed.
- Y. Inoue and A. Kimura, Adv. Microb. Physiol., 1995, 37, 177–227 CrossRef CAS PubMed.
- E. Racker, J. Biol. Chem., 1951, 190, 685–696 CAS.
- S. Ghosh, P. Chakraborty, P. Saha, S. Acharya and M. Ray, RSC Adv., 2014, 4, 23251–23261 RSC.
- T. Bock and B. W. Müller, Pharm. Res., 1994, 11, 589–591 CrossRef CAS.
- M. Ray, J. Halder, S. K. Dutta and S. Ray, Int. J. Cancer, 1991, 47, 603–609 CrossRef CAS.
- A. Pal, D. Talukdar, A. Roy, S. Ray, A. Mallick, C. Mandal and M. Ray, Int. J. Nanomed., 2015, 10, 3499–3518 CAS.
- J. R. Tennant, Transplantation, 1964, 2, 685–694 CrossRef CAS PubMed.
- A. Boyum, Scand. J. Immunol., Suppl., 1976, 5, 9–15 CrossRef.
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, Exp. Cell Res., 1988, 175, 184–191 CrossRef CAS PubMed.
- A. H. Ding, C. F. Nathan and D. J. Stuehr, J. Immunol., 1988, 141, 2407–2412 CAS.
- L. Henderson, E. Jones, T. Brooks, A. Chetelat, P. Ciliutti, M. Freemantle, C. A. Howard, J. Mackay, B. Phillips, S. Riley, C. Roberts, A. K. Wotton and E. J. van de Waart, Mutagenesis, 1997, 12, 163–167 CrossRef CAS PubMed.
- M. Ghosh, A. Chakraborty and A. Mukherjee, J. Appl. Toxicol., 2013, 33, 1097–1110 CrossRef CAS PubMed.
- R. Savic, L. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615–618 CrossRef CAS PubMed.
- S. Pande and R. M. Crooks, Langmuir, 2011, 27, 9609–9613 CrossRef CAS PubMed.
- I. Nemet, D. Vikic-Topic and L. V. Defterdarovic, Bioorg. Chem., 2004, 32, 560–570 CrossRef CAS PubMed.
- S. Ghosh, D. Ghosh, P. K. Bag, S. C. Bhattacharya and A. Saha, Nanoscale, 2011, 3, 1139–1148 RSC.
- S. Ghosh, S. C. Bhattacharya and A. Saha, Anal. Bioanal. Chem., 2010, 397, 1573–1582 CrossRef CAS PubMed.
- S. Ghosh and A. Saha, Nanoscale Res. Lett., 2009, 4, 937–941 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomedicine, 2012, 7, 5577–5591 CrossRef PubMed.
- S. Ghosh, A. Priyam and A. Saha, J. Nanosci. Nanotechnol., 2009, 9, 6726–6735 CrossRef CAS PubMed.
- K. Amin and R. M. Dannenfelser, J. Pharm. Sci., 2006, 95, 1173–1176 CrossRef CAS PubMed.
- J. S. Ma, W. J. Kim, J. J. Kim, T. J. Kim, S. K. Ye, M. D. Song, H. Kang, D. W. Kim, W. K. Moon and K. H. Lee, Nitric Oxide, 2010, 23, 214–219 CrossRef CAS PubMed.
- K. E. Iles and H. J. Forman, Immunol. Res., 2002, 26, 95–105 CrossRef CAS PubMed.
- T. Cruz, R. Gaspar, A. Donato and C. Lopes, Pharm. Res., 1997, 14, 73–79 CrossRef CAS.
- J. S. Kuo, M.-S. Jan and Y.-L. Lin, J. Controlled Release, 2007, 120, 51–59 CrossRef CAS PubMed.
- N. Bhattacharyya, A. Pal, S. Patra, A. K. Haldar, S. Roy and M. Ray, Int. Immunopharmacol., 2008, 8, 1503–1512 CrossRef CAS PubMed.
- A. M. Schrand, J. J. Schlager, L. Dai and S. M. Hussain, Nat. Protoc., 2010, 5, 744–755 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23477h |
| This journal is © The Royal Society of Chemistry 2016 |