
A three-dimensional co-culture of HepG2 spheroids and fibroblasts using double-layered fibrous scaffolds incorporated with hydrogel micropatterns

Abstract
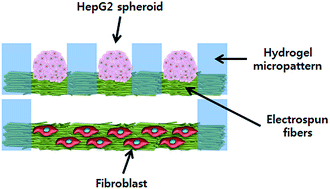
We developed a novel methodology of constructing a three-dimensional (3D) heterotypic co-culture system based on double-layered fibrous scaffolds incorporated with hydrogel micropatterns. The combination of electrospinning and hydrogel patterning generated micropatterned fibrous scaffolds consisting of poly(ethylene glycol) (PEG) hydrogel micropatterns and polycarprolactone (PCL) fibers. The thickness of the hydrogel micropatterns and fiber matrices, which were in the ranges of 100 to 250 μm and 20 to 80 μm, respectively, could be controlled by the volume of the hydrogel precursor solution and the collection time used for electrospinning. Because the resultant micropatterned fibrous scaffolds were obtained as a free-standing and bidirectionally-porous sheet, they could be stacked in a double-layered structure in which each scaffold contains different cell types for co-culture studies. As a model system, double-layered scaffolds for the co-culture of HepG2 and fibroblast cells were constructed by placing HepG2-containing scaffolds on top of fibroblast-containing scaffolds. The micropatterned fibrous scaffolds were demonstrated to be suitable for the culture of both HepG2 and fibroblasts cells. In addition, by controlling the micropattern size, HepG2 spheroids of uniform size (187.2 ± 10.7 μm) were formed in the top layer and used for the co-culture studies. According to the co-culture experiment, enhanced albumin secretion was observed from the co-cultured HepG2 cells compared with the single-cultured HepG2 cells, suggesting that micropatterned fibrous scaffolds are a promising tool that can be applied to heterotypic co-culture systems in various tissue engineering applications.
 Please wait while we load your content...
Please wait while we load your content...

