
Phenylalanine iminoboronates as new phenylalanine hydroxylase modulators†
Francesco Montalbano‡
,
João Leandro‡,
Gonçalo D. V. F. Farias,
Paulo R. Lino,
Rita C. Guedes,
João B. Vicente,
Paula Leandro* and
Pedro M. P. Gois*
Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003, Lisboa, Portugal. E-mail: pedrogois@ff.ulisboa.pt; aleandro@ff.ulisboa.pt; Web: http://www.ff.ul.pt/∼pedrogois/index.html Fax: +351 217946470; Tel: +351 217946400
First published on 6th November 2014
Abstract
Herein we report the discovery of new modulators of human phenylalanine hydroxylase (hPAH) inspired by the structure of its substrate and regulator L-phenylalanine. These new hPAH modulators were simply prepared in good-to-excellent yields and excellent diastereoselectivities, based on a boron promoted assembly of L-phenylalanine, salicylaldehyde and aryl boronic acids. Iminoboronate 8, prepared with L-phenylalanine, para-methoxy-salicylaldehyde and phenyl boronic acid, was identified as the most efficient hPAH modulator, with an apparent binding affinity nearly identical to the natural allosteric activator L-phenylalanine.
Introduction
Phenylketonuria (PKU) is the most frequent disorder of amino acid metabolism with an overall incidence of one in 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 births and is characterized by intolerance to nutritional intake of L-phenylalanine (L-Phe).1,2 Untreated, PKU leads to a severe psycho-motor impairment due to the toxic effect of increased L-Phe levels in the central nervous system that has been recently shown to self-assemble into toxic amyloid-like fibrils suggesting an amyloid etiology for PKU.3,4 The low levels of L-Phe-derived biosynthesized neurotransmitters also contributes to negative clinical outcome.1,5 In most cases, PKU is related with deficient activity of phenylalanine hydroxylase (PAH) exhibiting kinetic and conformational defects imposed by mutations in the PAH gene.1,5 Human PAH (hPAH) belongs to the family of aromatic amino acid hydroxylases and catalyzes the hydroxylation of L-Phe to L-tyrosine (L-Tyr) in the presence of the cofactors (6R)-L-erythro-5,6,7,8-tetrahydrobiopetrin (BH4, Scheme 1) and a non-heme mononuclear iron ion, with dioxygen as co-substrate, which is the first step of the catabolic degradation of L-Phe.5 To avoid L-Phe accumulation up to neurotoxic levels, PKU patients are forced to stringently hold on to an L-Phe-free diet, which often results in malnutrition and neurologic problems.5,6 Alternative strategies to treat PKU are now emerging, such as dietary supplementation with the natural cofactor BH4, which is able to act as a pharmacological chaperone.7 Unfortunately, BH4 is often used in high amounts per dose (20 mg kg−1 body weight) and patients with more severe phenotypes of PAH deficiency are unresponsive to this molecule.6,7 Therefore, the discovery of small molecule modulators of hPAH remains as a very important and challenging topic of research. In this context, compounds I and II (Scheme 1) were recently disclosed and shown to improve hPAH stability and the protein in vitro and in vivo steady-state levels.8,9
000 births and is characterized by intolerance to nutritional intake of L-phenylalanine (L-Phe).1,2 Untreated, PKU leads to a severe psycho-motor impairment due to the toxic effect of increased L-Phe levels in the central nervous system that has been recently shown to self-assemble into toxic amyloid-like fibrils suggesting an amyloid etiology for PKU.3,4 The low levels of L-Phe-derived biosynthesized neurotransmitters also contributes to negative clinical outcome.1,5 In most cases, PKU is related with deficient activity of phenylalanine hydroxylase (PAH) exhibiting kinetic and conformational defects imposed by mutations in the PAH gene.1,5 Human PAH (hPAH) belongs to the family of aromatic amino acid hydroxylases and catalyzes the hydroxylation of L-Phe to L-tyrosine (L-Tyr) in the presence of the cofactors (6R)-L-erythro-5,6,7,8-tetrahydrobiopetrin (BH4, Scheme 1) and a non-heme mononuclear iron ion, with dioxygen as co-substrate, which is the first step of the catabolic degradation of L-Phe.5 To avoid L-Phe accumulation up to neurotoxic levels, PKU patients are forced to stringently hold on to an L-Phe-free diet, which often results in malnutrition and neurologic problems.5,6 Alternative strategies to treat PKU are now emerging, such as dietary supplementation with the natural cofactor BH4, which is able to act as a pharmacological chaperone.7 Unfortunately, BH4 is often used in high amounts per dose (20 mg kg−1 body weight) and patients with more severe phenotypes of PAH deficiency are unresponsive to this molecule.6,7 Therefore, the discovery of small molecule modulators of hPAH remains as a very important and challenging topic of research. In this context, compounds I and II (Scheme 1) were recently disclosed and shown to improve hPAH stability and the protein in vitro and in vivo steady-state levels.8,9
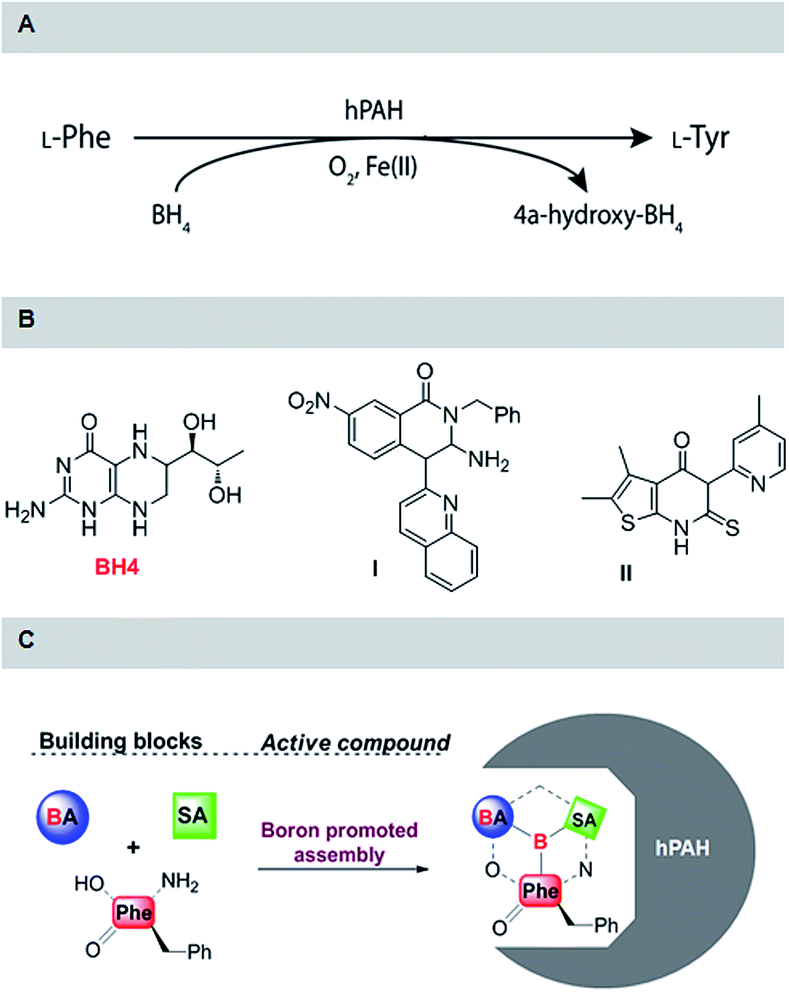 | ||
| Scheme 1 (A) hPAH catalysed hydroxylation of L-Phe; (B) structures of compounds that were shown to improve the stability of hPAH; (C) boron promoted assembly of salicylaldehyde (SA), L-phenylalanine (L-Phe) and boronic acid (PBA). | ||
Human PAH is a homotetrameric enzyme finely regulated by BH4 and L-Phe.10 The cofactor inhibits the enzyme rendering a more stable form. Pre-incubation with the substrate L-Phe activates (∼2.5-fold activation) the enzyme. Although this activation mechanism is not yet fully comprehended and there is no agreement on whether it is caused by L-Phe binding to an allosteric site or by homotropic activation.11–14,1,15,16
Regardless of its action mode, hPAH activation by L-Phe is the most important physiological mechanism to protect the organism from damage by increased levels of L-Phe. Therefore it is rather surprisingly that L-Phe has not been explored as the key structural motif to develop new modulators for this protein envisaging improving enzyme function.9,17 Recently we have demonstrated that boron may be efficiently used to assemble complex molecular structures that may be readily tuned for optimal interaction with a biological target (Scheme 1).18,19 Based on this, we envisioned that by incorporating L-Phe as one of the assembly components, we could simply generate useful structures to modulate hPAH activity and probe the enzyme active site.
Results
To test this idea, iminoboronates 1, 2, and 3 depicted in Scheme 2, were prepared using salicylaldehyde (SA), phenylboronic acid (PBA), L-Phe, L-leucine (L-Leu) and L-alanine (L-Ala) respectively as assembly components. The reaction was conducted in water at 90 °C for 20 h, and this simple protocol afforded the expected compounds in good-to-excellent yields and excellent diastereoselectivities. Once prepared, compounds 1–3 were readily evaluated for their effect on the activity of tetrameric wild-type hPAH, employing three experimental conditions (ESI Scheme S1†): (i) condition I involved pre-incubation of hPAH with substrate and compound to evaluate the competition between the iminoboronate and L-Phe (‘substrate-activated’ condition); (ii) condition II was performed with no pre-incubation step, the substrate and iminoboronate being added simultaneously at time zero (‘non-activated’ condition); and (iii) condition III involved pre-incubation with the tested iminoboronate alone, to establish its ability to pre-activate the enzyme, mimicking L-Phe-promoted pre-activation (‘compound-activated’ condition). Pre-activation by each iminoboronate was thus calculated as the ratio between the activity in condition III and condition II. Blank assays with each iminoboronate alone and omitting L-Phe were performed to rule out L-Phe release from the iminoboronates bearing this moiety and consequent conversion to L-Tyr. | ||
| Scheme 2 Biological activity of hPAH in the presence of iminoboronates containing L-phenylalanine (1), L-leucine (2) and L-alanine (3). The enzymatic assays correspond to the substrate activated (■), non-activated (□) and compound activated conditions (▨). Control assays were performed in the presence of DMSO, phenylboronic acid (PBA) and salicylaldehyde (SA). aC0.5, concentration for half-maximal binding. Data are presented as mean ± SD. Statistical significance is given by *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (n = 3), for the relative fold activation between compound-activated (II) and non-activated condition (III). | ||
Very gratifyingly, as shown in Scheme 2, the iminoboronate 1 was able to modulate the activity of hPAH, activating the enzyme by 1.5-fold (P < 0.05) in the absence of L-Phe, while competing with the substrate L-Phe. Very differently, the iminoboronates 2 and 3, respectively prepared with L-Leu and L-Ala, failed to activate hPAH. Taken together with the fact that the individual components SA and PBA were also unable to activate the enzyme, these results clearly suggest that the observed effect was most probably due to the incorporation of L-Phe into the structure of heterocycle 1. Due to the high demands of the hPAH activity assay, we devised an experimental approach to determine the compounds' binding kinetics, evaluating their effect on the protein's thermal denaturation profiles, employing differential scanning fluorimetry (DSF, see ESI†). By fluorescence-monitored thermal denaturation profiles at different compound concentrations, it was determined that 1 binds to hPAH with an apparent binding affinity of C0.5 of 10.6 ± 0.9 μM, while the iminoboronates 3, prepared with L-Ala, did not reveal any measurable affinity to hPAH (Scheme 2 and ESI†).
Based on these encouraging results, we embarked on a modification campaign to optimize the structure of compound 1. Therefore, following the aforementioned methodology, the iminoboronates 4, 5 and 6 were simply prepared combining L-Phe, salicylaldehyde and para substituted phenyl boronic acids. As shown in Scheme 3, compound 4, bearing a methyl at the boronic acid aromatic para position, demonstrated a profile indicative of competition with the substrate as observed for 1, though with only marginal activation ability. Very differently, compound 5, prepared with 4-fluorophenylboronic acid, markedly activated hPAH (1.5-fold; P < 0.001), while exhibiting an apparent binding affinity (C0.5 of 14.4 ± 2.9 μM) comparable to the one observed when using 1. The introduction of a methoxide substituent in compound 6 resulted in a clear inhibition (Scheme 3).
 | ||
| Scheme 3 Biological activity of hPAH in the presence of iminoboronates containing L-phenylalanine and para substituted phenyl boronicacids: methyl (4), fluoro (5) and methoxide (6). The enzymatic assays correspond to the substrate activated (■), non-activated (□) and compound activated conditions (▨). Control assays were performed in the presence of DMSO. aC0.5, concentration for half-maximal binding. Data are presented as mean ± SD. Statistical significance is given by *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (n = 3), for the relative fold activation between compound-activated (II) and non-activated condition (III). | ||
Then, we evaluated the impact of substituents at the salicylaldehyde component with the synthesis of compounds 7 and 8 (Scheme 4). Very gratifyingly, although the introduction of a methyl at the para position of the salicylaldehyde had a marginal impact on the enzyme activation, the introduction of a methoxide in compound 8, clearly improved the hPAH activation by 1.8-fold (conditions II and III in Scheme 4; P < 0.0001), maintaining a high apparent binding affinity (C0.5 of 14.8 ± 4.9 μM). Compound 8 displayed a 1.7 h t1/2 in buffer (see ESI†). As shown is Scheme 4, the combination of para-methoxy-salicylaldehyde with different aromatic and vinylboronic acids 9–13 (Scheme 4) did not improve the activation previously observed with compound 8, except for a slight increase (1.3-fold; P < 0.001) observed for the 2-bromophenylboronic acid substituent in compound 13.
 | ||
| Scheme 4 Biological activity of hPAH in the presence of iminoboronates containing L-phenylalanine and substituents at the salicylaldehyde (7 and 8) and para-methoxy-salicylaldehyde in combination with aromatic and vinylboronic acids (9 to 13). The enzymatic assays correspond to the substrate activated (■), non-activated (□) and compound activated conditions (▨). Control assays were performed in the presence of DMSO. aC0.5, concentration for half-maximal binding. Data are presented as mean ± SD. Statistical significance is given by *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (n = 3), for the relative fold activation between compound-activated (II) and non-activated condition (III). | ||
Aiming at a rationalization of the observed biological activity when using substrate L-Phe or compounds 1, 3, 6, 8, and to get insight into their potential binding and interactions with hPAH, we performed detailed in silico molecular docking studies in the active site of hPAH using the GOLD 5.1 software (Fig. 1). Very interestingly, compounds 1 (Fig. 1B) and 8 (Fig. 1E and F), which elicit a ‘competitive-activator’ profile, adopt similar poses inside hPAH active site, with the phenyl ring in the same position of the phenyl ring of L-Phe, showing only a small ring rotation. These compounds likely establish π–π interactions with the imidazole group of His285 at an average distance of 3.1 Å. This is particularly important as interactions of thiophyl and alkyl lateral chains of the substrate analogues 3-(2-thienyl)-L-alanine (THA; 3.8 Å) and norleucine (NLE; 3.9 Å) respectively, with His285 were reported as governing substrate binding affinity.20 In addition, the compound 8 methoxy moiety oxygen is predicted to form a hydrogen bond with Ser251 (1.9 Å), a residue that is also known to establish a hydrogen bond with the dihydroxypropyl side chain of the cofactor BH4 (see Fig. S5 in ESI†).21 Other hydrophobic interactions are also observed between these compounds and Phe254, another residue essential for BH4 binding. Differently, compound 6 which markedly inhibits hPAH, shows a completely different pose inside the active site (Fig. 1D), displaying no interactions with Ser251 and adopting a pose shifted to the right side of the pocket, though being unable to establish any interaction with the final part of the binding pocket. Finally compound 3 prepared with L-Ala, displayed no important interactions inside the binding pocket (Fig. 1C), corroborating the importance of the benzyl group for recognition. In fact, the benzyl group of compound 8 adopts a perfect position inside the binding pocket, positioning the phenyl ring in a conformation close to the iron (Fig. 1F). The superposition of the crystallographic structure of hPAH complexed with BH4 and NLA (PDB code 1MMT) and the docking poses of compounds 1 and 8 revealed that these compounds are almost completely overlapped inside the binding site. The phenyl ring of compound 8 overlaps the alkyl chain of NLA and probably governs compound affinity and activation. The methoxy moiety of compound 8 occupies the BH4 binding site explaining the slight decrease of hPAH activity observed for the L-Phe pre-activated conditions (see Fig. S6 in ESI†). It also points to a reversible binding of the compounds, as BH4 and L-Phe are still able to bind to the protein, with concomitant production of L-Tyr (Scheme 1 and 4). The estimate of the binding energies (scores) of compounds 1 and 8 calculated with Goldscore fitness function revealed that these compounds show a strongest affinity to the hPAH (scores = 62 and 60) compared to L-Phe (score = 49). The binding parameters of the compounds determined by DSF (Fig. S1 and S2†) are in the low micromolar range (10–14 μM), comparable with the apparent binding of L-Phe (16.3 ± 6.3 μM) determined by the same method.
 | ||
| Fig. 1 Best docking poses obtained for L-Phe (A), compounds 1 (B), 3 (C), 6 (D) and 8 (E). (F) Possible interactions between compound 8 (yellow) and hPAH residues at the active site (green), resulting from docking of the compound onto the hPAH structure (PDB code 1MMT). | ||
In comparison with molecules I and II, which exhibit very promising results in the stabilization of hPAH, these iminoboronates, namely compound 8, are able to improve directly the enzyme activity by a pre-activation mechanism and to predispose the hPAH enzyme to accommodate the natural substrate.
Conclusions
In summary, in this study we have developed for the first time a series of iminoboronates that are able to directly increment hPAH activity by a pre-activation mechanism similar to the one induced by the substrate L-Phe. To this end, we have chosen to study the compounds on the fully active wild-type tetrameric hPAH, in the perspective of a future application of these iminoboronates in the activation of clinically relevant variants. Since missense mutations often originate functionally impaired variants, the most effective iminoboronates are expected to significantly increase the variant hPAH enzymatic activity to functionally relevant levels. The studied compounds were very efficiently prepared using a boron promoted assemblage of L-Phe with salicylaldehydes and aryl boronic acids. The resulting iminoboronates featuring the L-Phe (1, 4–6 and 8) structure were shown to interact with hPAH, while iminoboronates prepared with L-Leu (2) and L-Ala (3) did not. The L-Phe based iminoboronates (1, 5 and 8) exhibit apparent binding constants in the micromolar range (10–14 μM), which are comparable with the apparent binding constant of L-Phe (16.3 ± 6.3 μM). In this study, compound 8, prepared with L-Phe, para-methoxy-salicylaldehyde and phenyl boronic acid, was identified as the most effective activator of hPAH improving the enzyme activity by 1.8-fold (P < 0.0001), maintaining a high apparent binding affinity (C0.5 of 14.8 ± 4.9 μM). Docking studies performed with these compounds, corroborated all experimental observations and revealed that iminoboronates 8 establishes important interactions either with the substrate and BH4 recognition sites.Experimental section
Escherichia coli TOP 10 and the prokaryotic expression vector pTrcHis were obtained from Invitrogen (Carlsbad, CA). The cofactor (6R)-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4), L-Phe, Hepes were from Sigma (St. Louis, MO, USA). Ascorbic acid was obtained from Merck (Darmstadt, Germany). Unless stated otherwise, all reagents were of analytical grade.General procedure for preparation of boron heterocycles using water as a solvent
A round bottom flask equipped with a magnetic stirrer was charged with amino acid (2.0 equiv.), aldehyde (1.5 equiv.) and distilled water (2.0 mL). This suspension was stirred at 90 °C for 1 h after which the boronic acid (0.41 mmol) was added, the mixture was then stirred at 90 °C for 20 h. The reaction mixture, which appears as a biphasic composition of precipitate and a supernatant liquid, was filtered and the solid retained in the filter was then washed with water followed by hexane. The desired compound was recovered with dichloromethane, which was subsequently removed under reduced pressure.Expression and purification of recombinant wild-type hPAH protein
Recombinant wild-type hPAH protein was expressed in E. coli as a fusion protein with the hexa-histidyl tag (6xHis-(pep)EK-hPAH) as described.22 Cells were grown at 37 °C, protein expression was induced by addition of 1 mM isopropyl-β-D-thio galactoside (IPTG), and the cells were harvested after 3 h. After a first purification step by immobilized metal-affinity chromatography, the tetramers were isolated by size exclusion chromatography, using a HiLoad Superdex 200 HR column (1.6 cm × 60 cm, GE-Healthcare) and a mobile phase containing 20 mM Na–Hepes, 200 mM NaCl, pH 7 pumped at a flow rate of 0.7 mL min−1.Enzymatic activity assays
The hPAH activity was measured essentially as previously described22 in a 200 μL final volume reaction mixture, containing 100 μM L-Phe, 0.1 M Na–Hepes, pH 7, 0.1 mg mL−1 catalase, 5 μg of recombinant wild-type hPAH tetramers, 100 μM of each compound or 1% DMSO (vehicle control). After 4 minutes of pre-incubation, 100 μM (NH4)2Fe(II)SO4 was added and, unless otherwise stated, the reaction was started by addition of 75 μM BH4 (together with 5 mM ascorbic acid) after 1 minute incubation with the iron (condition I in ESI Scheme S1;† ‘substrate-activated’ condition). To study the specific activity of the non-activated hPAH, 100 μM L-Phe and 100 μM of each compound were added together with 75 μM BH4 at the start of the hydroxylation reaction (condition II in ESI Scheme S1;† ‘non-activated’ condition). To evaluate pre-activation of the enzyme by the compound, hPAH was pre-incubated 4 minutes with each compound whereas the L-Phe substrate was only added at the start of the reaction, together with 75 μM BH4 at the start of the reaction (condition III in ESI Scheme S1;† ‘compound-activated’ condition). Pre-activation by each iminoboronate was thus calculated as the ratio between the activity in condition III and condition II. Blank reactions where the substrate L-Phe was omitted were also made for each compound. The amount of L-Tyr produced after 1 min was quantified by a HPLC method23 using a LiChroCART® 250-4 LiChrospher® 60 RP-select B (5 μm) column (Merck KGaA, Darmstadt, Germany), a 5% ethanol mobile phase pumped at 0.7 mL min−1 and fluorimetric detection (λexc = 274 nm and λem = 304 nm). Specific activities are presented as mean ± SEM obtained from three independent experiments. Tests for statistical significance were performed using 1-way ANOVA with *P < 0.05; **P < 0.01; ***P < 0.001 and ****P < 0.0001.Differential scanning fluorimetry
Differential scanning fluorimetry (DSF) was performed in a C1000 Touch thermal cycler equipped with a CFX96 optical reaction module (Bio Rad). For all fluorescence measurements, samples containing purified recombinant wild-type hPAH tetramers at 100 μg mL−1 in 20 mM Na–Hepes, 200 mM NaCl, pH 7, 2.5-fold Sypro Orange (Invitrogen; 5000-fold commercial stock solution), 1% DMSO (unless otherwise stated) and 100 μM of each compound were incubated at 20 °C for 10 minutes. The PCR plate was sealed with Optical-Quality Sealing Tape (Bio-Rad) and centrifuged at 500× g for 1 min. The DSF assay was carried out by increasing the temperature from 20 to 90 °C, with a 1 s hold time every 0.2 °C and fluorescence acquisition using the FRET channel. Control experiments in the absence of DMSO and/or compounds were routinely performed in each microplate. Data were processed using CFX Manager Software V3.0 (Bio-Rad) and the GraphPad Prism 6. Temperature scan curves were fitted to a biphasic dose–response function and the Tm values were obtained from the midpoint of the first and second transitions. To monitor the binding properties of the regulatory and catalytic domain towards each compound, DSF assays were run in the presence of increasing compound concentrations (0–2.56 mM) or 1% DMSO (vehicle control). C0.5 values provide apparent binding affinities and are best-fit parameters obtained from the effect of the compound on the contribution of the regulatory domain to the overall unfolded process (compounds 1, 5 and 8) or on the melting temperature of the first transition (Tm,1) (compound 3).Docking studies
All calculations were performed on iMed.ULisboa scientific cluster. GOLD (version 5.2)24 was used for docking calculations and Molecular Operating Environment (MOE) software (version 2013.10)25 were used to build and optimize the structures of iminoboronates molecules and for enzyme structure refinement. In the present study, the crystal structure of hPAH in complex with the physiological cofactor (6R)-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4) and the substrate analogue L-norleucine (NLE) at a resolution of 2.0 Å (PDB code 1MMT)20 was employed in the docking calculations. The ternary hPheOH–Fe(II)·BH4·NLE structure comprises 308 residues, 149 structural water oxygen atoms, one NLE molecule, one BH4 molecule and one sulphate ion. The physiological cofactor (6R)-L-erythro-5,6,7,8-tetrahydrobiopterin (BH4), the substrate analogue NLE and all crystallographic water molecules were removed from the coordinate set using MOE software. Hydrogen atoms were added to this reduced crystal structure and the protein was protonated to pH = 7. The protein was then submitted to restrained molecular mechanics refinement using the AMBER99 force field implemented in MOE software. The final structure of the hPAH protein was used for the docking calculations. To establish our docking procedure, a preliminary validation study was carried out redocking NLE to the refined protein structure. The performance of molecular docking protocol was evaluated by comparing the redocked binding poses of NLE with the experimental X-ray (PDB code 1MMT). The redocked pose agrees well with the X-ray crystallized pose (RMSD ≤ 2.0 Å). After this validation, the iminoboronates structures were docked into the active site of hPAH using the Gold Software with the goldscore scoring mode. The following GOLD parameters were employed: 1000 runs, population size of 100, 100.000 genetic algorithm operations, and 5 islands at normal time speed up setting, were conducted for each compound. In the docking process, the protein conformation was fixed while the docked ligand was flexible. The best binding pose for each molecule was saved for further analysis. The 20 top-ranked compounds were visually inspected with PyMol.26Acknowledgements
Fundação para a Ciência e Tecnologia (PTDC/QUI-QUI/118315/2010; Pest-OE/SAU/UI4013/2011; P. M. P. Gois is a FCT Investigator) is thanked for financial support.Notes and references
- M. I. Flydal and A. Martinez, IUBMB Life, 2013, 65, 341–349 CrossRef CAS PubMed
.
- J. J. Mitchell, Y. J. Trakadis and C. R. Scriver, Genet. Med., 2011, 13, 697–707 CrossRef CAS PubMed
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8, 701–706 CrossRef CAS PubMed
- V. Singh, R. K. Rai, A. Arora, N. Sinha and A. K. Thakur, Sci. Rep., 2014, 4, 3875 Search PubMed
- J. Underhaug, O. Aubi and A. Martinez, Curr. Top. Med. Chem., 2012, 12, 2534–2545 CrossRef CAS
- F. J. van Spronsen and G. M. Enns, Mol. Genet. Metab., 2010, 99(suppl. 1), S90–S95 CrossRef CAS PubMed
- A. Martinez, A. C. Calvo, K. Teigen and A. L. Pey, Prog. Mol. Biol. Transl. Sci., 2008, 83, 89–134 CAS
- R. Torreblanca, E. Lira-Navarrete, J. Sancho and R. Hurtado-Guerrero, ChemBioChem, 2012, 13, 1266–1269 CrossRef CAS PubMed
- A. L. Pey, M. Ying, N. Cremades, A. Velazquez-Campoy, T. Scherer, B. Thony, J. Sancho and A. Martinez, J. Clin. Invest., 2008, 118, 2858–2867 CrossRef CAS PubMed
- T. Flatmark and R. C. Stevens, Chem. Rev., 1999, 99, 2137–2160 CrossRef CAS PubMed
- J. P. Abita, S. Milstien, N. Chang and S. Kaufman, J. Biol. Chem., 1976, 251, 5310–5314 CAS
- J. Leandro, P. Leandro and T. Flatmark, Biochim. Biophys. Acta, 2011, 1812, 602–612 CrossRef CAS PubMed
- T. Solstad and T. Flatmark, Eur. J. Biochem., 2000, 267, 6302–6310 CrossRef CAS
- J. Tipper and S. Kaufman, J. Biol. Chem., 1992, 267, 889–896 CAS
- J. Li, U. Ilangovan, S. C. Daubner, A. P. Hinck and P. F. Fitzpatrick, Arch. Biochem. Biophys., 2011, 505, 250–255 CrossRef CAS PubMed
- M. Thórólfsson, B. Ibarra-Molero, P. Fojan, S. B. Petersen, J. M. Sanchez-Ruiz and A. Martinez, Biochemistry, 2002, 41, 7573–7585 CrossRef PubMed
- S. Santos-Sierra, J. Kirchmair, A. M. Perna, D. Reiss, K. Kemter, W. Roschinger, H. Glossmann, S. W. Gersting, A. C. Muntau, G. Wolber and F. B. Lagler, Hum. Mol. Genet., 2012, 21, 1877–1887 CrossRef CAS PubMed
- F. Montalbano, P. M. Cal, M. A. Carvalho, L. M. Goncalves, S. D. Lucas, R. C. Guedes, L. F. Veiros, R. Moreira and P. M. Gois, Org. Biomol. Chem., 2013, 11, 4465–4472 CAS
- F. Montalbano, N. R. Candeias, L. F. Veiros, V. Andre, M. T. Duarte, M. R. Bronze, R. Moreira and P. M. Gois, Org. Lett., 2012, 14, 988–991 CrossRef CAS PubMed
- O. A. Andersen, A. J. Stokka, T. Flatmark and E. Hough, J. Mol. Biol., 2003, 333, 747–757 CrossRef CAS PubMed
- O. A. Andersen, T. Flatmark and E. Hough, J. Mol. Biol., 2002, 320, 1095–1108 CrossRef
- P. Leandro, I. Rivera, M. C. Lechner, I. T. de Almeida and D. Konecki, Mol. Genet. Metab., 2000, 69, 204–212 CrossRef CAS PubMed
- R. Kand'ar and P. Zakova, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3926–3929 CrossRef PubMed
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed
- Molecular Operating Environment (MOE), 2013.2010, Chemical Computing Group Inc, Montreal Canada, 2012, http://www.chemcomp.com Search PubMed
- W. L. Delano, The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, USA., 2002 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Full description of compounds characterization, enzymatic activity assays, differential scanning fluorimetry and docking studies. See DOI: 10.1039/c4ra10306h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |