
Human hair-derived graphene-like carbon nanosheets to support Pt nanoparticles for direct methanol fuel cell application
Guiqin Sun,
Lihui Zhou,
Jinxia Li*,
Jing Tang and
Ying Wang
School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: lijinxia@ecust.edu.cn; Tel: +86 21 64253847
First published on 20th August 2015
Abstract
In this paper, a novel graphene-like carbon nanosheet (HGCN) was synthesized using human hair as a carbon source and ferric chloride (FeCl3) as a graphitic catalyst, which had high specific surface area (1427 m2 g−1), multimodal pore system and favorable graphitic structure. Moreover, the HGCN carbon framework is doped with heteroatoms of nitrogen and sulfur. For methanol fuel cell application, the hybrid catalyst of HGCN supported Pt nanoparticles (Pt/HGCN) with a homogeneous distribution of 3.2 ± 1.0 nm, was further prepared through facile in situ chemical co-reduction. Compared with commercial Pt/XC-72 catalyst, the Pt/HGCN hybrids possess much larger electrochemically active surface areas and higher electrochemical stability towards electro-oxidation of methanol. The results indicate that HGCN is a promising alternative carbon support material for electrocatalysts in direct methanol fuel cells.
1. Introduction
The direct methanol fuel cell (DMFC) is attracting widespread attention and interest for its potential application in portable and small devices such as mobile phones, laptops, cameras.1,2 It has the advantages of high specific energy density, environmental friendliness and easy operation.3–7 Platinum (Pt) or platinum-based catalysts are the most effective electro-catalysts for oxidation and reduction processes. However, the low stability of the catalyst due to the aggregation of Pt nanoparticles has limited the performance of DMFCs in commercial application. In order to improve the activity and utilization of platinum catalysts, great efforts have been devoted to exploring new supporting materials for the loading of Pt.Unique properties of the carbon materials (i.e. large surface area and high electrical conductivity) make them as the best candidate for supporting materials.8,9 Various carbon-based materials have been used as catalyst support for DMFCs, such as carbon nanotubes,10–13 carbon nanofibers,14,15 mesoporous carbon.16,17 Recently, graphene has aroused interest as potential support owing its high electron transfer rate, large surface area and good conductivity.18–20 Compared with traditional carbon materials, the two-dimensional planar structure of graphene allows both the edge planes and basal planes to interact with the catalyst nanoparticles. Ji et al.21 synthesized graphene supported platinum nanoclusters and found that PtNCs/graphene has significantly higher electrocatalytic activity and stability for methanol electro-oxidation compared to the normal Vulcan XC-72 and graphite supported Pt nanoclusters.
Heteroatoms such as N, S doping into carbon materials offer a good choice to increase the catalytic performance of Pt-based catalysts in DMFC.22,23 Heteroatoms bonded with carbon framework can introduce defect sites due to different bond length. Liu et al.24 fabricated N-doped ordered mesoporous graphitic arrays with the pore-size distribution of 3.8 nm and found that moderate nitrogen content provided a high surface area and a graphitic framework, leading to high electrocatalytic activity, excellent long term stability, and resistance to methanol crossover effects for the ORR compared to commercial Pt/C. Zhang et al.25 utilize aniline to form nitrogen-doped carbon layer to prevent the aggregation among graphene (NCL-RGO). The Pt/NCL-RGO reveals larger electrochemical surface area and better dispersal of Pt nanoparticles comparing to Pt/RGO, which leads to much higher electrocatalytic activity and better stability of the catalyst.
Herein, a novel graphene-like carbon nanosheet (HGCN) was synthesized using human hair as a novel, low-cost and easily available precursor and FeCl3 as activating agent and graphitic catalyst. One advantage of choosing hair as carbon source is that N and S can be formed in situ in the carbon framework during the high temperature carbonization process. The other benefit is that N and/or S doping can easily form highly conductive carbon and significantly improve the electrochemical properties of carbon materials. Using a simple in situ chemical co-reduction method, the small and homogeneous Pt nanoparticles were deposited on the surface of HGCN. The electrocatalytic performance of Pt/HGCN was characterized by cyclic voltammetry (CV) and chronoamperometry (CA) methods.
2. Experimental
2.1. Materials and apparatus
Human hair was supplied by a local hair salon. Ferric chloride (FeCl3), ethylene glycol ((CH2OH)2) and acetone (CH3COCH3) were purchased from Sinopharm Chemical Reagent Co., Ltd (China). Chloroplatinic acid (H2PtCl6·6H2O), sodium borohydride (NaBH4) and Nafion® 117 solution were purchased from Sigma-Aldrich. Vulcan XC-72 was purchased from Cabot Corporation in the United States. All chemicals used were of analytical grade and the solutions were prepared with doubly distilled water.Nitrogen adsorption isotherm was recorded at 77 K using ASAP-2020 instrument (Micromeritics, USA). X-ray diffraction (XRD) patterns were performed on a D/max 2550 diffractometer (Rigaku, Japan) operating with CuKα radiation (λ = 0.154 nm, 40 kV, 100 mA, 2θ = 5–80°). The transmission electron microscopy (TEM) images were taken on a JEM-2100 microscopy at an acceleration voltage of 200 kV (JEOL, Japan). And the statistical average dimension of Pt catalyst was estimated from bright-field TEM images. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALAB 250Xi spectrometer (Thermo Scientific) with AlKα X-ray source for excitation. Sample charging was calibrated by setting the binding energy of the C 1s peak to 284.6 eV as reference. The actual Pt content was determined by inductively coupled plasma-atomic emission spectrometry (ICP, VISTA-MPX, Austria). Raman spectra were taken on a laser Raman microscope (Iuvia reflerx, Renishaw).
2.2. Synthesis of HGCN
In a typical synthesis, the collected hair fibers were cut into small pieces (0.5–1 cm), cleaned with water, ethanol and acetone, and then dried at 80 °C. Thoroughly dried hair was heated at 300 °C for 2 h under N2 flow to generate a partially carbonized material (char). 1 g char was mixed with 6 g FeCl3 in 30 mL ethanol and stirred for 3 h at the room temperature, and then stirred at 80 °C for 2 h to evaporate partial ethanol, finally dried at 100 °C in a conventional oven. Next, the dried sample was carbonized at 900 °C for 2 h with a heating rate of 5 °C min−1 in a horizontal tube furnace under N2 atmosphere. The black product was thoroughly washed with 2 mol L−1 HCl solution and deionized water to ensure it was neutral and dried in an oven at 80 °C for 12 h. The synthesized carbon material is denoted as HGCN. The hair-derived carbon support (denoted as HC) was also prepared in the absence of FeCl3 via the similar procedure for comparison purpose.2.3. Preparation of Pt/HGCN
The Pt nanoparticles were deposited on the supporting materials via an in situ chemical co-reduction according to the reference with some modifications.26 Typically, 378.3 mg NaBH4 and 5 mg NaOH were dissolved in 250 mL distilled water, and this solution was used as a reducing reagent. 50 mg HGCN was ultrasonicated in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethylene glycol/water solution (v/v) for 30 min, following by the addition of 1 mL of 0.0772 mol L−1 H2PtCl6 solution. The suspension was ultrasonicated vigorously for additional 30 min at room temperature before 10 mL of the above NaBH4/NaOH solution (0.04 mol L−1) was added. Then it was magnetically stirred for 2 h at 50–55 °C. The resulting slurry was filtered, washed thoroughly with water, and dried at 80 °C for 12 h, yielding black powder (denoted as Pt/HGCN). For comparison, the same Pt loading procedure was adopted to synthesize Pt on Vulcan XC-72 carbon (denoted as Pt/XC-72).
:1 ethylene glycol/water solution (v/v) for 30 min, following by the addition of 1 mL of 0.0772 mol L−1 H2PtCl6 solution. The suspension was ultrasonicated vigorously for additional 30 min at room temperature before 10 mL of the above NaBH4/NaOH solution (0.04 mol L−1) was added. Then it was magnetically stirred for 2 h at 50–55 °C. The resulting slurry was filtered, washed thoroughly with water, and dried at 80 °C for 12 h, yielding black powder (denoted as Pt/HGCN). For comparison, the same Pt loading procedure was adopted to synthesize Pt on Vulcan XC-72 carbon (denoted as Pt/XC-72).
2.4. Electrochemical measurements
The electrochemical measurements were carried out on a CHI 660 electrochemical workstation (Shanghai Chenhua, China) at room temperature with a conventional three-electrode system. The Ag/AgCl electrode and Pt wire were used as reference electrode and counter electrode, respectively. The working electrode was a glassy carbon (GC) electrode with diameter of 3 mm. The catalytic performance was studied by cyclic voltammograms (CV) and chronoamperometry (CA). CV curves were conducted in 0.5 mol L−1 H2SO4 solution with or without 1.0 mol L−1 CH3OH at a scan rate of 50 mV s−1. CA curves were carried out in 0.5 mol L−1 H2SO4 solution with 1 mol L−1 CH3OH at room temperature, with the electrode potential fixed at 0.6 V (vs. Ag/AgCl) for 1000 s. The stability of the catalysts was investigated by CV with 100 continuous voltammetric cycles at a scan rate of 50 mV s−1 between −0.2 to 1.0 V. High-purity nitrogen atmosphere was bubbled for 15 min before measurement.Prior to preparation of modified electrode, glassy carbon (GC) electrode was mechanically polished to a mirror finish with 0.05 μm alumina slurry, then ultrasonically washed with 1:1 HNO3 (v/v), NaOH (1.0 mol L−1), ethanol and redistilled water for 30 s, respectively. All the working electrodes were prepared with the same procedure as follows: 4 mg of catalyst was dispersed in 1 mL of ethanol solution containing 50 μL of Nafion (5 wt%), sonicated for 30 min, and then 5 μL of the dispersion was dropped onto the pretreated GC surface and dried for several hours at room temperature.
3. Results and discussion
3.1. Characterization of HGCN
Fig. 1a shows the nitrogen adsorption–desorption isotherms of HGCN and HC. The HGCN sample exhibits a typical type-I curve at very low relative pressure and a distinct hysteresis loop for the desorption branch under higher relative pressures from 0.5 to 1.0. The initial steep increase of the curve at very low relative pressure reveals the presence of micropores. The hysteresis loop indicates the existence of meso/macroporous characteristics, which are hypothesized to be formed by the randomly piled carbon nanosheets.27 However, for HC sample without FeCl3 activation, a small amount of gas adsorption is demonstrated and the data of specific surface area and pore volume could not be obtained, showing the non-porous structure of HC. Correspondingly, the pore size distribution curve of HGCN was determined by the adsorption branch of loop using Barrett–Joyner–Halenda method (Fig. 1b), which exhibits a large quantity of micropores (<2 nm) and a wide meso/macro-multimodal pore size distribution. The average BJH pore size is determined to be around 8.8 nm according to the adsorption branch. In addition, Brunauer–Emmett–Teller (BET) surface area and pore volume of HGCN are 1427 m2 g−1 and 0.53 cm3 g−1, respectively. | ||
| Fig. 1 (a) Nitrogen adsorption–desorption isotherms of the HGCN and HC, (b) pore size distribution curve of HGCN, (c) XRD patterns of the HGCN and HC, (d) Raman spectra of HGCN and HC. | ||
Fig. 1c shows wide-angle powder X-ray diffraction (XRD) of HC and HGCN materials. It can be clearly seen that HC sample possesses two diffraction peaks at 2θ of 24.6° and 43.4°, corresponding to the (002) and (100) lattice planes of a typical carbon. There is no obviously sharp peak in the HGCN pattern. The peak intensity at 24.6° was low and the peak at 43.4° disappeared, describing the amorphous structure of HGCN. However, XRD can only be used to characterize the long-range order of crystalline materials. While the short-range order in micro regions and slight structural changes of carbon materials can be further characterized by Raman spectra and HRTEM images. The Raman spectroscopy results of HC and HGCN are shown in Fig. 1d. The two peaks obtained at 1352 cm−1 and 1593 cm−1 were corresponded to the two main characteristic absorptions of D band (∼1350 cm−1) and G band (∼1580 cm−1) for carbon materials. The intensity ratio of ID and IG can be used as an indicator for disorder degree of carbon materials. The ID/IG ratio of HGCN is determined to be 2.50, much lower than that of HC (2.90). This result suggests that HGCN possesses a lower degree of disorder, which should be ascribed to the catalytic graphitization of FeCl3.
Low-magnification TEM and HR-TEM images of the HC and HGCN samples are shown in Fig. 2. The low-magnification image of HC (Fig. 2a) shows that thick and dense carbon fragment is obtained after carbonization of the hair without any treatments. Compared with HC, HGCN sample looks much thinner and more transparent (Fig. 2b). HC and HGCN samples are further characterized using HR-TEM, which are shown in Fig. 3c and d. By comparison, some graphitic carbon structures in micro regions can be clearly observed (Fig. 3d, red circles), revealing high degree of short-range order attributed to the catalytic graphitization of FeCl3.
 | ||
| Fig. 2 (a and b) Low-magnification TEM images of HC and HGCN, (c and d) HR-TEM images of HC and HGCN. | ||
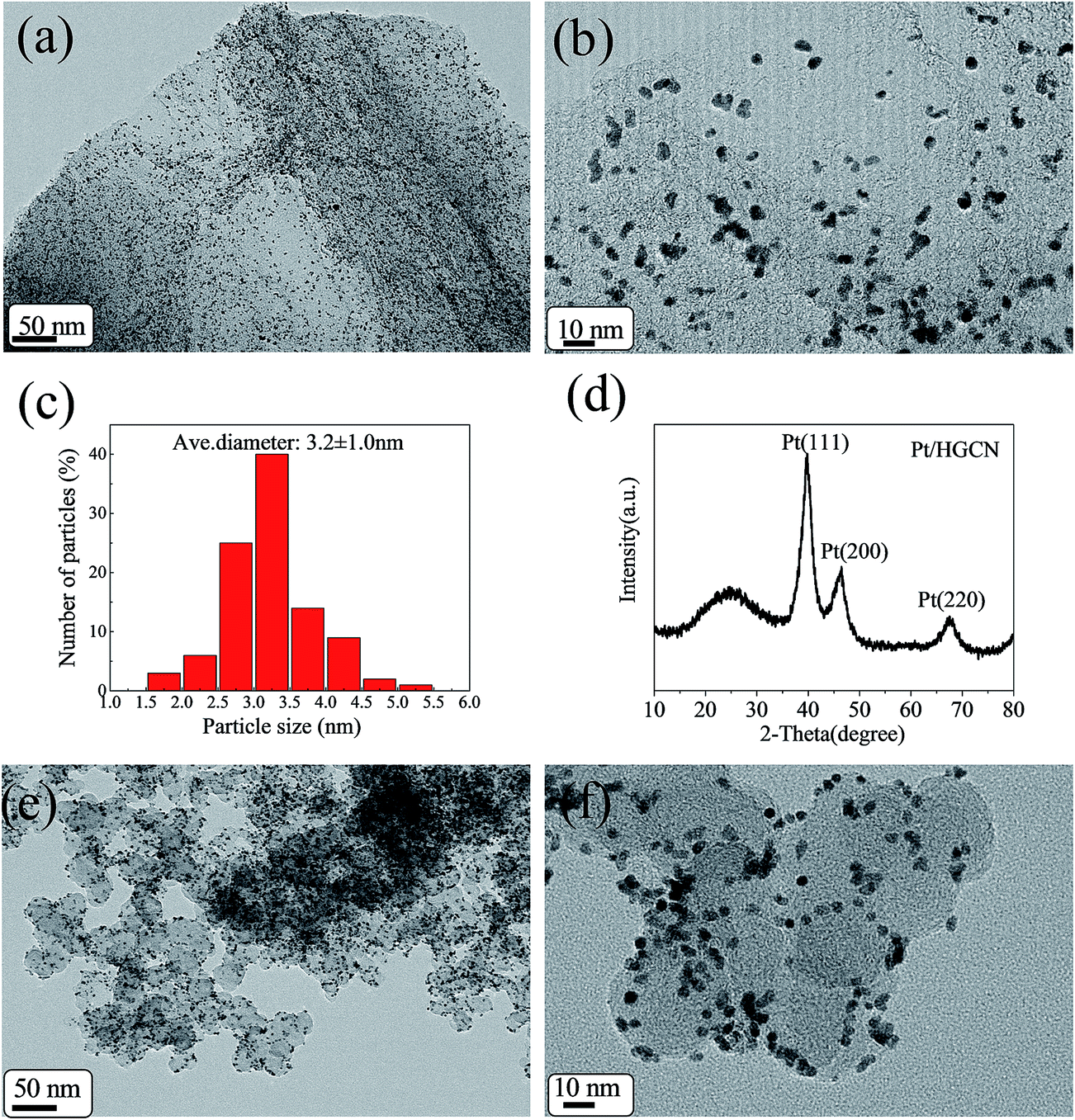 | ||
| Fig. 3 (a) Low-magnification TEM and (b) HR-TEM images of Pt/HGCN, (c) histograms of Pt size on Pt/HGCN, (d) XRD pattern of Pt/HGCN, (e) low-magnification TEM and (f) HR-TEM images of Pt/XC-72. | ||
Add it all up, HGCN with high surface area and multimodal pore size as well as favorable graphitic structure was successfully prepared when FeCl3 existed in the system. Thus we speculated that FeCl3 played an important role in the formation of the unique HGCN structure. Wang et al.28 has reported that the Fe components can form a carburized phase in the heating process, and the formation and decomposition process of the carburized phase can lead to the formation of graphene-like nanosheets. When carbonization at 900 °C under a N2 atmosphere, the chemical activation of FeCl3 may make the carbon source have a porous structure, and meanwhile, the Fe components can catalyze graphitization of the carbon source.29 In addition, it is important to mention here that the presence of meso/macropores in the interconnected carbon nanosheets offers the possibility of fast ion transfer by reducing the transportation path and favors the penetration of electrolyte to realize better performance of the electrode material.30,31
3.2. Characterization of morphology and structure of Pt/HGCN composite
Considering the high surface area, HGCN is a promising candidate as the supporting material to load Pt nanoparticles. In this work, a simple in situ chemical co-reduction method was used to deposit Pt nanoparticles on the surface of HGCN. The synthesis process of Pt/HGCN was shown in Scheme 1. | ||
| Scheme 1 The synthesis process of Pt/HGCN. | ||
Fig. 3a and b are TEM images of the Pt/HGCN nanocomposites with the different magnifications. As shown in Fig. 3a, it can be clearly seen that the Pt nanoparticles (black dots in the image) are uniformly and densely loaded on the HGCN surface. These particles are uniformly distributed on the basal plains and the edges of the carbon nanosheets, which can be ascribed to the surface functional groups of HGCN. Usually, the surface functional groups such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN and C–S–C serve as anchoring sites for the metal nanoparticles. Moreover, the oxygen functionalities, especially the carboxylic acids, provide active sites for the nucleation and growth of metal nanoparticles.32 In addition, the formation of very thin carbon nanosheets and multimodal pore structure can create many defects, such as edges and vacancies, which are much favorable for the immobilization of Pt nanoparticles.33 The size of Pt nanoparticles as revealed in HRTEM is shown in Fig. 3c. In addition, Fig. 3c shows the histograms of Pt nanoparticles size determined by statistical analysis of 200 randomly chosen isolated Pt particles from the HR-TEM image. According to the statistical survey results, the average diameter of Pt nanoparticles was 3.2 ± 1.0 nm.
O, CN and C–S–C serve as anchoring sites for the metal nanoparticles. Moreover, the oxygen functionalities, especially the carboxylic acids, provide active sites for the nucleation and growth of metal nanoparticles.32 In addition, the formation of very thin carbon nanosheets and multimodal pore structure can create many defects, such as edges and vacancies, which are much favorable for the immobilization of Pt nanoparticles.33 The size of Pt nanoparticles as revealed in HRTEM is shown in Fig. 3c. In addition, Fig. 3c shows the histograms of Pt nanoparticles size determined by statistical analysis of 200 randomly chosen isolated Pt particles from the HR-TEM image. According to the statistical survey results, the average diameter of Pt nanoparticles was 3.2 ± 1.0 nm.
Fig. 3d shows the X-ray diffraction (XRD) pattern of Pt/HGCN. The broad peak at 2θ of 24.3° is due to the (002) plane of the hexagonal structure of the HGCN carbon support. Moreover, the diffraction peaks observed at 39.8°, 46.7° and 67.6° can be indexed to the (111), (200) and (220) planes of the face-centered cubic (fcc) structure of Pt, respectively, revealing that a crystalline structure of the loaded Pt nanoparticles. These results indicated the successful formation of Pt nanoparticles on HGCN through the reduction of H2PtCl6 by using NaBH4 and (CH2OH)2. The peak corresponding to the (111) plane is more intense than the others, indicating that the (111) plane is the dominating orientation. The average crystallite size for the Pt nanoparticles can be estimated from the broadening of (111) diffraction peak using Scherrer equation d = 0.94λ/β(2θ)cosθ, where d is the average particle size (nm), λ is the wavelength of the X-ray (1.54056 Å), θ is the angle at the maximum of the peak and β(2θ) is full width of the diffraction peak at half maximum in radians (FWHM). By this equation, the average crystallite size of Pt is estimated to be 3.3 nm, which is in good agreement with the result obtained from TEM.
X-ray photoelectron spectroscopy (XPS, Fig. 4) was employed to further analyze the compositions and atom binding states of Pt/HGCN. From Fig. 4a, the deconvolution of C 1s spectrum contains three major components, corresponding to the presence of different carbon functionalities, such as CC (284.7 eV), C–N/C–O/C–S (286.0 eV) and CO/CN (288.8 eV).34 As seen in Fig. 4b, the deconvolution of N 1s spectrum yields four peaks: pyridinic-N (398.1 eV), pyrrolic-N (399.1 eV), graphitic-N (401.0 eV) and pyridinic N-oxide (403.2 eV) respectively. This suggests that nitrogen is indeed integrated into the carbon matrix as highly active pyridinic and pyrrolic nitrogen species.35,36 Similarly, peaks located at 162.6, 163.9 and 165.4 eV of S 2p (Fig. 4c) spectrum can be assigned to S0 (sulfur), C–S–C (thiophenic) and SO (sulfoxide) bonds, indicating that S is also doped into the carbon molecular scaffolds. Finally, Fig. 4d shows the Pt 4f XPS results of Pt/HGCN, three valences of Pt could be identified on doublet binding energy of Pt 4f7/2 and Pt 4f5/2. The main doublet at 71.3 eV (Pt 4f7/2) and 74.6 eV (Pt 4f5/2) are the characteristic of metallic Pt. Peaks located at 72.2 (Pt 4f7/2) and 75.4 eV (Pt 4f5/2) correspond to Pt2+, while the weakest of the two at 73.2 (Pt 4f7/2) and 76.5 eV (Pt 4f5/2) correspond to Pt4+. According to the quantitative analysis results of XPS, Pt/HGCN sample, on average, contains about 82.7% carbon, 5.3% oxygen, 5.5% nitrogen, 1.4% sulphur, 5.1% Pt (atomic%). It is reported that the interaction between the Pt nanoparticles and carbon support can modulate electronic properties of Pt catalysts due to strong π–d hybridization and further enhance electrocatalytic activity.33 Especially, the interaction between Pt and the N-doped carbon support may be further enhanced because of the hybridization of nitrogen lone pair electrons with the π electrons in the carbon. In addition, the quaternary nitrogen peaked at 402.5 eV can improve the electrical conductivity of carbon materials. Many previous researches have also reported that the S-doped carbon usually play a crucial role in the stabilization of Pt particles and electrocatalytic performance.37,38
 | ||
| Fig. 4 XPS high-resolution scans of (a) C 1s, (b) N 1s, (c) S 2p, (d) Pt 4f electrons of Pt/HGCN. | ||
3.3. Electrochemical characterizations of the Pt/HGCN electrodes
As known, Vulcan XC-72 carbon black material is regarded as the best commercial electrocatalyst support and is used by 80% of electrocatalyst. Herein, the electrochemical properties of Pt/HGCN with the Pt content of 16.0% (measured by ICP) was investigated and compared with that of Pt/XC-72 with the Pt content of 16.7%. Fig. 5 presents CV curves of Pt/HGCN and Pt/XC-72 catalysts carried out in an N2-saturated 0.5 mol L−1 H2SO4 solution at room temperature with a potential range from −0.2 and 1.0 V at a scan rate of 50 mV s−1 vs. Ag/AgCl. Both Pt/HGCN and Pt/XC-72 have hydrogen desorption–adsorption peaks occurring from −0.20 V to 0.16 V. From Fig. 5, we can see that Pt/HGCN exhibits much larger double layer capacitance than that of Pt/XC-72 (red dotted line), which should be ascribed to the presence of small micropores and relative poor conductivity of HGCN. Small micropores will lead the low utilization of surface area and increase the double layer capacitance.39 Meanwhile, the conductivity of the HGCN materials is not as good as that of commercial XC-72, which also affects the electron transfer, resulting in the larger double layer capacitance. The electrochemical active surface area (ECSA) values can be obtained from the hydrogen desorption peaks after subtraction of the double layer capacitance and displayed by the dash area in the CVs, according to the following equation:40| ECSA = QH/([Pt] × 0.21) |
 | ||
| Fig. 5 CV curves of (a) Pt/XC-72 and (b) Pt/HGCN at a scan rate of 50 mV s−1 between −0.2 and 1.0 V vs. Ag/AgCl in 0.5 mol L−1 H2SO4 solution. | ||
The methanol electro-oxidation measured by CVs is presented in Fig. 6a. The electrocatalytic experiment was carried out within a potential range from 0 to 1.0 V (vs. Ag/AgCl) at 50 mV s−1 in a nitrogen saturated 0.5 mol L−1 H2SO4 and 1.0 mol L−1 CH3OH solution. The cycling was repeated until steady CV curves were obtained and recorded. The currents for Pt/HGCN and Pt/XC-72 were normalized by their respective Pt loadings and ECSA to obtain the current densities. All cyclic voltammograms showed a similar methanol oxidation current peak in the forward scan (If) and an oxidation peak in the reverse scan (Ir) corresponding to the removal of the residual carbonaceous species formed in the forward scan. The forward peak current density is an important indicator with regard to the methanol electro-oxidation reaction. In Fig. 6a, the peak current density of Pt/HGCN (140.0 mA cm2 mg−1pt) is 1.63 times higher than that of Pt/XC-72 (86.1 mA cm2 mg−1pt). However, compared with Pt/XC-72, there is an obvious current generated prior to the onset of methanol oxidation reduction for Pt/HGCN sample, which should be due to the large double layer capacitance of HGCN. After subtraction of background current, the forward peak current densities on the Pt/HGCN and commercial Pt/XC-72 are 118.1 mA cm2 mg−1pt and 85.5 mA cm2 mg−1pt, respectively, indicating that the activity of the Pt/HGCN is still higher than that of the commercial Pt/XC-72 (as shown in Fig. 6b). These results are consistent with the ECSA results above. In summary, these improvements might be ascribed to the strong interaction between Pt nanoparticle and HGCN support with N and S doping and multimodal-pore structure, which can provide lots of ion adsorption sites, modulate electronic properties of Pt catalysts, promote the fast ion transport, resulting in the enhanced electrocatalytic activity.18,33
 | ||
| Fig. 6 (a) Original CV curves of Pt/HGCN and Pt/XC-72 at a scan rate of 50 mV s−1 between 0 and 1.0 V vs. Ag/AgCl in 0.5 mol L−1 H2SO4 + 1.0 mol L−1 CH3OH solution (b) normalized CV curves of Pt/HGCN and Pt/XC-72 after subtracting of background current (c) chronoamperometric curves of the Pt/HGCN and Pt/XC-72 in 0.5 mol L−1 H2SO4 + 1.0 mol L−1 CH3OH solution at 0.6 V. | ||
The stability of the Pt/HGCN electrode was investigated by performing CA at a potential of 0.6 V vs. Ag/AgCl in a solution containing 0.5 mol L−1 H2SO4 and 1.0 mol L−1 CH3OH under nitrogen saturation for 1000 s, as shown in Fig. 6c. For comparison, the same set of experiments was also performed using Pt/XC-72. In all two cases, the current densities drop over time, but eventually stabilize. However, the stable current density of 26.85 mA cm−2 mg−1pt obtained with Pt/HGCN catalyst after 1000 s is about 3.7 times higher than the value of 7.13 mA cm−2 mg−1pt on the Pt/XC-72 catalyst. This demonstrated that HGCN could enhance the activity and stability of the Pt incorporated onto the HGCN compared to Pt incorporated onto the Vulcan XC-72 substrate. In combination with the CV results, it can be concluded that the Pt/HGCN catalyst displayed enhanced electrocatalytic performance toward methanol oxidation reaction as compared with a commercial Pt/XC-72 catalyst.
4. Conclusion
A heteroatom doped graphene-like carbon nanosheets with a large surface area and suitable pore size were successful synthesized, which are much favorable for stable loading of small sized Pt particles. Compared with commercial Pt/XC-72 catalysts, the Pt/HGCN hybrids possess much larger electrochemically active surface area, and higher electrochemical stability towards electro-oxidation of methanol. Considering the facile preparation and reasonable cost, HGCN are an alternative carbon support material for electrocatalysts in direct methanol fuel cells.Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (Project No. 21305046) and Project of Shanghai Colleges and Universities Experimental Technology Team Construction Plan (YJ0114206).References
- A. S. Aricò, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 1, 133–161 CrossRef.
- J. N. Tiwari, R. N. Tiwari, G. Singh and K. S. Kim, Nano Energy, 2013, 2, 553–578 CrossRef CAS PubMed.
- S. Wasmus and A. Küver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef CAS.
- A. Veziroglu and R. Macario, Int. J. Hydrogen Energy, 2011, 36, 25–43 CrossRef CAS PubMed.
- W. Li, X. Wang, Z. Chen, M. Waje and Y. Yan, Langmuir, 2005, 21, 9386–9389 CrossRef CAS PubMed.
- Q. H. Huang, F. F. Tao, L. L. Zou, T. Yuan, Z. Q. Zou, H. F. Zhang, X. G. Zhang and H. Yang, Electrochim. Acta, 2015, 152, 140–145 CrossRef CAS PubMed.
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS PubMed.
- S. Tang, G. Sun, J. Qi, S. Sun, J. Guo, Q. Xin and M. H. Geir, Chin. J. Catal., 2010, 31, 12–17 CrossRef CAS.
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS PubMed.
- S. Yin, P. K. Shen, S. Song and S. P. Jiang, Electrochim. Acta, 2009, 54, 6954–6958 CrossRef CAS PubMed.
- T. Zhou, H. Wang, J. Key, S. Ji, V. Linkov and R. Wang, RSC Adv., 2013, 3, 16949–16953 RSC.
- M. M. Hasani-Sadrabadi, E. Dashtimoghadam, F. Majedi, S. Wu, A. Bertsch, H. Moaddel and P. Renaud, RSC Adv., 2013, 3, 7337–7346 RSC.
- S. J. Guo, S. J. Dong and E. K. Wang, Adv. Mater., 2010, 22, 1269–1272 CrossRef CAS PubMed.
- G. S. Chai, S. B. Yoon and J. S. Yu, Carbon, 2005, 4, 3028–3031 CrossRef PubMed.
- H. X. Huang, S. X. Chen and C. Yuan, J. Power Sources, 2008, 175, 166–174 CrossRef CAS PubMed.
- X. Bo, J. C. Ndamanisha, J. Bai and L. Guo, Talanta, 2010, 82, 85–91 CrossRef CAS PubMed.
- G. S. Chai, S. B. Yoon, J. S. Yu, J. H. Choi and Y. E. Sung, J. Phys. Chem. B, 2004, 108, 7074–7079 CrossRef CAS.
- Y. T. Kim, M. A. Matin and Y. U. Kwon, Carbon, 2014, 66, 691–698 CrossRef CAS PubMed.
- S. Y. Yin, Y. Y. Zhang, J. H. Kong, C. J. Zou, C. M. Li, X. H. Lu, J. Ma, F. Y. C. Boey and X. D. Chen, ACS Nano, 2011, 5, 3831–3838 CrossRef CAS PubMed.
- M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher and J. T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067–14069 CrossRef CAS PubMed.
- K. Ji, G. Chang, M. Oyam, X. Z. Shang, X. Liu and Y. B. He, Electrochim. Acta, 2012, 85, 84–89 CrossRef CAS PubMed.
- C. Jin, T. C. Nagaiah, W. Xia, B. Spliethoff, S. Wang, M. Bron, W. Schuhmann and M. Muhler, Nanoscale, 2010, 2, 981–987 RSC.
- S. Chen, Z. D. Wei, L. Guo, W. Ding, L. Dong, P. K. Shen, X. Qi and L. Li, Chem. Commun., 2011, 47, 10984–10986 RSC.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- X. Zhang, W. Yuan, J. Duan, Y. F. Zhang and X. Liu, Microelectron. Eng., 2015, 141, 234–237 CrossRef CAS PubMed.
- J. W. Chen, C. P. Jiang, X. Yang, L. Feng, E. B. Gallogly and R. L. Wang, Electrochem. Commun., 2011, 13, 314–316 CrossRef CAS PubMed.
- J. K. Ou, Y. Z. Zhang, L. Chen, H. Y. Yuan and D. Xiao, RSC Adv., 2014, 4, 63784–63791 RSC.
- L. Wang, C. G. Tian, H. Wang, Y. G. Ma, B. L. Wang and H. G. Fu, J. Phys. Chem. C, 2010, 114, 8727–8733 CAS.
- L. Sun, C. G. Tian, M. T. Li, X. Y. Meng, L. Wang, R. H. Wang, J. Yin and H. G. Fu, J. Mater. Chem. A, 2013, 1, 6462–6470 CAS.
- S. Song, S. Yin, Z. Li, P. K. Shen, R. Fu and D. Wu, J. Power Sources, 2010, 195, 1946–1949 CrossRef CAS PubMed.
- H. Chang, S. H. Joo and C. Pak, J. Mater. Chem., 2007, 17, 3078–3088 RSC.
- B. Xiong, Y. K. Zhou, Y. Y. Zhao, J. Wang, X. Chen, R. O'Hayre and Z. P. Shao, Carbon, 2013, 52, 181–192 CrossRef CAS PubMed.
- Y. Shen, Z. H. Zhang, R. R. Long, K. J. Xiao and J. Y. Xi, ACS Appl. Mater. Interfaces, 2014, 6, 15162–15170 CAS.
- H. Zhong, C. Deng, Y. Qiu, L. Yao and H. Zhang, J. Mater. Chem. A, 2014, 2, 17047–17057 CAS.
- J. K. Ou, Y. Z. Zhang, L. Chen, H. Y. Yuan and D. Xiao, RSC Adv., 2014, 4, 63784–63791 RSC.
- K. J. Antony, R. Shanmugam, R. Mahalakshmi and B. Viswanathan, Indian J. Chem., 2010, 49, 9–17 Search PubMed.
- C. L. Sun, H. H. Lee, J. M. Yang and C. C. Wu, Biosens. Bioelectron., 2011, 26, 3450–3455 CrossRef CAS PubMed.
- D. P. He, C. Zeng, C. Xu, N. C. Cheng, H. G. Li, S. C. Mu and M. Pan, Langmuir, 2011, 27, 5582–5588 CrossRef CAS PubMed.
- Z. L. Wang, X. B. Zhang, X. J. Liu, M. F. Lv, K. Y. Yang and J. Meng, Carbon, 2011, 49, 161–169 CrossRef CAS PubMed.
- Y. Li, W. Gao, L. Ci, C. Wang and P. M. Ajayan, Carbon, 2010, 48, 1124–1130 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |