
Effect of the graphene oxide reduction method on the photocatalytic and electrocatalytic activities of reduced graphene oxide/TiO2 composite†
H. Al-Kandaria,
A. M. Abdullah*b,
S. Al-Kandaric and
A. M. Mohamedc
aDepartment of Health Environment, College of Health Sciences, PAAET, P. O. Box 1428, Faiha 72853, Kuwait
bCenter for Advanced Materials, Qatar University, Doha, P. O. Box 2713, Qatar. E-mail: abubakr_2@yahoo.com; bakr@qu.edu.qa; Tel: +974-44035672
cChemistry Department, Kuwait University, P. O. Box 5969, Safat, 13060, Kuwait
First published on 19th August 2015
Abstract
Graphene oxide (GO) was synthesized from commercial graphite using a modified Hummers' method. Three different methods were used to prepare reduced GO/TiO2 composites. GO was (i) initially impregnated over TiO2 (GOTi) and then reduced using a stream of hydrogen gas at 450 °C (H2RGOTi), (ii) reduced using hydrazine hydrate solution (HH) in a 1000 W microwave oven (HHRGO) and then loaded on the TiO2 (HHRGOTi) or (iii) hydrothermally reduced (RGO) then loaded on TiO2 (RGOTi). Different characterization techniques were used e.g. X-ray photoelectron spectroscopy (XPS), X-ray diffraction patterns (XRD) and UV-Vis, Fourier transform infrared (FT-IR) and Raman spectroscopy. The effect of the GO reduction method on the photocatalytic activity of the aforementioned composites towards the degradation of phenol in the presence and absence of (i) UV and (ii) H2O2 was examined. High phenol degradation rates were achieved using the RGOTi photocatalyst, compared to the TiO2 nanoparticles, under UV illumination. On the other hand, the H2RGOTi composite has shown the highest electrocatalytic activity towards the oxygen reduction reaction under UV illumination.
1. Introduction
The rapid expansion of industrialization and population growth has resulted in the production of a huge amount of wastewater. Phenol is one of the various refractory organic contaminants found in wastewater which requires special attention because of its toxic effects to humans and the environment.1–5 The presence of phenol has been confirmed in many different industrial wastewaters e.g. chemical and petrochemical industries. The traditional water treatment techniques, including activated adsorption, chemical oxidation and biological digestion, have not been successful in meeting the World Health Organization (WHO) standards.3,6,7 Therefore, new treatment approaches for phenolic compounds in wastewater based on advanced oxidation processes (AOPs) were developed which can achieve higher efficiencies than the currently used ones. An example of AOPs in wastewater treatment is the use of a combination of hydrogen peroxide and ultraviolet radiation in the presence of TiO2. This system has proven to be efficient in wastewater treatment.8,9 Unlike chlorine, which is widely used worldwide, hydrogen peroxide is an eco-oxidant that does not generate carcinogenic residues and plays an important role in green chemistry.Graphene and reduced graphene oxide (RGO) have attracted attention due to the excellent electrical, mechanical, thermal and optical properties that can be transformed into potential energy and environmental applications e.g. electronics, super capacitors, sensors, solar cells, wastewater treatment and gas storage.10,11 In addition, it has been shown that RGO can amend the photocatalytic and electrocatalytic properties of several semiconductors e.g. ZnO, WO3 and TiO2 when it forms composites with them;12–22 however, the effect of the different types of graphene oxide's reduction methods on their catalytic properties has never been studied previously. So, the main objective of this work is (i) investigating how the reduction methods of graphene oxide affect the photocatalytic and electrocatalytic behaviours of RGO/TiO2 composites. The reason for the probable reduction method – dependent catalytic behaviour cannot be attributed only to the change in the band gap energy of the different composites but also due to the difference in the amount and types of the oxygen functional groups on the RGO surface and the changes in the surface areas as the reduction methods for graphene oxide (GO) differ. Furthermore, the different stabilities of some oxidants or their radicals e.g. H2O2 or HO˙ on the surfaces of some semiconductors/GO composites compared to others can be an additional sound reason for the differences in their catalytic properties. To investigate this point, four different composites of TiO2/graphene oxide or its reduced form were prepared and their photocatalytic and electrocatalytic behaviours were studied: graphene oxide/TiO2 (GOTi), hydrazine hydrate-reduced graphene oxide/TiO2 (HHRGOTi), hydrogen-reduced graphene oxide/TiO2 (H2RGOTi) and thermally reduced graphene oxide/TiO2 (RGOTi).
Other objectives for this work can be summarized as follows: (ii) chemical characterization of the as-prepared composites using bulk and surface analytical methods, (iii) electrochemical characterization of the same composites towards the oxygen reduction reaction (ORR) in an acidic medium (to target the 2-electron oxygen reduction mechanism which produces H2O2 that is needed for the AOPs) and (iv) photochemical characterization for these composites towards the phenol oxidation in the presence and absence of H2O2 under UV illumination.
2. Experimental
2.1. Synthesis
GO powder was prepared using a modified Hummers' method.23 First, 2 g of graphite powder (200 mesh, 99.9999%, Alfa Aesar A Johnson Matthy Company), 1 g of sodium nitrate (99.995%, Sigma-Aldrich) and 50 mL of concentrated sulphuric acid (98%, BDH) were mixed and vigorously stirred at 0 °C for 30 min using an ice water bath. Then, 6 g of potassium permanganate crystals (99.0%, Sigma-Aldrich) were added slowly to the solution while stirring. The ice water bath was then removed, and the mixture was stirred at 35 °C for 30 min. When the mixture became a pasty dark green colour with the evolution of gas bubbles, a 100 mL of distilled water was added slowly, and the temperature of the mixture was kept below 100 °C for 15 min. The diluted mixture acquired, at this stage, a brown colour. After this, the mixture was treated with a 100 mL of hydrogen peroxide (30%, Sigma-Aldrich) and stirred until it turned out to be a bright yellow suspension. Finally, the suspension was filtrated and washed with an excess amount of warm distilled water and was dried overnight under vacuum at 60 °C.Loading of GO on the TiO2 support was carried out using an impregnation technique in which a suspension of dispersed GO and an appropriate amount of deionized water/absolute ethanol were sonicated for 1 h. Commercial TiO2 nanoparticles (particle size: 21 nm, from Sigma Aldrich) were then added, and the suspension was stirred for 12 h at 280 rpm using a rotary evaporator. Finally, the solvents were evaporated by heating under vacuum at 60 °C, and then the dried powder was crushed.
The HHRGOTi composite was prepared by reducing GO using hydrazine hydrate (HH) and then loading it on the TiO2 according to the following recipe; an appropriate amount of GO suspension was added to a mixture of 20 mL of deionized water and 50 μL of hydrazine hydrate (25%, Sigma-Aldrich) until a yellowish brown suspension was formed. Then, it was heated in a 1000 W domestic microwave oven in two cycles (20 s on and 10 s off) for a total time of 60 s. Then, 10 mL of absolute ethanol and TiO2 powder were added to the black suspension, and was stirred for 24 h at room temperature. Finally, the suspension was centrifuged at 6000 rpm for 25 min, washed with deionized water and then with ethanol and dried overnight under vacuum at 60 °C. To obtain only HHRGO without the TiO2 support, the same procedure was followed except adding TiO2 was excluded from the procedure.
The H2RGOTi composite was prepared by reducing the TiO2-supported GO (GOTi) in a quartz reactor using a flow of H2 gas at a rate of 100 mL min−1 for 30 min and at a temperature of 450 °C. The sample was cooled at room temperature in the reactor and stored under vacuum. The same procedure was followed to obtain an unsupported H2RGO powder except the starting material was unsupported GO.
TiO2-supported thermally reduced GO (RGOTi) was prepared in a Teflon-lined stainless steel autoclave as follows: TiO2 was added to a mixture of deionized water/absolute ethanol and sonicated for 30 min. Afterwards, the GO suspension was added under vigorous stirring, and the pH was adjusted to 3.5 using ammonia solution and nitric acid. The suspension was transferred to a Teflon-lined stainless steel autoclave that was operated at a 120 °C for 24 h. Finally, the suspension was centrifuged, washed with 1 M HCl and deionized water and dried at 80 °C for 24 h.
The loading percentage of the carbonaceous materials on the TiO2 support was 0.1% for all photocatalytic experiments.
2.2. Characterization
IR spectra were measured for KBr – supported test samples (<1 wt%) at room temperature over a frequency range of 4000–400 cm−1 at a 4.0 cm−1 resolution with a scanning rate of 2 mm s−1, using a 6300 type A JASCO FT-IR spectrometer.X-ray diffraction (XRD) patterns were obtained with a Bruker D8 diffractometer equipped with a Lynxeye detector using CuKα radiation (λ = 15.406 × 10−2 nm). The diffractometer was operated at 40 kV and 40 mA. The data were collected between 10 and 80° with a scan step of 0.015° and a step time of 0.2 s. For phase identification purposes, the present investigation adopted the automatic JCPDS library search and match. The following computer software packages were used: Standard SERACH and DIFFRACT AT software (Australia).
Raman spectra were recorded on an InVia Raman microspectrometer working under macro conditions (f = 3 cm) with an excitation line of 532 nm. The samples were measured directly (without any pre-treatment), and the laser power was ∼2 mW (in all cases). The spectra were obtained from an average spectrum of 5 different registration of the Raman spectrum of the same sample (parameters of each registration: spectral range was from 100–3500 cm−1; number of accumulations was 3 and exposure time was 10 s).
UV-Vis diffuse reflectance spectra (DRS) were measured for the KBr – supported test samples (<1 wt%) at room temperature in the range 200–800 nm at a resolution of 0.05 nm using a Cary 5000 UV-Vis-NIR (Agilent, Australia) with an integrating sphere accessory.
X-ray Photoelectron Spectroscopy (XPS) was conducted using a Thermo Scientific ESCALAB-250Xi spectrometer. The radiation source was a monochromatic AlKα operating at a power of 300 W (15 kV, 20 mA). The vacuum in the analysis chamber was lower than 7 × 10−9 bar during all measurements. The in situ reduction was carried out in a high-pressure gas cell housed in the preparation chamber. The binding energies were referenced to the C 1s peak of the carbon contamination at 284.8 eV within an experimental error of ±0.2 eV.
The surface areas were measured using an automatic ASAP 2010 Micromeritics sorpometer (USA) which is equipped with an outgassing platform and an online data acquisition and handling system. It was operated using various computer-run methods to analyse the adsorption data.
For the electrocatalytic measurements, 1 mg of the composite was dispersed in 1 mL of 1% Nafion (diluted from 5 wt% Nafion using isopropyl alcohol, both from Sigma-Aldrich) and then sonicated for 2 hours. 5 μL of the dispersion was casted on a glassy carbon electrode (GC) that was held upside down and was allowed to dry for 2 hours. The electrode was electrochemically characterized towards ORR in the presence and absence of UV radiation (300 watt) using the linear sweep voltammetry (LSV) technique at a scan rate of 50 mV s−1 in an oxygen-saturated 0.5 M H2SO4 solution. During the LSV measurements, the potential was swept from the more to the less noble direction. The counter and reference electrodes were a Pt wire and a Ag/AgCl electrode, respectively. For comparison, a deaerated 0.5 M H2SO4 solution (purged with Ar gas for 30 min) was used. During the measurements, Ar gas was flowed continuously above the solution to prevent the diffusion of O2 to the deaerated solution.
2.3 Photocatalytic reactions
A phenol solution of 20 mg L−1 was prepared in deionized water. In each test, 0.1 g of one of the prepared catalysts was added to 100 mL of phenol solution and mixed using a magnetic stirrer in the dark for 30 min until the adsorption equilibrium was reached. Then, the suspension was illuminated using a high pressure mercury lamp (300 W) emitting ultraviolet radiation with a maximum radiation peak of 365 nm. No pH adjustment of the phenol solution was performed. The distance between the solution and the radiation source was 4.5 cm. The time at which the ultraviolet lamp was turned on was considered “time zero” the beginning of the experiment. Samples were taken at 10 min intervals from the reaction vessel and filtered to remove the suspended composite particles using nylon filter paper of a pore size 0.4 μm. The concentration of the phenol was determined using the UV-Vis spectrophotometer with a UV absorbance range of 190 to 400 nm, and the 269 nm absorbance corresponded to the maximum absorption of phenol.3. Results and discussion
3.1 Characterization
Fig. 1a shows the FT-IR spectrum of the initial graphite powder. A peak located at 3436 cm−1 corresponds to the absorbed water while the peak at 1631 cm−1 can be assigned either to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration band24 or to the deformation of water molecules.25 After the graphite powder oxidation process using the modified Hummers' method, the FT-IR spectrum of the produced GO powder was measured as shown in Fig. 1b. A red shift in the CC peak was observed in which the peak was shifted from 1631 to 1621 cm−1. The absorption band between 1720 and 1736 cm−1 corresponds to the carboxylic and carbonyl groups.26–28 Additionally, the absorption peaks at 1382–856 cm−1 are ascribed to epoxy and alkoxy groups.29–33 A broad band between 3900–3500 cm−1 is due to the O–H stretching vibrations of C–OH groups (from hydroxyl and/or carboxylic groups) and intercalated water molecules.24,32,34 These results confirm that the oxidation of graphite powder took place and the functional groups on the GO surface are mainly composed of hydroxyl, epoxy, carbonyl and carboxylic groups.35 However, the graphite oxidation process is incomplete, and this was confirmed by the peak at 1621 cm−1, which corresponds to the remaining sp2 character.36 After reducing the GO either by hydrogen gas (Fig. 1c) or hydrazine hydrate (Fig. 1d), the number of the oxygen functional groups decreased drastically37 and a new absorption peak at approximately 1566 cm−1 was detected that may be attributed to the skeletal vibration of the graphene sheets.13,36,38–40 Fig. 1e and f compare the FT-IR spectra of TiO2 and TiO2-supported GO (GOTi), respectively, in which a strong broad absorption band at low frequency (below 1000 cm−1) was measured. This band was attributed to the vibration of Ti–O–Ti in TiO2, similar to that of pure commercial P25-TiO2.18,29,39,41 The absorption band at 1633 cm−1 can be attributed to the Ti–O–Ti stretching vibration25 or to the deformation of water molecules. No absorption peak corresponding to the GO was observed. This is most likely due to the low amount of GO in the composite materials (0.1%).
C stretching vibration band24 or to the deformation of water molecules.25 After the graphite powder oxidation process using the modified Hummers' method, the FT-IR spectrum of the produced GO powder was measured as shown in Fig. 1b. A red shift in the CC peak was observed in which the peak was shifted from 1631 to 1621 cm−1. The absorption band between 1720 and 1736 cm−1 corresponds to the carboxylic and carbonyl groups.26–28 Additionally, the absorption peaks at 1382–856 cm−1 are ascribed to epoxy and alkoxy groups.29–33 A broad band between 3900–3500 cm−1 is due to the O–H stretching vibrations of C–OH groups (from hydroxyl and/or carboxylic groups) and intercalated water molecules.24,32,34 These results confirm that the oxidation of graphite powder took place and the functional groups on the GO surface are mainly composed of hydroxyl, epoxy, carbonyl and carboxylic groups.35 However, the graphite oxidation process is incomplete, and this was confirmed by the peak at 1621 cm−1, which corresponds to the remaining sp2 character.36 After reducing the GO either by hydrogen gas (Fig. 1c) or hydrazine hydrate (Fig. 1d), the number of the oxygen functional groups decreased drastically37 and a new absorption peak at approximately 1566 cm−1 was detected that may be attributed to the skeletal vibration of the graphene sheets.13,36,38–40 Fig. 1e and f compare the FT-IR spectra of TiO2 and TiO2-supported GO (GOTi), respectively, in which a strong broad absorption band at low frequency (below 1000 cm−1) was measured. This band was attributed to the vibration of Ti–O–Ti in TiO2, similar to that of pure commercial P25-TiO2.18,29,39,41 The absorption band at 1633 cm−1 can be attributed to the Ti–O–Ti stretching vibration25 or to the deformation of water molecules. No absorption peak corresponding to the GO was observed. This is most likely due to the low amount of GO in the composite materials (0.1%).
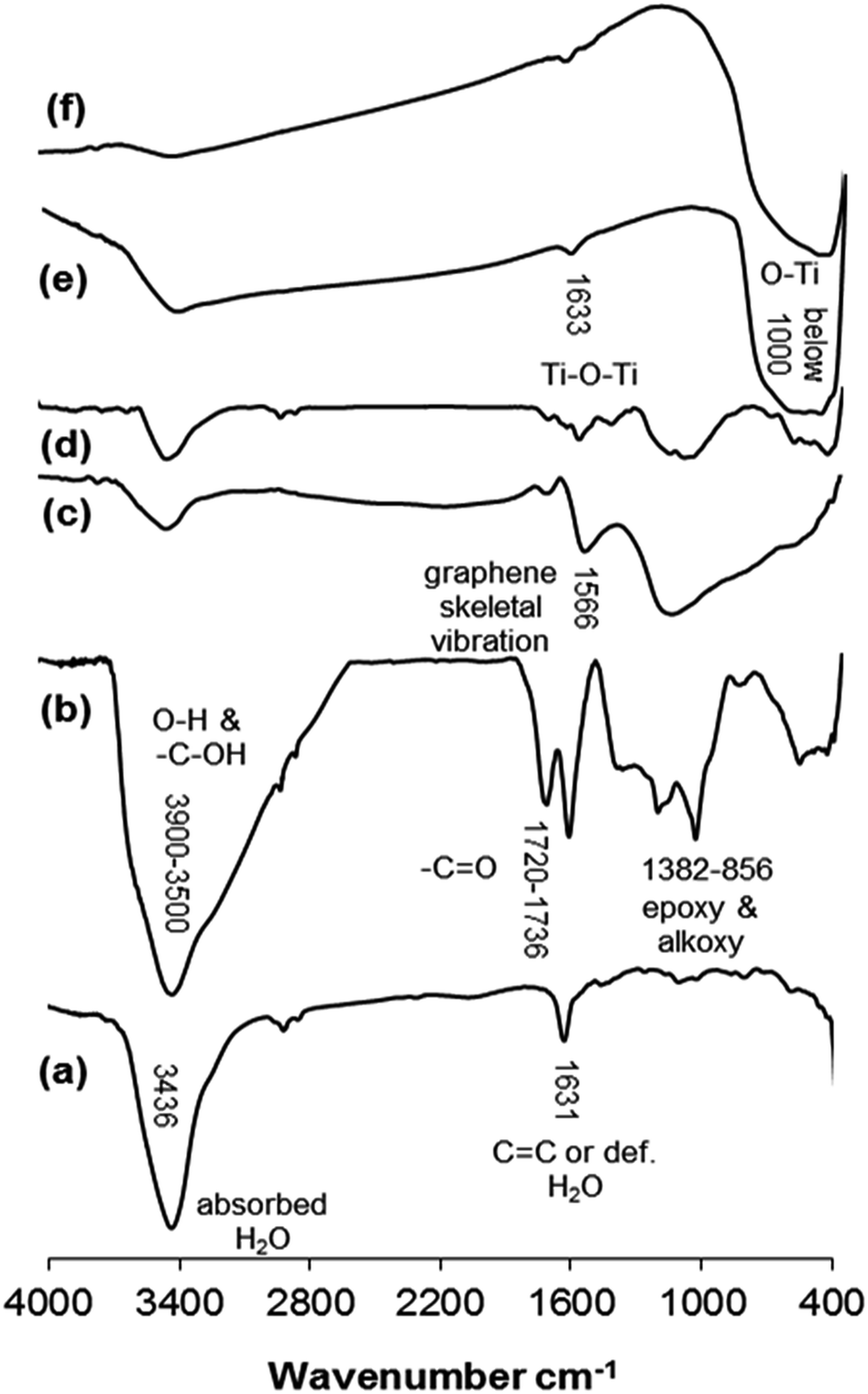 | ||
| Fig. 1 FT-IR spectra of (a) graphite, (b) GO, (c) H2RGO, (d) HHRGO, (e) Ti, (f) GOTi. | ||
Fig. 2 displays the XRD patterns of the unsupported (a) graphite, (b) as-prepared GO, (c) H2RGO, and (d) HHRGO. The graphite powder in Fig. 2a shows a typical diffraction peak at 2θ = 26.6° with an interlayer spacing of 3.35 Å that corresponds to the 002 facet. This peak disappeared upon oxidation, and a new peak at 2θ = 11.6° corresponds to the interlayer spacing of 7.78 Å was appeared as shown in Fig. 2b. The increase in the d-spacing is due to the presence of oxygen functional groups that facilitated the hydration and exfoliation of the graphene sheet in aqueous media and confirms the oxidation of the graphite powder.30,42 After reduction of GO with hydrogen (Fig. 2c), the diffraction peaks at 2θ = 11.6 and 26.6° disappeared and a new broad peak at 2θ = 25.8° with a corresponding d-spacing of 3.53 Å was recorded. This peak was also observed by Thema et al. upon reduction of GO using hydrazine hydrate.37 The XRD pattern of HHRGO (Fig. 2d) also showed the disappearance of the diffraction peaks at 26.6 and 11.6° and the presence of a new small broad peak at 2θ = 13.0° with a d-spacing of 6.80 Å. From these observations, we can conclude that the structure of HHRGO is different from H2RGO, and the reduction process was not complete in both cases because the d-spacing is higher than that of the parent graphite powder. In addition, based on the d-spacing, the extent of reduction for H2RGO is higher than HHRGO. Most likely, in the case of HHRGO, the small peak at 2θ = 13.0° is due to the formation of a new structure of GO with a lower oxygen content because its corresponding d-spacing (6.80 Å) lies within the range of 6.3 to 9.0 Å for GO28,38,43 that is prepared from graphite using the Hummers' method.23 Additionally, HHRGO has an amorphous structure that is consistent with other reports in the literature.42,44,45
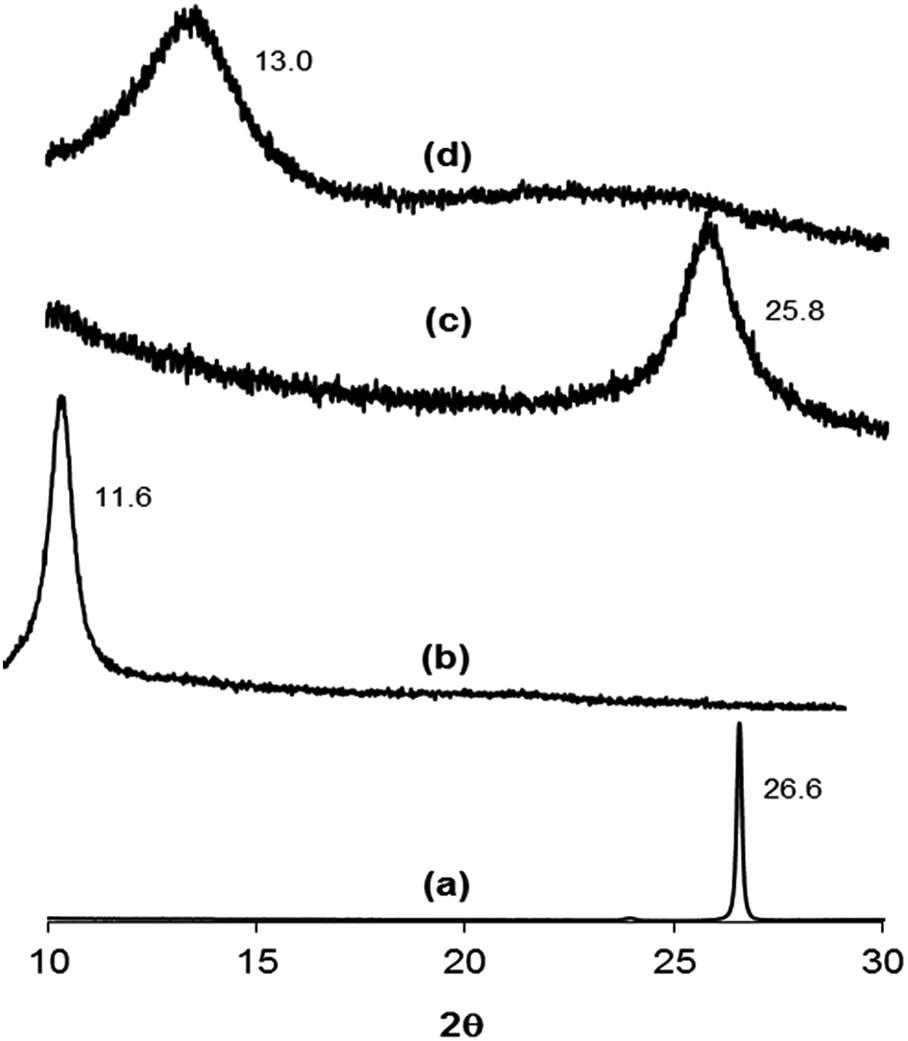 | ||
| Fig. 2 XRD patterns of (a) graphite, (b) GO, (c) H2RGO, (d) HHRGO. | ||
Fig. 3 shows the XRD patterns of (a) TiO2, (b) GOTi, (c) H2RGOTi, (d) HHRGOTi, and (e) RGOTi. The XRD patterns of all composites look similar to that of the neat TiO2 (Fig. 3a), which consists of a mixture of anatase (A) and rutile (R) phase with no detected diffraction pattern for the carbon material. Similar results were observed in previous reports.13,15,19,25,41,46 Therefore, we conclude that the addition of carbon species did not change the crystalline phases of TiO2, and the disappearance of their diffraction patterns may be attributed to: (1) their low percentage in the composite materials (0.1%), (2) their low diffraction intensity (below the detection limit of the instruments) or (3) their overlapping patterns with the anatase phase pattern at 2θ value of 25.4°.13,16–19,47,48
 | ||
| Fig. 3 XRD patterns of (a) TiO2, (b) GOTi, (c) H2RGOTi, (d) HHRGOTi, (e) RGOTi. | ||
Raman spectroscopy was used to distinguish between the ordered and disordered crystal structures of carbon. Spectra that were measured at 532 nm contained the D band, which is a disordered mode that is related to the presence of sp3 defects. This results from a breathing mode of κ-point phonons of A1g symmetry. The G band is assigned to the E2g phonon of C sp2 atoms, while the 2D band corresponds to the overtone of the D band and appears in the second-order Raman spectra of the crystalline graphite (free of disorder).33,49–52 Furthermore, the ratio of the intensities of the D and G band (ID/IG) can be used as an indication of the amount of functionalization in a carbon material. A high ID/IG ratio means a high degree of disorder, i.e., a high degree of functionalization.53,54 Fig. 4 shows the Raman spectra of (a) graphite, (b) GO, (c) H2RGO, and (d) HHRGO. As shown in Fig. 4a, the spectrum of the graphite exhibited a strong G band at 1567 cm−1, a very weak D band at 1321 cm−1 and a 2D band at 2689 cm−1. Upon graphite oxidation (Fig. 4b), the D and G bands widened and shifted towards higher wave numbers at 1339 cm−1 and 1588 cm−1, respectively. The blue shift in the G band is attributed to the existence of isolated double bonds that resonate at higher frequency than the G band in graphite.55,56 The decrease in the G band intensity and the increase of the D band confirms a reduction in the size of the in-plane sp2 domains due to the incorporation of functional groups that form sp3 bonds in the carbon network.33,57 In addition, broad peaks at approximately 2903 and 3173 cm−1 were detected that correspond to a D + G combination mode that is induced by a disorder50 and an overtone of the G line (G′), respectively.
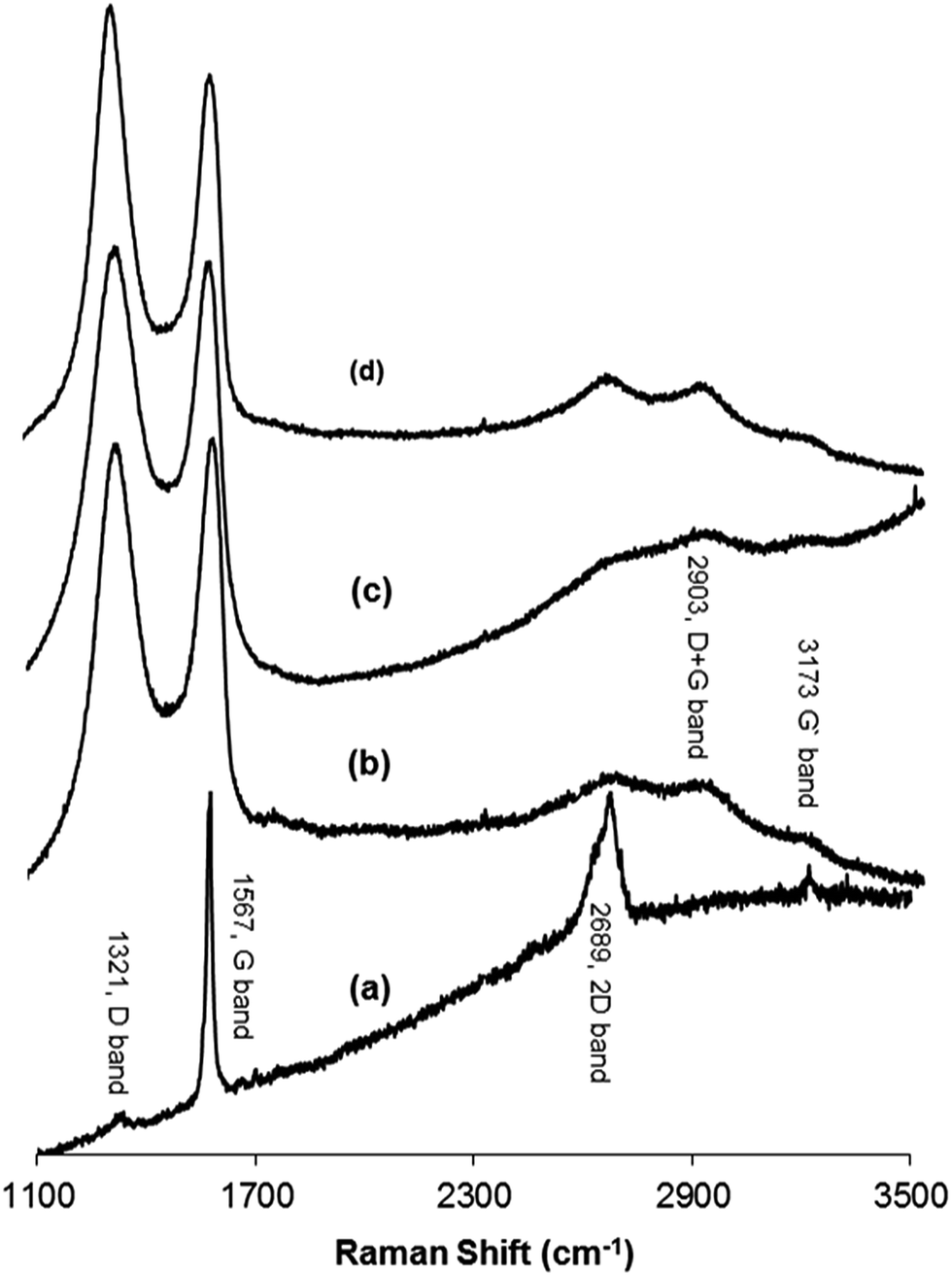 | ||
| Fig. 4 Raman spectra at 532 nm of (a) graphite, (b) GO, (c) H2RGO450, (d) HHRGO. | ||
When GO was reduced by hydrogen (Fig. 4c), the D band location did not change while the G band was shifted to a lower frequency region (1579 cm−1), confirming the reduction process.47,58 A decrease in the relative intensity of the D band was observed, which resulted in a decrease in the D/G intensity ratio of H2RGO (0.87) compared to the GO (0.90). These changes suggest an increase in the sp2 domain (i.e., more graphitization due to the reduction process), and the results prove that the structure of H2RGO is more ordered with less defects than the structure of GO.53 However, for HHRGO (Fig. 4d), the D and G bands shifted slightly to a lower frequency and the ratio of the D/G bands (1.04) was increased compared to the GO (0.90). These changes are attributed to the decrease in the average size of the sp2 domain where new graphitic domains were created that are higher in number and smaller in size than those present in GO.33,51,59 Therefore, we conclude that the structures of H2RGO and HHRGO are different, and the structure of the H2RGO is more ordered than the HHRGO. This was further supported by the results obtained from the XRD (compare Fig. 2c and d). According to Pimenta et al.,50 the crystalline size can be determined by eqn (1):
 | (1) |
 | ||
| Fig. 5 Raman spectra at 532 nm of (a) Ti, (b) GOTi, (c) H2RGOTi, (d) HHRGOTi. | ||
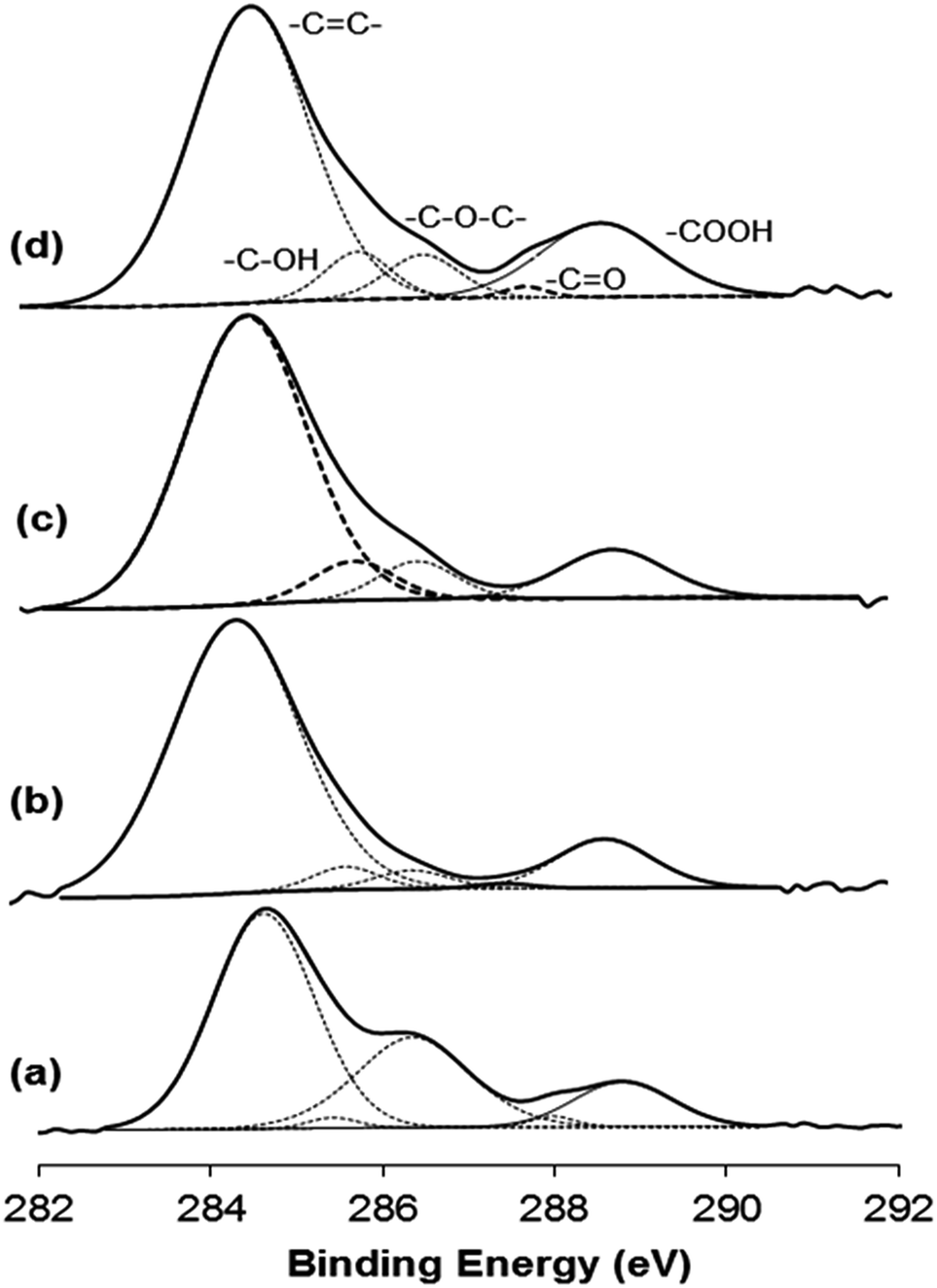
Fig. 6 reveals the C region of the XPS spectra of graphite, GO, H2RGO and HHRGO. Graphite exhibited a strong spectral line at 284.68 eV for carbon and a small broad peak at 285.33 eV for some adsorbed species on the sample's surface that likely represent water molecules (Fig. 6a). In the GO spectrum (Fig. 6b), the peak with a binding energy of 284.64 eV can be attributed to the C–C and CC bonds, while peaks centered at binding energies of 285.49, 288.60, 287.52 and 288.69 eV can be assigned to the –C–OH, –C–O–C–, –C–CO and –COOH, respectively.61 This proves the successfulness of the oxidation process of graphite. During the in situ reduction using hydrogen at 450 °C for 30 min, carboxylic, carbonyl and epoxy groups were reduced while hydroxyl groups were increased (Fig. 6c). This clearly confirms a considerable degree of reduction of GO, but the reduction process was incomplete when small amounts of oxygen functional groups were still detected. The same situation was observed during the reduction of GO with hydrazine hydrate (Fig. 6d), but the extent of reduction when hydrogen gas was used was more than when hydrazine hydrate was employed. These results are consistent with the results obtained from the XRD and the Raman spectra.
 | ||
| Fig. 6 Carbon region of XPS spectra of (a) graphite, (b) GO, (c) H2RGO, (d) HHRGO. | ||
Fig. 7 shows the XPS spectra of (a) GOTi, (b) H2RGOTi, (c) HHRGOTi and (d) RGOTi. In Fig. 7a, the same functional groups are shown as for unsupported GO (shown in Fig. 6b). However, after reduction (Fig. 7b), the peak corresponding to a carboxylic acid group did not disappear. In fact, its intensity was increased compared to the corresponding spectra shown for the reduced GOTi. This is opposite to what was observed for unsupported RGO (Fig. 6c). To explain this observation, the XPS of commercial TiO2 was measured and subjected to a hydrogen flow at 450 °C for 1 h (Fig. 8). It is obvious from the carbon region in the XPS spectra of TiO2 that the carboxylic acid groups exist in commercial TiO2, and they were not reduced after their exposure to the stream of H2 gas at 450 °C for 1 h (Fig. 8a). A small shift of 0.75 eV for the Ti 2p3/2 (458.66 eV) and Ti 2p1/2 (465.34 eV) spin orbit coupling peaks to higher binding energies after exposure to hydrogen gas at 450 °C was observed (Fig. 8b). However, the spacing between them remains the same before and after reduction with hydrogen (5.68 eV). Therefore, it can be concluded that the carboxylic acid groups are strongly bonded to the TiO2 most likely through the oxygen of the TiO2. As expected, the oxidation number of titanium did not change because the reduction of Ti(IV) to lower valence state(s), such as Ti2O3, does not commence at temperatures less than 1000 °C.62 Furthermore, the reduction process can even promote the bonding between TiO2 and carboxylic acid groups because it has been observed that the amount of carboxylic groups is increased after reduction compared to GOTi.
 | ||
| Fig. 7 Carbon region of XPS of (a) GOTi, (b) H2RGOTi, (c) HHRGOTi, (d) RGOTi. | ||
 | ||
| Fig. 8 XPS of the carbon (on left) and Ti regions (on right) for commercial TiO2 (a) before and (b) after hydrogen reduction using a stream of H2 gas at 450 °C for 1 h. | ||
An etching experiment using argon bombardment was carried out for GOTi to obtain information regarding which material is the top layer. It is clear from Table 1 that GO is on the surface of TiO2 because the amount of Ti increased while the amount of GO decreased as the etching time increases.
| Titanium (%) | Etching time (s) | Etching depth (nm) | Carbon (%) |
|---|---|---|---|
| 43.19 | 15 | 1 | 4.71 |
| 47.60 | 40 | 3 | 0.90 |
| 48.34 | 65 | 5 | 0.68 |
| 49.51 | 90 | 7 | 0.34 |
| 50.09 | 125 | 10 | 0.31 |
| 50.36 | 230 | 20 | 0.23 |
The surface areas of the different prepared and used materials in this study are summarized in Table 2. It was observed that surface area of graphite is increased from 20.5 to 40.3 m2 g−1 upon its oxidation, and it is further increased when the GO is reduced by hydrazine hydrate or hydrogen gas. However, the surface area of H2RGO is 343 m2 g−1, which is more than that of HHRGO (256.4 m2 g−1). There were no significant changes in the BET surface areas of the TiO2, GOTi and the reduced GOTi composites. Similar observations have been noted by other researchers.18,26
| Material | BET (m2 g−1) |
|---|---|
| Graphite | 20.5 |
| GO | 40.3 |
| H2RGO | 343.0 |
| HHRGO | 256.4 |
| TiO2 | 51.1 |
| GOTi | 50.0 |
| RGOTi | 50.2 |
| H2RGOTi | 47.7 |
| HHRGOTi | 50.9 |
| RGO | 49.1 |
Fig. 9 displays the linear sweep voltammograms for (a) GC, (b) TiO2/GC (Ti) (c) GOTi/GC and (d) H2RGOTi/GC electrodes in (i & ii) oxygen and (iii & iv) argon-saturated 0.5 M H2SO4 at 25 °C in presence (ii & iv) and absence (i & iii) of 300 watt UV radiation. The potential sweep started from the more noble direction where a very small current flows to the less noble one where the over potential increases and the oxygen reduction reaction is expected to start. This assisted in the determination of the ORR onset potential accurately. The UV-radiation affects the oxygen reduction reaction significantly as seen from the difference between the black and the red E-I curves in Fig. 9. Generally, it is reported that the mechanism of ORR on TiO2 surface under UV radiation follows a series of single electron steps as follows: (e: electron, h: hole, cb: conduction band and vb: valence band):63
| TiO2 + hν → ecb− + hvb+ | (2) |
| O2 + ecb− → O2˙− | (3) |
| H+ + O2˙− → HO˙2 (at pH < 4.8) | (4) |
| 2HO˙2 → H2O2 + O2 | (5) |
| HO˙2 + ecb− + H+ → H2O2 | (6) |
 | ||
| Fig. 9 Linear Sweep Voltammograms (LSVs) at a scan rate of 50 mV s−1 for (a) GC, (b) Ti/GC, (c) GOTi/GC and (d) H2RGOTi/GC electrodes in (i & ii) oxygen and (iii & iv) argon-saturated 0.5 M H2SO4 at 25 °C in the presence (ii & iv) and absence (i & iii) of 300 watt UV radiation. | ||
The best onset reduction potential (the most noble) for ORR is for GC (−350 mV), but the highest current at any given potential was observed for H2RGO/Ti (e.g., −1.4 × 10−4 A at a potential of −0.6 V). Furthermore, only H2RGOTi exhibited a high ORR rate under UV illumination at any potential. This was attributed to the lowering in the band gap energy of TiO2 when composited with reduced graphene oxide. Table 3 shows the band gap energies for the different catalysts and composites that were used in this work.
| Catalysts | Wavelength (nm) | Band gap energy |
|---|---|---|
| Ti | 393 | 3.16 |
| GOTi | 403 | 3.08 |
| RGOTi | 391 | 3.11 |
| HHRGOTi | 414 | 3.0 |
| H2RGOTi | 410 | 3.03 |
The UV radiation boosted the ORR kinetics for Ti (P25-TiO2) at less noble potentials but decreased it at the more noble potentials (Fig. 9b). On the contrary, the results were reversed for GOTi/GC in which the kinetics of the ORR was enhanced as the potential was scanned in the less noble direction. For the GC, both behaviors were observed in which the UV radiation only enhanced the ORR kinetics between −0.390 and −0.565 V. At potentials more noble than −0.390 V and less noble than −0.565, the ORR kinetics were retarded in the presence of UV radiation compared to when UV radiation was absent. Furthermore, the GOTi (Fig. 9c) did not show any significant difference between the ORR kinetics in the presence or absence of UV radiation at small overpotential. However, the noticed enhancement in the ORR kinetics at high overpotentials is attributed to the reduction of GO in the GOTi composite, i.e., the composite changed from GOTi to RGOTi (similar to Fig. 9d plot ii). This can be confirmed by comparing plots ii and iv in Fig. 9c where in absence of O2 (plot iv of Fig. 9c), the current started to increase at a potential of −0.53 V. This is almost the same potential at which the current in the presence of UV started to be higher than the corresponding potential in the absence of UV. Fig. 9 does not show the results for HHRGOTi/GC or RGOTi/GC because those electrocatalytic results were not comparable with those of GC and the H2RGOTi electrodes. This indicated that the reduction method of graphene oxide affected significantly the electrocatalytic behavior of the composite. It is worthy to mention that the measurements were repeated with the UV lamps of different powers, and the resulting trends did not change.
3.2 Photocatalytic reactions
In all cases, a 0.1% loading of carbonaceous species was used in the composite. Higher loading percentages of the “black” carbonaceous material increased its light absorption and consequently decreased the photocatalytic activity of TiO2. No detectable degradation of phenol was observed in the absence of TiO2 in any of the as-prepared composites. The kinetic plots corresponding to the degradation of a 20 mg L−1 of phenol (in water) in presence of (i) commercial TiO2 nanoparticles, (ii) GOTi, (iii) HHRGOTi, (iv) H2RGOTi or (v) RGOTi photocatalysts and in absence of H2O2 are plotted against UV irradiation time in Fig. 10.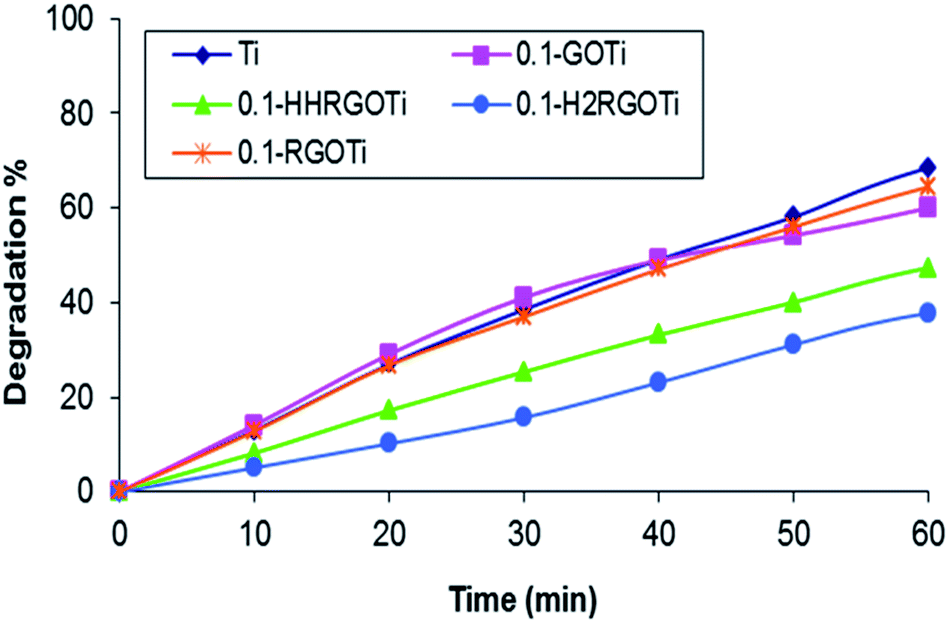 | ||
| Fig. 10 Kinetics of phenol degradation (20 mg L−1) under 300 W UV illumination in the presence of (a) Ti, (b) GOTi, (c) H2RGOTi, (d) HHRGOTi and (e) RGOTi catalysts. | ||
The degradation % represents (C0 − Ct/C0) * 100, where C0 is the remaining concentration of phenol after adsorption equilibrium before UV illumination. The Ct represents the remaining phenol concentration after UV illumination for a specific time. We observed that the phenol degradation is increased with UV irradiation time for all catalysts. The best phenol degradation percentage (fastest rate) was obtained in the case of TiO2, then RGOTi catalysts and the lowest degradation rate was observed for the H2RGOTi catalyst. However, the difference in the performance between TiO2 and RGOTi is small under the experimental conditions studied. A 38.5% conversion of phenol was observed after 30 min of UV illumination in the presence of commercial TiO2 and was increased to 58.1% after 50 min. For RGOTi, 37.0% of phenol degradation was achieved after 30 min of UV irradiation and was increased to 56.0% after 50 min. Different results were obtained with the addition of 70 μL H2O2 (Fig. 11). A faster phenol degradation was attained using RGOTi (Fig. 11e) than using the commercial TiO2 (Fig. 11a) at all times. For example, 42.6% and 71.1% conversions were obtained after 30 min and 50 min UV illumination respectively in the presence of RGOTi with addition of H2O2 compared to 31.8% and 66.0% conversions in case of TiO2 under same experimental conditions. Furthermore, in case of RGOTi at 30 and 60 min, phenol degradation rate is higher by 15% and 27%, respectively, compared to the experiment in which H2O2 was not added (Fig. 10).
 | ||
| Fig. 11 Kinetics of phenol degradation (20 mg L−1) under 300 W UV illumination in the presence of (a) Ti, (b) GOTi, (c) H2RGOTi, (d) HHRGOTi and (e) RGOTi catalysts. A 70 μL of H2O2 was used with all catalysts. | ||
Upon addition of 70 μL of the H2O2 and keeping constant all other experimental conditions, comparative studies were carried out between TiO2 and RGOTi with different phenol concentrations (see Fig. S1 and S2 in the ESI†). For both TiO2 and RGOTi, phenol degradation rate decreased with increasing the phenol concentration. However, for 40 mg L−1 phenol, the decrease in the degradation rate was more obvious for the commercial TiO2 than for the RGOTi. For example, after 60 min of UV illumination, a 39.2% conversion was attained in case of TiO2 while a 52.5% conversion was obtained in the case of RGOTi. It is worthy to mention here that, although H2RGOTi and HHRGOTi have the lowest band gap energies and consequently were expected to have the best photocatalytic behaviour compared to TiO2, the opposite exactly happened where they have shown the worst photocatalytic performance. This confirms that the band gap energy is not the only controlling factor when comparing between the different photocatalysts shown here. Furthermore, the surface area of the catalysts (see Table 3) is the controlling factor when H2O2 is absent but in presence of it, RGOTi had the highest photocatalytic activity. The reason for this is attributed to the more stability of H2O2 and its radicals in presence of RGOTi compared to TiO2.
The effect of H2O2 concentration on the rate of phenol degradation was studied (see Fig. S3 in the ESI†). A little higher degradation rate was observed upon adding 100 μL H2O2. The addition of 70 μL and 40 μL of H2O2 had nearly the same effect on the rate of phenol degradation before 40 min. At time higher than 40 min, phenol degradation rate was decreased in case of using 40 μL of H2O2.
It is worth mentioning that the kinetics curves in case of the bare TiO2 and the different composites, except RGOTi, (Fig. 11) have two slopes: one at the beginning of the experiment up to 30 minutes and another one after 30 min. The slope after 30 min is higher than the one before where a kind of activation for the catalyst has taken place after 30 min. In case of RGOTi, the kinetics curves are always linear (i.e. one slope) as can be seen in Fig. 11, S2 and S3.†
The effect of the UV radiation power was also studied using 0.1 g of RGOTi in the presence of 70 μL H2O2. Changing the UV lamp power from 150 to 300 to 500 W did not change the rate of phenol degradation on the RGOTi catalyst (see Fig. S4 in the ESI†).
4. Conclusions
Four composites with different band gap energies were prepared using (a) commercial TiO2 and (b) one of the following synthesized carbonaceous materials: GO, H2RGO, HHRGO or RGO. The hydrogen reduction method resulted in a more ordered graphene structure that has a higher surface area compared with the thermally and chemically reduced GO.Chemical and physical characterization techniques documented many differences between GO that were reduced using different reduction methods.
In terms of the measured oxygen reduction currents, H2RGOTi composite has shown a superior electrocatalytic behaviour towards ORR under UV illumination compared with GC and the other reduced graphene oxide/TiO2 composites. At −0.6 V SCE, the highest rate for ORR followed this order: H2RGOTi/GC > GOTi/GC ≈ Ti/GC > GC which might be useful for the in situ generation of H2O2 during an advanced oxidation process.
Also, the effect of the GO reduction method on the photocatalytic activity of the aforementioned composites towards the degradation of phenol in the presence and absence of (i) UV and (ii) H2O2 was examined. Although the band gap energy for HHRGOTi and H2RGOTi are the lowest (3 and 3.03 eV) compared to TiO2 (3.16 eV), high phenol degradation rates were achieved using TiO2 the RGOTi photocatalyst (3.11 eV) under UV illumination in absence and presence of H2O2, respectively. This proved that the band gap energy is not the only controlling parameter for the photocatalysts used for phenol degradation. In fact the reason that RGOTi has the best photocatalytic behavior in presence of H2O2 can be attributed to the number and type of oxygen functional groups on the surface of RGOTi in addition to the stability of H2O2 and its radicals on the surfaces of RGOTi compared to TiO2 and the other prepared photocatalysts.
Acknowledgements
This work was supported and co-funded by the Kuwait Foundation for Advancement of Sciences (KFAS) through project No. 2012-1405-01 and the Public Authority for Applied Education and Training (PAAET) through research project No. HS-12-02. Research Title: Photocatalytic Oxidative Removal of Phenolic Compounds from Wastewater Using Ozone and Hydrogen Peroxide Produced by Advanced Electrodes.References
- Z. Wu, Y. Cong, M. Zhou, Q. Ye and T. Tan, Korean J. Chem. Eng., 2002, 19, 866–870 CrossRef CAS.
- A. Ortiz-Gomez, B. Serrano-Rosales, M. Salaices and H. de Lasa, Ind. Eng. Chem. Res., 2007, 46, 7394–7409 CrossRef CAS.
- M. Tian, G. Wu, B. Adams, J. Wen and A. Chen, J. Phys. Chem. C, 2008, 112, 825–831 CAS.
- J. Yang, J. Dai, C. Chen and J. Zhao, J. Photochem. Photobiol., A, 2009, 208, 66–77 CrossRef CAS PubMed.
- M. Pera-Titus, V. García-Molina, M. A. Baños, J. Giménez and S. Esplugas, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS PubMed.
- J. Yan, W. Jianping, B. Jing, W. Daoquan and H. Zongding, Biochem. Eng. J., 2006, 29, 227–234 CrossRef CAS PubMed.
- W. Z. Tang and A. Huren, Chemosphere, 1995, 31, 4171–4183 CrossRef CAS.
- L. H. Jack, J. Catal., 2003, 216, 455–460 CrossRef.
- M. A. O. Badmus, T. O. K. Audu and B. U. Anyata, Afr. J. Biotechnol., 2007, 6, 238–242 CAS.
- A. Wei, J. Wang, Q. Long, X. Liu, X. Li, X. Dong and W. Huang, Mater. Res. Bull., 2011, 46, 2131–2134 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, L. Gao, B. Liu, C. Jiang and H.-M. Cheng, Carbon, 2009, 47, 493–499 CrossRef CAS PubMed.
- V. Štengl, S. Bakardjieva, T. M. Grygar, J. Bludská and M. Kormunda, Chem. Cent. J., 2013, 7, 1–12 CrossRef PubMed.
- S. Liu, H. Sun, S. Liu and S. Wang, Chem. Eng. J., 2013, 214, 298–303 CrossRef CAS PubMed.
- C. H. Kim, B.-H. Kim and K. S. Yang, Carbon, 2012, 50, 2472–2481 CrossRef CAS PubMed.
- H. Zhao, F. Su, X. Fan, H. Yu, D. Wu and X. Quan, Chin. J. Catal., 2012, 33, 777–782 CrossRef CAS.
- D. Wang, X. Li, J. Chen and X. Tao, Chem. Eng. J., 2012, 198–199, 547–554 CrossRef CAS PubMed.
- F. Wang and K. Zhang, J. Mol. Catal. A: Chem., 2011, 345, 101–107 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380–386 CrossRef PubMed.
- N. R. Khalid, E. Ahmed, Z. Hong, L. Sana and M. Ahmed, Curr. Appl. Phys, 2013, 13, 659–663 CrossRef PubMed.
- F. S. Omar, H. Nay Ming, S. M. Hafiz and L. H. Ngee, Int. J. Photoenergy, 2014, 2014, 8 CrossRef.
- M. K. Kavitha, S. C. Pillai, P. Gopinath and H. John, J. Environ. Chem. Eng., 2015, 3, 1194–1199 CrossRef CAS PubMed.
- Y. H. Ng, A. Iwase, N. J. Bell, A. Kudo and R. Amal, Catal. Today, 2011, 164, 353–357 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. Hu, Y. Wang, J. Chai, R. Gao, Z. Yang, E. S.-W. Kong and Y. Zhang, Sensor. Actuat. B-Chem., 2012, 163, 107–114 CrossRef CAS PubMed.
- H. Liu, X. Dong, X. Wang, C. Sun, J. Li and Z. Zhu, Chem. Eng. J., 2013, 230, 279–285 CrossRef CAS PubMed.
- H.-L. Ma, Y. Zhang, Q.-H. Hu, D. Yan, Z.-Z. Yu and M. Zhai, J. Mater. Chem., 2012, 22, 5914–5916 RSC.
- M. Z. Kassaee, E. Motamedi and M. Majdi, Chem. Eng. J., 2011, 172, 540–549 CrossRef CAS PubMed.
- T. Kyotani, K.-y. Suzuki, H. Yamashita and A. Tomita, Tanso, 1993, 160, 255–265 CrossRef CAS.
- J. Guo, S. Zhu, Z. Chen, Y. Li, Z. Yu, Q. Liu, J. Li, C. Feng and D. Zhang, Ultrason. Sonochem., 2011, 18, 1082–1090 CrossRef CAS PubMed.
- V. Loryuenyong, K. Totepvimarn, P. Eimburanapravat, W. Boonchompoo and A. Buasri, Adv. Mater. Sci. Eng., 2013, 1–5 CrossRef PubMed.
- J. Song, X. Wang and C.-T. Chang, J. Nanomater., 2014, 1–6 Search PubMed.
- Y.-p. Zhang, J.-j. Xu, Z.-h. Sun, C.-z. Li and C.-x. Pan, Prog. Nat. Sci., 2011, 21, 467–471 CrossRef.
- J. Oh, N. D. Luong, T. Hwang, J. Hong and J. Nam, ICCM-18, Jeju Island, South Korea, 2011 Search PubMed.
- M. E. Achaby, F. Z. Arrakhiz, S. Vaudreuil, E. M. Essassi and A. Qaiss, Appl. Surf. Sci., 2012, 258, 7668–7677 CrossRef PubMed.
- H. Al-Kandari, A. M. Abdullah, A. M. Mohammad and S. Al-Kandari, ECS Trans., 2014, 61, 13–26 CrossRef PubMed.
- Y. Yao, S. Miao, S. Liu, L. P. Ma, H. Sun and S. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef CAS PubMed.
- F. T. Thema, M. J. Moloto, E. D. Dikio, N. N. Nyangiwe, L. Kotsedi, M. Maaza and M. Khenfouch, J. Chem., 2013, 2–6 Search PubMed.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS PubMed.
- K. Zhou, Y. Zhu, X. Yang, X. Jiang and C. Li, New J. Chem., 2011, 35, 353–359 RSC.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2271–2288 CrossRef CAS PubMed.
- Z. Qianqian, B. Tang and H. Guoxin, J. Hazard. Mater., 2011, 198, 78–86 CrossRef PubMed.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS PubMed.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- C. Fu, Y. Kuang, Z. Huang, X. Wang, Y. Yin, J. Chen and H. Zhou, J. Solid State Electrochem., 2011, 15, 2581–2585 CrossRef CAS.
- H. M. Hassan, V. abdelsayed, A. E. R. S. Khder, K. M. AbouZeid, J. Terner, M. S. El-Shall, S. I. Al-Resayes and A. A. El-Azhary, J. Mater. Chem., 2009, 19, 3832–3837 RSC.
- D. F. Wang, D. Chen, G. X. Ping, C. Wang, H. Z. Chen and K. Y. Shu, Adv. Mater. Res., 2012, 430–432, 1005–1008 CAS.
- X. Zhou, T. Shi, J. Wu and H. Zhou, Appl. Surf. Sci., 2013, 287, 359–368 CrossRef CAS PubMed.
- Z. Li, B. Gao, G. Z. Chen, R. Mokaya, S. Sotiropoulos and G. L. Puma, Appl. Catal., B, 2011, 110, 50–57 CrossRef CAS PubMed.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238–242 CrossRef CAS PubMed.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1290 RSC.
- C. Xu, J. Wang, L. Wan, J. Lin and X. Wang, J. Mater. Chem., 2011, 21, 10463–10471 RSC.
- C. Casiraghi, S. Pisana, K. S. Novoselov, A. K. Geim and A. C. Ferrari, Appl. Phys. Lett., 2007, 91 Search PubMed.
- O. Akhavan, ACS Nano, 2010, 4, 4174–4180 CrossRef CAS PubMed.
- M. Alexander, V. Roumen, S. Koen, V. Alexander, Z. Liang, T. Gustaaf van, V. Annick and H. Chris van, Nanotechnology, 2008, 19, 305604 CrossRef PubMed.
- Z. Zhang, W. Yang, X. Zou, F. Xu, X. Wang, B. Zhang and J. Tang, J. Colloid Interface Sci., 2012, 386, 198–204 CrossRef CAS PubMed.
- W. Zhang, X. Shi, Y. Zhang, W. Gu, B. Li and Y. Xian, J. Mater. Chem. A, 2013, 1, 1745–1753 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- T. S. Sreeprasad, S. M. Maliyekkal, K. P. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS PubMed.
- T. N. Lambert, C. A. Chavez, B. Hernandez-Sanchez, P. Lu, N. S. Bell, A. Ambrosini, T. Friedman, T. J. Boyle, D. R. Wheeler and D. L. Huber, J. Phys. Chem. C, 2009, 113, 19812–19823 CAS.
- U. Balachandran and N. G. Eror, J. Solid State Chem., 1982, 42, 276–282 CrossRef CAS.
- K. Zhang, K. C. Kemp and V. Chandra, Mater. Lett., 2012, 81, 127–130 CrossRef CAS PubMed.
- C. Hauf, R. Kniep and G. Pfaff, J. Mater. Sci., 1999, 34, 1287–1292 CrossRef CAS.
- H. Sheng, H. Ji, W. Ma, C. Chen and J. Zhao, Angew. Chem., Int. Ed., 2013, 52, 9686–9690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra13065d |
| This journal is © The Royal Society of Chemistry 2015 |