
Advantageous asymmetric ketone reduction with a competitive hydrogenation/transfer hydrogenation system using chiral bifunctional iridium catalysts†
Junki Moritani,
Yoshihito Kayaki and
Takao Ikariya*
Department of Applied Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, O-okayama 2-12-1-E4-1, Meguro-ku, Tokyo 152-8552, Japan. E-mail: tikariya@apc.titech.ac.jp
First published on 23rd October 2014
Abstract
Hydrogenation of aromatic ketones with chiral bifunctional amidoiridium complexes proceeds in preference to transfer hydrogenation in methanol, in which the pressurised hydrogen can suppress unintended racemisation of the alcoholic product, leading to enhanced enantioselectivity.
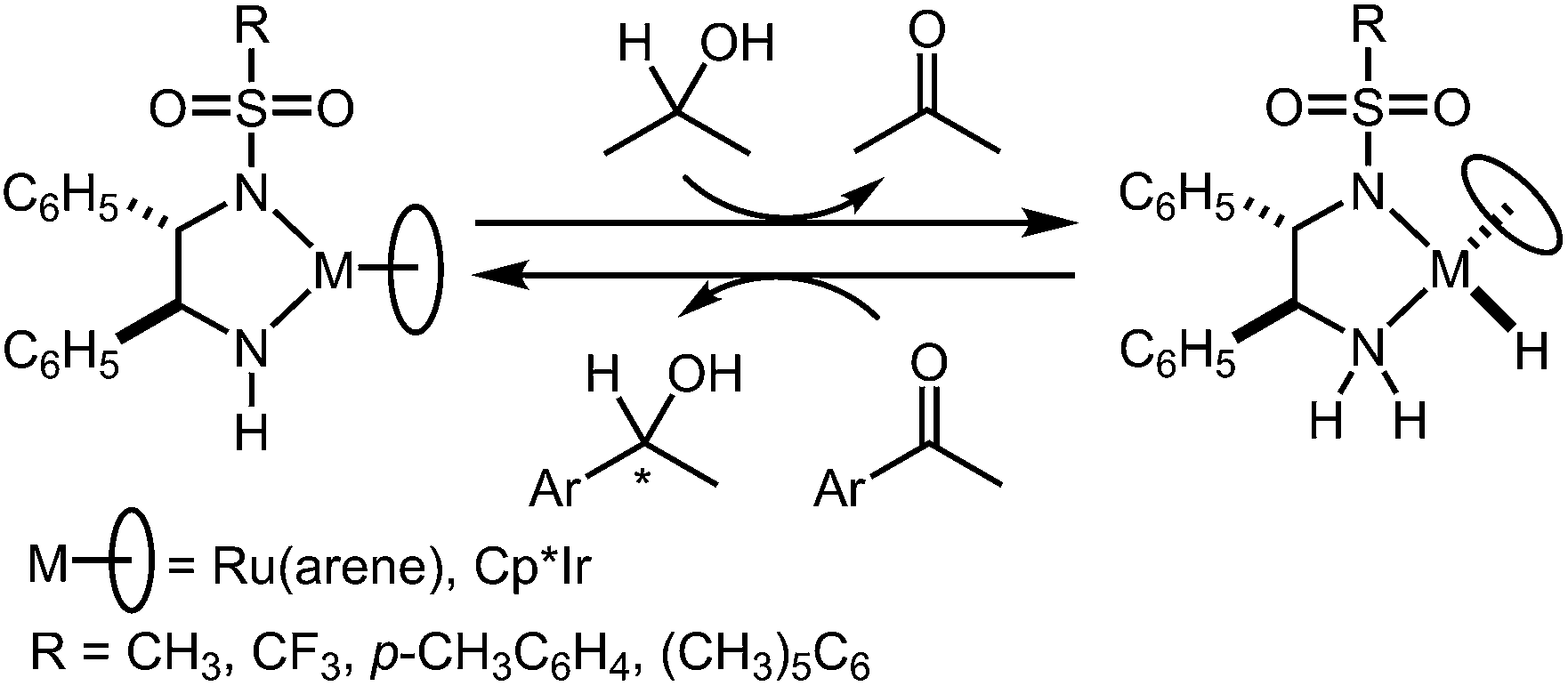
Asymmetric reduction of ketones to chiral alcohols is an established and extensively utilised transformation in organic synthesis.1 Noyori, Ikariya and co-workers have developed a series of well-defined metal/NH bifunctional Ru(η6-arene) catalysts bearing Tsdpen [Tsdpen = N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine] (1), for the asymmetric transfer hydrogenation (ATH) of aromatic and propargylic ketones using 2-propanol2 or formic acid–triethylamine (5
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)3 as a reaction medium and a hydrogen source (Scheme 1). Analogous Cp*M-Tsdpen and -Tscydn complexes [Cp* = η5-1,2,3,4,5-pentamethylcyclopentadienyl; M = Rh, Ir; Tscydn = N-(p-toluenesulfonyl)-1,2-cyclohexanediamine]4 have also been demonstrated to catalyse the ATH of aromatic ketones with 2-propanol, while these complexes were less effective for asymmetric H2-hydrogenation (AH) of the unsaturated substrates in aprotic solvents. In contrast, some cationic amine equivalents, Ru(OTf)(Tsdpen)(η6-p-cymene), [Cp*Rh(Tsdpen)]+ and [Cp*Ir(Tscydn)(CH3CN)]+, which can be synthesised by treatment of the amido complex with a stoichiometric amount of TfOH or by a chloro(amine) catalyst precursor with silver salts, are found to be highly effective for the AH of either ketones or imines.5–8 A related Cp*RuCl(cod)/diamine/KOH catalyst, which is isoelectronic to 1 but inert to transfer hydrogenation from 2-propanol, is highly effective for the hydrogenation of ketones in alcohols.9 In this reaction, the alcoholic solvents are proposed to participate in the H2 activation through the formation of a hydrogen-bonding network (Scheme 2).10,11
:2)3 as a reaction medium and a hydrogen source (Scheme 1). Analogous Cp*M-Tsdpen and -Tscydn complexes [Cp* = η5-1,2,3,4,5-pentamethylcyclopentadienyl; M = Rh, Ir; Tscydn = N-(p-toluenesulfonyl)-1,2-cyclohexanediamine]4 have also been demonstrated to catalyse the ATH of aromatic ketones with 2-propanol, while these complexes were less effective for asymmetric H2-hydrogenation (AH) of the unsaturated substrates in aprotic solvents. In contrast, some cationic amine equivalents, Ru(OTf)(Tsdpen)(η6-p-cymene), [Cp*Rh(Tsdpen)]+ and [Cp*Ir(Tscydn)(CH3CN)]+, which can be synthesised by treatment of the amido complex with a stoichiometric amount of TfOH or by a chloro(amine) catalyst precursor with silver salts, are found to be highly effective for the AH of either ketones or imines.5–8 A related Cp*RuCl(cod)/diamine/KOH catalyst, which is isoelectronic to 1 but inert to transfer hydrogenation from 2-propanol, is highly effective for the hydrogenation of ketones in alcohols.9 In this reaction, the alcoholic solvents are proposed to participate in the H2 activation through the formation of a hydrogen-bonding network (Scheme 2).10,11
 | ||
| Scheme 1 Hydrogen transfer from alcohols to ketones based on interconversion between amido and hydrido(amine) complexes. | ||
 | ||
| Scheme 2 Heterolysis of molecular hydrogen by cationic amine species. | ||
Herein, we disclose that the catalytic AH of ketones with Cp*Ir[(S,S)-RSO2dpen] [RSO2 = Ms (CH3SO2): 2a, Ts: 2b] is accelerated by the support of methanol or ethanol without any modification of the amidoiridium complexes. Furthermore, it was experimentally demonstrated that the AH conditions can overcome the intrinsic reversibility of the competitive hydrogen transfer reaction between ketone substrates and alcohol products, leading to higher enantioselectivities rather than the products obtained by ATH.
Our initial efforts concentrated on the stoichiometric reaction of amidoiridium complexes 2a–b with H2 as shown in Table 1. As mentioned in above, 2b bearing (S,S)-TsDPEN ligand did not readily react with atmospheric pressure of H2 in aprotic THF (entry 1), and hydridoiridium complexes 3a and 3b were formed in yield of 37% and 20% respectively, even under pressurised hydrogen (30 atm) after 6–7 h (entries 2 and 3).

On the other hand, treatment of amidoiridium complexes 2a and 2b in excess CD3OD provided the corresponding deuterated hydridoiridium complexes, 3a and 3b, in excellent yields within 10 min (Scheme 3). In the 2H NMR spectra, the total integral intensity of signals attributed to the coordinating amine on 3a or 3b formed is almost two times as much as that of the deuterido ligand, indicating that both amine protons are rapidly exchanged with the OD group of the CD3OD solvent. The deuterido ligand was exclusively derived from CD3OD, but unfortunately, the resulting dehydrogenated formaldehyde (CD2O) was not detected in the methanolic reaction mixtures by NMR spectroscopy, possibly due to formation of hemiacetal species including oligomers.12
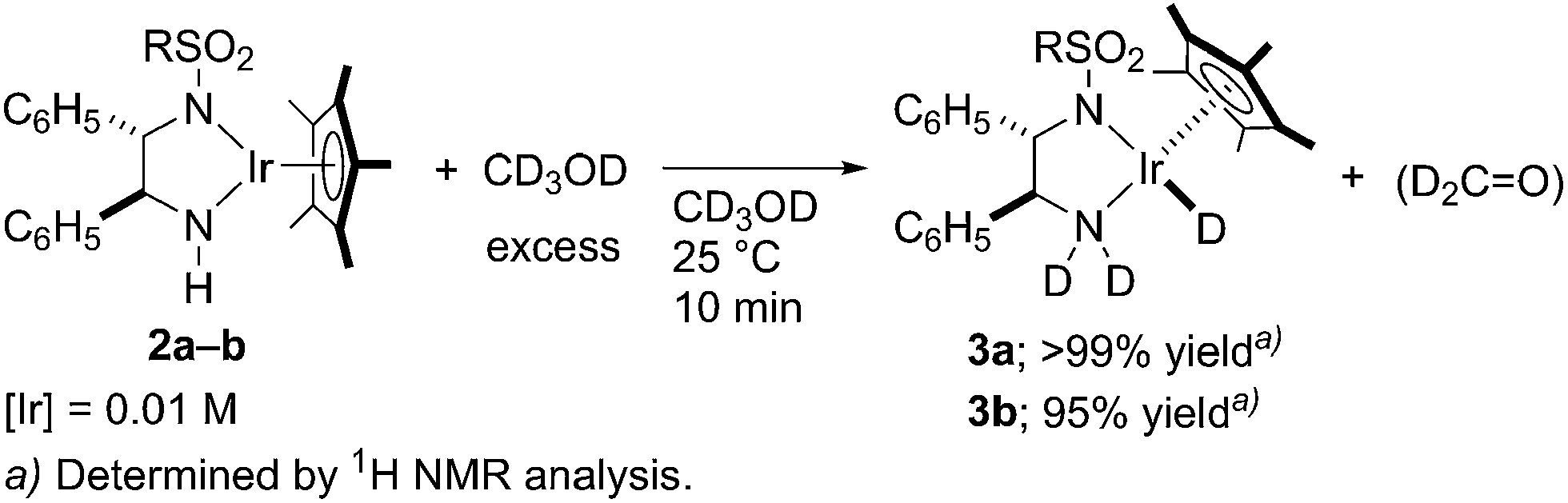 | ||
| Scheme 3 Deuterium transfer from methanol-d4 to amidoiridium 2. | ||
Although the amido complexes 2 are highly susceptible to the hydrogen transfer from methanol, it was anticipated that alcoholic solvents could assist the H2 heterolysis with 2 in similar to the above-mentioned mechanism in Scheme 2, and would operate in favour of catalytic AH without any modification of catalyst structure. In order to evaluate the relative rate of H2-hydrogenation and transfer hydrogenation using methanol, deuterium labelling reactions of 4′-methylacetophenone (4) catalysed by the amidoiridium complexes 2 with a substrate/catalyst molar ratio (S/C) of 100 were conducted in CD3OD (1 M) under hydrogen pressure (30 atm) at 30 °C. The conversion of the substrate 4 and yield of 1-(4′-methylphenyl)ethanol (5) were determined based on signals of the methyl protons (Ha and Hb) on the aromatic ring of 4 and 5 (2.37 ppm and 2.28 ppm in CD3OD, respectively) in 1H NMR spectra of the reaction mixture containing triphenylmethane as an internal standard. The deuterium content at the methine hydrogen (Hc) in the product 5 was confirmed by comparing the integral intensity, which enables to discriminate the Hc atoms delivered from CD3OD and H2. As summarised in Table 2, when 2a was used as the catalyst (entry 1), 5 was obtained in almost quantitative yield (>99%) with 98% ee (S). In this reaction, the %D value at Hc of 5 was 42%, indicating that 58% of the ketone 4 was reduced via the AH in preference to the ATH from CD3OD. The reaction conducted in CH3OD gave 5 without noticeable deuterium incorporation into Hc (entry 2). Consequently, it assures that the AH/ATH estimation under these conditions is not influenced by a potent reversible exchange between the alcoholic proton with the iridium hydride species. The reduction rate dramatically changes depending on the sulfonyl groups on the DPEN ligand of the bifunctional catalysts. The reaction using the amido complex 2b with a Ts substituent gave 5 in 60% yield with 98% ee (S) under the identical reaction conditions (entry 3), whereas 2c having a bulky pentamethylbenzenesulfonyl group proved to be catalytically less active for the reduction (entry 4). The moderate Hc-deuteration (48% D) in the Tsdpen–Ir system also corroborates the AH in conjunction with the ATH.
|
|||||
|---|---|---|---|---|---|
| Entry | Cat | % conva | % yielda | % eeb | H/D at Hc |
| a Determined by 1H NMR analysis.b Determined by HPLC analysis using a Chiralcel OD-H column.c CH3OD was used as solvent.d n.d. = not determined. | |||||
| 1 | 2a | >99(Ha) | >99(Hb) | 98 | 58/42 |
| 2c | 2a | 99(Ha) | 99(Hb) | 98 | >99/— |
| 3 | 2b | 66(Ha) | 60(Hb) | 98 | 52/48 |
| 4d | 2c | <5(Ha) | <5(Hb) | n.d. | n.d. |

When the ATH reaction of 4 using the effective bifunctional catalyst 2a was performed in CH3OH under argon atmosphere under otherwise identical conditions, the chiral alcohol 5 was produced in lower yield and enantioselectivity of 85% and 90% ee, compared to the outcome of the AH/ATH reaction. Elongation of the reaction time to 72 h to complete consumption of 4 resulted in 5% loss in optical purity of 5 (entries 1 and 2 of Table 3), because the reversibility in hydrogen transfer between alcohols and ketones is known to deteriorate the enantioselectivity as the reaction time passes. Notably, the simultaneous AH and ATH system holds excellent levels of 97–98% ee during 3–24 h reactions (entries 3–5), suggesting that the pressurised H2 would make the ATH pathway irreversible. In fact, the deuterium content of 32% at the Hc of 5 after the 3 h reaction was slightly increased to 40% after 6 h, and a further H/D exchange was mostly hampered under the AH conditions. In the ATH in methanol, formaldehyde is generated and can act as the hydrogen acceptor in the reverse hydrogen transfer from the product chiral alcohol; however, the AH reaction will lead to complete consumption of the aldehyde. Therefore, hydrogen pressure is important to maintain the excellent optical purity of the product.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Time, h | % conva | % yielda | % eeb | H/D at Hc |
| a Determined by 1H NMR analysis and GC analyses.b Determined by HPLC analysis using a Chiralcel OD-H column.c ATH reaction without H2.d Acetophenone was used as substrate. | ||||||
| 1c | CH3OH | 24 | 84 | 85 | 90 | — |
| 2c | CH3OH | 72 | 97 | >99 | 85 | — |
| 3 | CD3OD | 3 | 57(Ha) | 57(Hb) | 97 | 68/32 |
| 4 | CD3OD | 6 | 84(Ha) | 78(Hb) | 98 | 60/40 |
| 5 | CD3OD | 24 | >99(Ha) | >99(Hb) | 98 | 58/42 |
| 6 | C2D5OD | 24 | >99(Ha) | >99(Hb) | 98 | 47/53 |
| 7 | IPA-d8 | 24 | 56(Ha) | 65(Hb) | 98 | 0/100 |
| 8d | THF | 24 | 11 | 8 | 82 | — |

The ATH/AH reaction of 4 also proceeded in ethanol-d6 to give 5 in almost quantitative yield with 98% ee in similar to the reaction in CD3OD, while increase of the H/D ratio at Hc was observed as a result of enhancement of the ATH from the alcoholic solvent (entry 6). On the contrary, the AH reaction was not promoted in 2-propanol-d8 to give 5 by net ATH in 65% yield and with 98% ee (S) (entry 7). According to the H2 heterolysis via an ion-pair intermediate composed of a cationic amine complex and an alkoxide counter anion depicted in Scheme 2, it may be more difficult to activate molecular hydrogen in less acidic 2-propanol (pKa = 17.1) than methanol (pKa = 15.2) or ethanol (pKa = 15.9). Actually, hydrogenation of acetophenone was unsuccessful in aprotic THF in the presence of H2 (30 atm), affording 1-phenylethanol in yield of 8% (entry 8).
The beneficial feature of this hydrogenation guided by methanol was successfully verified by the reaction of diacetylbenzenes, for which few ATH or AH catalysts have been reported.13 As shown in Table 4, 1,4-diacetylbenzene was almost completely reduced to the corresponding (S,S)-diol with an excellent enantioselectivity of 99.7% ee and a dl/meso ratio of 96/4 (entry 1), whereas the ATH in methanol resulted in a moderate conversion of 43%, affording a small amount (1% yield) of the diol in addition to a mono-alcohol product, (S)-1-acetyl-4-(1-hydroxyethyl)benzene (41% yield, 84% ee). The AT/ATH reaction of 1,3-diacetylbenzene also furnished (S,S)-1,3-bis(1-hydroxyethyl)benzene with 99.5% ee, as well (entry 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | % conva | % yield (diol)a | % eeb | dl/mesob |
| a Determined by 1H NMR analysis.b Determined by GC analysis using a CP-Chirasil-Dex CB column.c ATH reaction in a methanol solution (0.5 M) without H2. | |||||
| 1 | 1,4-Diacetylbenzene | 100 | >99 | 99.7 | 96/4 |
| 2c | 1,4-Diacetylbenzene | 43 | 1 | — | — |
| 3 | 1,3-Diacetylbenzene | 100 | 99 | 99.5 | 95/5 |

In conclusion, the formation of bifunctional hydridoiridium complexes via heterolytic cleavage of H2 using amidoiridium 2 becomes competitive to hydrogen transfer from alcoholic hydrogen donors by employment of primary alcohols as the solvent. The simultaneous hydrogenation favourably influences the outcome of stereoinduction, owing to overcome undesired decrease in optical purity of the chiral alcohol products arising from the inherent reversibility of the hydrogen transfer reaction between ketones and alcohols. In comparison with the original ATH, this AH/ATH system is advantageous to accelerate the ketone reduction with maintaining the excellent enantiomer discrimination ability of the bifunctional Ir catalysts without any modification of the molecular structure.
Acknowledgements
This work is supported by JSPS KAKENHI Grant number 22225004, 24350079, and 26621043, and partly supported by the GCOE Program.Notes and references
- (a) J.-i. Ito and H. Nishiyama, Tetrahedron Lett., 2014, 55, 3133–3146 CrossRef PubMed; (b) R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS PubMed; (c) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- (a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7573 CrossRef CAS; (b) K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS; (c) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (d) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS.
- (a) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS; (b) K. Murata, K. Okano, M. Miyagi, H. Iwane, R. Noyori and T. Ikariya, Org. Lett., 1999, 1, 1119–1121 CrossRef CAS; (c) K. Okano, K. Murata and T. Ikariya, Tetrahedron Lett., 2000, 41, 9277–9280 CrossRef CAS; (d) M. Watanabe, K. Murata and T. Ikariya, J. Org. Chem., 2002, 67, 1712–1715 CrossRef CAS PubMed.
- (a) K. Mashima, T. Abe and K. Tani, Chem. Lett., 1998, 27, 1199–1200 CrossRef; (b) K. Mashima, T. Abe and K. Tani, Chem. Lett., 1998, 27, 1201–1202 CrossRef; (c) K. Murata, T. Ikariya and R. Noyori, J. Org. Chem., 1999, 64, 2186–2187 CrossRef CAS.
- (a) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724–8725 CrossRef CAS PubMed; (b) C. A. Sandoval, T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata and R. Noyori, Chem.–Asian J., 2006, 1–2, 102–110 CrossRef PubMed; (c) T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori and K. Murata, Org. Lett., 2007, 9, 255–257 CrossRef CAS PubMed; (d) T. Ohkuma, N. Utsumi, M. Watanabe, K. Tsutsumi, N. Arai and K. Murata, Org. Lett., 2007, 9, 2565–2567 CrossRef CAS PubMed; (e) C. A. Sandoval, F. Bie, A. Matsuoka, Y. Yamaguchi, H. Naka, Y. Li, K. Kato, N. Utsumi, K. Tsutsumi, T. Ohkuma, K. Murata and R. Noyori, Chem.–Asian J., 2010, 5, 806–816 CrossRef CAS PubMed; (f) N. Arai, H. Sato, N. Utsumi, K. Murata, K. Tsutsumi and T. Ohkuma, Org. Lett., 2013, 15, 3030–3033 CrossRef CAS PubMed.
- (a) C. Li and J. Xiao, J. Am. Chem. Soc., 2008, 130, 13208–13209 CrossRef CAS PubMed; (b) C. Li, C. Wang, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2008, 130, 14450–14451 CrossRef CAS PubMed; (c) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967–6969 CrossRef CAS PubMed.
- (a) Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2009, 131, 3593–3600 CrossRef CAS PubMed; (b) C. S. Letko, Z. M. Heiden and T. B. Rauchfuss, Eur. J. Inorg. Chem., 2009, 4927–4930 CrossRef CAS.
- S. Shirai, H. Nara, Y. Kayaki and T. Ikariya, Organometallics, 2009, 28, 802–809 CrossRef CAS.
- M. Ito, M. Hirakawa, K. Murata and T. Ikariya, Organometallics, 2001, 20, 379–381 CrossRef CAS.
- M. Ito and T. Ikariya, Chem. Commun., 2007, 5134–5142 RSC.
- (a) C. Hedberg, K. Källström, P. I. Arvidsson, P. Brandt and P. G. Andersson, J. Am. Chem. Soc., 2005, 127, 15083–15090 CrossRef CAS PubMed; (b) T. Otsuka, A. Ishii, P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 9600–9603 CrossRef CAS PubMed.
- (a) S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2013, 52, 10253–10255 CrossRef CAS PubMed; (b) A. L. Balashov, S. M. Danov, V. L. Krasnov, A. Y. Chernov and T. A. Ryabova, Russ. J. Gen. Chem., 2002, 72, 744–747 CrossRef CAS.
- (a) X. Zhou, X. Wu, B. Yang and J. Xiao, J. Mol. Catal., 2012, 357, 133–140 CrossRef CAS PubMed; (b) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675–2676 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07854c |
| This journal is © The Royal Society of Chemistry 2014 |