
Synthesis of γ-Fe2O3@SiO2@polypyrrole core/shell/shell nanospheres with flexible controllability of electromagnetic properties†
Yajie Zhanga,
Zhiming Zhang*a,
Shicong Xub,
Liangmin Yu*a,
Yunze Longb and
Qunwei Tangc
aKey Laboratory of Marine Chemistry Theory and Technology, Ministry of Education, Ocean University of China, Qingdao 266100, China. E-mail: zzmcyj@ouc.edu.cn; yuyan@ouc.edu.cn
bCollege of Physics & Collaborative Innovation Center for Low-Dimensional Nanomaterials and Optoelectronic Devices, Qingdao University, Qingdao 266071, China
cInstitute of Materials Science and Engineering, Ocean University of China, Qingdao 266100, China
First published on 23rd December 2015
Abstract
A new anchoring method by –SO3H groups was proposed to prepare γ-Fe2O3@SiO2@polypyrrole (γ-Fe2O3@SiO2@PPy) nanospheres with core/shell/shell structure. In the reaction process involved, the sulfonic-functionalized γ-Fe2O3@SiO2 (γ-Fe2O3@SiO2–SO3H) acts as the core for the polymerization of pyrrole. And PPy can be anchored to form on the surface of γ-Fe2O3@SiO2 to form γ-Fe2O3@SiO2@PPy nanospheres with the help of the sulfonic groups on the surface of γ-Fe2O3@SiO2. Moreover, flexible controllability of the conductivity and saturation magnetization of the resulting γ-Fe2O3@SiO2@PPy nanospheres can be realized through this method. The resulting electromagnetic γ-Fe2O3@SiO2@PPy nanospheres show a maximum conductivity of 53 S cm−1 and saturation magnetization of 8.17 A m2 kg−1. The method reported here may provide an efficient way to realize the best impedance matching between complex permeability and complex permittivity.
Introduction
Electromagnetically functionalized micro/nanostructures of conducting polymers have triggered growing interest due to their specific applications in electromagnetic interference shielding and microwave absorption.1–3 It has been reported that microwave absorbing properties can be enhanced via the complementation of dielectric loss and magnetic loss. The electromagnetically functionalized composites composed of magnetic nanoparticles (MNPs) and conducting polymers not only have dielectric loss and magnetic loss, but also a synergistic effect of the dielectric loss and magnetic loss.4,5 So it is very interesting to obtain electromagnetically functionalized conducting polymers. Early studies on preparing electromagnetically functionalized micro/nanostructures of conducting polymers mainly focussed on a two-step method (TSM) including preparation of polyaniline (PANI) nanotubes containing Fe3O4 NPs6 and coaxial PANI/γ-Fe2O3 nanofibers7 by an in situ chemical oxidation polymerization in the presence of Fe3O4 NPs or γ-Fe2O3 nanoneedles. A remaining problem on this aspect is the aggregation of MNPs, leading to unhomogeneous distribution in conducting polymer system. By addressing this issue, we have developed a simple chemical one-step method (COSM) in our previous study to prepare PANI/γ-Fe2O3 nanofibers.8 This COSM has successfully realized the one-step synthesis of electromagnetically functionalized composite nanofibers, in which FeCl3 can act as the oxidant for the aniline polymerization reaction and as a source of FeIII for the precipitation of γ-Fe2O3 magnets, allowing for a simultaneous preparation of the conducting polymer and magnetic γ-Fe2O3 as well as homogeneous distribution of magnetic γ-Fe2O3 NPs in the hybrids. More recently, an improved COSM (ICOSM) has also been created to prepare uniform electromagnetic PPy/γ-Fe2O3 nanospheres having high electrical conductivity without compromising the magnetic properties.9 However, it is hard to regulate the electrical conductivity and saturation magnetization independently for this ICOSM.It is noted that the use of core/shell structure has been an efficient way for improving the microwave absorption properties in recent years.10–12 Especially, the MNPs/conducting polymer core/shell structure has also been reported.1,13 Moreover, it is possible to control the magnetism and conductivity independently by regulating the dosage of MNPs cores and the polymerization condition of conducting polymer shell, respectively. However, the emulsifiers or/and stabilizers were introduced in order to conducting polymer coated well on the magnetic nanoparticles. The weak interactions between the magnetic nanoparticles and conducting polymers make it hard to ensure the magnetic nanoparticles to be coated well.
It is well known that silica layer formed on the surface of MNPs can screen the magnetic dipolar attraction between MNPs, favoring the dispersion of MNPs in liquid media and protects them from etching in an acidic environment.14 Furthermore, the silica surface is active to be grafted by specific functional groups such as amidogen,15,16 sulfonic acid group,17–19 or be modified by chitosan20 or poly(N-vinylpyrrolidone)21 to anchor the polymerization reaction. Moreover, it is noted that silica is one of the most popular wave-transmitting materials.22,23 The superior characteristics of silica inspire us to design a sandwiched γ-Fe2O3@SiO2@PPy core/shell/shell structure. In details, the silica-coated γ-Fe2O3 (γ-Fe2O3@SiO2) core/shell composite spheres is prepared and then modified with sulfonic acid groups (–SO3H), which are subsequently employed to guide the oxidation polymerization of pyrrole monomers on the γ-Fe2O3@SiO2 surface. Through this, the γ-Fe2O3@SiO2@PPy core/shell/shell nanospheres were prepared successfully and the weak interfacial interactions between the inorganic γ-Fe2O3 MNPs and organic PPy, as well as the poor wave-transmissivity may be improved efficiently at the same time.
Experimental
Materials
The chemicals used in this work, such as FeCl3·6H2O, FeSO4·7H2O, tetraethoxysilane (TEOS), p-toluenesulfonic acid (p-TSA), 3-mercaptopropyltrimethoxysilane (MPTS) and pyrrole, were all of analytical purity provided by the Shanghai Sinopharm Chemical Reagent Co., and were used as received without further purification. Deionized water was used in all experiments.Preparation of functional Fe3O4 NPs
Fe3O4 NPs were prepared by chemical precipitation method according to previous literature.14 In details: 54.06 g of FeCl3·6H2O (0.2 mol) and 27.80 g of FeSO4·7H2O (0.1 mol) were dissolved in 440 mL of deionized water. The obtained transparent solution was degassed with nitrogen flow and then 60 mL of NaOH (16.67 M) was dropped into the above solution within 1 h under vigorous agitation. After reaction at 1 h at 20 °C and subsequent heating at 90 °C for 1 h, the resulting black precipitates were collected and thoroughly rinsed with deionized water and ethanol and dried at 50 °C in vacuum for 12 h.The as-synthesized Fe3O4 NPs were modified by citric acid to get highly dispersed and chemically stable aqueous magnetic fluids (MFs).24 Briefly, 1 g of Fe3O4 NPs and 1.7 g of citric acid were dispersed in 200 mL of deionized water under supersonic agitation for 1 h. The pH value of the reagent was adjusted to 4.8 with hydrochloric acid, and the reaction was allowed to proceed with stirring for 12 h at room temperature. Subsequently, the modified Fe3O4 NPs were collected and thoroughly rinsed with deionized water/acetone mixture to remove the residual citric acid, the volume of the mixture was 100 mL and the volume ratio of water to acetone was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. Finally, the modified Fe3O4 NPs were dispersed in 100 mL of deionized water for further use.
:4. Finally, the modified Fe3O4 NPs were dispersed in 100 mL of deionized water for further use.
Preparation of γ-Fe2O3@SiO2 (FS) core/shell nanospheres
20 mL of Fe3O4 MFs (10 mg mL−1) was mixed with 80 mL of ethanol and 2.4 mL of 28 wt% aqueous ammonia. After ultrasonic agitation for 1 h, 2 mL of TEOS was added dropwise to the above dispersion and the reaction was proceeded under supersonic stirring for 2 h. The obtained product was collected by magnetic separation and rinsed with concentrated HCl, deionized water and ethanol three times, respectively, and then dried at 50 °C for 4 h. Finally, the γ-Fe2O3@SiO2 nanospheres were heated at 300 °C for 5 h in air. The obtained γ-Fe2O3@SiO2 was designated as FS.Sulfonic-functionalization of γ-Fe2O3@SiO2 (FSH) core/shell nanospheres
The FS nanospheres were functionalized with –SO3H according to previous literature18 and the approach employed was shown in Scheme 1. The FSH nanospheres were achieved by reacting MPTS with the silanol terminal groups to introduce mercapto groups on the surface of silica followed by oxidation with H2O2 resulting in covalently bounded –SO3H groups. Briefly, 1 g of FS nanospheres were dispersed in a mixture containing 30 mL of acetic acid and 4 mL of H2O2 (30 wt%) under supersonic agitation. Subsequently, 2 g of MPTS were added and the reaction was allowed to proceed at 80 °C for 3 h. After that, the precipitates were collected by magnetic separation and washed with ethanol three times, and then dried at 50 °C for 12 h. The obtained γ-Fe2O3@SiO2–SO3H was designated as FSH. | ||
| Scheme 1 Schematic illustration for synthesizing sulfonic-functionalization FS nanospheres. | ||
Preparation of γ-Fe2O3@SiO2@PPy (FSHPx-y) core/shell/shell nanospheres
The typical processes for γ-Fe2O3@SiO2@PPy (FSHPx-y) were as follows: stoichiometric FSH nanospheres were dispersed in 55 mL of aqueous solution containing pyrrole and p-TSA under supersonic agitation. Under mechanical stirring at a speed of 800 rpm in an ice-water bath, 20 mL of FeCl3 solution was added dropwise to the dispersion within 1 h for the in situ oxidative polymerization of pyrrole monomers. The reaction system was kept at 0–4 °C and stirred for 6 h. Finally, the black products were collected and rinsed with deionized water and ethanol, and dried at 50 °C for 12 h.As a reference, γ-Fe2O3@SiO2@PPy nanocomposites (assigned as FSPx-y) using unfunctionalized FS seeds were also prepared as a comparison to investigate the effect of –SO3H on the morphology and conductivity of the γ-Fe2O3@SiO2@PPy composites. In FSHPx-y or FSPx-y, x and y referred to the grams of FSH (or FS) and the batch number of different pyrrole/p-TSA/FeCl3·6H2O molar ratios (Table 1), respectively.
| Sample | FSH or FS (g) | Pyrrole (mmol) | Pyrrole/p-TSA/FeCl3·6H2O (molar ratio) | Water solution (mL) |
|---|---|---|---|---|
| FSHP0.6-1 | 0.6 | 5 | 1:0:4 |
75 |
| FSHP0.6-2 | 0.6 | 5 | 1:1:4 |
75 |
| FSHP0.6-3 | 0.6 | 5 | 1:2:4 |
75 |
| FSHP0.6-4 | 0.6 | 5 | 1:1:2 |
75 |
| FSHP0.05-2 | 0.05 | 5 | 1:1:4 |
75 |
| FSHP0.1-2 | 0.1 | 5 | 1:1:4 |
75 |
| FSHP0.2-2 | 0.2 | 5 | 1:1:4 |
75 |
| FSHP1.0-2 | 1.0 | 5 | 1:1:4 |
75 |
| FSP1.0-2 | 1.0 | 5 | 1:1:4 |
75 |
Characterization
Field emitting scanning electron microscope (SEM, JSM-6700F), energy dispersive X-ray (EDX) and transmission electron microscope (TEM, JEM-200CX and FEI TECNAI G2 F20) were used to study the morphologies of the γ-Fe2O3@SiO2–SO3H and γ-Fe2O3@SiO2@PPy nanocomposites. The thermogravimetric (TG) analysis was carried out on a SETSYS Evolution TGA (Setaram) from room temperature to 800 °C at a heating rate of 10 °C min−1 and a dynamic dry air flow. FTIR spectra (KBr pellet, Perkin-Elmer System), X-ray photoelectron spectroscopy (XPS, PHI-5702) and X-ray diffraction (XRD, RINT2000 wide angle goniometer) were used to characterize the chemical structures of the resulting nanocomposites. The magnetization curves at different magnetic field (−800 to 800 kA m−1) were measured at room temperature by an extracting sample magnetometer (Lakeshore 7404). The conductivity at room temperature was measured using four-probe method with a KEITHLEY 2400 multimeter and a KEITHLEY 2182A nanovoltmeter.Results and discussion
The whole procedure to prepare γ-Fe2O3@SiO2@PPy nanospheres is illustrated in Scheme 2. First, the Fe3O4 NPs were prepared by chemical precipitation method and modified by citric acid to get MFs. Then, SiO2 shell was coated on the Fe3O4 NPs surface through a sol–gel process. Subsequently, the achieved Fe3O4@SiO2 nanospheres were transformed into γ-Fe2O3@SiO2 by heating in air. After reacting MPTS with the γ-Fe2O3@SiO2 and further oxidation with H2O2, the –(CH2)3SO3H groups were successfully grafted onto SiO2 surface. Finally, pyrrole monomer polymerized on the surface of γ-Fe2O3@SiO2–SO3H nanospheres by in situ oxidation polymerization, generating core/shell/shell γ-Fe2O3@SiO2@PPy nanospheres. | ||
| Scheme 2 A schematic diagram for the fabrication process of γ-Fe2O3@SiO2@PPy nanospheres. | ||
Morphologies and formation mechanism
Typical SEM and TEM images of γ-Fe2O3@SiO2–SO3H, γ-Fe2O3@SiO2@PPy were employed to investigate the morphologies of the as-prepared samples. SEM image (Fig. 1a) indicates that the resultant γ-Fe2O3@SiO2–SO3H indeed have a nanosphere in shape, and TEM image (Fig. 1a′) confirms the γ-Fe2O3@SiO2–SO3H nanospheres possess a well-defined core/shell structure. Compared with γ-Fe2O3@SiO2–SO3H nanospheres, well-defined γ-Fe2O3@SiO2@PPy core/shell/shell three-layer spherical structure can be observed clearly by SEM and TEM images (Fig. 1b and b′), which indicate the polymerization of pyrrole monomers has occurred on the surface of SiO2 shell. | ||
| Fig. 1 SEM and TEM images of FSH (a and a′); FSHP1.0-2 (b and b′); FSP1.0-2 (c and c′). The blue arrow represents the bare FS two-layer structure and the red arrow represents the freely deposited PPy particles. | ||
It is noteworthy to mention that the surface of SiO2 shell is hydrophilic, while pyrrole monomer is hydrophobic. In this fashion, the combination of SiO2 with PPy is a challenge by in situ polymerizing PPy in SiO2 solution. We can facilely realize the homogeneous coverage of PPy on γ-Fe2O3@SiO2 by anchoring PPy with modified γ-Fe2O3@SiO2–SO3H. As a reference, the unmodified γ-Fe2O3@SiO2 has also been utilized as the core to prepare γ-Fe2O3@SiO2@PPy nanospheres. As illustrated in TEM image of Fig. 1c′ the resulting γ-Fe2O3@SiO2@PPy composite contains both bare γ-Fe2O3@SiO2 two-layer structure (blue arrow) and freely deposited PPy particles (red arrow). In other words, the polymerization process isn't favoured on the unmodified silica surface. Therefore, –SO3H group is the key factor to influence the core/shell/shell morphology.
Energy dispersive X-ray spectroscopy (EDX) elemental mapping of the nanospheres also clearly displays the expected core/shell/shell structure.25 The different colour images shown in Fig. 2c–f indicate Fe-, Si-, C-, and N-enriched areas of the FSHP1.0-2 sample, respectively, which confirm the presence of both SiO2 and PPy serving as the shell of γ-Fe2O3 core. The images also confirm that the coating of SiO2 and PPy is uniform, and after the coating of PPy, the nanospheres agglutinate. The line profiles further confirm the three-layer structure (Fig. 2g) and the 38 nm thickness of the SiO2 shell and 46 nm thickness of the PPy shell were calculated accordingly, which in correspondence with the HAADF STEM image (Fig. 2b).
 | ||
| Fig. 2 (a) Schematic of γ-Fe2O3@SiO2@PPy nanospheres. (b) Typical HAADF STEM image of FSHP1.0-2. (c–f) Elemental mapping of three agglutinate γ-Fe2O3@SiO2@PPy nanospheres in FSHP1.0-2 with Fe, Si, C and N signals. (g) EDS line scanning profiles of FSHP1.0-2 across the map. The red line in (b) corresponds to the EDS line scanning region. | ||
The synthesis conditions, as summarized in Table 1, have significant impact on the morphologies of γ-Fe2O3@SiO2@PPy.
Fig. 3 shows the morphology evolution of γ-Fe2O3@SiO2@PPy with FSH dosage ranging from 19.2 to 83.1 wt% (mass fraction calculated by TGA data, ESI Table S1†). Apparently, enhancement of FSH dosage is beneficial to homogeneous distribution of γ-Fe2O3@SiO2@PPy nanospheres and to the reduction of γ-Fe2O3@SiO2@PPy diameter. However, we can't find apparent deviations by tuning molar ratio of pyrrole/p-TSA/FeCl3 from 1:0:4 to 1:1:2 while fixing FSH dosage of 0.6 g, yielding spherical morphology in all aspects, as shown in Fig. 4. Notably, the p-TSA dopant does not affect the morphologies of γ-Fe2O3@SiO2@PPy composites.
 | ||
| Fig. 3 SEM images of (a) FSHP0.05-2, (b) FSHP0.1-2, (c) FSHP0.2-2, (d) FSHP0.6-2 and (e) FSHP1.0-2. | ||
 | ||
| Fig. 4 SEM images of (a) FSHP0.6-1, (b) FSHP0.6-2, (c) FSHP0.6-3 and (d) FSHP0.6-4. | ||
A possible formation mechanism behind the aforementioned results for γ-Fe2O3@SiO2@PPy nanospheres is illustrated in Scheme 3. It has been widely investigated that the morphologies of PPy composites highly depend on seed templates' shape in the polymerization system of pyrrole monomers, such as particulates, nano/microspheres and nano-needles.20,26,27 As has demonstrated in previous work,26 the micelles consisting of p-TSA and pyrrole can be formed in the reaction solution resulting from the hydrophobic pyrrole adhering to the oil-wet side of p-TSA and the presence of hydrophilic –SO3H group in the p-TSA, thus the pyrrole/p-TSA supermolecules serve as the “soft-templates” in the formation of PPy-p-TSA nanostructures. Herein, the outer –SO3H groups are hydrophilic and the inner γ-Fe2O3@SiO2–(CH2)3– structure is hydrophobic (Scheme 2), therefore the γ-Fe2O3@SiO2–SO3H nanosphere has a micelle-like structure (micelle-like A). In the absence of p-TSA, pristine pyrrole diffuses into the inner oil-wet side of micelle-like A to form micelle-like B. In the presence of p-TSA, micelle-like C occurs when the oil-wet side of p-TSA adheres to micelle-like A, the subsequently added pyrrole diffuses into micelle-like C to form micelle-like D. B and D are believed as the templates to form γ-Fe2O3@SiO2@PPy nanospheres. Due to fact that the solubilized pyrrole molecules are oxidized by FeCl3, the polymerization reaction is believed to proceed in the interface of micelles and water.28 In our experiments, it is obvious that the micelle-like B and D mainly act as the template judged from the morphology of the as-prepared γ-Fe2O3@SiO2@PPy. In case of the decided amount of pyrrole monomers and polymerization condition used in the reaction systems, the amount of the pyrrole monomers distributed to every γ-Fe2O3@SiO2–SO3H core decrease when the amount of γ-Fe2O3@SiO2–SO3H cores increase. As a result, the thickness of PPy shell decreases and thus leads to smaller diameter and more sphere-like of the γ-Fe2O3@SiO2@PPy nanospheres (Fig. 3a–e). On the other hand, when the amount of Fe2O3@SiO2–SO3H cores is decided, there's no apparent deviation on the morphologies (Fig. 4a–d).
 | ||
| Scheme 3 Schematic illustration of the formation mechanism of the γ-Fe2O3@SiO2@PPy nanospheres. | ||
The typical SEM and TEM images of FSHP1.0-2 (Fig. 1b and b′) show that these nanospheres are aggregated, which has also been indicated by dynamic light scattering (DLS) data (ESI Fig. S1†). The mean hydrodynamic diameter of 858 nm and polydispersity (PDI) of 0.34 suggest the nanospheres are not monodispersed.29 The aggregation of the nanospheres may result from the relatively high Hamaker constant reported for PPy30 and the interaction between Fe3+ and –OH (from –SO3H on γ-Fe2O3@SiO2–SO3H surface) in the reaction system. While some steric stabilizers such as poly(N-vinylpyrrolidone)31 and chitosan20 were used in the PPy coating process, which would prevent the latex particles from aggregating, it can lead to the low and uncontrollable conductivity of the resultant composites. Herein, without the steric stabilizer, the PPy coated nanospheres have a propensity to aggregate, and on the other hand, the huge amounts of iron ions in the reaction system may lead micelle-like B or D to aggregate, and the pyrrole monomers residual in the solution are oxidized by Fe3+ to polymerize between the micelles, which responsible for the agglutination of the nanospheres.
Structural characterization
Fig. 5 shows the thermo gravimetric analysis (TGA) curves of FS, FSH, FSHP1.0-2 and PPy. It's reasonable to consider that the variation of residual mass fraction at 800 °C between FSH and FS is attributed to the propylsulfonic thermal degradation in FSH (Fig. 5a and b). There are two obvious stages in the process of thermal decomposition for pure PPy (Fig. 5d). The first stage in the range of 30–180 °C results from the evaporation of some impurities such as oligomers and absorbed water, the other one at above 330 °C is caused by the degradation of PPy chains, and it has decomposed completely above 550 °C. In contrast with the pure PPy, the temperature corresponding to maximum decomposition rate of FSHP1.0-2 is higher than that of pure PPy, which indirectly indicates that the PPy shell indeed has some interaction with the FSH cores, thus the thermal stability of FSHP composites has been improved. The contents of magnetic FSH cores in FSHPx-2 and FSHP0.6-y samples have also been determined by TGA data (ESI Fig. S2, Table S1†), which are responsible for the variation of magnetism of the samples. | ||
| Fig. 5 TG curves of FS (a), FSH (b), FSHP1.0-2 (c), PPy (d). | ||
X-photoelectron spectroscope (XPS) is utilized to determine the molecular structures. Fig. 6 shows the full XPS spectrums of the as-prepared FS and FSH. In comparison with unfunctionalized FS (Fig. 6a), the characteristic peak occurs at 169.1 eV is attributed to the sulfur atom from the silane coupling agent for the functionalized FSH (Fig. 6b). The peak centred at 168.6 eV corresponding to –SO3H group32,33 is detected as shown in the S 2p high-resolution spectrum for the functionalized FSH (inset of Fig. 6), while the peak near 164 eV due to the –SH group34,35 is undetected. This result further demonstrates the mercapto groups have been oxidized completely to sulfonic ones. The S/Si atomic ratio of ∼0.4 determined by XPS data manifests the surface of silica is enriched with sulfonic groups.32 In addition, there is also a substantial increase in the relative intensity of C 1s peak compared with the untreated FS. The increase in the relative intensity of C 1s peak can be attributed to the propyl chain from MPTS grafted to silica. In the case of the untreated FS, the C 1s peak mainly results from adventitious carbon contamination. Besides, for both FS and FSH, the typical peaks at 155.1 and 103.5 eV are attributed to Si 2s, Si 2p, respectively, while the peaks corresponding to characteristic doublets of Fe 2p3/2 and 2p1/2 core-level spectra in iron oxide, cannot be observed. This phenomenon can be explained by the fact that the SiO2 layer thickness (38 nm) exceeds the detecting depth of XPS measurement.
 | ||
| Fig. 6 XPS spectra of (a) FS, (b) FSH. The inset shows the expanded spectra of S 2p. | ||
The grafting of –SO3H groups was also confirmed by FTIR spectra. As shown in Fig. 7b, FS shows the absorption bands in the range of 3654 cm−1 and 1260–1000 cm−1 assigned to SiO–H and Si–O coordination, respectively. Other intense bands at 1635 cm−1 (O–H bending vibration), 966 cm−1 (Si–O stretching vibration), 806 cm−1 (Si–O–Si bending vibration) and 450 cm−1 (Si–O–Si rocking vibration) are also observed for FS.19 For FSH (Fig. 7c), the characteristic peaks of –SO3H group attributed to O–H stretching vibration band centred at 3400 cm−1 and the bands at 2940, 2880 cm−1 (C–H stretching vibration) and 1460–1420 cm−1 (C–H bending vibration) related to the C–H and C–C vibrational modes of the propyl group36 can be observed clearly. The peaks assigned to SO2 are overlapped by the intense band of Si–O at 1260–1000 cm−1. FTIR spectra indicate that the sulfonic-functionalization of γ-Fe2O3@SiO2 was well done. In the FTIR spectrum of FSHP1.0-2 (Fig. 7d), the characteristic absorption bands at 1545, 1453, 1310, 1166, 1037, and 897 cm−1 attributed to the coated of PPy are similar to those of PPy/γ-Fe2O3 synthesized.11,26 The peaks at 1545 and 1453 cm−1 are ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N stretching vibrations. The peaks at 1313 and 1186 cm−1 correspond to the C–H in-plane vibration. The peaks at 1037 and 885 cm−1 are attributed to the C–H in-plane bending and the ring deformation, respectively. These results indicate that the structure of the main PPy chain in the γ-Fe2O3@SiO2@PPy nanospheres prepared is identical to those of the PPy/Fe3O4 nanostructures prepared previously.37
C and C–N stretching vibrations. The peaks at 1313 and 1186 cm−1 correspond to the C–H in-plane vibration. The peaks at 1037 and 885 cm−1 are attributed to the C–H in-plane bending and the ring deformation, respectively. These results indicate that the structure of the main PPy chain in the γ-Fe2O3@SiO2@PPy nanospheres prepared is identical to those of the PPy/Fe3O4 nanostructures prepared previously.37
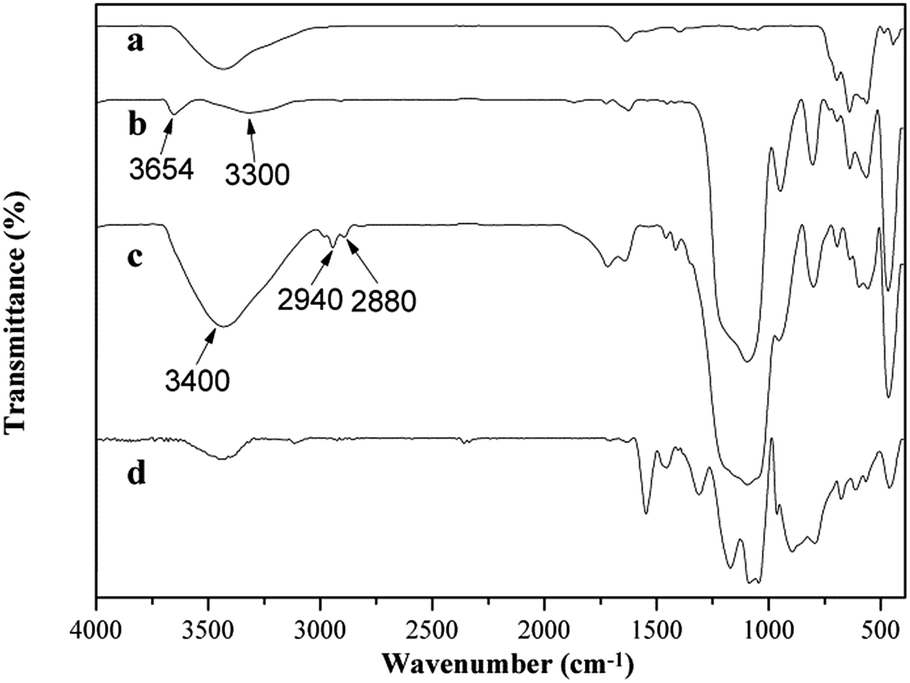 | ||
| Fig. 7 FTIR spectra of (a) γ-Fe2O3, (b) FS, (c) FSH and (d) FSHP1.0-2. | ||
Fig. 8 shows the XRD patterns of FSHP1.0-2, FSH, γ-Fe2O3 synthesized and standard γ-Fe2O3. The sharp peaks at 2θ = 30.2°, 35.6°, 43.3°, 53.7°, 57.3°, and 62.9°are observed in the synthesized γ-Fe2O3 (Fig. 8c), which match well with the standard peaks of γ-Fe2O3 (JCPDS file no. 39-1346, Fig. 8d). Apart from the peaks attributed to γ-Fe2O3, a broad diffraction peak centred at 2θ = 23.5° attributed to the amorphous SiO2 (Fig. 8b) and a broad diffraction peak centred at 2θ = 23.5°assigned to PPy (Fig. 8a) are observed in the XRD patterns of FSH and FSHP1.0-2.11,38
 | ||
| Fig. 8 XRD patterns of (a) FSHP1.0-2, (b) FSH, (c) as-synthesized γ-Fe2O3, and (d) standard γ-Fe2O3 (JCPDS no. 39-1346). | ||
Electrical and magnetic properties
It has been demonstrated that magnetic and electrical properties can be successfully integrated into a single COSM.8 Subsequently, the conductivity and magnetization can be enhanced simultaneously by controlling the amounts of FeCl2 ([FeII]/[FeIII] ratio < 0.046) for ICOSM.9 However, the conductivity and saturation magnetization decrease negatively simultaneously when [FeII]/[FeIII] ratio is higher than 0.046. By controlling the synthesis conditions, as summarized in Table 1, an electrical conductivity ranging from 17 to 53 S cm−1 (Fig. 9) and a high saturation magnetization in a range of 2.18–8.17 A m2 kg−1 (Fig. 10) can be realized using γ-Fe2O3@SiO2–SO3H cores. It's worth mention that the conductivities maintain a high level (50–60 S cm−1) even the γ-Fe2O3@SiO2–SO3H mass fraction reaches 83.1 wt%, which can be attributed to the complete encapsulation of γ-Fe2O3@SiO2–SO3H cores by PPy shell. By contrast, the conductivity of the FSP1.0-2 sample obtained by using unfunctionalized γ-Fe2O3@SiO2 as seeds is as low as 8 S cm−1, which is one magnitude lower than that of FSHP1.0-2 (55 S cm−1). Till now, we can conclude that the merits on high conductivity and high magnetization can also be integrated into a single nanosphere using the method reported here. Moreover, we can realize the independent control of electrical conductivity and saturation magnetization of the electromagnetically functionalized conducting polymer. | ||
| Fig. 9 Effect of the polymerization condition on the conductivity and saturation magnetization of FSHP0.6-y samples. | ||
 | ||
| Fig. 10 Effect of the FSH mass fraction on the conductivity and saturation magnetization of FSHPx-2 samples. | ||
Fig. 11 shows the dependence of magnetism (M) on applied magnetic field (H) for FSHP0.6-y samples and FSHPx-2 samples. All the γ-Fe2O3@SiO2@PPy nanocomposites exhibit superparamagnetic behavior (i.e., no hysteretic loop). The saturation magnetization (Ms) of the γ-Fe2O3@SiO2@PPy composites increases with an increase of the FSH content (Fig. 11b). And when the dosage of FSH cores was decided, the obtained γ-Fe2O3@SiO2@PPy composites with different polymerization conditions have slightly different contents of FSH, and the Ms of γ-Fe2O3@SiO2@PPy composites is directly proportional to the contents of FSH (Fig. 11a).
 | ||
| Fig. 11 Dependence of the magnetism on the applied magnetic field at room temperature: (a) FSHP0.6-y samples with different polymerization conditions; (b) FSHPx-2 samples with different FSH dosages. FSH mass fractions were given based on TGA data. | ||
Conclusions
Electromagnetic functionalized γ-Fe2O3@SiO2@PPy core/shell/shell nanospheres have been successfully realized by in situ polymerization of pyrrole monomers in the presence of spherical γ-Fe2O3@SiO2–SO3H templates. A prerequisite of realizing the γ-Fe2O3@SiO2@PPy nanospheres is to anchor PPy shell onto SiO2 surface by –SO3H groups, allowing for the homogeneously spherical morphology and high conductivity of the γ-Fe2O3@SiO2@PPy composites at high γ-Fe2O3@SiO2–SO3H dosage. In particularly, the conductivity as high as 17–53 S cm−1 and saturation magnetization of 2.18–8.17 A m2 kg−1 can be independently tuned by controlling polymerization conditions and γ-Fe2O3@SiO2–SO3H dosages.Acknowledgements
This project was supported by the National Natural Science Foundation of China (No. 41476059) and National Science and Technology Support Program (2012BAB15B02).References
- B. Zhang, Y. Du, P. Zhang, H. Zhao, L. Kang, X. Han and P. Xu, J. Appl. Polym. Sci., 2013, 130, 1909–1916 CrossRef CAS.
- F. Xu, L. Ma, Q. Huo, M. Gan and J. Tang, J. Magn. Magn. Mater., 2015, 374, 311–316 CrossRef CAS.
- C. Cui, Y. Du, T. Li, X. Zheng, X. Wang, X. Han and P. J. Xu, J. Phys. Chem. B, 2012, 116, 9523–9531 CrossRef CAS PubMed.
- Y. F. Zhu, Q. Q. Ni, Y. Q. Fu and T. Natsuki, J. Nanopart. Res., 2013, 15, 1988–1998 CrossRef PubMed.
- O. Akman, Z. Durmus, H. Kavas, B. Aktas, U. Kurtan, A. Baykal and H. Sözeri, Phys. Status Solidi A, 2013, 210, 395–402 CrossRef CAS.
- Z. Zhang and M. Wan, Synth. Met., 2003, 132, 205–212 CrossRef CAS.
- Z. Zhang, M. Wan and Y. Wei, Nanotechnology, 2005, 16, 2827–2832 CrossRef CAS.
- Z. Zhang, J. Deng, J. Shen, M. Wan and Z. Chen, Macromol. Rapid Commun., 2007, 28, 585–590 CrossRef CAS.
- Z. Zhang, Q. Li, L. Yu, Z. Cui, L. Zhang and G. A. Bowmaker, Macromolecules, 2011, 44, 4610–4615 CrossRef CAS.
- J. W. Liu, R. C. Che, H. J. Chen, F. Zhang, F. Xia, Q. S. Wu and M. Wang, Small, 2012, 8, 1214–1221 CrossRef CAS PubMed.
- Y. J. Chen, F. Zhang, G. G. Zhao, X. Y. Fang, H. B. Jin, P. Gao, C. L. Zhu, M. S. Cao and G. Xiao, J. Phys. Chem. C, 2010, 114, 9239–9244 CAS.
- G. Wang, Y. Chang, L. Wang and C. Liu, Adv. Powder Technol., 2012, 23, 861–865 CrossRef CAS.
- W. C. Zhou, X. J. Hu, X. X. Bai, S. Y. Zhou, C. H. Sun, J. Yan and P. Chen, ACS Appl. Mater. Interfaces, 2011, 3, 3839–3845 CAS.
- Y. H. Deng, C. C. Wang, J. H. Hu, W. L. Yang and S. K. Fu, Colloids Surf., A, 2005, 262, 87–93 CrossRef CAS.
- M. Chehimi, M. M. Chehimi, D. Mordenti, M. Bri and M. Delamará, J. Mater. Chem., 1998, 8, 2185–2193 RSC.
- C. Perruchot, M. M. Chehimi, M. Delamar and F. Fievet, Surf. Interface Anal., 1998, 26, 689–698 CrossRef CAS.
- T. Dai, X. Yang and Y. Lu, Mater. Lett., 2007, 61, 3142–3145 CrossRef CAS.
- C. H. TangM.S. thesis, Beijing University of Chemical Technology, China, 2011.
- L. C. Fonseca, R. Faez, F. F. Camilo and M. A. Bizeto, Microporous Mesoporous Mater., 2012, 159, 24–29 CrossRef CAS.
- X. Yang, T. Dai and Y. Lu, Polymer, 2006, 47, 441–447 CrossRef CAS.
- L. Hao, C. Zhu, C. Chen, P. Kang, Y. Hu, W. Fan and Z. Chen, Synth. Met., 2003, 139, 391–396 CrossRef CAS.
- J. H. Zhu, S. Y. Wei, N. Haldolaarachchige, D. P. Young and Z. H. Guo, J. Phys. Chem. C, 2011, 115, 15304–15310 CAS.
- Y. Yang, Z. W. Li, C. P. Neo and J. Ding, J. Phys. Chem. Solids, 2014, 75, 230–235 CrossRef CAS.
- B. Liu, D. P. Wang and W. H. Huang, J. Funct. Mater., 2007, 38, 1074–1077 CAS.
- P. C. Naha, A. A. Zaki, E. Hecht, M. Chorny, P. Chhour, E. Blankemeyer, D. M. Yates, W. R. Witschey, H. I. Litt, A. Tsourkas and D. P. Cormode, J. Mater. Chem. B, 2014, 2, 8239–8248 RSC.
- H. M. Xiao, W. D. Zhang, M. X. Wan and S. Y. Fu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4446–4453 CrossRef CAS.
- X. Li, M. Wan, Y. Wei, J. Shen and Z. Chen, J. Phys. Chem. B, 2006, 110, 14623–14626 CrossRef CAS PubMed.
- B. J. Kim, S. G. Oh, M. G. Han and S. S. Im, Synth. Met., 2001, 122, 297–304 CrossRef CAS.
- A. L. Brown, P. C. Naha, V. Benavides-Montes, H. I. Litt, A. M. Goforth and D. P. Cormode, Chem. Mater., 2014, 26, 2266–2274 CrossRef CAS PubMed.
- M. M. Chehimi, S. F. Lascelles and S. P. Armes, Chromatographia, 1995, 41, 671–677 CAS.
- C. Mangeney, M. Fertani, S. Bousalem, M. Zhicai, S. Ammar, F. Herbst, P. Beaunier, A. Elaissari and M. M. Chehimi, Langmuir, 2007, 23, 10940–10949 CrossRef CAS PubMed.
- M. D. González, P. Salagre, E. Taboada, J. Llorca, E. Molins and Y. Cesteros, Appl. Catal., B, 2013, 136, 287–293 CrossRef.
- M. M. Nasef and H. Saidi, Appl. Surf. Sci., 2006, 252, 3073–3084 CrossRef CAS.
- Z. C. Liu, Q. G. He, P. Hou, P. F. Xiao, N. Y. He and Z. H. Lu, Colloids Surf., A, 2005, 257, 283–286 CrossRef.
- R. Lenigk, M. Carles, N. Y. Ip and N. J. Sucher, Langmuir, 2001, 17, 2497–2501 CrossRef CAS.
- Q. Qu, Q. Gu, Z. Gu, Y. Shen, C. Wang and X. Hu, Colloids Surf., A, 2012, 415, 41–46 CrossRef CAS.
- W. D. Zhang, H. M. Xiao, L. P. Zhu, S. Y. Fu and M. X. Wan, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 320–326 CrossRef CAS.
- F. Lv, L. Fu, E. P. Giannelis and G. Qi, Solid State Sci., 2014, 34, 49–55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25576g |
| This journal is © The Royal Society of Chemistry 2016 |