
Design of an n-type low glass transition temperature radical polymer†
Teng
Chi
a,
Siddhartha
Akkiraju
b,
Zihao
Liang
b,
Ying
Tan
b,
Ho Joong
Kim
b,
Xikang
Zhao
b,
Brett M.
Savoie
 *b and
Bryan W.
Boudouris
*ab
*b and
Bryan W.
Boudouris
*ab
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA
bCharles D. Davidson School of Chemical Engineering, Purdue University, 480 Stadium Avenue, West Lafayette, Indiana 47907, USA. E-mail: bsavoie@purdue.edu; boudouris@purdue.edu
First published on 17th February 2021
Abstract
We document the design, synthesis, and characterization of the first low glass transition temperature, n-type (i.e., preferentially-reduced) radical polymer. Specifically, a macromolecule composed of a polysiloxane backbone that bears galvinoxyl radical pendant groups, poly[2,6-di-tert-butyl-4-((3,5-di-tert-butyl-4-(λ1-oxidaneyl)phenyl)(4-((3-(methoxydimethylsilyl)propoxy) methyl)phenyl)methylene)cyclohexa-2,5-dien-1-one] (PGMS), was created as our calculations predicted that the galvinoxyl radical molecular structure would facilitate radical–radical aggregation. In turn, this suggested that charge transport would be rapid in these systems, which would lead to large solid-state electronic conductivity values. After the design and successful synthesis of the PGMS radical polymers, their optical, spin, thermal, and electrochemical properties were evaluated in full. These experiments backed the idea that PGMS has a low glass transition temperature and robust electrochemical behavior. Furthermore, when a PGMS macromolecule was cast into a thin film, a solid-state conductivity of 10−2 S m−1 was achieved, and this was despite the fact that only ∼36% of the pendant groups contained a galvinoxyl radical. This high conductivity appears to be a direct result of the radical–radical aggregation that occurs due to the molecular design of the galvinoxyl radical species. Therefore, this work highlights the import of developing next-generation open-shell entities for solid-state radical polymer conductors, and it provides a clear path forward for creating high conductivity, non-conjugated conducting macromolecules.
Introduction
Nonconjugated macromolecules bearing stable radical pendent groups (i.e., radical polymers) are an emerging class of electronically-active materials that are less frequently employed in device applications relative to their more established π-conjugated polymer counterparts.1–6 From a macromolecular design standpoint, they are intriguing due to the inherently decoupled parameters associated with their macromolecular backbone, which dictates the macroscopic thermomechanical properties, and their redox-active pendant groups that dictate their optoelectronic and electrochemical behavior. In fact, the initial intrigue and evaluation of radical polymers came about due to the rapid redox reactions involved with their pendant groups; as such, most of the application focus of these materials was centered on electrolyte-based systems.7–10 Thus, radical polymers were frequently utilized in energy storage applications due to the high density of redox-active species present along the pendant groups of the macromolecules.9–22 Whereas, only over the last handful of years have the solid-state electrical conductivity properties of radical polymers been evaluated in full.8,23–28In many of the early solid-state electrical conductivity evaluation efforts, the conductivity of a model radical polymer, poly (2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PTMA), was quantified by multiple groups with the a highest value of ∼10−4 S m−1 being reported.23,26,29,30 This solid-state electrical conductivity was improved due to an improved radical polymer design in that poly(2,3-bis(2′,2′,6′,6′-tetramethylpiperidinyl-N-oxyl-4′-oxycarbonyl)-5-norbornene) (PTNB) had a higher radical content than what was typically observed in the PTMA-based macromolecular design case, and a thin film of this material achieved a solid-state electronic conductivity of 7 × 10−3 S m−1.31 Hindering the charge transport of both of these materials was the relatively high glass transition temperature (Tg) associated with PTMA and PTNB, and the lack of a thermal processing window that was above the glass transition temperature but below the onset degradation temperature of the materials. To overcome this limitation, a low-Tg radical polymer, poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl) (PTEO), was synthesized, and a solid-state electronic conductivity of ∼20 S m−1 was observed at room temperature.32 This highlighted that creating macromolecules that allowed for relatively long degrees of radical–radical coupling through space within the solid-state was critical for rapid charge transport, and that large degrees of crystallinity and/or large conjugation lengths were not required for high conductivity values to be had. Due to this change in polymer design archetype and the continued improvements in the design of radical polymers, open-shell macromolecules have been successfully implemented in myriad electronic systems recently including photovoltaic (PV) devices,33 organic electrochromic devices,34 rewritable memory units,35 and organic transistors.36,37 Despite showing solid performance in all of these applications, a key hindrance still exists in these materials, and this is the fact that all of the solid-state electronic applications using radical polymers reported to date have relied on preferentially-oxidized (i.e., p-type) radical groups. From a historical perspective, this is sensible as the majority of radical polymers are p-type in nature, and the most oft-used open-shell groups are those of the nitroxide class.38–47 This draws a parallel to the conjugated polymer literature where hole-transporting macromolecules dominated the research landscape; however, it was clear in the conjugated polymer regime previously, and it is clear in the radical polymer community now, that developing preferentially-reduced (i.e., n-type) open-shell macromolecules will be of critical importance in the near future.3,8,48,49
To address this gap, here we design, synthesize, and characterize the electronic and electrochemical properties of the first low glass transition temperature, n-type (i.e., preferentially-reduced) radical polymer. Specifically, we synthesized a radical polymer with a flexible polysiloxane backbone bearing galvinoxyl radical moieties, poly[2,6-di-tert-butyl-4-((3,5-di-tert-butyl-4-(λ1-oxidaneyl)phenyl)(4-((3-(methoxydimethylsilyl)propoxy)methyl)phenyl)methylene)cyclohexa-2,5-dien-1-one] [poly(galvinoxyl methyl siloxane), PGMS]. To accomplish this objective, semi-empirical quantum chemistry calculations were first introduced to establish the potential charge transport benefits in moving to a galvinoxyl radical relative to the oft-used, yet highly-localized, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) radical. These calculations predicted that the galvinoxyl radical, when successfully incorporated into a polymer with a flexible macromolecular backbone, would have promising charge conduction properties. Then, PGMS was synthesized by coupling a protected radical group to a polysiloxane macromolecular backbone, and the closed-shell pendant groups were converted to the active open-shell groups through simple oxidation after the coupling reaction occurred. Despite the addition of a relatively bulky open-shell group to the polysiloxane chain, the glass transition temperature of the PGMS macromolecules synthesized remained low (i.e., below room temperature); however, the bulkiness of the protected galvinoxyl groups did limit the ultimate coupling efficiency of the radical-bearing pendant groups to the main chain. Thus, relatively high, but not record-setting, conductivity values (∼10−2 S m−1) were had when evaluating the solid-state charge transport ability of the PGMS thin films. Moreover, we anticipate that this initial value can be pushed to even higher values if higher loadings of radical groups can be incorporated into the polymer backbone. Therefore, this work is the first report of an n-type, low glass transition temperature radical polymer and its associated electronic conductivity, and it highlights the crucial fact that next-generation open-shell groups like the galvinoxyl radical possess the potential to achieve relatively high conductivities in the solid state.
Results and discussion
To guide the macromolecular design, the conformers of PGMS with various radical loadings (i.e., 0%, 25%, 50%, 75% and 100% of galvinoxyl radical species present on the repeat units) were characterized using CREST, a metadynamics-based search algorithm recently developed by Grimme and co-workers.50,51 CREST yields the relative energetics (ΔE) and configuration of each conformer, which we have used to assay the expected spatial distribution of galvinoxyl radicals and the favorability of intra-chain charge transfer at each loading. Although these evaluations were performed on isolated chains, the trends illustrate several potential mechanisms for the strong density dependence of conductivity on the radical loading. First, we observe a monotonic increase in the polymer radius of gyration (Rg) as the radical loading increases, which saturates at high loadings (Fig. 1a). In particular, at low loadings, there is sufficient free volume about the backbone to accommodate additional galvinoxyl radicals, while at the higher loadings, crowding constrains the configurations that the galvinoxyl radicals can adopt, and this leads to a saturation of Rg. This saturation suggests a strong loss of configurational entropy as the loading increases, signifying a potential synthetic challenge to obtaining complete loading of the radical in this system.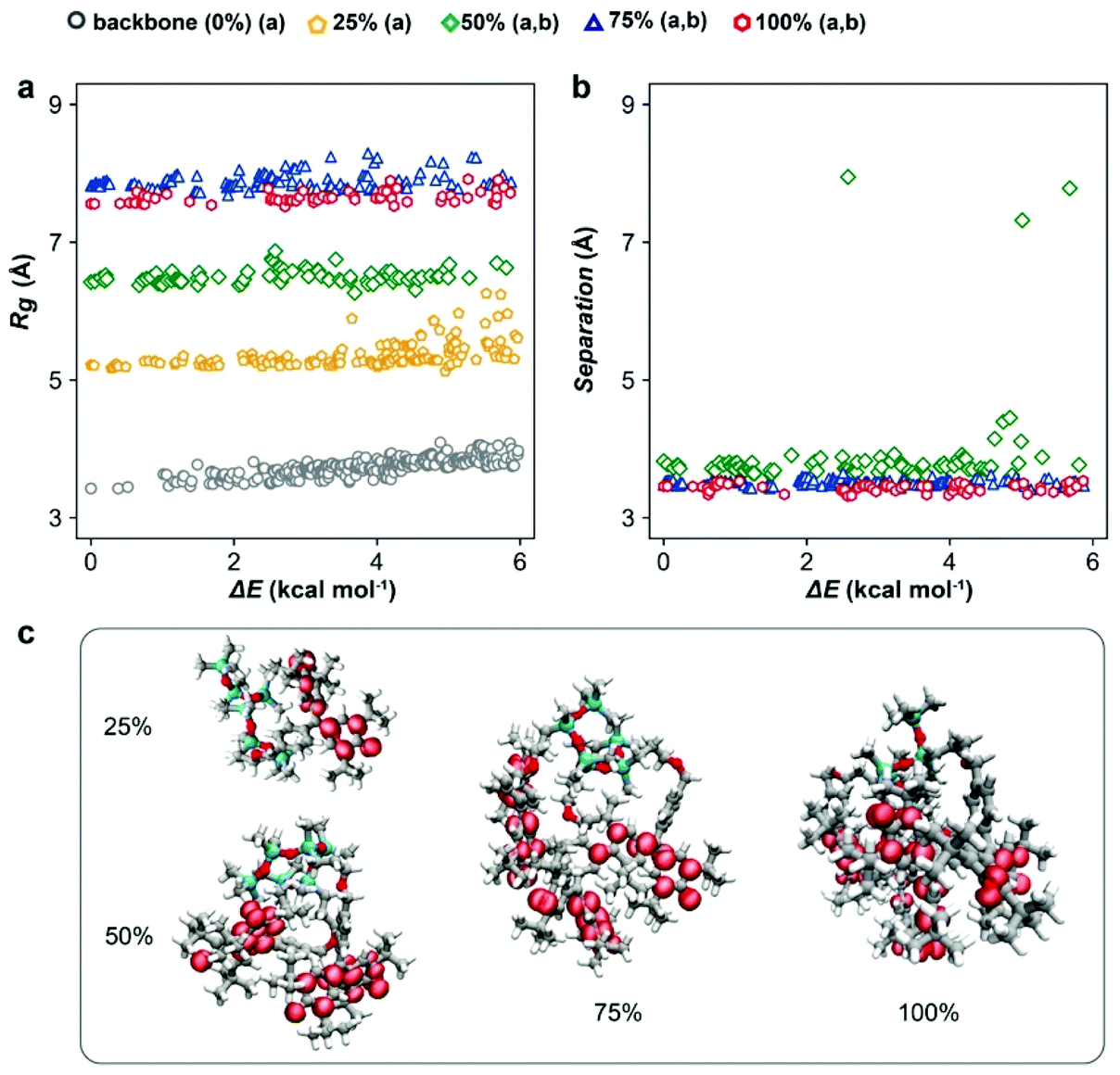 | ||
| Fig. 1 (a) Radius of gyration of the polymer and the relative energy (ΔE) of each conformer with various radical densities in the 6 kcal mol−1 energy window computed using CREST. (b) Average galvinoxyl radical nearest-neighbor distance (NND) and relative energy of the PGMS conformers with 50%, 75% and 100% radical densities in the 6 kcal mol−1 energy window computed using CREST. (c) Molecular structures and spin density visualization of the lowest energy conformer at each radical density, calculated at the GFNn–xTB level of theory. Atom representation: C – gray; O – red; Si – cyan; H – white. | ||
Comparing the average nearest-neighbor distances (NNDs) between galvinoxyl radical groups at each loading (Fig. 1b) also suggests that intra-chain transport is promoted by strong galvinoxyl radical–galvinoxyl radical interactions. Specifically, at all loadings above 25% (i.e., loadings with two or more galvinoxyl radicals per polysiloxane chain) the galvinoxyl radical NNDs are comparable. This is because the polymer favors configurations that allow the galvinoxyl radical moieties to interact, even at low loadings where conformers with larger NNDs are possible. Nevertheless, we observe a small number of conformations for the 50% radical loading system where the galvinoxyl sites exhibit large separations. When translated into the condensed phase, the occurrence of such conformations along the backbone would severely disrupt intrachain charge transport. Although in the condensed phase intra-chain open-shell aggregation competes with other complex interchain interactions and packing effects, these results suggest that intra-chain charge transport is a potentially significant charge transport channel at radical loadings above 25%. This aggregation behavior is also evident from the spin-density isosurfaces calculated for the lowest energy conformer at each loading (Fig. 1c). That is, we observe the spin density is delocalized throughout the systems, with radical aggregation evident at loadings >25%. These data indicate several positive design features when translating this macromolecular architecture to the experimental side as the increase of radical density and aggregation should favor rapid charge transfer.
These positive computational results encouraged the synthesis route shown in Scheme 1. Here, 4-bromo-2,6-di-tert-butylphenol (1) was first protected by trimethylsilyl chloride (TMS-Cl) under basic conditions to generate (4-bromo-2,6-di-tert-butylphenoxy)trimethylsilane (3),52–54 and methyl-4-(hydroxymethyl)benzoate (2) was treated with triethyl amine and TMS-Cl in sequence to form methyl-4-(((trimethylsilyl)oxy)methyl)benzoate (4).55,56 Using a lithium-halogen exchange, a strong nucleophile was generated using n-butyl lithium, and it underwent a nucleophilic substitution followed by a nucleophilic addition reaction.57 The intermediate was quenched by KOH followed by HCl to give 2,6-di-tert-butyl-4-((3,5-di-tert-butyl-4-hydroxyphenyl)(4-(hydroxymethyl) phenyl)methylene)cyclohexa-2,5-dien-1-one (5) in one pot.57–61
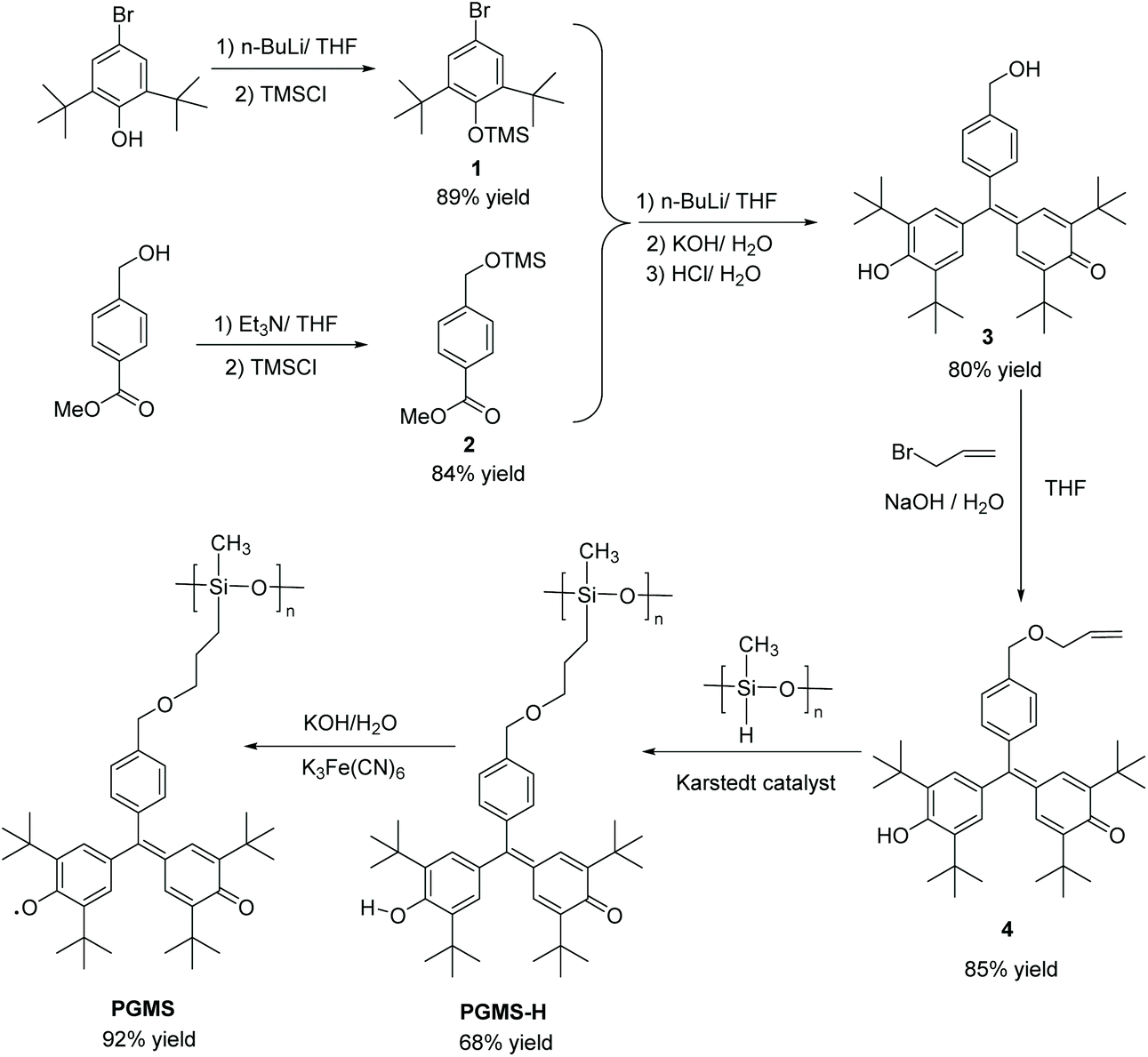 | ||
| Scheme 1 Synthetic pathway for PGMS radical polymers. | ||
Then, the alkene functional group was successfully introduced (i.e., to form 6) by a substitution reaction under basic conditions.32,62,63 Next, this precursor molecule was bound to the polysiloxane backbone through a Karstedt reagent-catalyzed coupling reaction to form the PGMS-H.64–66 These compounds were characterized by 1H NMR spectroscopy and 13C NMR spectroscopy (Fig. S1–S3†), and they were found to be of high purity. Finally, the PGMS open-shell macromolecule was created through a straightforward oxidation reaction.67
To evaluate any potential effects of molecular weight on the final properties of the materials, three polysiloxane parent polymers were utilized with number-average degrees of polymerization of 4, 24, and 39 repeat units in the main chain, respectively. Thus, three different molecular weights of PGMS were synthesized, and their properties were compared to those predicted by the computational results. All of the closed-shell polymers (i.e., the three different molecular weights of PGMS-H synthesized) showed similar ultraviolet–visible (UV–Vis) light absorption spectra with a peak absorption at 400 nm, and their absorption profiles are independent of the number of repeat units present (Fig. 2). After oxidation to the functional open-shell PGMS species, the peak at 400 nm decreased while a new peak at 480 nm emerged, and this is consistent with the absorption spectrum of previously-reported galvinoxyl radical-containing materials.16,60,67 In the samples with more repeat units, PGMS-24 and PGMS-39, there was an additional local absorption maximum observed around a wavelength of 600 nm, which is likely due to the relatively lower solubility of this higher molecular weight sample and the subsequent aggregation of the materials in solution.
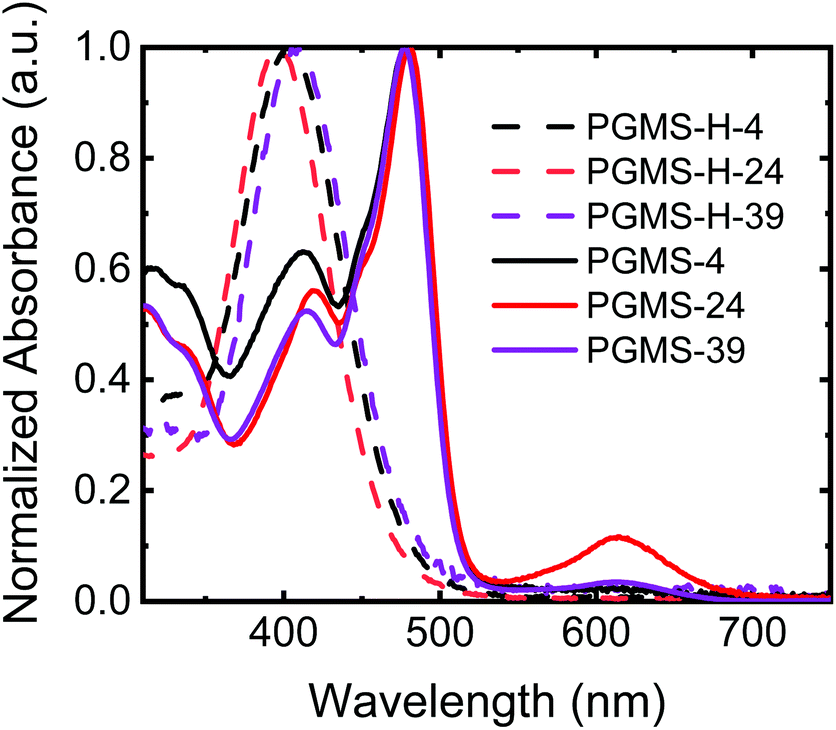 | ||
| Fig. 2 The normalized UV–Vis light absorption spectra of the closed-shell PGMS-H polymers and the open-shell PGMS radical polymers with different molecular weights when the polymers were dissolved in THF. | ||
The electron paramagnetic resonance (EPR) spectrum of the galvinoxyl radical small molecule showed its characteristic signal, with two symmetric large peaks and two minor peaks (Fig. 3a). Then, when the galvinoxyl radical moieties were coupled into a single polymer chain for the radical polymer samples, a Lorentzian signal was observed in the EPR spectra (Fig. 3b). This is consistent with intrachain radical–radical interactions, and these data highlight the ability to couple the small molecule galvinoxyl radical precursor material to the polysiloxane chains. Moreover, PGMS macromolecules with the larger molecular weights showed the same shape and position of EPR peaks as PGMS-4, which suggests that there was little impact of molecular weight with respect to the coupling reaction (over the limited molecular weight range evaluated here). The radical content of all the PGMS polymers was between 13% and 36% of the maximum value, and larger radical loadings were had when larger excesses of 4 (i.e., the vinyl-containing small molecule precursor) were used in the coupling reaction. The conformer search simulation provides a potential explanation for this relatively low radical content. The number of energetically accessible conformers significantly decreased when more pendent groups were coupled to the backbone, which indicated that it would be difficult to couple the galvinoxyl pendent groups to every repeating unit in the polymer chain. Thus, it is likely that the low radical content is due to steric interactions impacting the coupling reaction, and it is not due to the final oxidation step as this step has been used in an efficient manner previously.16,67,68 Therefore, the steric interactions of the chosen coupling reaction appear to limit the maximum radical content present in the macromolecules to ∼36% of the maximum possible loading (i.e., only one open-shell unit per every two or three repeat units was achieved as opposed to the maximum possible radical loading of one open-shell species per repeat unit).
 | ||
| Fig. 3 The normalized EPR spectra of (a) the small molecule galvinoxyl radical in toluene and (b) PGMS-4, PGMS-24, and PGMS-39 radical polymers in toluene. | ||
The reversible reduction of the galvinoxyl radical in PGMS (Fig. 4a) was confirmed using cyclic voltammetry (CV) with a peak-to-peak width of 0.26 V vs. an Ag/AgCl reference electrode. The results of PGMS-39 are shown in Fig. 4b due to the higher solution stability (i.e., a lower degree of solubility in the electrolyte) of the higher molecular weight sample relative to the two lower molecular weight polymers. We note that the lower molecular weight PGMS samples behaved in a similar manner to PGMS-39 in the first a few redox cycles, but they dissolved into the solvent rapidly. A notable shift in the position of the redox reaction (−0.71 V) in comparison to the literature was observed, which is potentially attributed to the lack of organic base additives oft-utilized in the CV of galvinoxyl moiety, as this known to affect the redox peak position.9,16 A satellite oxidative peak at +0.12 V more closely coincides with that of galvinolate oxidation to galvinoxyl neutral radical in the oxidative scans reported in the literature,9,16 which may arise from internal charge transfer between swollen yet undissolved higher molecular weight species. Overall, the redox stability of PGMS in organic electrolytes combined with the useful redox potentials of this n-type radical polymer hints that, with well-contemplated molecular design and a changeable molecular weight, galvinoxyl-bearing non-conjugated polymer may be utilized in charge (and ion) conducting applications.
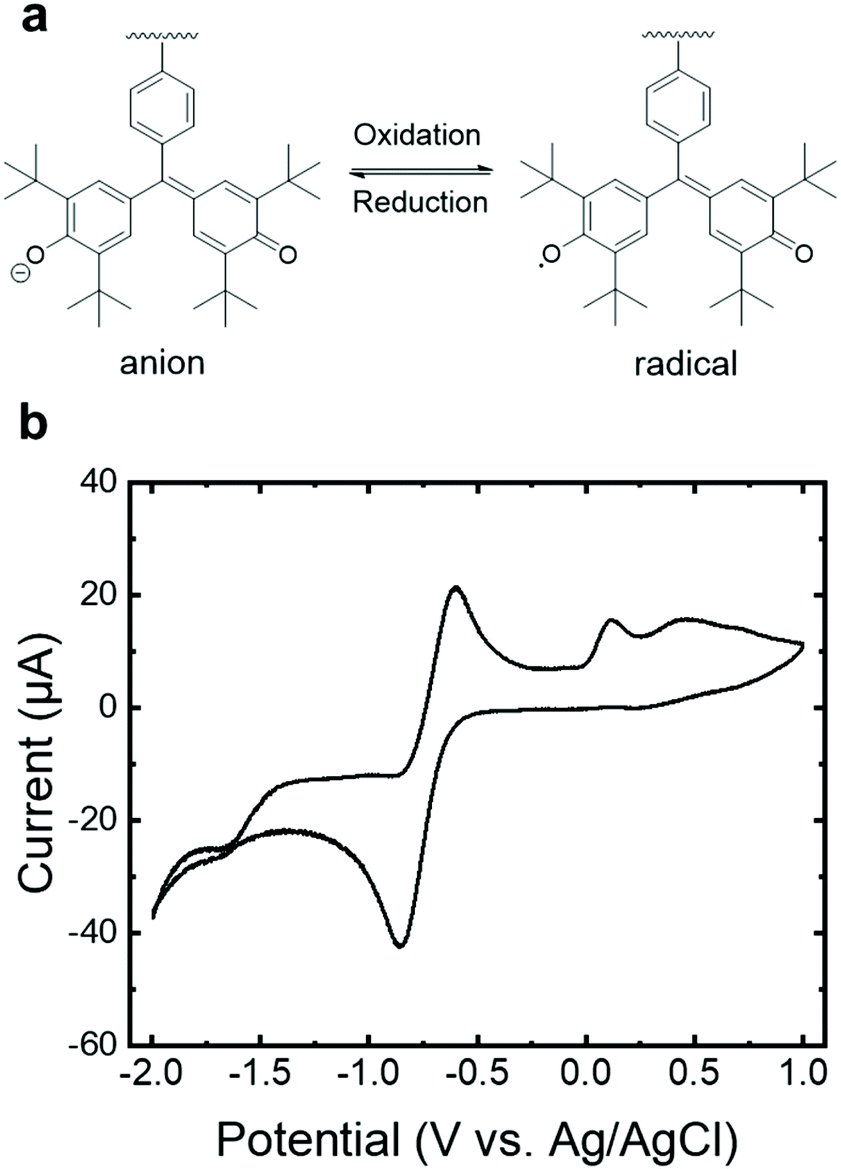 | ||
| Fig. 4 (a) The reversible redox reaction of PGMS. (b) Cyclic voltammogram of PGMS-39 (radical content 29%) on a gold working electrode using platinum as the counter electrode, recorded at a scan rate of 0.1 V s−1 in 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in N,N-dimethylformamide (DMF). The voltammogram shown was obtained after 5 conditioning cycles. | ||
Despite the relatively bulky, redox-active galvinoxyl radical being present on the side chain of the PGMS macromolecules, the radical polymers maintained a relatively low glass transition temperature value (Tg ∼−20 °C for PGMS-4, Fig. S4†), which is consistent with the flexible backbone of the polysiloxane parent polymer. Because PGMS-4 had a relatively low glass transition temperature, it was brought forward for electronic conductivity testing experiments. Fig. 5 shows the impact of temperature on the electronic conductivity of two different PGMS-4 samples with different radical loadings. In particular, the radical content of different batches of the PGMS-4 polymer were manipulated by changing the relative amount of oxidant to the number of closed-shell repeat units in PGMS-H-4 during the final synthetic step. Thus, PGMS-4-13 and PGMS-4-36 represent a PGMS-4 macromolecule with 13% and 36% radical content on average (where 100% would be 1 open-shell unit on every repeat unit of every radical polymer chain in the batch), respectively (Fig. S5†).
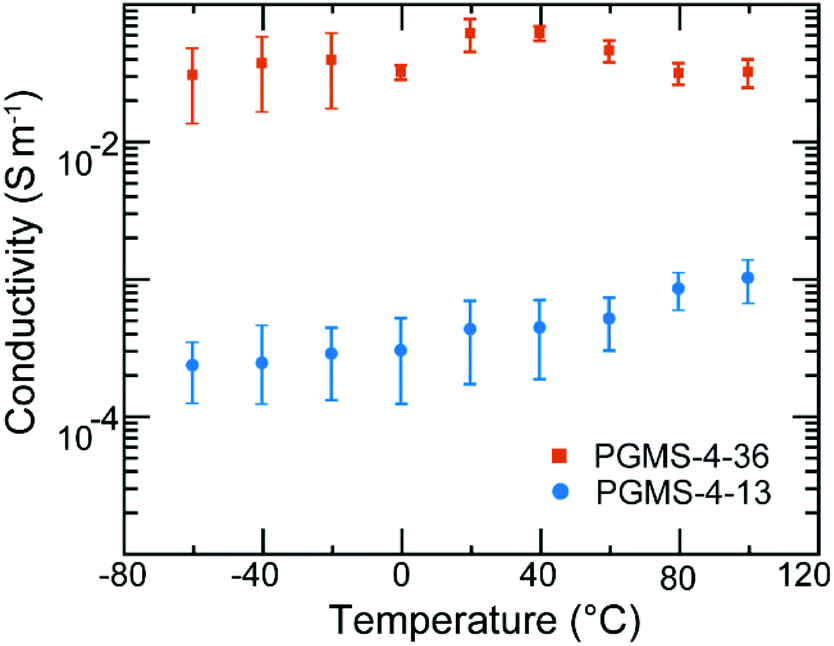 | ||
| Fig. 5 Electronic conductivity of PGMS as a function of temperature for a 400 nm channel length. The conductivity of the thin films did not change significantly despite an increase in temperature of >100 K. An increase in radical content increased the conductivity of the thin films from PGMS-4-13 to PGMS-4-36. Each data point represents the average of 4 different PGMS thin film measurements and the error bars represent the standard deviation from the average. | ||
Aside from the difference in radical loading, we note that the PGMS-4 samples with different radical content showed little difference in their thermal and electrochemical behavior. In the electrical conductivity experiments, thin films were cast into device structures where the channels had lengths of 400 nm and widths of 1 mm (Fig. S6a†) such that the in-plane conductivity of the PGMS thin films could be evaluated. Once cast, the thin films were immediately placed in a vacuum probe station, and the temperature was reduced to well below the Tg of PGMS. In these measurements, the two PGMS samples showed relatively high conductivities of 10−4 and 10−2 S m−1. Moreover, both PGMS samples maintained these high conductivity values when the temperature was increased well beyond their glass transition temperature (Fig. S6b and S6c†). Thus, it appears that the relatively high mobility of the PGMS chains and their ability to support polymer chain rearrangement allowed for the thin films to find local order from the processing method applied.
As the glass transition temperature of PGMS is ∼40 °C lower than room temperature, locally ordered domains, which facilitate electronic communication between open-shell groups, form readily as the solid-state film is cast. Thus, there was no large spike in conductivity due to local domain formation as we have seen in previous radical polymer systems.32 Moreover, PGMS thin films demonstrated electronic conductivity properties that were weakly temperature-dependent after the annealing process, and this is consistent with our previous results regarding radical polymers.31,32 While the electronic conductivity of PGMS is only a weak function of temperature, it is highly dependent on radical content as there is a two order of magnitude jump in conductivity among different PGMS thin films. Such an increase in conductivity could originate from both the increase in the number of charge carriers as well as the charge transfer capability. The latter factor is intrinsically affected by the former factor in this system. That is, as we increase the radical content, more domains with enhanced radical–radical interactions occur such that there is an increased possibility of charge hopping events, and thus, a likely increase in charge mobility. In this manner, PGMS is similar to other high-performing radical polymers, such as PTEO, which relies on high radical content and formation of local order for high electronic conductivity.32 However, it should be stressed that the radical–radical aggregation of the galvinoxyl open-shell group predicted by theory, and noted in the UV–Vis absorption spectra above, is critical for these materials to achieve high conductivity values. This is because the relative loading of galvinoxyl radicals in these materials is quite low relative to what has been achieved in nitroxide-based radical polymers (e.g., PTEO). In fact, if these low loadings were present in nitroxide-based radical polymers, they would appear as electrical insulators. Thus, these data highlight that the design of the radical functionality is critical in terms of end-use performance. Moreover, we anticipate that even higher electronic conductivity values could be had with these materials if higher radical content were achieved, as higher radical loadings would facilitate rapid redox reactions between the pendant groups and allow for a corresponding increase in the macroscopic electronic conductivity. Ultimately, while the electronic properties of PGMS are advantageous due to its temperature-independence; its dependence on radical content must be considered for future solid-state applications.
Conclusions
A first-of-its-kind n-type, low glass transition temperature polymer, PGMS, which bears pendant galvinoxyl radical groups was designed. Specifically, it was synthesized by attaching functionalized galvinoxyl small molecules to a polysiloxane backbone through a simple coupling chemistry to achieve different molecular weights that mirrored those of the commercially-available parent closed-shell polymers. By comparing the UV–Vis absorption, we discovered that molecular weight did not impact the optical absorption of the closed-shell derivatives (PGMS-H), but the higher molecular weight PGMS polymers aggregated in solution, which altered their light absorption behavior. Relative to the EPR spectrum of the small molecule galvinoxyl radical analog, there was clear evidence of intrachain radical–radical interactions for the PGMS polymers. Moreover, there appeared to be no impact on the radical–radical coupling as a function of molecular weight. Because of the presence of the open-shell groups, clear and reversible redox peaks were present, and they illustrated the potential of these materials in next-generation applications. To this point, relatively high, temperature-independent conductivity (∼10−2 S m−1) values were achieved in the solid state, as had been predicted by our theoretical calculations. These high electronic conductivity values are unique as they occur at relatively low radical loadings along the polymer pendant groups, as was initially suggested by our calculations. This highlights the import of moving to next-generation open-shell groups in the application of radical polymers in solid-state devices, and it brings forward the idea that nitroxide-based radical polymers may have properties that are quite different from other classes of radical polymers, despite the large amount of attention paid to these first-generation materials to date. In summary, this work highlights the crucial fact that less frequently studied open-shell groups, like the galvinoxyl radical, will be key in moving diverse sets of radical polymer-based organic electronic devices forward in the future.Experimental
Materials and general experimental procedures
The poly(methyl siloxane) macromolecule with 24 repeat units, on average, (product code: HMS 991) was purchased from Gelest, Inc., and it was used as received. All other chemicals were purchased from Sigma-Aldrich, and all chemicals were used as received. All ultraviolet–visible (UV–Vis) light spectroscopy data were collected with the wavelength range of 330 nm ≤ λ ≤ 800 nm by a Cary 60 spectrometer. Glass transition temperature values were evaluated using a TA Instruments Q20 Series differential scanning calorimeter (DSC). The sample was sealed in Tzero hermetic pans and annealed at 70 °C under a nitrogen gas purge and then cooled to −60 °C before the trace shown, which begin at −60 °C and ended at 70 °C, was obtained. The data were collected at scan rate of 10 °C min−1. Electron paramagnetic resonance (EPR) data were obtained by Bruker EPR-EMX spectrometer. PGMS samples were dissolved in toluene at a concentration of 1 mg mL−1 and small molecule galvinoxyl radicals were prepared in the same way as a standard. In these experiments, 0.2 mL of solution were added to the EPR tubes, and the data were collected at room temperature. All nuclear magnetic resonance (NMR) spectroscopy data were collected using a Bruker AV-III-400-HD NMR spectrometer. The concentration of the molecules in deuterated chloroform was ∼5%, by weight.Synthesis of (4-bromo-2,6-di-tert-butylphenoxy) trimethylsilane (Compound 3)
4-Bromo-2,6-di-tert-butylphenol (10 g, 35 mmol, 1) was added to a 250 mL round bottom flask with 100 mL of anhydrous THF. The flask was cooled to −78 °C, and n-butyl lithium (2.5 M, 21 mL, 52 mmol) was injected dropwise. After stirring for 1 h, trimethylsilyl chloride (TMS-Cl) (7.6 mL, 60 mmol) was added slowly, and the reaction was slowly warmed to room temperature. After another 1 hour, the reaction was stopped, and the product was precipitated in hexane to form white crystals with 80% yield. 1H NMR (400 MHz, CDCl3): δ 7.32 (s, 2H), 1.38 (s, 18H), 0.40 (s, 9H).Synthesis of methyl 4-(((trimethylsilyl)oxy)methyl)benzoate (Compound 4)
Methyl 4-(hydroxymethyl)benzoate (4 g, 24 mmol, 2) was added to a 250 mL round bottom flask with 125 mL anhydrous THF. The flask was cooled to 0 °C, and then triethyl amine (6.6 mL, 48 mmol) was added. TMS-Cl (4.6 mL, 36 mmol) was injected dropwise. After the reaction was stirred for 1 h, the ice bath was removed, and the reaction warmed to room temperature. After 24 h, all the inorganic white solid was removed via filtration, and the organic residue was concentrated using a rotary evaporator. The mixture was purified by column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 1:1 v/v) to give a solid with 85% yield. 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.4 Hz, 2H), 7.41 (dd, J = 8.1, 0.5 Hz, 2H), 4.77 (s, 2H), 3.93 (s, 3H), 0.19 (s, 9H).
:ethyl acetate = 1:1 v/v) to give a solid with 85% yield. 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 8.4 Hz, 2H), 7.41 (dd, J = 8.1, 0.5 Hz, 2H), 4.77 (s, 2H), 3.93 (s, 3H), 0.19 (s, 9H).
Synthesis of 2,6-di-tert-butyl-4-((3,5-di-tert-butyl-4-hydroxyphenyl)(4-(hydroxymethyl) phenyl)methylene)cyclohexa-2,5-dien-1-one (Compound 5)
In this reaction, 3 (4.85 g, 13.58 mmol) was dissolved in 20 mL of anhydrous THF and added to a Schlenk flask under a nitrogen environment. The flask was cooled to −78 °C, and n-butyl lithium (2.5 M, 15 mmol) was injected dropwise. After the reaction stirred for 30 min, 4 (1 g, 6.11 mmol) dissolved in THF was added, and the reaction color changed rapidly. The flask was warmed slowly to room temperature, and the reaction was allowed to stir for 24 h. Next, KOH (2.3 g, 41 mmol) in 40 mL water was added to the reaction slowly, and the reaction was stirred for another 24 h. To neutralize the reaction, an HCl (2 M) solution was added dropwise until the reaction turned orange. The organic mixture was washed with brine three times followed by an extraction with diethyl ether. The mixture was purified by column chromatography. (hexane:ethyl acetate = 10:1 v/v) to give a solid with 50% yield. 1H NMR (400 MHz, CDCl3) δ: 7.40 (t, J = 6.0 Hz, 2H), 7.29–7.26 (m, 2H), 7.23 (t, J = 2.5 Hz, 1H), 7.13 (t, J = 2.2 Hz, 1H), 7.01 (d, J = 3.0 Hz, 2H), 5.50 (d, J = 3.3 Hz, 1H), 4.80 (t, J = 4.7 Hz, 1H), 1.79 (t, J = 5.8 Hz, 1H), 1.40 (s, 18H), 1.28 (s, 9H), 1.25–1.22 (m, 9H).
Synthesis of 4-((4-((allyloxy)methyl)phenyl)(3,5-di-tert-butyl-4-hydroxyphenyl)methylene)-2,6-di-tert-butylcyclohexa-2,5-dien-1-one (Compound 6)
Tetrabutylammonium hydrosulfate (0.05 g, 0.15 mmol), allyl bromide (0.33 mL, 4 mmol), and 5 mL of THF were added to a 25 mL round bottom flask. A NaOH solution (3 mL, 50% by weight), was injected to the reaction next and the solution was stirred for 10 minutes. A solution of 5 (0.45 g, 0.8 mmol) in 3 mL of THF was then added dropwise into the mixture. The reaction was stirred for 48 h at room temperature. The organic phase was washed with brine three times followed by an extraction with ethyl acetate. The mixture was purified by column chromatography (hexane:ethyl acetate = 5:1 v/v) to give a solid with 45% yield. 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 1.1 Hz, 1H), 7.25–7.22 (m, 2H), 7.13 (d, J = 2.6 Hz, 1H), 7.02 (s, 2H), 6.00 (ddt, J = 17.2, 10.4, 5.7 Hz, 1H), 5.50 (s, 1H), 5.38–5.30 (m, 1H), 5.26–5.22 (m, 1H), 4.61 (s, 2H), 4.10 (ddd, J = 6.1, 3.8, 2.3 Hz, 2H), 1.40 (s, 18H), 1.28 (s, 9H), 1.24 (s, 9H).
Synthesis of poly(galvinoxyl-H methyl siloxane) (PGMS-H)
To begin, 6 (500 mg, 0.89 mmol) was dissolved with 1 mL of toluene and added to a 15 mL sealed tube. Then, poly(methyl siloxane) (5 mg, 0.05 mmol) dissolved in 1 mL of toluene was added. Next, Karstedt's catalyst in xylene solution (with Pt ∼2%, 128 mg, 150 μL) was added. After three freeze–pump–thaw cycles, the sealed tube was slowly heated to 100 °C. After 24 h, the organic mixture was washed with DI water three times followed by a dilution with ethyl acetate. The polymer was purified by precipitation from hexane to give a soft material in 68% yield.Synthesis of poly(galvinoxyl methyl siloxane) (PGMS)
In this reaction, KOH (60 mg, 1.07 mmol), K3Fe(CN)6 (120 mg, 0.36 mmol), and water (3 mL) were added to a 20 mL flask. PGMSH (15 mg) was dissolved with 3 mL diethyl ether and added to the flask. This two-phase reaction was vigorously stirred in dark condition. After 50 min, the reaction was stopped, and the color of the reaction solution had changed from red to purple. The organic phase was washed with DI water 10 times followed by a concentrating the solution using a rotary evaporator. The overall yield of the polymer was ∼37%.Cyclic voltammetry
Cyclic voltammetry data of PGMS samples were collected using a VersaSTAT 3 (Princeton Applied Research) workstation with a three-electrode setup. PGMS was mixed with an equal mass of an electrochemically-inert polymer, poly(vinylidene fluoride) (PVDF), whose weight-average molecular weight was nominally 534 kg mol−1, as reported by the vendor. These solids were stirred in DMF at 50 °C for 30 min. The homogeneous solution was then spun-coat onto a gold working electrode deposited on a glass substrate. The organic electrolyte (0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in DMF) was deoxygenated by bubbling nitrogen through the solution for 30 min prior to the measurement. The CV plots were obtained at a scan rate of 100 mV s−1, and the data shown are from the fifth scan as the first four scans were used to condition the films.Electronic conductivity measurements
Silicon substrates with a thermally-grown silicon dioxide layer were cleaned with a piranha solution [H2O2 (30%, by weight):H2SO4 (96%, by weight) in water, 1:3, by volume)] for 20 min, and the substrates were then rinsed with DI water. Then, they were baked for 100 °C for 60 s to remove any residual water. Positive poly(methyl methacrylate) (PMMA) (495 kg mol−1, A4 resist) was spun-coat for 60 s at 3000 rpm to create an ∼200 nm-thick film. Electron-beam (e-beam) lithography (Raith e-Line) was used to pattern channels with lengths and widths of 400 nm and 1 mm, respectively. The parameters for the e-beam lithography were an energy of 20 keV, an aperture of 60 μm, and an area dose 250 μC cm−2. Afterwards, the substrates were developed in methyl isobutyl ketone:isopropyl alcohol (MIBK:IPA) (1:3, by volume) for 60 s and rinsed with isopropanol. Then, 5 nm of Ti and 50 nm of Au were evaporated onto the substrates using a thermal evaporator. PMMA was then removed through ultrasonication in acetone for 20 min. PGMS was dissolved in chloroform and cast from a concentrated solution onto the channels to create films. PGMS-4-13 thin films had an average thickness of ∼20 μm while PGMS-4-36 thin films had an average thickness of ∼6 μm, as measured using a Dektak profilometer. While drop-casting can cause film uniformity issues, this was not observed here as the standard deviation from the average values of the film thicknesses were <15%. A scanning electron microscope (SEM) was used to image the substrates using a Raith SEM with an accelerating voltage of 20 keV.
Current–voltage (I–V) measurements were acquired by sweeping voltages across the range of −1 V ≤ V ≤ + 1 V and recording the current values. The measurements were performed under vacuum in a PS100 Lakeshore probe station with a Keithley 2400 source meter. Temperature measurements were performed from −60 °C to 100 °C with a step size of 20 °C. Each temperature was held for 30 min prior to any electrical conductivity measurement to ensure that the sample had reach thermal equilibrium at the given temperature.
Computational methods
Initial geometries were generated for the closed-shell polysiloxane precursor macromolecule and the galvinoxyl-containing radical polymers based on a universal force field (UFF)69 as implemented in Avogadro,70 followed by optimization at the semi-empirical GFN2–xTB level.71 Starting from these optimized structures, the conformer-rotamer ensemble sampling tool (CREST), as implemented within the xTB package, was used to identify all conformers within 6 kcal mol−1 of the lowest energy structure using the iMTD-GC algorithm. For the CREST calculations, an integration time step of 5 fs, and a total run time of 10 ps were used. Five parallel CREST trajectories were performed at each loading to facilitate comprehensive sampling. The trajectory that discovered the lowest energy conformer at each loading was used for further analysis and reported in the main text. The resulting conformers were further characterized based on the radius of gyration (Rg), the relative energy compared with the lowest energy conformer (ΔE), and the galvinoxyl nearest-neighbor distance (NND). The radius of gyration was calculated according to eqn (1).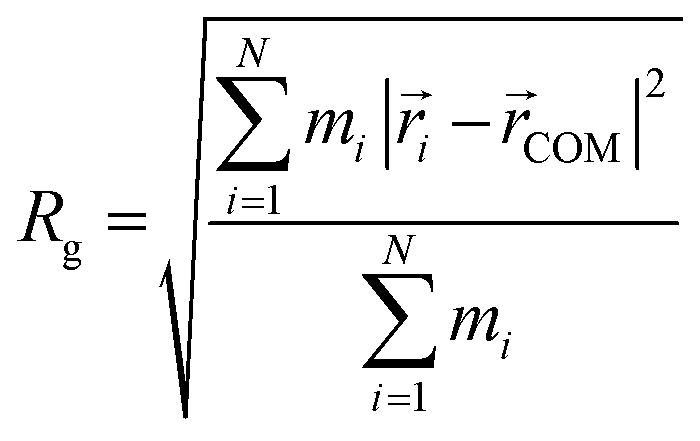 | (1) |
Here, mi is the atomic mass, 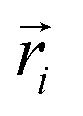 is the atomic position,
is the atomic position, 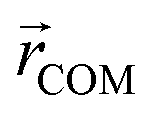 is the polymer center of mass, and the summations run over all polymer atoms. The energy difference (ΔE) was calculated as the single point energy (i.e., the total electronic energy calculated at the GFN2–xTB level) difference of each conformer with the lowest energy conformer at each radical loading. The galvinoxyl–galvinoxyl NND was calculated as the average over the NND for each galvinoxyl in the polymer. For 25% loading, only a single galvinoxyl is included in the polymer, and thus, the NND metric is not reported.
is the polymer center of mass, and the summations run over all polymer atoms. The energy difference (ΔE) was calculated as the single point energy (i.e., the total electronic energy calculated at the GFN2–xTB level) difference of each conformer with the lowest energy conformer at each radical loading. The galvinoxyl–galvinoxyl NND was calculated as the average over the NND for each galvinoxyl in the polymer. For 25% loading, only a single galvinoxyl is included in the polymer, and thus, the NND metric is not reported.
Conflicts of interest
The authors declare that there are no conflicts.Acknowledgements
The work of Y. T. and B. M. S. was made possible through the Air Force Office of Scientific Research (AFOSR) under support provided by the Organic Materials Chemistry Program (Grant number: FA9550-19-1-0271, Program Manager: Dr Kenneth Caster), and we gratefully thank the AFOSR for this support. Support for the other efforts on this project was kindly provided by the National Science Foundation (NSF) through the Polymers Program (Award number: 1554957, Program Manager: Dr Andrew Lovinger), and we are extremely appreciative of this support as well. We thank Kangying Liu for many fruitful discussions.Notes and references
- T. Kurosaki, K. W. Lee and M. Okawara, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 3295–3310 CrossRef CAS.
- A. J. Wingate and B. W. Boudouris, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1875–1894 CrossRef CAS.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- K. Zhang, M. J. Monteiro and Z. Jia, Polym. Chem., 2016, 7, 5589–5614 RSC.
- F. Li, S. Wang, Y. Zhang and J. L. Lutkenhaus, Chem. Mater., 2018, 30, 5169–5174 CrossRef CAS.
- C. Liedel, A. Moehle, G. D. Fuchs and C. K. Ober, MRS Commun., 2015, 5, 441–446 CrossRef CAS.
- K. Oyaizu, T. Kawamoto, T. Suga and H. Nishide, Macromolecules, 2010, 43, 10382–10389 CrossRef CAS.
- D. Yuan, W. Liu and X. Zhu, Chem, 2021, 7, 333–357 Search PubMed.
- T. Jähnert, B. Häupler, T. Janoschka, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2013, 214, 2616–2623 CrossRef.
- Y. Sasada, R. Ichinoi, K. Oyaizu and H. Nishide, Chem. Mater., 2017, 29, 5942–5947 CrossRef CAS.
- H. Nishide, K. Koshika and K. Oyaizu, Pure Appl. Chem., 2009, 81, 1961–1970 CAS.
- K. Nakahara, K. Oyaizu and H. Nishide, Chem. Lett., 2011, 40, 222–227 CrossRef CAS.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- H. Nishide, S. Iwasa, Y. J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Ugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23, 751–754 CrossRef CAS.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S.-K. Jung, H.-D. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682 CrossRef.
- D. R. Nevers, F. R. Brushett and D. R. Wheeler, J. Power Sources, 2017, 352, 226–244 CrossRef CAS.
- C. Friebe and U. S. Schubert, in Electrochemical Energy Storage, Springer, 2017, pp. 65–99 Search PubMed.
- S. Wang, A. M. G. Park, P. Flouda, A. D. Easley, F. Li, T. Ma, G. D. Fuchs and J. L. Lutkenhaus, ChemSusChem, 2020, 13, 2371–2378 CrossRef CAS.
- T. Jähnert, B. Häupler, T. Janoschka, M. D. Hager and U. S. Schubert, Macromol. Rapid Commun., 2014, 35, 882–887 CrossRef.
- L. Rostro, A. G. Baradwaj and B. W. Boudouris, ACS Appl. Mater. Interfaces, 2013, 5, 9896–9901 CrossRef CAS.
- S. Mukherjee and B. W. Boudouris, Organic Radical Polymers: New Avenues in Organic Electronics, Springer, 2017 Search PubMed.
- J. Lutkenhaus, Science, 2018, 359, 1334 LP – 1335 CrossRef.
- Y. Zhang, A. Park, A. Cintora, S. R. McMillan, N. J. Harmon, A. Moehle, M. E. Flatté, G. D. Fuchs and C. K. Ober, J. Mater. Chem. C, 2018, 6, 111–118 RSC.
- T. W. Kemper, R. E. Larsen and T. Gennett, J. Phys. Chem. C, 2015, 119, 21369–21375 CrossRef CAS.
- S. Wang, A. D. Easley and J. L. Lutkenhaus, ACS Macro Lett., 2020, 9, 358–370 CrossRef CAS.
- S. Wang, F. Li, A. D. Easley and J. L. Lutkenhaus, Nat. Mater., 2019, 18, 69–75 CrossRef CAS.
- A. D. Easley, L. M. Vukin, P. Flouda, D. L. Howard, J. L. Pena and J. L. Lutkenhaus, Macromolecules, 2020, 53, 7997–8008 CrossRef CAS.
- M. E. Hay, S. H. Wong, S. Mukherjee and B. W. Boudouris, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 1516–1525 CrossRef CAS.
- Y. Joo, V. Agarkar, S. H. Sung, B. M. Savoie and B. W. Boudouris, Science, 2018, 359, 1391–1395 CrossRef CAS.
- L. Zheng, S. Mukherjee, K. Wang, M. E. Hay, B. W. Boudouris and X. Gong, J. Mater. Chem. A, 2017, 5, 23831–23839 RSC.
- J. He, S. Mukherjee, X. Zhu, L. You, B. W. Boudouris and J. Mei, ACS Appl. Mater. Interfaces, 2018, 10, 18956–18963 CrossRef CAS.
- Y. Yonekuta, K. Susuki, K. Oyaizu, K. Honda and H. Nishide, J. Am. Chem. Soc., 2007, 129, 14128–14129 CrossRef CAS.
- S. H. Sung, N. Bajaj, J. F. Rhoads, G. T. Chiu and B. W. Boudouris, Org. Electron., 2016, 37, 148–154 CrossRef CAS.
- H. Tokue, K. Kakitani, H. Nishide and K. Oyaizu, Chem. Lett., 2017, 46, 647–650 CrossRef CAS.
- K. Oyaizu, Y. Ando, H. Konishi and H. Nishide, J. Am. Chem. Soc., 2008, 130, 14459–14461 CrossRef CAS.
- A. Cintora, H. Takano, M. Khurana, A. Chandra, T. Hayakawa and C. K. Ober, Polym. Chem., 2019, 10, 5094–5102 RSC.
- K.-A. Hansen and J. P. Blinco, Polym. Chem., 2018, 9, 1479–1516 RSC.
- F. Li, Y. Zhang, S. R. Kwon and J. L. Lutkenhaus, ACS Macro Lett., 2016, 5, 337–341 CrossRef CAS.
- Y. Zhang, A. M. Park, S. R. McMillan, N. J. Harmon, M. E. Flatté, G. D. Fuchs and C. K. Ober, Chem. Mater., 2018, 30, 4799–4807 CrossRef CAS.
- F. Li, D. N. Gore, S. Wang and J. L. Lutkenhaus, Angew. Chem., 2017, 129, 9988–9991 CrossRef.
- M. Khodeir, B. Ernould, J. Brassinne, S. Ghiassinejad, H. Jia, S. Antoun, C. Friebe, U. S. Schubert, Z. Kochovski and Y. Lu, Soft Matter, 2019, 15, 6418–6426 RSC.
- T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007, 1730–1732 RSC.
- S. Wang, A. D. Easley, R. M. Thakur, T. Ma, J. Yun, Y. Zhang, C. K. Ober and J. L. Lutkenhaus, Chem. Sci., 2020, 11, 9962–9970 RSC.
- C. Liedel and C. K. Ober, Macromolecules, 2016, 49, 5884–5892 CrossRef CAS.
- Y. Lu, J.-Y. Wang and J. Pei, Chem. Mater., 2019, 31, 6412–6423 CrossRef CAS.
- B. Esser, Org. Mater., 2019, 1, 63–70 CrossRef CAS.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS.
- R. M. Lucente-Schultz, V. C. Moore, A. D. Leonard, B. K. Price, D. V. Kosynkin, M. Lu, R. Partha, J. L. Conyers and J. M. Tour, J. Am. Chem. Soc., 2009, 131, 3934–3941 CrossRef CAS.
- P. M. Lahti, Y. Liao, M. Julier and F. Palacio, Synth. Met., 2001, 122, 485–493 CrossRef CAS.
- M. Otaki and H. Goto, Macromolecules, 2019, 52, 3199–3209 CrossRef CAS.
- R. M. Franzini and E. T. Kool, J. Am. Chem. Soc., 2009, 131, 16021–16023 CrossRef CAS.
- G.-J. ten Brink, I. W. C. E. Arends, M. Hoogenraad, G. Verspui and R. A. Sheldon, Adv. Synth. Catal., 2003, 345, 1341–1352 CrossRef CAS.
- M. Iyoda, K. Sato and M. Oda, Tetrahedron Lett., 1987, 28, 625–628 CrossRef CAS.
- B. Kirste, W. Harrer and H. Kurreck, J. Am. Chem. Soc., 1985, 107, 20–28 CrossRef CAS.
- Z. Shi, J. Wang, M. Teraguchi, T. Aoki and T. Kaneko, Polymers, 2019, 11, 1877 CrossRef CAS.
- T. Kaneko, H. Tatsumi, T. Aoki, E. Oikawa, H. Yoshiki, N. Yoshioka, E. Tsuchida and H. Nishide, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 189–198 CrossRef CAS.
- E. Fukuzaki, N. Takahashi, S. Imai, H. Nishide and A. Rajca, Polym. J., 2005, 37, 284–293 CrossRef CAS.
- A. Franzke and A. Pfaltz, Chem. – Eur. J., 2011, 17, 4131–4144 CrossRef CAS.
- A. G. Baradwaj, L. Rostro and B. W. Boudouris, Macromol. Chem. Phys., 2016, 217, 477–484 CrossRef CAS.
- B. D. Karstedt, US PatUS3775452A, 1973 Search PubMed.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- C. M. Downing and H. H. Kung, Catal. Commun., 2011, 12, 1166–1169 CrossRef CAS.
- S. Mukherjee and B. W. Boudouris, Mol. Syst. Des. Eng., 2017, 2, 159–164 RSC.
- M. Miyasaka, T. Yamazaki and H. Nishide, Polym. J., 2001, 33, 849–856 CrossRef CAS.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, DSC, SEM, current–voltage curves and radical content calculations. See DOI: 10.1039/d0py01645d |
| This journal is © The Royal Society of Chemistry 2021 |