DOI:
10.1039/D3TB02693K
(Paper)
J. Mater. Chem. B, 2024,
12, 1905-1925
Injectable and self-healable nano-architectured hydrogel for NIR-light responsive chemo- and photothermal bacterial eradication†
Received
13th November 2023
, Accepted 26th January 2024
First published on 27th January 2024
Abstract
Hydrogels with multifunctional properties activated at specific times have gained significant attention in the biomedical field. As bacterial infections can cause severe complications that negatively impact wound repair, herein, we present the development of a stimuli-responsive, injectable, and in situ-forming hydrogel with antibacterial, self-healing, and drug-delivery properties. In this study, we prepared a Pluronic F-127 (PF127) and sodium alginate (SA)-based hydrogel that can be targeted to a specific tissue via injection. The PF127/SA hydrogel was incorporated with polymeric short-filaments (SFs) containing an anti-inflammatory drug – ketoprofen, and stimuli-responsive polydopamine (PDA) particles. The hydrogel, after injection, could be in situ gelated at the body temperature, showing great in vitro stability and self-healing ability after 4 h of incubation. The SFs and PDA improved the hydrogel injectability and compressive strength. The introduction of PDA significantly accelerated the KET release under near-infrared light exposure and extended its release validity period. The excellent composites’ photo-thermal performance led to antibacterial activity against representative Gram-positive and Gram-negative bacteria, resulting in 99.9% E. coli and S. aureus eradication after 10 min of NIR light irradiation. In vitro, fibroblast L929 cell studies confirmed the materials’ biocompatibility and paved the way toward further in vivo and clinical application of the system for chronic wound treatments.
Introduction
Wound healing presents an intricate challenge in modern medicine, necessitating a comprehensive approach due to its dynamic and spatiotemporal nature.1 This process encompasses overlapping and interacting hemostasis, inflammation, proliferation, and remodeling phases.2 Damage to the human skin by pathogenic factors disrupts its integrity, rendering the epidermal site susceptible to infection and inflammation, hindering wound repair, and potentially leading to septicemia.3,4 Chronic wound problems have been progressively growing due to the increasing number of elderly and the prevalence of lifestyle diseases. Despite the availability of numerous therapeutic options, their efficacy remains limited.5 Consequently, there is an urgent need to develop innovative materials to optimize chronic wound treatment outcomes.6 Chronic wounds typically occur in patients with comorbidities, and these wounds take more than three weeks to heal and impede the transition from the inflammatory to the proliferation phase.7 Persistent inflammation within the wound microenvironment, triggered by factors like a bacterial infection, may prompt fibroblasts to enter a senescent state.8 The presence of senescence-like fibroblasts hampers the recruitment of sufficient neutrophils, forming a biofilm – a self-generated polysaccharide matrix produced by bacteria.9 This biofilm acts as a shield, protecting bacteria from pharmacological treatment and neutrophil attacks, thus perpetuating the chronic state of the wound.10 Given the intricate and multifaceted nature of the wound-healing process, an ideal wound dressing should efficaciously ameliorate the diverse impediments encountered during wound healing. In addition to the timely delivery of anti-inflammatory agents, bacterial-infected injuries represent a significant challenge.11 Moreover, the recent surge in bacterial drug resistance has emerged as a formidable menace to human health, necessitating the exploration of novel antibacterial modalities.3 A cutting-edge non-invasive therapeutic approach known as photo-thermal therapy (PTT) has emerged, demonstrating remarkable potential in conferring antibacterial attributes through photo-thermal agents.12,13
Hydrogels gained popularity in this field due to their oxygen permeability capacity, wound exudate absorption, and moisture retention properties. Generally, hydrogels can be prepared by the physical or chemical cross-linking process.14,15 The physical cross-linking of hydrogels entails the creation through hydrophobic interactions triggered by environmental stimuli (e.g., temperature) acting upon the water-soluble polymer chain.16,17 Thermo-sensitive hydrogels such as Pluronic-F127 (PF127) are a class of thermo-reversible gels with stimuli-responsive polymer content that triggers self-assembly during temperature changes. PF127 is a well-known material approved by medical agencies for drug delivery applications that helps control drug release from the matrix.18 PF127 exhibits a lower critical solution temperature (LCST) and has a phase transition in the 20–25 °C temperature range, depending on the concentration and addition of other components.19 However, the physically gelated hydrogels exhibit low stability and resistance to degradation. For example, Shah et al. developed an injectable platform made of PF127, hyaluronic acid, and pullulan, which degraded 100% within less than 24 hours.20
Besides, the shape of traditional hydrogel dressings, which come in sheets and require pre-application preparation, results in insufficient coverage of wounds with irregular contours and incomplete penetration into deeper wounds.21 On the other hand, hydrogel dressings represent significant advancements in treating atypical wounds.22 Nevertheless, these materials suffer from prolonged gelation time, lack mechanical strength, and an absence of natural extracellular matrix (ECM)-like components. The introduction of additional ion cross-linked components such as sodium alginate (SA) can improve the stability of the system by prolonging the degradation time, improving the mechanical strength, and entrapping polydopamine (PDA) particles and short-filaments (SFs), preventing their escape and deaccelerate drug release, targeting more sustained kinetics. Importantly, the addition of SA can also elevate the gelation temperature (Tgel) of PF127 closer to the human body's natural temperature, making the system suitable for biomedical applications.23
Many hydrogels with embedded fibrous membranes have been reported in the literature. Electrospinning is one of the most promising techniques to fabricate fibers for regenerative medicine and wound healing. It can be used to produce biomaterial mimicking the properties of the ECM components, which is essential for enhancing the efficacy of diverse therapeutic approaches and improving hydrogel biocompatibility.24 In this frame, Jang et al. presented the fabrication of a 3D nanofiber-reinforced hydrogel composite prepared using layer-by-layer electrospinning. Compared to pure hydrogel, the structure enhanced compressive strength and stiffness by 221% and 434%, respectively.25 However, due to the limited interfacial interaction between the fibrous mat and the hydrogel matrix, these composites may experience delamination, leading to an unstable structure. Moreover, such structures are unsuitable for further injectability in personalized wound dressing preparation. To address these limitations, notable progress has been achieved in developing hydrogels incorporating SFs by integrating segmented fibers within the hydrogel network.26–30 These SFs-embedded hydrogels possess the dual capability of effectively recapitulating the ECM and being injectable through small-size needles, meeting the requirements for clinical applications.31
Timely delivery of anti-inflammatory drugs can quickly regulate the levels of various inflammatory factors and normalize the inflammatory response of chronic wounds with persisting inflammation.9,32 Furthermore, introducing non-steroidal anti-inflammatory drugs (NSAIDs) such as ketoprofen (KET) presents an adjunctive therapeutic wound approach, achieved through modulating macrophage polarization and facilitating the healing of chronic wounds.33 The ideal drug delivery system (DDS) should exhibit adjustable and controllable drug release kinetics to induce efficacious and safe therapeutic outcomes. Introducing anti-inflammatory agents, such as KET, allows the composite of an on-demand drug delivery system (DDS) when combined with a photo-responsive agent.34,35 Photo-responsive particles can accelerate drug release from the carrier via localized heat generation during NIR light irradiation.36–38 For this reason, the addition of photo-thermal particles like PDA can be exploited during PPT, simultaneously providing a more efficient drug delivery and antibacterial properties to the system. PDA stands out due to its photo-absorbing capability, straightforward manufacturability, remarkable photodegradation resistance, advantageous biodegradation propensity, and biocompatibility as an analog to naturally occurring melanin.39 Thus, integrating PDA enables the fabrication of stimuli-responsive materials with applications in bacteria eradication and drug delivery.
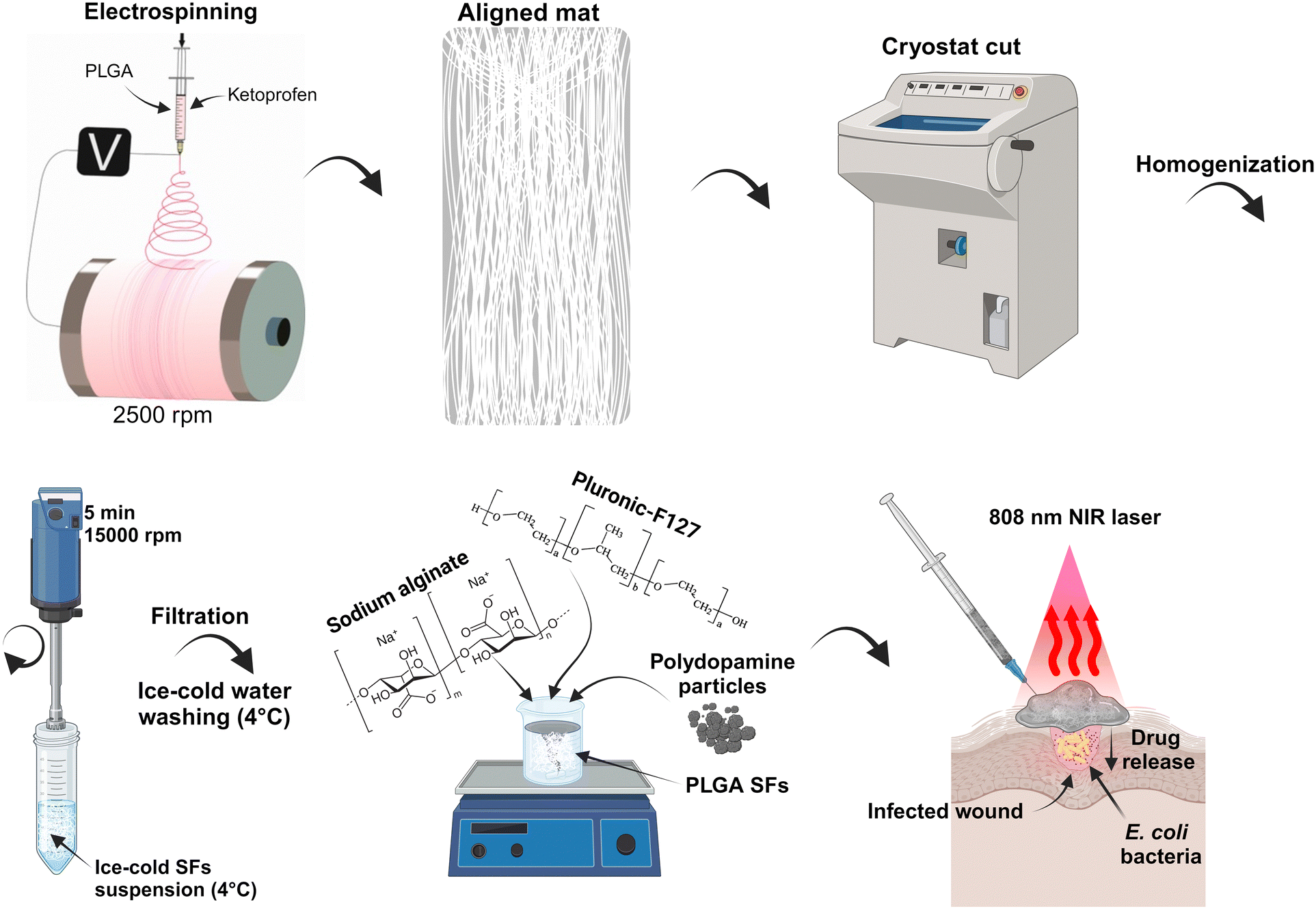
In this study, we present a development of injectable, in situ-forming hydrogel with on-demand drug delivery and antibacterial properties. More in detail, we prepared thermo-sensitive interpenetrating hydrogels made of PF127 and SA with excellent stability.40 Adding drug-containing SFs and photo-thermal PDA increased the local temperature due to NIR light–heat conversion and allowed the eradication of E. coli and S. auereus. Moreover, repeated NIR irradiations accelerated the KET release due to temperature elevation, resulting in gradual drug release kinetics. Additionally, we presented a novel mechanical approach for developing SFs containing active agents with low loss of active molecule content. In Scheme 1, the procedure of the material preparation to its possible application is presented. Briefly, the electrospinning technique was used to prepare the aligned poly(D,L-lactide-co-glycolide) (PLGA) mat on a high-speed rotatory drum collector. The mat was frozen and cut into stripes using the cryostat equipment. The polymeric stripes were homogenized, washed with deionized water, and lyophilized. The SFs powder was mixed with PF127/SA hydrogel, and PDA particle powder was added. The final material can possibly be injected onto the skin surface, covering the irregular wound and providing delivery of anti-inflammatory KET and anti-bacterial properties.
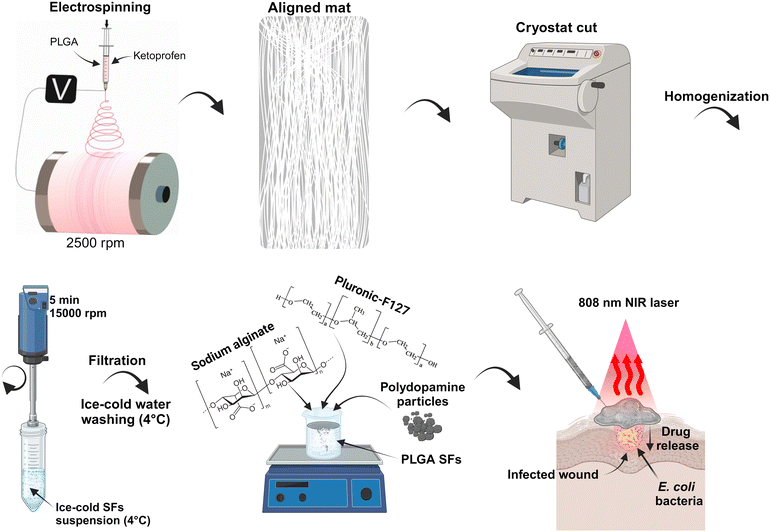 |
| | Scheme 1 Illustration representing the development of the platform and its application. | |
Materials and methods
Materials
Poly(D,L-lactide-co-glycolide) (PLGA, PDLG 5010 with lactide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide 50:50, Corbion Purac, Netherlands); 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, Acros Organics, USA); ketoprofen (Sigma-Aldrich, Poland, >98% (TLC)); Tissue-Tek® O.C.T. compound (Sakura Finetek, USA); HEPES; sodium alginate (Sigma-Aldrich, Poland, alginic acid sodium salt from brown algae, low viscosity); pluronic F-127 (Sigma-Aldrich, Poland); dopamine hydrochloride (Sigma-Aldrich, Poland); tris HCl (Sigma-Aldrich, Poland); bovine serum albumin (BSA) (Sigma-Aldrich, Poland); hexamethyldisilazane (HMDS) (Sigma-Aldrich, Poland); phosphate buffer saline (PBS) (Sigma-Aldrich, Poland); Triton X (Sigma-Aldrich, Poland); DAPI (Sigma-Aldrich, Poland); and L929 murine fibroblasts (Sigma-Aldrich, Poland); Dulbecco's modified Eagle's medium (DMEM) (Gibco Invitrogen, USA); fetal bovine serum (FBS) (Gibco Invitrogen, USA); penicillin–streptomycin (PS) (Gibco Invitrogen, USA); and EDTA-trypsin (Gibco Invitrogen, USA); Alexa Fluor 488 Phalloidin (Thermo-Fisher Scientific, USA); live/dead assay (Thermo-Fisher Scientific, USA); lysogeny broth (LB) and lysogeny agar (LB agar) (A&A Biotechnology, Poland); Gram-negative bacteria Escherichia coli (E. coli) ATCC 25922 and Gram-positive bacteria Staphylococcus aureus (S. aureus) ATCC 6538 (Pol-AURA, Poland).
:glycolide 50:50, Corbion Purac, Netherlands); 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, Acros Organics, USA); ketoprofen (Sigma-Aldrich, Poland, >98% (TLC)); Tissue-Tek® O.C.T. compound (Sakura Finetek, USA); HEPES; sodium alginate (Sigma-Aldrich, Poland, alginic acid sodium salt from brown algae, low viscosity); pluronic F-127 (Sigma-Aldrich, Poland); dopamine hydrochloride (Sigma-Aldrich, Poland); tris HCl (Sigma-Aldrich, Poland); bovine serum albumin (BSA) (Sigma-Aldrich, Poland); hexamethyldisilazane (HMDS) (Sigma-Aldrich, Poland); phosphate buffer saline (PBS) (Sigma-Aldrich, Poland); Triton X (Sigma-Aldrich, Poland); DAPI (Sigma-Aldrich, Poland); and L929 murine fibroblasts (Sigma-Aldrich, Poland); Dulbecco's modified Eagle's medium (DMEM) (Gibco Invitrogen, USA); fetal bovine serum (FBS) (Gibco Invitrogen, USA); penicillin–streptomycin (PS) (Gibco Invitrogen, USA); and EDTA-trypsin (Gibco Invitrogen, USA); Alexa Fluor 488 Phalloidin (Thermo-Fisher Scientific, USA); live/dead assay (Thermo-Fisher Scientific, USA); lysogeny broth (LB) and lysogeny agar (LB agar) (A&A Biotechnology, Poland); Gram-negative bacteria Escherichia coli (E. coli) ATCC 25922 and Gram-positive bacteria Staphylococcus aureus (S. aureus) ATCC 6538 (Pol-AURA, Poland).
Methods
Preparation and characterization of electrospun filaments.
The electrospinning polymer solution was prepared by dissolving 9% (w/w%) PLGA in a HFIP solvent. Subsequently, KET was introduced into the solution at a final concentration of 15% (w/w) relative to PLGA. Electrospinning was performed using a 15 kV positive voltage applied to a cut and blunt 24G needle. The flow rate of the electrospinning solution was maintained at 800 μL h−1 throughout the entire process. To fabricate the aligned PLGA mat, the fibers were collected on a grounded rotating drum collector spinning at 2500 rpm, positioned 15 cm away from the needles’ tip. The electrospinning process was carried out at a temperature of 22 °C and a humidity level of approximately 50–60%. As a reference, a PLGA mat without KET was prepared under the same conditions. The resulting nanofibrous mats were sectioned into 1 × 2 cm rectangles and frozen in Tissue-Tek. Subsequently, samples were cut using a cryostat perpendicular to the alignment into 5 μm stripes. The obtained stripes were homogenized for 5 minutes at 15000 rpm (PT2500E, Kinematica) and washed at least 3 times with distilled water by centrifugation, leading to SFs production.
Polydopamine synthesis.
The PDA particles were synthesized according to the method presented by Bernsmann et al. using the dopamine hydrochloride oxidation process.41 The dopamine hydrochloride was immersed in the tris buffer solution (pH = 8.5) and left stirring to form PDA particles for 24 hours. The suspension was moved into the dialyzing bag and put into a beaker filled with 1.5 L of MilliQ water. The dialyzing process was conducted for 7 days to remove all possible cytotoxic dopamine leftovers. The particles were freeze-dried and stored in powder form.
Preparation of hydrogel composite and stability test.
The physically blended hydrogels consisting of thermo-gelated PF127 and calcium-ion-crosslinked SA were prepared by creating an interpenetrating polymeric network. A physical combination of PF127 and SA was prepared using the weight-to-volume ratios (w/v%) of PF127/SA, specifically 17.5%/1.5%. The solution concentration was selected based on the Chou et al. research.40 First, the separated water solutions of PF127 and SA were prepared. PF127 formulations were prepared according to the cold method by Schmolka.42 Briefly, PF127 was dissolved in deionized water at a temperature under 4 °C and continuous stirring. The solutions were equilibrated undisturbed for a minimum of 24 h at 4 °C. The SA was dissolved in water at 37 °C under continuous stirring, cooled, and mixed with PF127 stock solution to obtain the final concentration of PF127/SA equal to 17.5%/1.5% (w/v%), respectively. The material was mixed with lyophilized SFs powder and PDA particles using an ultrasonic bath until a homogenous suspension was prepared. The final composition was 17.5%/1.5%/0.5%/1% of PF127/SA/PDA/SF, respectively.
Sol–gel transition analysis and stability of hydrogel.
Sol–gel phase transitions of aqueous blends were attained by the tube-inversion method using a water bath set to the desired temperature. The Tgel was tuned with different component concentrations, as mentioned above. The tube inversion method represents a procedural approach to determining the transition between sol and gel states by observing the system's flow or non-flow characteristics within an inverted vial. Briefly, 1 mL of each homogenous solution was placed in the vials and cooled at 4 °C for 1 hour. All the vials were immersed in a water bath with temperature control. The temperature was changed by 1 °C for 5 minutes, from 20 °C to 55 °C. To determine the gel–sol transition temperature and evaluate the construct stability at the desired temperature during laser irradiation (see below). Moreover, the stability of the hydrogel precursor suspension was investigated by determining the sedimentation rate over time in order to confirm that components such as SFs and PDA do not tend to sediment fast and agglomerate during storage.
Physiochemical characterization
Differential scanning calorimetry (DSC).
DSC analysis determined the polymeric nanofibers’ glass transition temperature (Tg) and the structurization effect on this parameter. The measurements were conducted using a PYRIS-1 DSC PerkinElmer calorimeter, and data were analyzed using OriginPro software. The materials were analyzed from 0 to 100 °C with a rate of 10 °C min−1 and the samples weighed approximately 2.5 mg. Using the intersection of tangents, Tg was determined in OriginPro software from the cooling curves.
Fourier-transform infrared spectroscopy (FTIR).
Chemical bonds were analyzed using Fourier Transform Infrared spectroscopy. The spectra were obtained between 400 and 4000 cm−1 with a resolution of 2 cm−1, and 12 scans were acquired for each sample with VERTEX 70 (Bruker) equipment. The cross-linked hydrogel samples were lyophilized before the measurement. The spectra of the PLGA mat, PLGA SFs containing KET, and dry KET powder were obtained, as well as spectra of single hydrogel components and hydrogel composite after the freeze-drying process.
Dynamic light scattering (DLS).
Size distribution (dynamic light scattering method) of PDA particles was measured using a Zetasizer Nano ZS, model ZEN3600 (Malvern Instruments, Malvern, UK). A concentration of 10 μg mL−1 PDA was used for the analysis. Each sample was measured in triplicates after sonication and 10 min of stabilization at 25 °C.
Microstructure.
The scanning electron microscope (SEM) and Field Emission Scanning Electron Microscope (FE-SEM) were utilized to observe the morphological characteristics of the hydrogels. Unorganized gels underwent a process of cross-linking, washing, and reaching a state of equilibrium through swelling at a temperature of 37 °C, after which they were subjected to freeze-drying in a lyophilizer for 24 hours. Subsequently, the PF127/SA, PF127/SA/SFs, and PF127/SA/PDA/SFs hydrogels were coated with a layer of gold and visualized using the SEM and FE-SEM.
Equilibrium swelling ratio (ESR), and moisture content (MC).
The ESR was performed at a physiological temperature of 37 °C in HEPES solution (pH = 7.4). The gelated and cross-linked PF127/SA, PF127/SA/SFs, and PF127/SA/SFs/PDA hydrogels were dried until an equilibrium dry weight was reached. Accurately weighted hydrogels were immersed with HEPES solution and placed at 37 °C. The change in mass was measured at predefined time point intervals. Results were recorded until the equilibrium in weight was achieved. The ESR parameter was calculated using the following formula:
| ESR = (Wd − Wf)/Wf × 100% |
where Wd is the initial weight of dry hydrogel, and Wf is the final weight of hydrogels at pre-defined time points. The MC was measured similarly using the hydrogel weight before and after dehydration as follows:
where Ww is the wet weight before dehydration and Wa is the dry weight after dehydration.
Compression tests.
The compression tests on hydrogel composites were conducted using the texture analyzer (CTX TA, Brookfield Ametek), featuring a 50 N load cell and maintaining a constant rate of 1 mm min−1. Compressive stress–strain profiles were recorded, and the compressive strength was defined as the stress at the point of sample fracture during the experiment. The compressive modulus was computed through linear regression applied to the stress–strain curve within the 10 to 15% strain range.
Injectability.
The PF127/SA solution pure or containing SFs or SFs/PDA was injected through a 24G needle (30 mm long) connected to the 1 mL syringe, using a texture analyzer with custom-made syringe holder. The 50 N load cell imparted uniform force to the syringe piston through a special custom-made connector. 500 μL of the solution was ejected with the 1 mm s−1 speed into the vial (atmospheric pressure). The apparatus recorded the force required to push the plunger.
Photo-thermal characterization.
The variation in temperature of the materials after the formation of hydrogel structure in response to infrared irradiation was quantified using an infrared thermal imaging apparatus (FLIR A655sc thermal camera) and NIR laser (Laser MDL-H-808/5W1, CNI Optoelectronics Tech). The thermal characterization was performed for the materials in a wet state, and the NIR laser power-ΔT dependence was investigated. Different concentrations of PDA were tested (0.25%, 0.5%, 0.75%, 1%, 1.5% wt to the hydrogel content) by an 808 nm NIR laser irradiation for a specified period. The temperatures were recorded every second using the FLIR studio program setup. The final material was optimized to reach >55 °C within 5 minutes of laser irradiation to effectively eradicate the bacteria and deliver the drug to the wound site on demand.
In vitro drug release studies and release kinetics.
Release studies of KET from materials before and after structurization into SFs and from PF127/SA structures were carried out. The unstructured mats were sectioned into rectangular pieces (1 cm × 1 cm) weighing approximately 10 mg and immersed in 1 mL phosphate-buffered saline (PBS) solution with a pH of 7.4. The solutions were collected at specified time points. Approximately 10 mg of lyophilized SFs were prepared and placed in 1 mL of the same releasing medium to determine the drug loss during material structuration and investigate the change in releasing kinetics. The solution was collected after centrifugation of the sample at 15000 rpm for 3 minutes. The drug release studies were performed in different conditions to investigate the temperature-dependent kinetics – room temperature, 37 °C, 46 °C, and 55 °C. At room temperature, samples were placed in 2 mL vials on the glass hot plate heated up to 35 °C to mimic the on-skin conditions as precisely as possible. The NIR irradiation delivery tests were performed at similar conditions to room temperature samples. Briefly, PF127/SA/PDA/SFs hydrogels were put into 2 mL vials and exposed to NIR laser irradiation for 10 minutes at every predetermined time points. Every releasing condition was performed in 3 repetitions for evaluation.
The KET concentration released at each time point was measured using a UV-spectrometer (Multiskan GO, Thermo Scientific). The 260 nm maximum absorption peak was determined using the reference KET standard used for the preparation of the materials. The final concentration was calculated according to the prepared calibration curve. The measurements for polymeric material were conducted against PBS blank samples. Meanwhile, measurements from hydrogel materials were conducted against the solution collected from the reference samples. The reference samples consisting of hydrogels devoid of drugs and immersed in PBS solution were applied to avoid any interferences from the hydrogel component.
In vitro cell studies
Cell culture and seeding.
L929 murine fibroblasts were seeded on cell culture dishes and cultured in DMEM supplemented with 10% FBS and 1% PS. Cells were incubated at 37 °C and 5% CO2, and the culture medium was refreshed every two days. When cells reached a confluence of ∼80%, they were detached from the culture dish, washed in PBS, and incubated for 3 minutes in 0.05% EDTA–trypsin at 37 °C and 5% CO2. Cells were centrifuged at 1200 rpm for 5 min, forming a cell pellet. The pellet was resuspended in a culture medium and seeded in 96-tissue culture plates at a density of 10000 cells per well. Cells were incubated for 24 hours in DMEM to allow an optimal attachment to the well bottom and used the following day for the indirect cytotoxicity test.
Sample preparation and collection of extracts.
PDA and alginate powder, as well as freeze-dried SFs, were sterilized under UV light for 30 min. Hydrogel preparation was carried out in sterile conditions using filtered HEPES and CaCl2. The resulting PF127/SA/PDA/SFs, PF127/SA/SFs, and PF127/SA were washed for 1 week in sterile HEPES and subsequently weighed. Samples were then incubated for 24 hours in a calculated volume of DMEM according to their weight as suggested by the ISO standard (0.2 g mL−1). Extracts were collected and filtered through a 0.22 μm filter and used to replace cell culture media in contact with the seeded cells. Fresh DMEM was used as a control condition.
Cell viability.
Cell viability was observed after staining the cells with a live/dead assay kit. Briefly, cells were washed with PBS and incubated for 10 minutes in a staining solution made of calcein (0.5 μL mL−1) – to stain alive cells in green – and ethidium homodimer (2 μL mL−1) – to stain dead cells in red. After three washing steps, cells were imaged with a confocal microscope (Leica TCS SP5 X). The percentages of cell viability were measured using ImageJ (Cell Counter plugin, National Institute of Health, USA).
Cell proliferation.
The proliferation of the cells was evaluated using a PrestoBlue assay. At time points 1, 3, and 7 days of culture, cells were incubated in a PrestoBlue solution (10% (v/v) in culture medium) for 1 hour at 37 °C and 5% CO2. Five replicates of each tested condition were used for the analysis. After 1 hour, the PrestoBlue solution in contact with the cells was collected in 100 μL aliquots, transferred to a 96-well plate, and measured with a fluorometer (Fluoroskan Ascent TM Microplate Fluorometer, Thermo Scientific) with 530 nm excitation and at 620 nm emission.
Cell morphology.
The morphology of the cells was visualized in confocal images of cells fixed at days 3 and 7 of culture. The staining protocols provides a first washing with PBS and fixing of the cells in 4% paraformaldehyde for 15 min at room temperature. Cells were washed in PBS and treated with 0.3% (v/v) Triton X-100 solution for 15 minutes. Subsequently, a solution of 1% (w/v) BSA in PBS was added for 30 minutes. Samples were incubated in 1:40 Alexa Fluor 488 Phalloidin in PBS for 40 minutes while protected from light. Lastly, the nuclei of the cells were stained with 1:500 DAPI solution in PBS for 10 min. Three final washing steps were performed, and cells were imaged with a confocal microscope (Leica TCS SP5 X).
Evaluation of antibacterial properties.
E. coli (ATCC 25922) and S. aureus (ATCC 6538) were cultured on lysogeny broth (LB) agar and isolated with a streak plate method. During the experiment, a bacteria colony was isolated and inoculated in 3 mL of fresh LB broth. The solution was incubated overnight in an orbital shaker at 37 °C.
Bacterial inactivation.
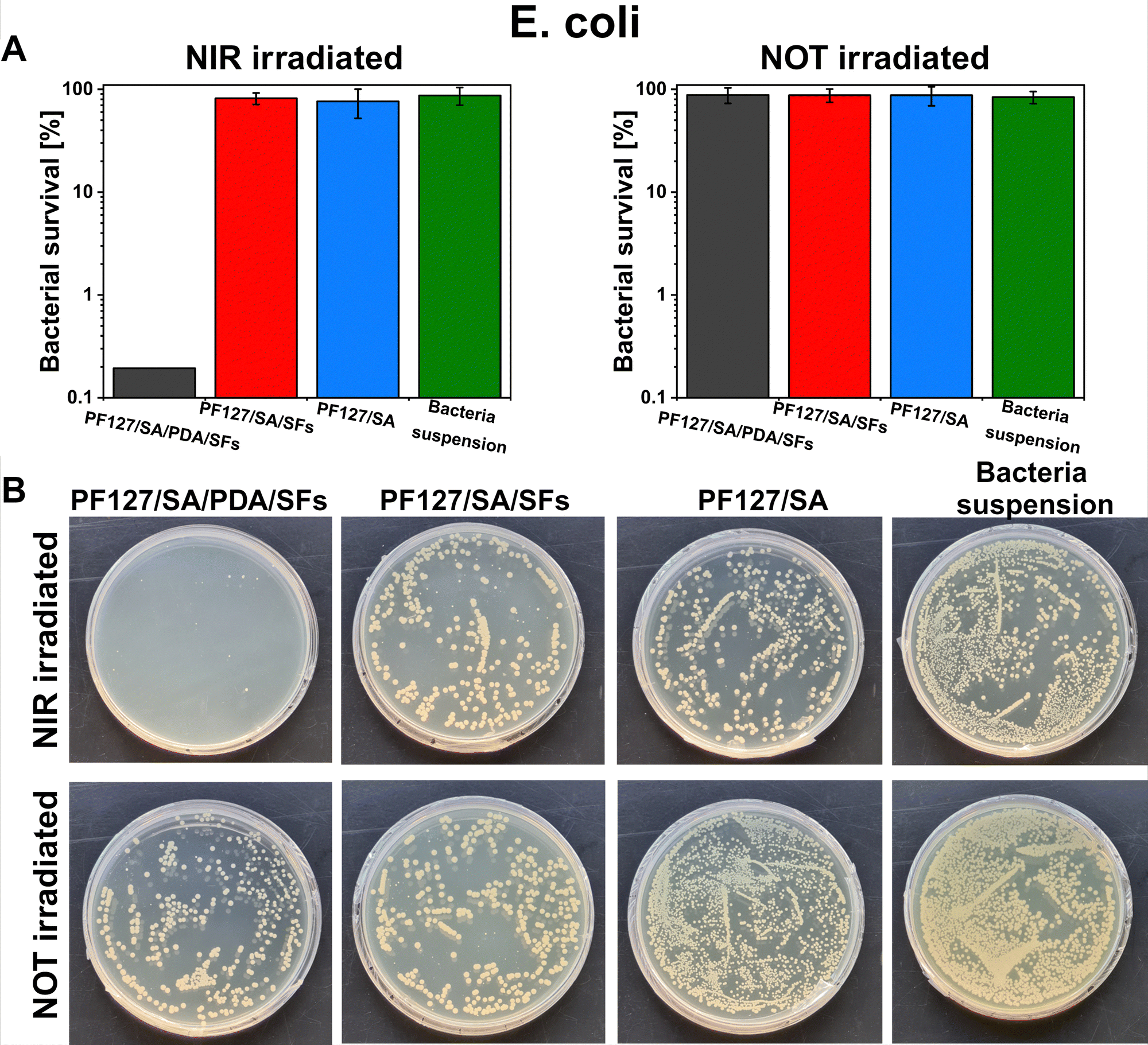
PDA and SA powder, as well as freeze-dried SFs, were sterilized for 30 minutes under UV light to decrease the bioburden. The hydrogels were prepared in filtered HEPES under the laminar hood using filtered CaCl2. Prepared PF127/SA/PDA/SFs, PF127/SA/SFs, and PF127/SA were placed in sterile flat-bottom 96-well plates for NIR irradiation. One colony of E. coli or S. aureus bacteria was isolated and incubated overnight in LB broth (3 mL). The day after, it was diluted in sterile PBS to obtain a dilution of 106 colony-forming units (CFU) ml−1. 100 μl of bacterial solution was then added to the samples in each well. After optimization of the experimental protocol, materials were exposed to NIR irradiation for 10 min in the case of E. coli and 20 min in the case of S. aureus. The pure bacterial suspension was tested as a positive control. At the same time, control samples were incubated at room temperature (RT) in the dark for 10 min with no irradiation. Materials were tested in triplicates. After the optimized test time, 100 μL of sterile PBS was added to each well, and the bacteria were gently resuspended. Then, 100 μL aliquots of the resulting diluted solution were transferred to a second 96-well plate and serially diluted. 3 technical repetitions of each dilution were carried out and plated on LB agar plates. Plates were incubated overnight at 37 °C, and the bacterial survival was investigated by counting the bacterial colonies. The plating technique limited the highest inactivation degree. It was detected at 3log units of CFU mL−1, allowing the detection of a survival rate of ≥0.1%. Finally, to be able to macroscopically observe the bacteria survival at 0.05%, after the test, the bacteria suspensions in contact with the materials were further diluted 10 times and spread evenly on representative LB agar plates with the help of a glass spreader. Samples were incubated overnight at 37 °C, and representative photos were captured the following day.
Statistical analysis
The tests were performed at least in n > 3 repetitions. All data were presented as mean ± standard deviation (SD). A one-way analysis of variance (ANOVA) and an analysis of variances (Turkey's test) was used to determine differences (p value ≤0.05: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Results and discussion
Herein, we crafted hydrogels, which are responsive to temperature changes, by combining PF127 and SA, resulting in highly stable formulations. The electrospun drug-containing polymer SFs were incorporated into the material, and by the addition of photo-responsive PDA, we could raise the local temperature through NIR light–heat conversion, effectively eliminating E. coli and S. aureus. Furthermore, repeated NIR treatments expedited the release of KET by raising the temperature, resulting in a gradual drug release pattern. The SFs were prepared using the cryocut technique. The end product could potentially be administered topically to cover irregular wounds, facilitating the delivery of the anti-inflammatory KET and offering antibacterial properties (Scheme 1).
Electrospun fiber characterization.
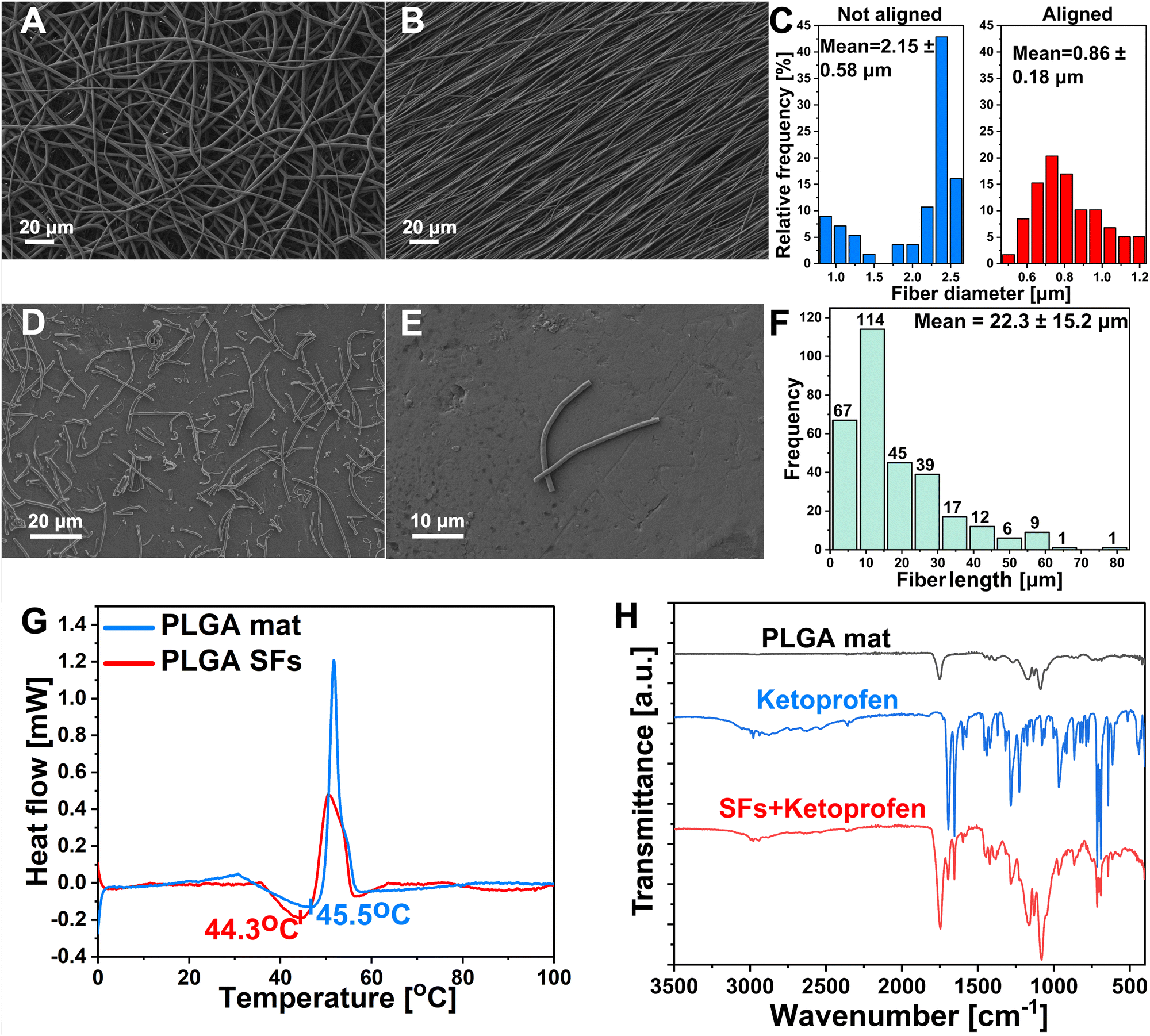
PLGA polymer was selected as the material for the segmented fibers preparation owing to its favorable biocompatibility, biodegradability, robust mechanical attributes, and Tg slightly above the body temperature.43,44 Recently, many studies reported different methodologies for the SFs preparation, including both mechanical and chemical approaches.45 Wang et al. proposed a chemical aminolysis of fibrous mat leading to amino-modified electrospun PLGA SFs with uniform distribution.31 Nevertheless, it is worth noting that the aminolysis process or other chemical modifications carry substantial danger for drug-containing materials. Wang et al. prepared silk fibroin fibers using the ultrasonication method. This study provided data about the impact of ultrasonic power and the influence of the medium on alterations in shape, surface characteristics, and structural transformations. However, the technique uses an acidic aqueous medium and necessitates an extended, ideal processing duration, potentially leading to drug loss in the formulation.46 On the other hand, Broda et al. proposed a novel approach for PCL SFs preparation. The method involves cryo-cutting of an aligned mat followed by sono-homogenization. SFs were characterized by excellent distribution and lack of agglomeration.26 It has been demonstrated that the lack of branching and highly aligned polymer mat makes it more straightforward to disperse into individual fibers, and fewer agglomerates are formed.47,48 Inspired by their work, we devised the idea of drug-releasing SFs. The electrospinning process was optimized to prepare aligned fibers free of beads and branching (Fig. 1(A)). The alignment process of the electrospun mat was conducted using a high-speed rotary collector (Fig. 1(B)). The alignment process improved the average fiber diameter distribution from 2.15 ± 0.58 μm (n > 100) before the alignment to 0.86 ± 0.18 μm (n > 100) after (Fig. 1(C)). The fibrous mat was cut into squares and sectioned using the cryostat, followed by homogenization, washing, and freeze-drying. Cryosectioned lyophilized polymeric single fibers were visualized under SEM. It can be seen that SFs showed undamaged structures with a lack of significant agglomerations (Fig. 1(D) and (E)). The average length distributions of SFs were measured. The fibers displayed short length (22.3 ± 15.2 μm (n > 250)) and proper uniformity (Fig. 1(F)). The DSC analysis was performed in the 0–100 °C temperature range to determine polymer Tg and structurization effect on this parameter. Tg is commonly characterized as the temperature at which a polymer transitions from a rigid, glassy state to a relatively compliant, rubbery state. During the glass transition, the disordered molecular chains within the amorphous region begin to disentangle, enhancing the mobility of the polymer chains and resulting in a softening of the rubbery substance.44,49 As Lappe et al. showed, Tg is a crucial parameter for controlling the drug release kinetics from the polymeric matrix.50 Exceeding the Tg significantly accelerates the release and burst phase while maintaining the temperature below Tg results in sustained release kinetics with lower initial burst. The DSC showed that the electrospun PLGA mat before cryo-cutting displayed slightly higher Tg (45.5 °C) than SFs prepared from the same material (44.3 °C) (Fig. 1(G)). Exceeding the aforementioned 44.3 °C temperature will be crucial in controlling the KET release from the SFs and the final photo-responsive hydrogel material. FTIR spectra of the PLGA mat, PLGA SFs containing KET, and KET were obtained (Fig. 1(H)). The spectrum of the PLGA mat showed characteristic peaks at 2947 cm−1 and 2998 cm−1 corresponding to CH, CH2, and CH3, functional group stretching vibrations, while the peak near 1752 cm−1 appears due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching. A bond located at 1089 cm−1 is associated with the symmetrical stretching of the CO, and the 1171 cm−1 is attributed to the asymmetrical stretching of the O–C–O bond.51,52 The FTIR spectra of pure KET exhibited characteristic symmetric carbonyl peaks at 1694 cm−1 and 1653 cm−1 due to the dimetric carboxylic and ketonic group stretching vibrations, respectively.53 In order to confirm the KET unchanged persistence in the PLGA structure after the structurization to the SFs, the spectra of SFs + KET were obtained. The pure drug's characteristic acid carbonyl stretching band appeared unchanged in the sample, and the spectra were the sum of the pure components. No differences were found in the PLGA and KET structures after the structurization.
O stretching. A bond located at 1089 cm−1 is associated with the symmetrical stretching of the CO, and the 1171 cm−1 is attributed to the asymmetrical stretching of the O–C–O bond.51,52 The FTIR spectra of pure KET exhibited characteristic symmetric carbonyl peaks at 1694 cm−1 and 1653 cm−1 due to the dimetric carboxylic and ketonic group stretching vibrations, respectively.53 In order to confirm the KET unchanged persistence in the PLGA structure after the structurization to the SFs, the spectra of SFs + KET were obtained. The pure drug's characteristic acid carbonyl stretching band appeared unchanged in the sample, and the spectra were the sum of the pure components. No differences were found in the PLGA and KET structures after the structurization.
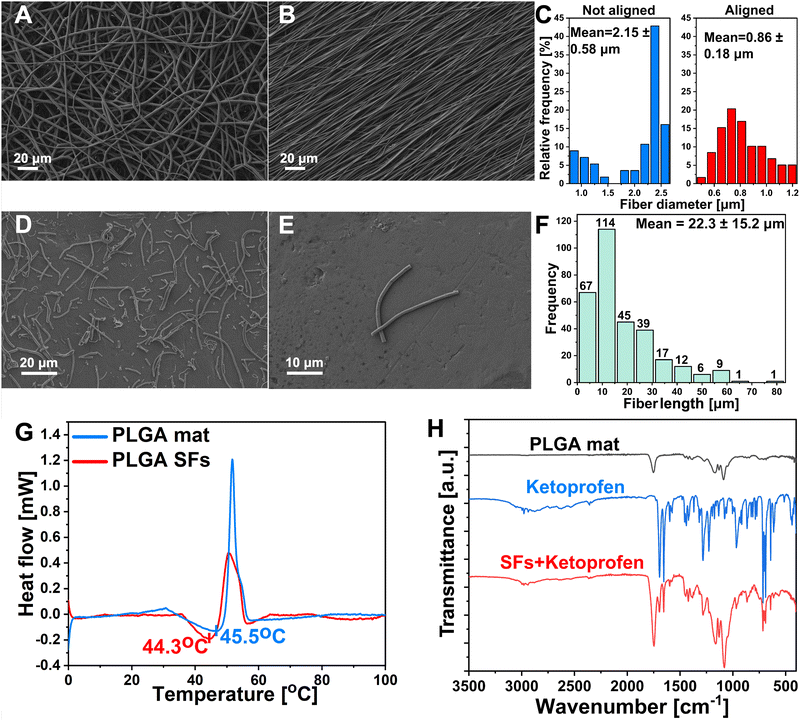 |
| | Fig. 1 Graphs representing characterization of electrospun material. (A) SEM image of PLGA mat as spun. (B) SEM image of aligned PLGA mat. (C) Fiber diameter of not aligned (2.15 ± 0.58 μm) and aligned fibers (0.86 ± 0.18 μm). (D) and (E) SEM images of SFs at different magnifications showing the structure of single filaments after structurization. (F) SFs length distribution of mean value 22.3 ± 15.2 μm. (G) DSC of polymeric material representing the Tg before structurization into SFs (45.5 °C) and after (44.3 °C). (H) FTIR of material before and after structurization containing KET. | |
Hydrogel characterization
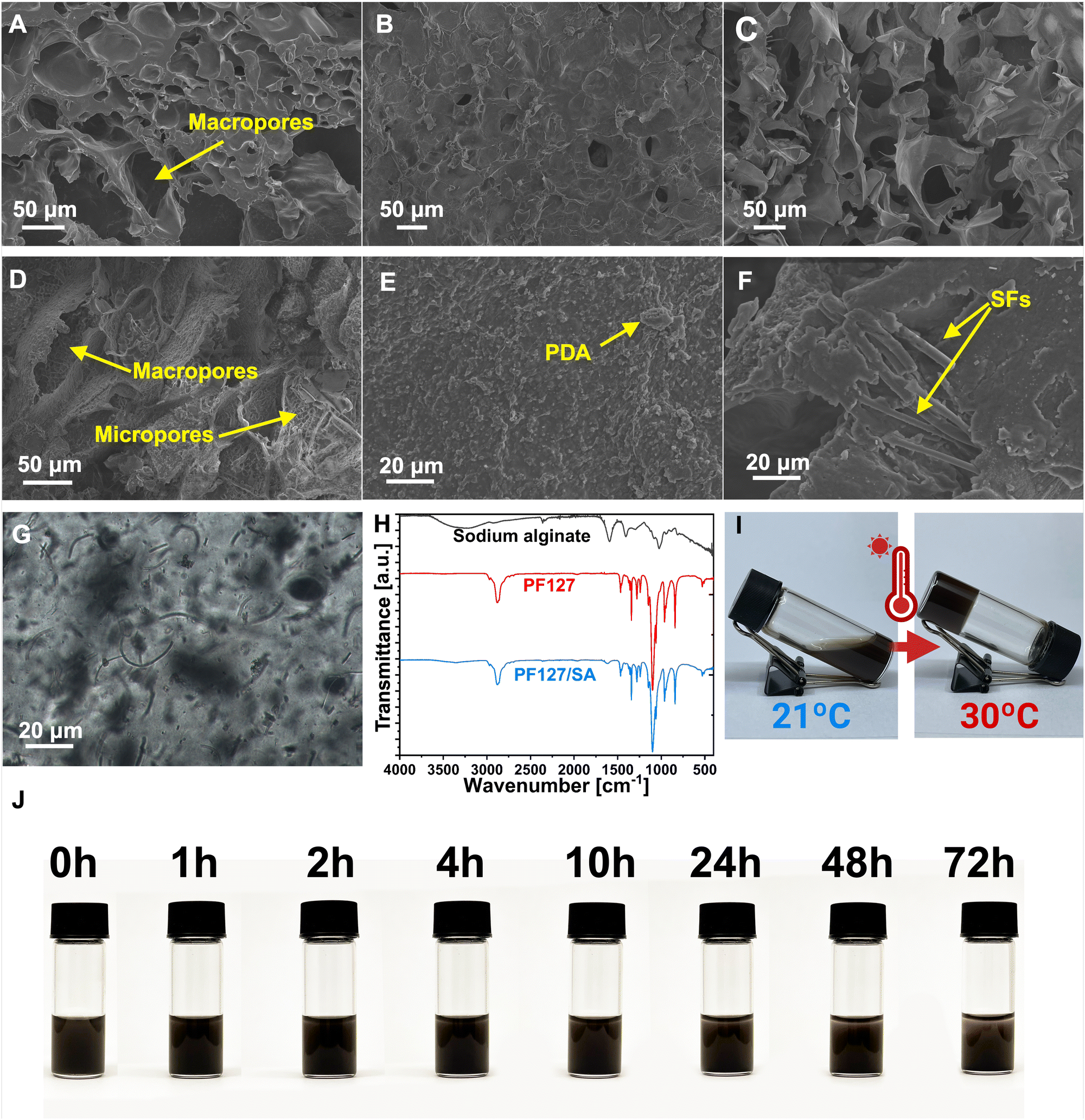
The impact of individual components on the PF127/SA hydrogel network, including SFs and PDA, was analyzed using SEM. The prepared injectable hydrogel exhibits a microscopically porous structure, crucial to retaining substantial water content and enhancing the SFs loading and dispersion. SEM images of PF127/SA showed a spongy surface with macro and micropores and wavy grooves (Fig. 2(A)). Specific irregularly interconnected structures and large pores within the network confirm the inter-chain interactions. Injectable hydrogels must possess a network of interconnected pores since a well-structured porous microarchitecture is a foundation for the permeation of oxygen, nutrients, and metabolites. The internal microporous structure mimics the ECM and plays a vital role in biological activity and tissue regeneration.54,55 The visual representations of PF127 displayed a surface morphology characterized by a loose and coarse texture featuring tiny pores (Fig. 2(B)). SA showed an even more rough and porous structure compared to the complex PF127/SA, confirming the crucial influence of SA to the final material characteristic morphology (Fig. 2(C)). The influence of PDA on the hydrogel structure was also investigated. The surface of the hydrogel became even more rough and irregular, increasing the microporosity of the construct (Fig. 2(D) and (E)). The water uptake of the hydrogels can be attributed to the filling of macropores, followed by the gradual accumulation of water in micropores. This process enables the effective retention of a significant amount of water for drug releases through diffusion.56 Production of hydrogel with added fillers requires uniform filler dispersion within the hydrogel structure. It is a crucial parameter impacting the mechanical characteristics of composites.57 The PF127/SA hydrogel, after cross-linking reinforced with SFs, was visualized using a SEM (Fig. 2(F)) and optical microscope (Fig. 2(G)). It can be seen that single fibers are homogeneously dispersed. Still, small agglomerations occurred during hydrogel formation due to the hydrophobic nature of PLGA that tends to accumulate in the hydrophilic hydrogel medium. The SFs concentration was chosen according to the Wang et al. research. They showed that a 1% w/w concentration of SFs compared to the hydrogel is optimal in terms of injectability of the material while maintaining sufficient mechanical properties.31
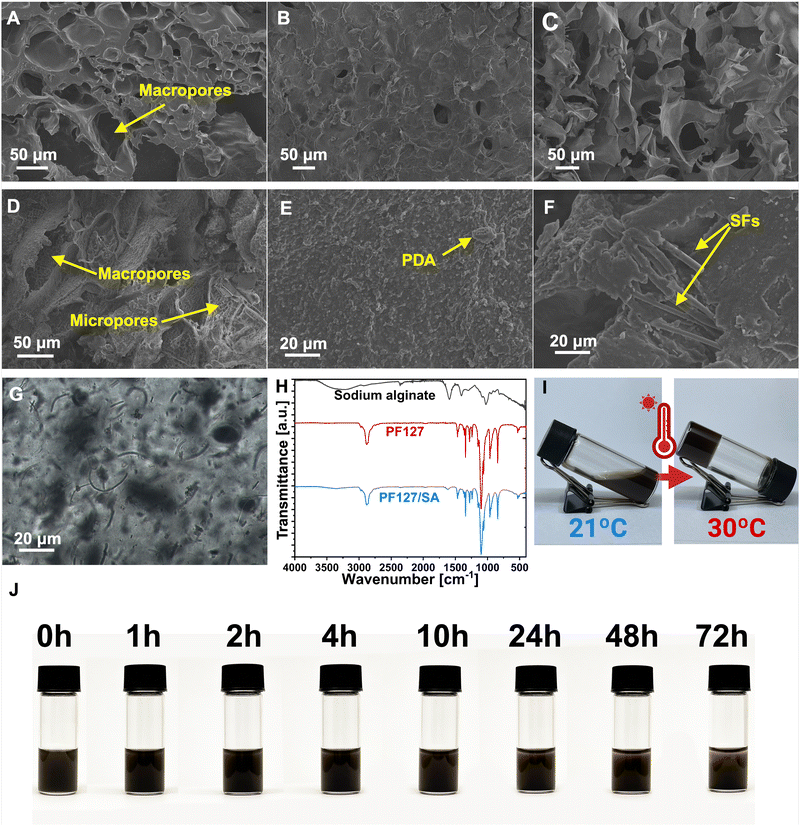 |
| | Fig. 2 Images and graphs characterizing the hydrogel composite. (A) SEM image of PF127/SA (B) SEM image of PF127. (C) SEM image of SA. (D) and (E) 1 – macro visualization of PF127/SA/PDA and 2 – micro visualization of the hydrogel surface structure. (F) SEM image of PF127/SA/SFs. (G) Optical microscope image of PF127/SA/SFs. (H) FTIR of hydrogel and each component. The characteristic peaks of PF127 and SA are found in the hydrogel structure, confirming the existence of an interpenetrating hydrogel network. (I) Sol–gel transition process of hydrogel when heated up from 21 °C to 32 °C. (J) Stability of PF127/SA/PDA/SFs suspension over time. The sedimentation process is significant after 24 h. | |
The FTIR spectra were utilized to analyze both the hydrogel and its individual components, providing insights into their structural characteristics and confirming the presence of an interpenetrating hydrogel network (Fig. 2(H)). In the case of SA, its spectrum revealed a broad peak at 3218 cm−1, corresponding to the stretching of hydroxyl groups (O–H), as well as distinctive bands at 1599 cm−1 and 1429 cm−1, attributed to the asymmetric and symmetric stretching of –COO– groups (carboxylate salts), respectively.58 Additionally, a sharp and broadband at 1111 cm−1 was observed, indicating stretching vibrations of C–O–C bonds. Furthermore, rocking bands at 964 cm−1 and 824 cm−1 were assigned to –CH2 stretching.59 The absorption peak at 2890 cm−1 was associated with the C–H stretching of the methylene functional group.60 In the PF127 spectrum, an absorption peak at 2886 cm−1 confirmed the –C–C alkanes vibrational stretching. The wavy peaks at 1281 cm−1 and 1241 cm−1 are associated with the twisting stretch of the –CH2 group. The presence of the methyl group –CH3 was confirmed by the peak around 1360 cm−1.61 The sharp bands detected at 1467 cm−1 and 1342 cm−1 were attributed to the stretching of the CH2 functional group. The absorption peaks at 1149 cm−1, 1101 cm−1, and 1060 cm−1 were described as stretching vibrations of C–O–C– linkage.62 The PF127/SA hydrogel spectrum showed peaks at 2886 cm−1, 1467 cm−1, 1342 cm−1, and 1102 cm−1 attributed to the PF127. Moreover, the characteristic SA peaks at 3351 cm−1, 2858 cm−1, and 1622 cm−1 were shifted due to the ionic cross-linking of the –COO– group of Ca2+ and alginate.61 The absorption peaks of PF127 and SA were found in the hydrogel structure, confirming the existence of an interpenetrating hydrogel network.
Physicochemical properties of hydrogels
Sol–gel transition analysis and stability.
The PF127/SA hydrogel exhibits thermo-reversible characteristics, remaining in a liquid state at room temperature and transforming into a gel when exposed to body temperature due to the PF127 presence – an amphiphilic, triblock polymer. The PF127 can form a self-assembled micelle structure in aqueous solutions. These micelles have the ability to pack together above the critical Tgel densely. In the composite PF127/SA hydrogel structure, SA fills the space between the pluronic micelles and plays a role as an inter-molecular crosslinker, forming a highly dense inter-micellar structure at temperatures about the Tgel. Moreover, the critical Tgel can be tailored by controlling the SA concentration with respect to the PF127.40,63 PF127/SA and PF127/SA/PDA/SFs hydrogels were immersed in a water bath with controlled temperature. Different PF127/SA ratios were tested during the optimization process to find the optimal conditions for on-skin gelated formulation (Table S1, ESI†). The chosen PF127/SA hydrogel exhibited the sol–gel transition at 27 °C, and PF127/SA incorporated with PDA and SFs showed a higher transition temperature equal to 30 °C (Fig. 2(I)). According to the literature, the incorporation of particles and SA into the PF127 structure can elevate Tgel. Dispersed particles impede the ability of PF127 micelles to create closely packed structures and inhibit their connectivity.64–66 The thermo-reversible properties of the injectable hydrogel were evaluated using the freeze–thaw technique. Injectable hydrogel solutions were subjected to a thawing cycle at 37 °C, followed by freezing at 4 °C and then thawing again at 37 °C. Notably, the hydrogel displayed in both cases consistent Tgel during the first and second thawing cycles. However, the ion cross-linking of the SA component by calcium chloride resulted in the loss of the thermo-reversible effect. Nevertheless, external cross-linkers are commonly employed to enhance the physical properties of alginate-based systems.67,68 The thermo-reversible attribute is crucial for the injectability of the system and in situ on-skin gelation to provide personalized irregular wound healing. Additionally, the pre-gel suspension stability was tested to evaluate the capacity for sedimentation and aggregation of SFs and PDA over time. The significant sedimentation becomes apparent after 24 h of suspension storage (Fig. 2(J)).
Equilibrium swelling ratio (ESR), moisture content (MC).
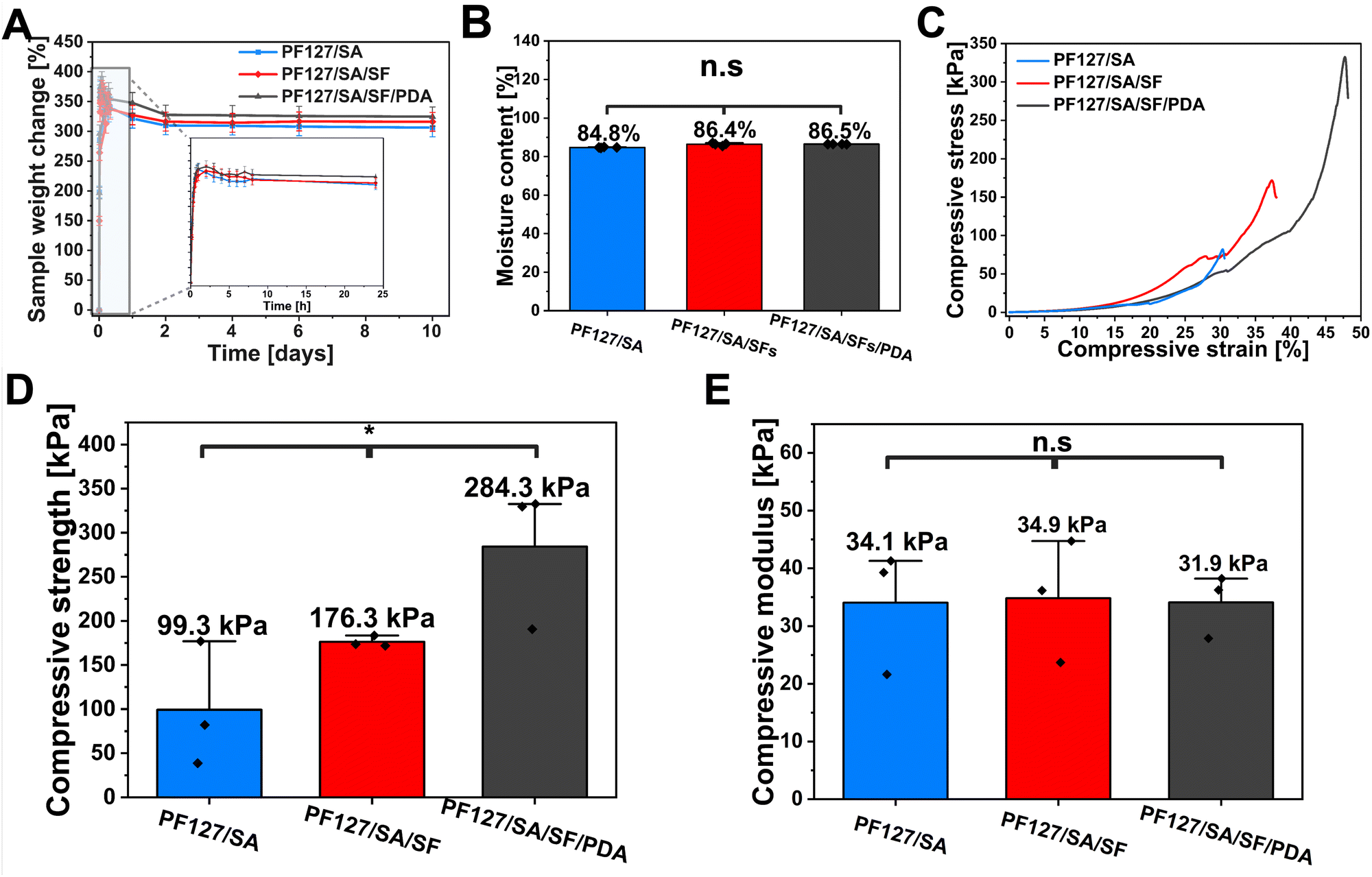
To evaluate the in vitro swelling rate and stability of injectable hydrogel, the fabricated samples were dehydrated and incubated in HEPES solution (pH 7.4) at 37 °C. According to the Yang et al. research, in which stimuli-responsive cryogels with controlled NO release and self-adaptivity were developed. Thanks to the cryogels’ microporosity, the material performed an excellent wound exudate absorption capacity compared to the traditional hydrogels, showing that swelling is crucial in determining the networks’ ability to transport nutrients, growth factors, drugs, and oxygen.69 Swelling is crucial in determining the networks’ ability to transport nutrients, growth factors, drugs, and oxygen. The amount of fluid absorbed by a three-dimensional injectable hydrogel is affected by the void spaces within the structure and the degree of self-assembly.70 The hydrogel was tested in three configurations: PF127/SA, PF127/SA/SFs, and PF127/SA/SFs/PDA, to evaluate each component's influence on the swelling ability. All the groups showed similar swelling properties (Fig. 3(A)). The control group of PFSA127/SA reached the 371% equilibrium swelling ratio after 60 minutes of incubation. Whereas the group containing SFs performed a slightly lower rate of 367% after 120 min. The group containing SFs and PDA – also displayed the highest swelling ability – 381% after 120 min. All the samples reached an equilibrium state quickly, within 120 min, and started losing weight over time. After 10 days of incubation, the samples lost 65%, 51%, and 56% of their initial weight for PF127/SA, PF127/SA/SFs, and PF127/SA/SFs/PDA groups, respectively. Nevertheless, the system showed good stability over incubation time. Stability can be attributed to the cross-linking of SA that caused the resistance to degradability. The initial water uptake can be associated with the porous hydrogel structure and water penetrating the pores. Then, the weight decreases due to biodegradation and washing out the PF127 component, which is not bound into the interpenetrating hydrogel network. The ANOVA curve analysis of ESR showed no significant differences between the samples (p value ≤0.05, n = 4). Specifically, the incorporation of SFs and PDA had a minimal impact on the rate of swelling and degradation of the hydrogels. In addition, moisture content was investigated (Fig. 3(B)). The hydrogel showed 84.8%, 86.4%, and 86.5% MC for PF127/SA, PF127/SA/SFs, and PF127/SA/SFs/PDA samples, respectively. No significant differences between samples were found (p value ≤0.05, n = 4). The moderate swelling ratio and good stability can play a role in preserving a moist wound environment and absorbing wound fluids, making the system suitable for a prolonged drug release system applied in wound healing.
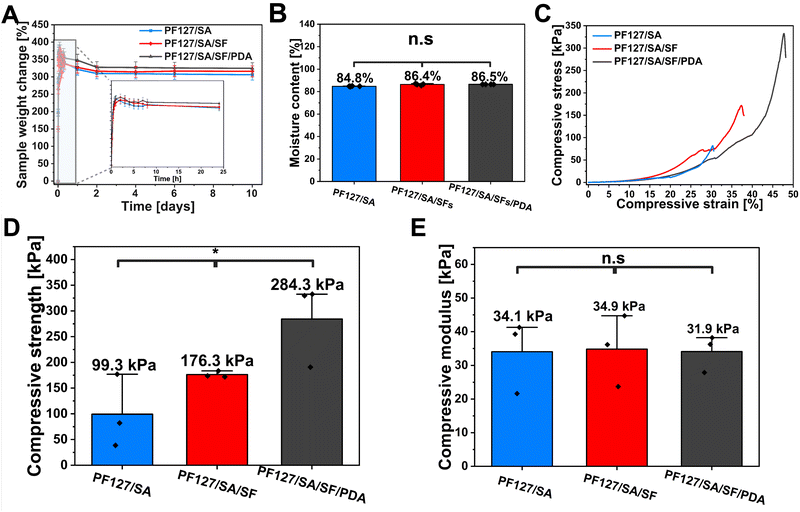 |
| | Fig. 3 Physical properties of the hydrogel. (A) Equilibrium swelling ratio and in vitro stability profile of PF127/SA, PF127/SA/SFs, and PF127/SA/SFs/PDA hydrogels. (B) Moisture content of hydrogels. (C) Compressive stress–strains curves of hydrogels and corresponding. (D) compressive strength and (E) average compressive modulus. | |
Mechanical tests.
One of the most essential properties of the hydrogels applied on the skin is the ability to endure the external pressure from the surrounding tissue during movement. Consequently, wound dressings must possess appropriate compressive strength to preserve structural integrity.71 The compression tests were conducted to evaluate the compressive resistance of cross-linked hydrogels and investigate the influence of each component on the compressive strength and compressive modulus. The introduction of SFs and PDA into the hydrogel network significantly (p value ≤0.05, n = 3) increased the compressive strength (284.3 kPa) compared to the hydrogel with SFs (176.3 kPa) and pure hydrogel (99.3 kPa) (Fig. 3(C) and (D)). Despite that, the reinforcement of hydrogel with SFs and PDA fillers increased the compressive strength – more pressure is needed for the hydrogel fracture. The PDA group showed the highest strength, possibly due to the hydrogen bonding between the SA component and PDA.72 Nevertheless, the analysis of the plots’ initial course showed no significant (p value ≤0.05, n = 3) differences between the pure hydrogel (34.1 kPa), hydrogel reinforced with SFs (34.9 kPa), and the material containing SFs and PDA (31.8 kPa) (Fig. 3(E)). The PF127/SA and PF127/SA/SFs showed higher SD values ±10.8 kPa and ±10.6 kPa, respectively, compared to the PF127/SA/SFs/PDA – ±5.5 kPa. The PF127/SA and PF127/SA/SFs showed a larger range of results and less reproducible measurements. Even though, generally, fillers increase the modulus of a composite, in this case, the higher hydrogel porosity of the PF127/SA/SFs/PDA (shown in the SEM images in Fig. 2(D) and (E)) possibly weakens the hydrogel.27
Self-healing abilities and injectability.
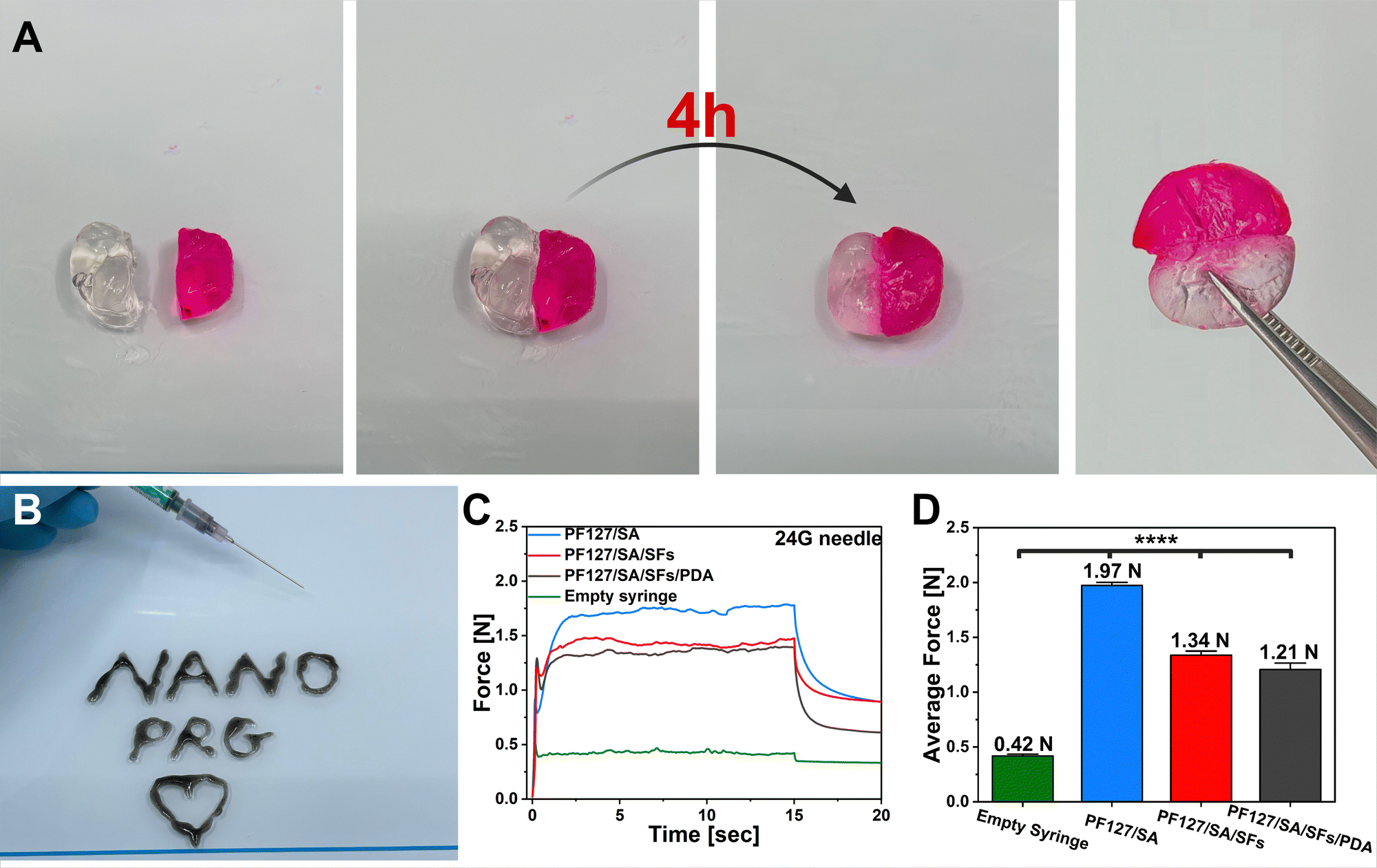
Subsequently, our focus turned to examining the self-healing ability of nanocomposite. The self-healing ability of hydrogel is a superior property in wound closure by prolonging the hydrogels’ viability and possibly preventing further wound infection. Moreover, it enables an automatic repair of damage sustained during use. In the context of prepared material, such damage could lead to undesirable PDA particles and SFs diffusion into surroundings.73,74 We assessed this capability through macroscopic self-healing experiments. Two disc-shaped hydrogels (one stained with rhodamine B) were cut using a scalpel, and their cut surfaces were brought into direct contact. Following a 4-hour incubation at room temperature, two differently colored segments successfully reconnected and could support their weight (Fig. 4(A)). This phenomenon can be attributed to the dynamic physical interactions (supramolecular assembly). Furthermore, under physiological temperature and rapid PF127 reaction to the temperature changes, the second cross-linking occurred as a result of the hydrophobic interactions among polymer chains of PF127 and SA.75,76 The injectability and in situ gelation behavior of tested hydrogel is crucial for personalized therapy. The composite injectability was evaluated using a 24G needle. As shown in Fig. 4(B), the PF127/SA/PDA/SFs suspension could easily pass through the needle without clogging and immediately be gelated when placed onto a glass hotplate (35 °C). The hydrogel can be shaped into desired structures, as presented in the photo. One of the critical parameters when designing injectable systems is the force needed for the syringe to eject its load. The experiment was conducted at atmospheric pressure at the needle outlet. The temporary plots of force needed to apply over time to eject the syringe content were obtained (Fig. 4(C)). The plot shows the initial build-up of the force, and the following plateau maintained constant, confirming that no agglomeration occurred. The average force needed to inject 0.5 mL of each hydrogel configuration was 1.97 N, 1.34 N, and 1.21 N for PF127/SA, PF127/SA/SFs, and PF127/SA/SFs/PDA, respectively (Fig. 4(D)). The hydrogel precursors showed significantly (p value ≤0.0001, n = 4) higher force needed for ejection compared with an empty syringe (0.42 N). The tests were done by injecting into the air. During in vivo clinical settings, the relevant injection forces typically fall below 20 N.77 The tests proved the injectability properties of composites and possible clinical applications. Incorporating SFs and PDA slightly decreased the force needed for injection compared to the pure hydrogel – 1.34 N and 1.21 N for the PF127/SA/SFs and PF12/SA/SFs/PDA samples, respectively. The investigation clearly showed that the final material can be injected effortlessly.
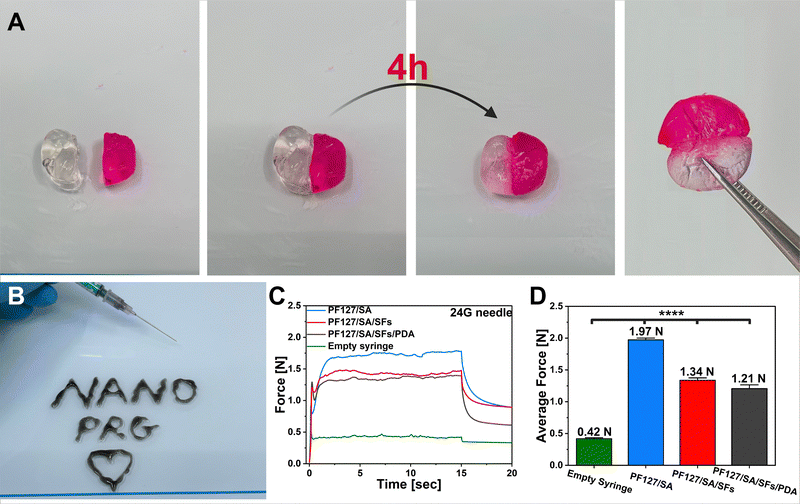 |
| | Fig. 4 Representation of hydrogel functionalities. (A) Self-healing abilities of the developed hydrogel (rhodamine B was used as a die). (B) Photo showing the injectability and in situ gelation properties of PF127/SA/SFs/PDA. (C) Time–force plot showing the force needed to eject 0.5 mL of different hydrogel precursor suspensions. (D) Average force needed to eject 0.5 mL of different hydrogel precursor suspensions. Significant differences were found between the pure hydrogel and hydrogel incorporated with fillers. | |
Photo-thermal characterization
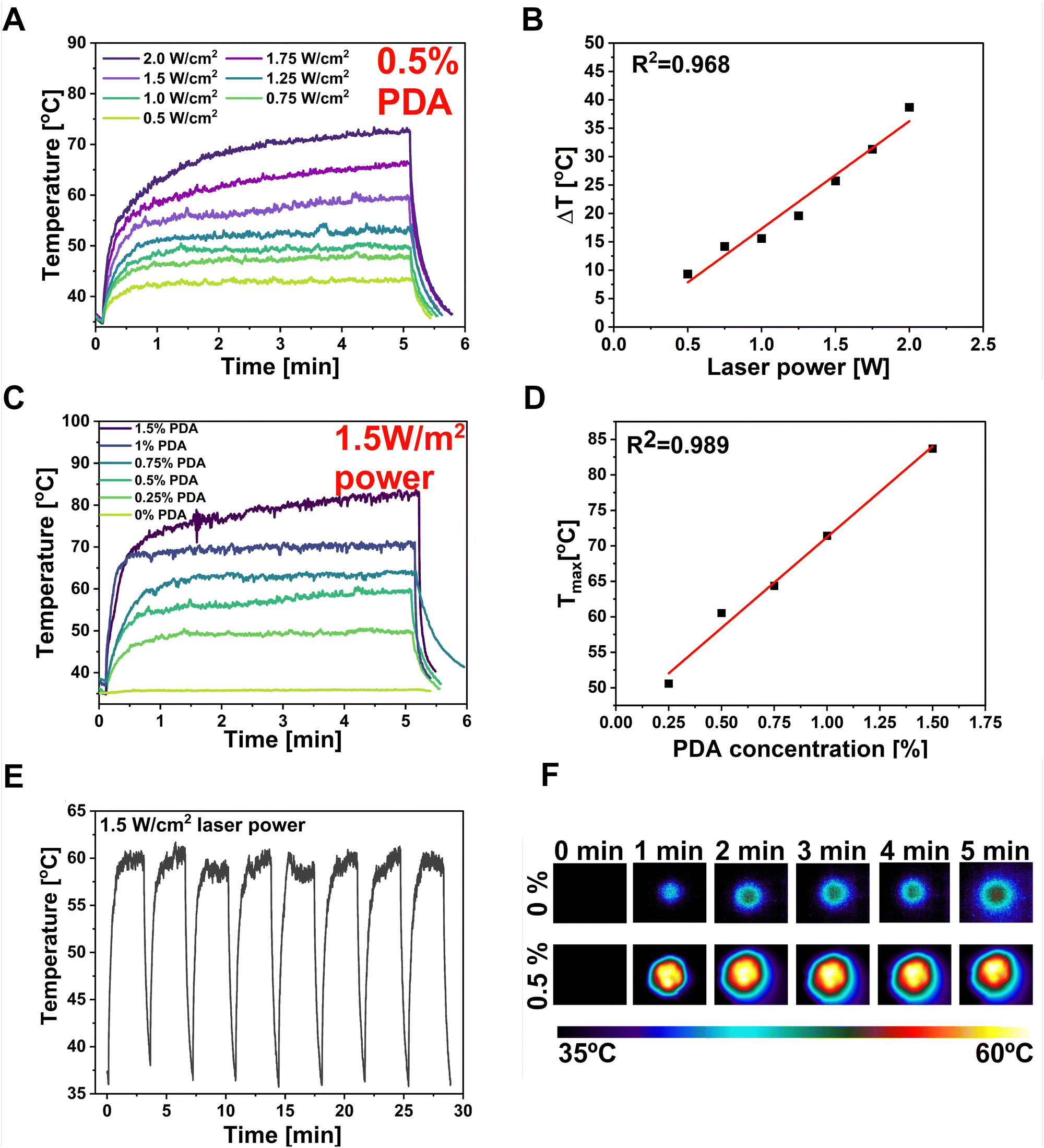
The PDA particles demonstrate strong absorption in the NIR region and can efficiently convert the NIR laser light into heat, increasing the local temperature.78 The DLS investigation of the PDA particles used in this study showed an average particle diameter of 1015 nm, with the polydispersity index (PDI) equal to 0.211 (Fig. S1, ESI†). The PDI diameter represents the size distribution within the sample, ranging from 0.0 (perfectly uniform) to 1.0 (highly varied samples with multiple-size populations).50 The sample has shown a PDI below 0.3, which can be attributed to a predominantly uniform particle population. According to Carmignani et al. research, they provided evidence that micro-sized particles (>900 nm) of PDA are preferable for PTT and controlled drug delivery due to the more effective light–heat conversion.79 Thus, the PF127/SA/SFs/PDA's behavior after NIR light stimulation was investigated by irradiating it with an 808 nm wavelength laser beam. The PDA incorporation provided a robust NIR absorption, resulting in great photo-responsivity of the hydrogel. Different laser intensities were used to record the maximum temperature the system can reach after 5 minutes with desired laser power (Fig. 5(A)). The time–temperature profiles at different intensity values of the laser beam were tested starting from 0.5 W cm−2 up to 2 W cm−2, showing rapid and significant temperature increase proportional to the laser beam power. The sample could be effectively heated up to 85 °C upon irradiation and cooled down to the starting temperature after the pump beam is turned off. The linear correlation (R2 = 0.968) was found between the ΔT shown by the hydrogel and the intensity of the laser light (Fig. 5(B)). The mentioned phenomenon can be explained by the fact that the heat power density generated by PDA is directly related to the square of the incident electric field.80 This ability of photo-thermal material can be used to optimize the material to reach the desired temperature optimal for bacteria eradication in infected wounds. The thermal behavior of hydrogel with different PDA concentrations was also investigated using constant laser power (1.5 W cm−2) (Fig. 5(C)). The graph showed that the maximum temperature of the hydrogel can be increased at the highest PDA concentration, reaching 85 °C when 1.5% w/w of PDA is added. The linear correlation (R2 = 0.989) between Tmax reached by the sample and PDA content was also proved (Fig. 5(D)). However, the optimal temperature for the designed material should take into account the necessity to overcome the Tg of the polymeric SFs to control the drug release while reaching the temperature range for antibacterial PTT (55–60 °C). Thus, optimal parameters in terms of material concentration (0.5% PDA w/w) and laser beam power (1.5 W cm−2) were selected to reach the desired temperature during the test of stability and repeatability of photo-thermal properties. So, the hydrogel was subjected to 8 cycles of laser irradiation-cooling (Fig. 5(E)). The test shows that the composite could be effectively heated after exposure to the laser and cooled quickly after the beam was turned off. In every cycle, the material reached a temperature between 59–61 °C within 3 min and dropped the temperature to the starting range within 1 min. The temperature images before and after 1, 2, 3, 4, 5 min of laser irradiation for optimized sample containing 0.5% PDA compared with reference hydrogel without PDA was shown in Fig. 5(F).
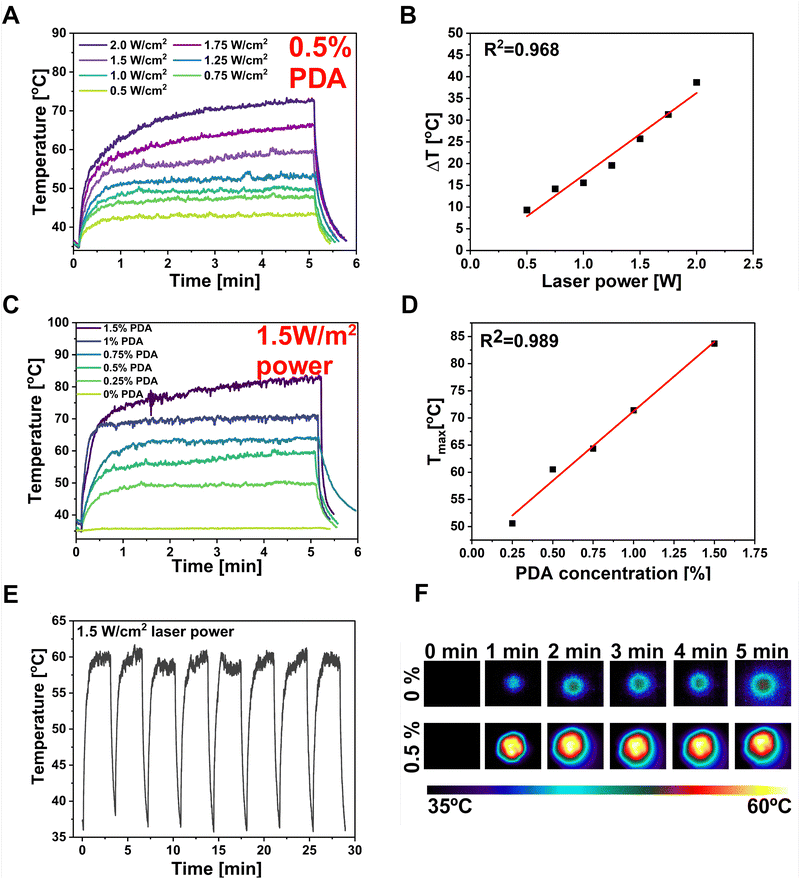 |
| | Fig. 5 Photo-thermal optimization of the material. (A) Temporal plots of different laser powers–temperature dependence over time using 0.5% PDA concentration. (B) Linear plot of laser power and Δtemperature dependence. (C) Temporal plots of PDA concentration–temperature dependence over time using 1.5 W laser power. (D) Linear plot of PDA concentration and Tmax dependence. (E) temporal plot of multiple NIR irradiation of platform representing its stability. The platform reaches same temperature every cycle and cools down quickly within 1 minute after laser is turned off. (F) Panel with thermal camera images showing the reference PF127/SA and PF127/SA/PDA sample temperature before and after 1, 2, 3, 4 and 5 min of laser irradiation. Samples were immersed in medium. | |
In vitro drug release studies and release kinetics
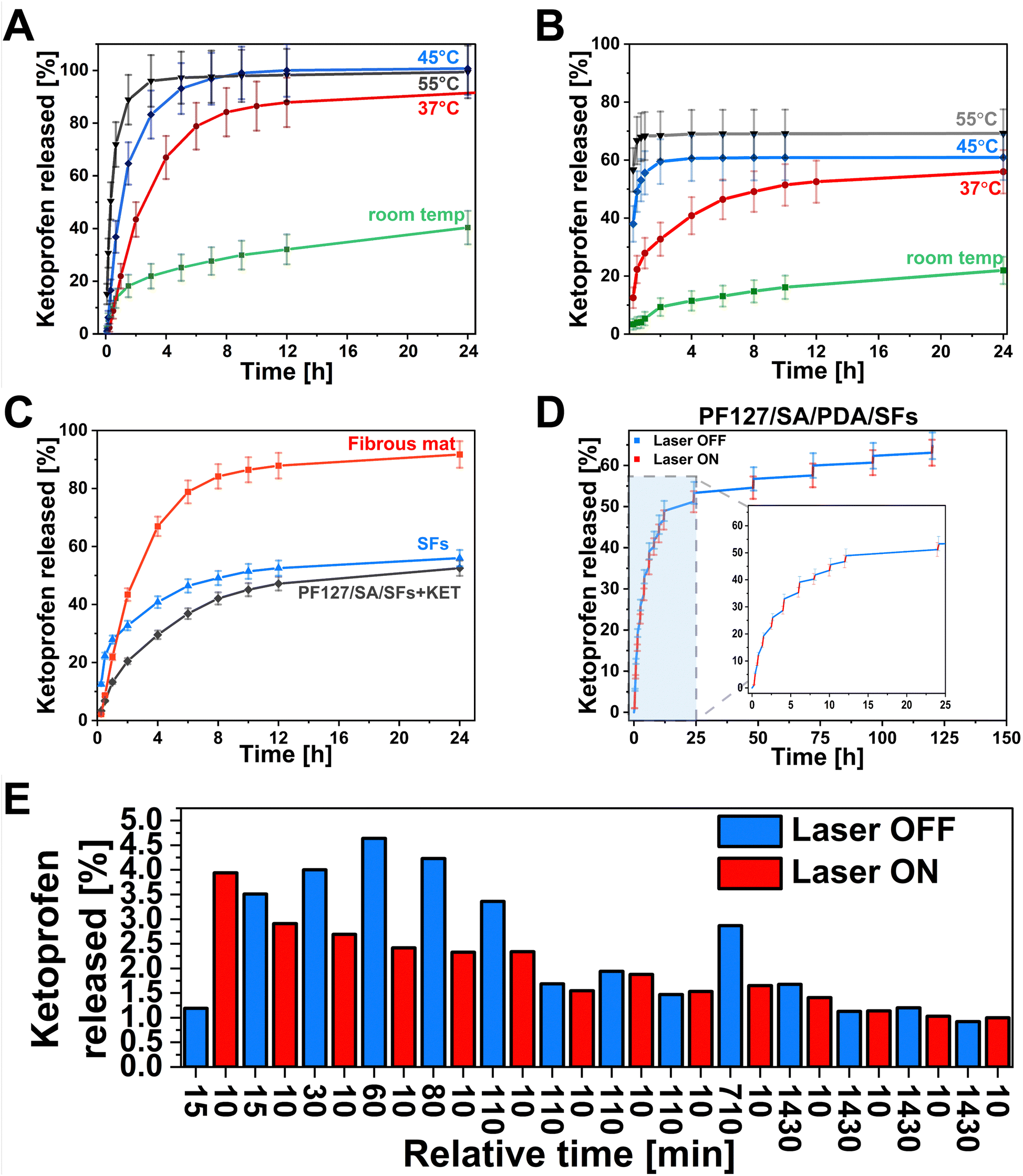
One of this study's main aims was to develop a smart drug delivery system with on-demand release properties. The hydrogel can facilitate controlled drug delivery by reacting to the external NIR light stimulation within the injured site in the range of the therapeutic index while minimizing nonspecific toxicity. The drug can be delivered precisely to the right site at the targeted time. This ability is crucial in chronic wounds when an elongated inflammatory state negatively affects wound healing. The pulsatile release of drugs can be adjusted by laser irradiation to align with the therapy requirements.81 Light as a stimulus offers distinctive benefits, including spatiotemporal precision, relative safety, and limited interference with cellular signaling pathways.82,83 For this purpose, the KET was selected as a small, non-steroidal anti-inflammatory drug. The KET release kinetics were tested in 4 different temperature conditions to emphasize the influence of laser on drug release from the polymeric material. We choose various temperatures such as room temperature, 37 °C, 45 °C, and 55 °C as crucial in understanding the temperature-mediated kinetics of drug release (Fig. 6(A) and (B)). At room temperature, the samples were placed on the glass hot plate heated to 35 °C to mimic the on-skin environment as precisely as possible. The temperature of 37 °C represents the physiological human body temperature, and the 45 °C is slightly above the Tg of prepared SFs – the temperature when release can be accelerated due to enhancing the polymeric chain mobility. Moreover, it is also considered a temperature level for performing the mild-temperature PTT.84 The 55 °C represents the desired temperature targeted during the laser exposure to reach antibacterial requirements.85,86 The drug release studies were conducted from the PLGA mat before structurization (Fig. 6(A)). Within 24 h, the cumulative release in different conditions showed 40%, 91%, 100%, and 99% at room temperature, 37 °C, 45 °C, and 55 °C, respectively. In the first 40 minutes of the study, 13%, 21%, 36%, and 72% of the drug, according to the increasing temperature, is released from the matrix. It can be observed the higher temperature leads to the more significant initial burst release. The cumulative release after 24 h at 55 °C was almost 2.5× higher than at room temperature. The influence on drug release behavior after material structurization into SFs was also investigated (Fig. 6(B)). In this case, the cumulative release after 24 h indicated the same temperature dependence, resulting in 22%, 56%, 61%, and 70% at room temperature, 37 °C, 45 °C, and 55 °C, respectively. Based on the release at high temperatures, a 30% lower release rate was observed from SFs, indicating the loss of the drug from the material due to SFs preparation methodology. In the first 40 minutes of the test, 4%, 21%, 53%, and 68% of the drug is released at room temperature, 37 °C, 45 °C, and 55 °C, respectively. The difference in initial release kinetics between the temperatures below the Tg (room temperature, 37 °C) and above (45 °C, 55 °C) is even more pronounced than from the fibrous mat. The results show promising possibilities for controlling the KET release using the temperature. Hydrogel incorporated with SFs offered an opportunity to control the release kinetics slightly (Fig. 6(C)). The cumulative release at room temperature showed 53%, 56%, and 92% for the PF127/SA/SFs, SFs, and fibrous mat, respectively. Although there were no significant differences between the SFs group and hydrogel/SFs in cumulative release ratio, the initial burst release within the first 40 min was significantly decreased compared to the fibrous mat (22%), SFs (21%), and PF127/SA/SFs (13%), due to the hydrogel-mediated control release. During the main drug delivery experiments, nano-hierarchical hydrogels filled with PDA and SFs immersed in the medium were irradiated by a laser beam in a cyclic pattern, with one cycle lasting 10 minutes and for a total of 14 cycles (Fig. 6(D)). Initial burst release causes difficulties in distinguishing the irritation and non-irritation phase. A similar amount of KET is released at each time point, with and without a laser beam (3–4% per time point) (Fig. 6(E)). As the time between each point increases, the differences are more visible. From 1 hour to 6 hours of the test, the irradiated period of 10 min released half of the drug compared to the non-irradiated. Over time, after 8 hours, the system releases a similar amount of KET at each time point. Towards the end, every 10 minutes of irradiation causes the discharge of substance equal to the 24-hour not irradiated period. The laser exposure accelerates the drug release from the hydrogel and increases the cumulative release (61.6%) compared to the non-irradiated sample (53%), in agreement with data reported in Fig. 6(A) and (B) that show increased release in case of higher temperature.
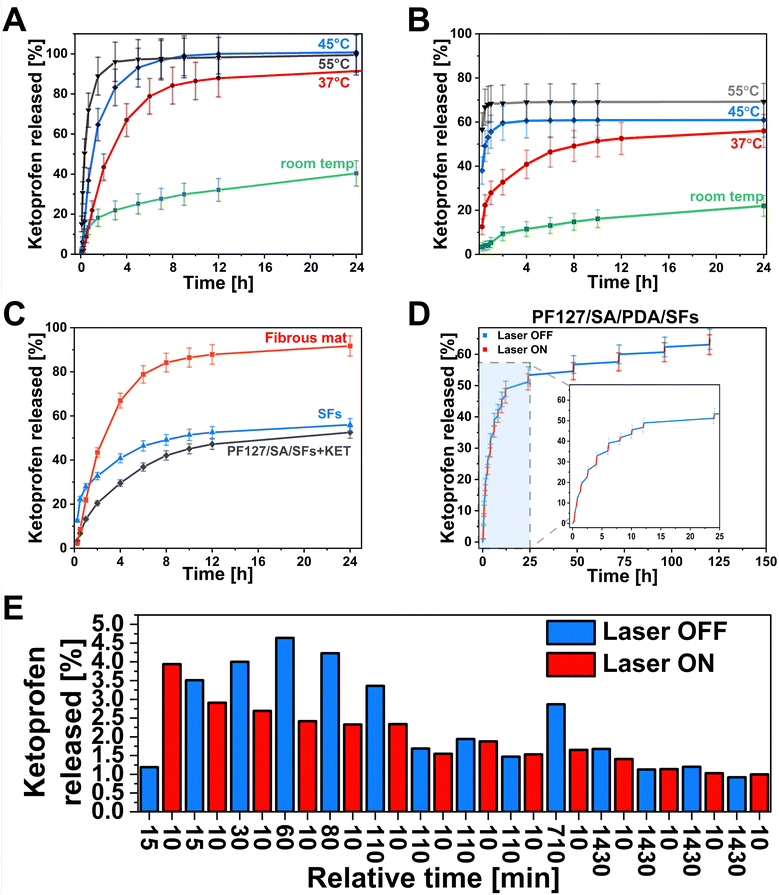 |
| | Fig. 6 Drug delivery curves from (A) fibrous mat before cutting showing the release kinetics depending on temperature – the higher the temperature is, the faster release from the matrix is. (B) SFs, after cutting, show the same dependence as mat before structurization. (C) Hydrogel influence on release kinetics. SFs incorporated into hydrogel decreased the initial burst release. (D) PF127/SA/PDA/SF hydrogel under NIR laser irradiation, representing the gradual release of KET after exposure to the laser light. (E) The bar graph representing KET release at each relative time point with and without laser irradiation. | |
In vitro cell studies
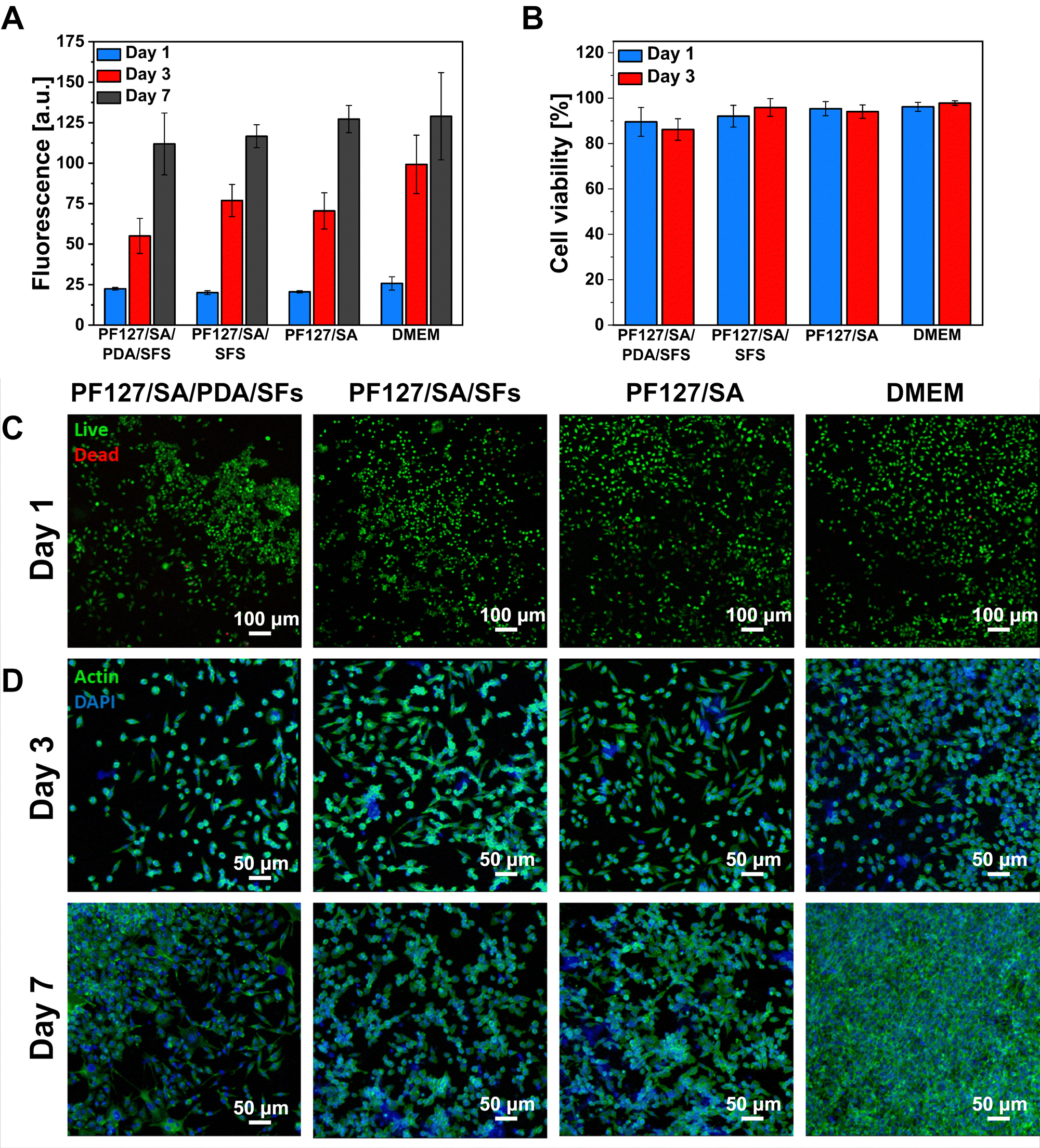
Indirect cytocompatibility of the proposed hydrogels was assessed to verify the safe use of the platform for biomedical applications. Following the ISO Standard, samples were incubated in a culture medium for 24 hours, and extracts were placed in contact with L929 fibroblasts cultured onto a tissue culture plate. The effect of the presence of a PDA and SFs in the hydrogel formulation was evaluated in terms of the response of cells cultured in contact with sample extracts. Results were then compared to the cell response provoked by extracts of pure alginate hydrogel and fresh DMEM. More in detail, cell viability, cell growth, and cell morphology were analyzed during the culture time at selected time points. Fig. 7(A) shows the proliferation rate of the cells cultured in contact with samples’ extracts. The bar chart shows linear cell growth at each time point, revealing no significant difference among the tested conditions (n = 5). No adverse effect of PDA or SFs presence in the hydrogel formulation is reported.87–89 To support the proliferation data, cells were stained with live/dead solution and visualized as green cells – if alive – or red cells – if dead. Live/dead images permit the visualization and calculation of the cell viability percentage, which was detected as >86% for each sample at both selected time points (n = 3, Fig. 7(B) and (C)). Results demonstrate the cytocompatibility of the proposed materials and their suitability for cell survival and growth with no adverse effect of PDA or SFs. The cell morphology was also investigated by staining the cells with actin/DAPI and visualizing them using a confocal microscope (Fig. 7(D)) at a later stage of culture. Cell cytoskeleton (marked in green color) appeared spread and elongated with the typical L929 fibroblast spindle shape at both selected time points, with no evident difference among the tested samples and control conditions (n = 3).
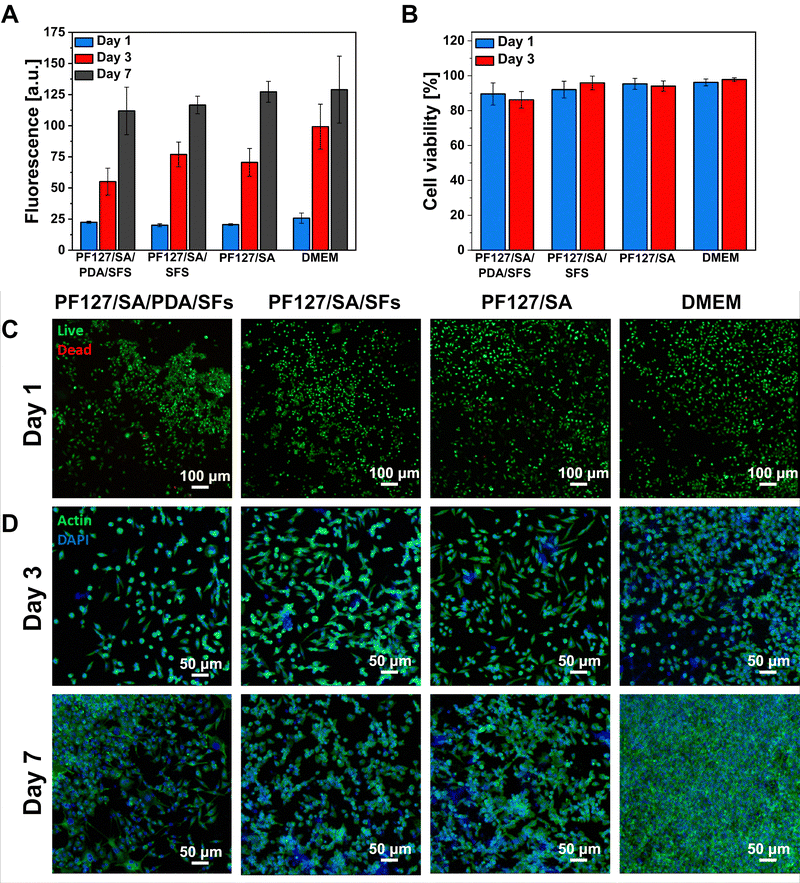 |
| | Fig. 7 Indirect cytotoxicity of PF127/SA/PDA/SFs, PF127/SA/SFs, and PF127/SA. (A) Cell proliferation was measured at days 1, 3, and 7 of culture, evidencing the cell growth during the culture time for all the tested conditions. (B) Percentage of viable cells extracted from the live/dead images, reporting cell viability >86% at each time point for all the samples. (C) Representative confocal images of alive (green) and red (dead) cells, showing the large majority of viable cells for all the conditions. (D) Cell morphology visualized as green cytoskeleton (actin staining) and blue nuclei (DAPI staining). | |
In vitro antibacterial properties
E. coli and S. aureus bacteria were selected to test the potential antibacterial properties of the proposed photothermal PDA-based material when exposed to NIR irradiation. Those strains were chosen because are the most commonly used to investigate bacteria inactivation and response and to study and assess the antibacterial properties of material.80 Additionally, the E. coli and S. aureus strains are frequently found as the most common pathogens of infected wounds, and they are biofilm formers.90,91 Moreover, finding effective therapies for multidrug-resistant Gram-negative bacteria such as E. coli – and Gram-positive bacteria – like S. aureus – has posed a significant challenge due to their unprecedented drug resistance.92 Guo et al. presented the development of physical double-network photo-thermal hydrogels with excellent antibacterial properties, which could eradicate E. coli and S. aureus strains almost 100% by 10 min NIR laser exposure (1.4 W m−2), targeting the 60 °C.93 According to our work, similar conditions (10–20 minutes of irradiation and 1.5 W m−2 laser power) were applied to target up to 60–66 °C and consequently reached highly efficient E. coli and S. aureus eradication (99.9%). The outstanding photothermal properties of PDA were evident after around 150 seconds under NIR irradiation (Fig. 5(E) and (F)). During 10 minutes of NIR light exposure, E. coli and S. aureus were inactivated to 3log units (the limit of detection) when in contact with the PF127/SA/SFs/PDA, thus reporting only 0.1% of survived bacteria (n = 3, Fig. 8(A) and 9(A)). Furthermore, in the representative agar plates prepared in the case of this testing condition, only very few bacteria colonies are visible, showing 99.95% of bacteria inactivation (n = 3, Fig. 8(B) and 9(B)). Results prove the effective bacteria eradication and antibacterial properties of the PF127/SA/SFs/PDA when exposed to NIR irradiations, in agreement with previous studies.72,88,93 On the other hand, no bacteria inactivation was reported in the case of PF127/SA/SFs/PDA not exposed to NIR light, as well as in the case of the other materials tested for both irradiated and not irradiated conditions. As expected, the E. coli and S. aureus suspension control conditions were not inactivated in the case of NIR irradiation nor not irradiation. Results highlight the beneficial photothermal properties of polydopamine for bactericidal performance towards representative Gram-negative (E. coli) and Gram positive (S. aureus) bacteria. Indeed, when exposed to NIR light, the PDA-incorporated hydrogel could reach a temperature of 60.1 °C after 10 min and 66.9 °C after 20 min of laser irradiation, damaging the bacterial membrane and provoking its death.94
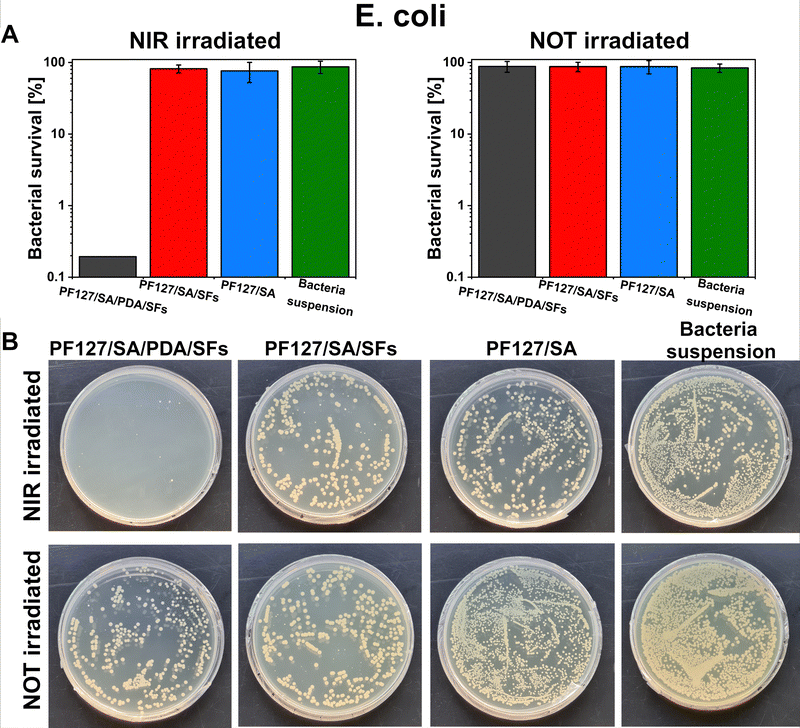 |
| | Fig. 8 Photothermal inactivation of E. coli in contact with PF127/SA/PDA/SFs irradiated under NIR light, (A) survival percentage of bacteria in contact with the proposed materials after 10 minutes of NIR irradiation or no irradiation (bacteria suspension was tested as a control condition), (B) representative photos of bacterial colonies grown on LB agar plates for each tested condition. Data are presented on a logarithmic scale as mean ± standard deviation. | |
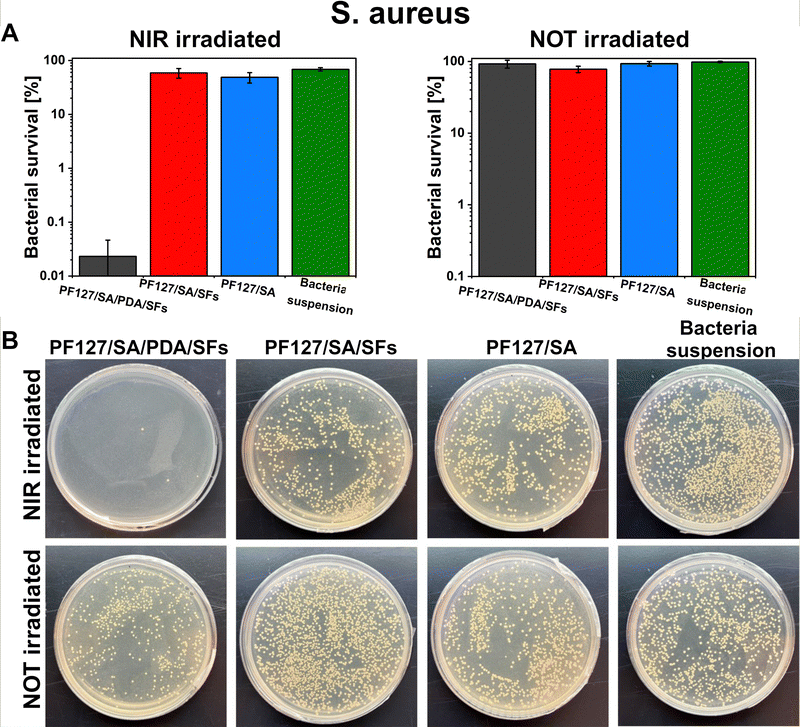 |
| | Fig. 9 Photothermal inactivation of S. aureus in contact with PF127/SA/PDA/SFs irradiated under NIR light, (A) survival percentage of bacteria in contact with the proposed materials after 20 min of NIR irradiation or no irradiation (bacteria suspension was tested as a control condition), (B) representative photos of bacterial colonies grown on LB agar plates for each tested condition. Data are presented on a logarithmic scale as mean ± standard deviation. | |
Conclusions
In this study, we presented the development and characterization of a versatile composite thermoresponsive hydrogel with antibacterial properties for potential use in wound healing applications and drug delivery. The cryo-cutting method was used to prepare the non-agglomerated and drug-containing SFs with optimal distribution. The hydrogel showed a significant ability to absorb water, which is vital for wound healing and drug delivery applications. Incorporating PDA particles effectively converted the NIR light into localized heat, featuring the on-demand drug release ability and effective Gram-positive and Gram-negative bacterial inactivation. The final material could reach a temperature 60 and 66 °C and eradicate 99.9% of E. coli and S. aureus, respectively. The hydrogel showed the ability to pass through a 24G needle with low force needed for injection. The self-healing abilities and suitable mechanical properties demonstrated suitability for further in vivo applications. Overall, the results indicated that the injectable, self-healing, in situ forming PF127/SA/SFs/PDA hydrogel is a potential candidate as an antibacterial and drug-delivery material, and it holds promise in the field of irregular wound treatment. However, the main limitations of hydrogels considered in clinical applications are their poor mechanical properties as well as weak drug-release properties. Therefore, in our study, the incorporation of polymeric SFs improved compressive strength and controlled the ketoprofen release from the hydrogel matrix. Nevertheless, it is important to note that even though NIR light poses minimal health risks and can penetrate deeply, there is a requirement for the advancement of materials that exhibit high energy efficiency when activated by low-energy light.95,96 Thus, we tried to optimize the material to be effective under as low laser beam power as possible while maintaining light–heat transition efficacy. However, the primary and critical constraint associated with such materials lies in their potential toxicity to nearby cells, which can result from overheating.95,96
Author contributions
Daniel Rybak: conceptualization, validation, investigation (SEM, FTIR, photo-thermal characterization, drug releases, ESR tests), methodology, writing – original draft, writing – review & editing. Chiara Rinoldi: validation, investigation (in vitro antibacterial and cell studies), methodology, writing – original draft, writing – review & editing. Paweł Nakielski: investigation (mechanical and injectability tests), formal analysis, supervision, writing – review & editing. Jingtao Du: investigation, resources. M. A. Bayan Haghighat: investigation (stability tests), writing – review & editing. Seyed Shahrooz Zargarian: methodology, writing – review & editing. Michał Pruchniewski: investigation (DLS), resources. Xiaoran Li: investigation, writing – review & editing, resources, supervision. Barbara Strojny-Cieślak: resources, writing – review & editing, supervision. Bin Ding: supervision, review & editing, project administration, funding acquisition. Filippo Pierini: conceptualization, writing – review & editing, supervision, formal analysis, project administration, funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Center for Research and Development (NCBiR) (WPC2/NanoHealer/2021) and the Ministry of Science and Technology of China (2021YFE0105100). The authors kindly thank Syed Ahmed Shah (Institute of Fundamental Technological Research, Polish Academy of Sciences, IPPT PAN, Warsaw, Poland) for assistance in the hydrogel preparation.
References
- M. Aragona, S. Dekoninck, S. Rulands, S. Lenglez, G. Mascré, B. D. Simons and C. Blanpain, Nat. Commun., 2017, 8, 14684 CrossRef PubMed
 .
.
- H. Li, B. Li, D. Lv, W. Li, Y. Lu and G. Luo, Adv. Drug Delivery Rev., 2023, 196, 114778 CrossRef CAS PubMed .
- Y. Liang, Y. Liang, H. Zhang and B. Guo, Asian J. Pharm. Sci., 2022, 17, 353–384 CrossRef PubMed .
- G. C. Gurtner, S. Werner, Y. Barrandon and M. T. Longaker, Nature, 2008, 453, 314–321 CrossRef CAS PubMed .
- R. Dong and B. Guo, Nano Today, 2021, 41, 101290 CrossRef CAS .
- S. Saghazadeh, C. Rinoldi, M. Schot, S. S. Kashaf, F. Sharifi, E. Jalilian, K. Nuutila, G. Giatsidis, P. Mostafalu, H. Derakhshandeh, K. Yue, W. Swieszkowski, A. Memic, A. Tamayol and A. Khademhosseini, Adv. Drug Delivery Rev., 2018, 127, 138–166 CrossRef CAS PubMed .
- B. K. Sun, Z. Siprashvili and P. A. Khavari, Science, 2014, 346, 941–945 CrossRef CAS PubMed .
- N. S. Gasek, G. A. Kuchel, J. L. Kirkland and M. Xu, Nat. Aging, 2021, 1, 870–879 CrossRef PubMed .
- Z. Xu, B. Liang, J. Tian and J. Wu, Biomater. Sci., 2021, 9, 4388–4409 RSC .
- Y. Chen, J. Cai, D. Liu, S. Liu, D. Lei, L. Zheng, Q. Wei and M. Gao, Regener. Biomater., 2022, 9, rbac019 CrossRef CAS PubMed .
- K. Kaur, S. Reddy, P. Barathe, V. Shriram, U. Anand, J. Proćków and V. Kumar, Front. Microbiol., 2021, 12, 747019 CrossRef PubMed .
- H. Kim, Y. R. Lee, H. Jeong, J. Lee, X. Wu, H. Li and J. Yoon, Smart Molecules, 2023, 1, e20220010, DOI:10.1002/smo.20220010 .
- K. Yang, S. Zhao, B. Li, B. Wang, M. Lan and X. Song, Coord. Chem. Rev., 2022, 454, 214330 CrossRef CAS .
- R. Narayanaswamy and V. P. Torchilin, Molecules, 2019, 24, 603, DOI:10.3390/molecules24030603 .
- W. Hu, Z. Wang, Y. Xiao, S. Zhang and J. Wang, Biomater. Sci., 2019, 7, 843–855 RSC .
- H. Deng, A. Dong, J. Song and X. Chen, J. Controlled Release, 2019, 297, 60–70 CrossRef CAS PubMed .
- R. Fan, Y. Cheng, R. Wang, T. Zhang, H. Zhang, J. Li, S. Song and A. Zheng, Polymers, 2022, 14, 2379, DOI:10.3390/polym14122379 .
- D. R. Devi, P. Sandhya and B. N. V. Hari, J. Pharm. Sci. Res., 2013, 5, 159–165 Search PubMed .
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. McAuley, Adv. Funct. Mater., 2021, 31, 2008123 CrossRef CAS .
- S. A. Shah, M. Sohail, M. U. Minhas, S. Khan, Z. Hussain, A. Mahmood, M. Kousar, H. E. Thu, M. Abbasi and M. U. R. Kashif, Int. J. Biol. Macromol., 2021, 185, 350–368 CrossRef CAS PubMed .
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS PubMed .
- Y. Xu, Q. Hu, Z. Wei, Y. Ou, Y. Cao, H. Zhou, M. Wang, K. Yu and B. Liang, Biomater. Res., 2023, 27, 36 CrossRef PubMed .
- B. Shriky, A. Kelly, M. Isreb, M. Babenko, N. Mahmoudi, S. Rogers, O. Shebanova, T. Snow and T. Gough, J. Colloid Interface Sci., 2020, 565, 119–130 CrossRef CAS PubMed .
- P. Nakielski, C. Rinoldi, M. Pruchniewski, S. Pawłowska, M. Gazińska, B. Strojny, D. Rybak, K. Jezierska-Woźniak, O. Urbanek, P. Denis, E. Sinderewicz, W. Czelejewska, J. Staszkiewicz-Chodor, M. Grodzik, Y. Ziai, M. Barczewska, W. Maksymowicz and F. Pierini, Small, 2022, 18, e2104971 CrossRef PubMed .
- J. Jang, J. Lee, Y.-J. Seol, Y. H. Jeong and D.-W. Cho, Composites, Part B, 2013, 45, 1216–1221 CrossRef CAS .
- S. K. Boda, S. Chen, K. Chu, H. J. Kim and J. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 25069–25079 CrossRef CAS PubMed .
- F. Mohabatpour, A. Karkhaneh and A. M. Sharifi, RSC Adv., 2016, 6, 83135–83145 RSC .
- Y. Shen, Y. Xu, B. Yi, X. Wang, H. Tang, C. Chen and Y. Zhang, Biomacromolecules, 2021, 22, 2284–2297 CrossRef CAS PubMed .
- T.-S. Jang, H.-D. Jung, H. M. Pan, W. T. Han, S. Chen and J. Song, Int. J. Bioprint., 2018, 4, 126 CrossRef PubMed .
- N. Han, J. Johnson, J. J. Lannutti and J. O. Winter, J. Controlled Release, 2012, 158, 165–170 CrossRef CAS PubMed .
- M. Wang, J. Du, M. Li, F. Pierini, X. Li, J. Yu and B. Ding, Biomater. Sci., 2023, 2383–2394, 10.1039/d2bm01891h .
- S. A. Eming, P. Martin and M. Tomic-Canic, Sci. Transl. Med., 2014, 6, 265sr6 Search PubMed .
- N. Halfter, E. Espinosa-Cano, G. M. Pontes-Quero, R. A. Ramírez-Jiménez, C. Heinemann, S. Möller, M. Schnabelrauch, H.-P. Wiesmann, V. Hintze and M. R. Aguilar, J. Funct. Biomater., 2023, 14, 160, DOI:10.3390/jfb14030160 .
- H. S. Kim, X. Sun, J.-H. Lee, H.-W. Kim, X. Fu and K. W. Leong, Adv. Drug Delivery Rev., 2019, 146, 209–239 CrossRef CAS PubMed .
- C. Huang, L. Dong, B. Zhao, Y. Lu, S. Huang, Z. Yuan, G. Luo, Y. Xu and W. Qian, Clin. Transl. Med., 2022, 12, e1094 CrossRef CAS PubMed .
- X. Tong, W. Pan, T. Su, M. Zhang, W. Dong and X. Qi, React. Funct. Polym., 2020, 148, 104501 CrossRef CAS .
- K. Liguori, I. Keenum, B. C. Davis, J. Calarco, E. Milligan, V. J. Harwood and A. Pruden, Environ. Sci. Technol., 2022, 56, 9149–9160 CrossRef CAS PubMed .
- M. A. Haghighat Bayan, Y. J. Dias, C. Rinoldi, P. Nakielski, D. Rybak, Y. B. Truong, A. L. Yarin and F. Pierini, J. Polym. Sci., 2023, 1–13 Search PubMed .
- X. Qi, Y. Huang, S. You, Y. Xiang, E. Cai, R. Mao, W. Pan, X. Tong, W. Dong, F. Ye and J. Shen, Adv. Sci., 2022, 9, e2106015 CrossRef PubMed .
- H.-Y. Chou, C.-C. Weng, J.-Y. Lai, S.-Y. Lin and H.-C. Tsai, Polymers, 2020, 12, 2138, DOI:10.3390/polym12092138 .
- F. Bernsmann, V. Ball, F. Addiego, A. Ponche, M. Michel, J. J. de, A. Gracio, V. Toniazzo and D. Ruch, Langmuir, 2011, 27, 2819–2825 CrossRef CAS PubMed .
- I. R. Schmolka, J. Biomed. Mater. Res., 1972, 6, 571–582 CrossRef CAS PubMed .
- S. Gurpreet, K. Tanurajvir, K. Ravinder and K. Anudeep, Int. J. Pharmacol. Pharm. Sci., 2014, 1(2), 30–42 Search PubMed .
- K. Park, A. Otte, F. Sharifi, J. Garner, S. Skidmore, H. Park, Y. K. Jhon, B. Qin and Y. Wang, Mol. Pharmaceutics, 2021, 18, 18–32 CrossRef CAS PubMed .
- U. A. Shakil, S. B. Abu Hassan, M. Y. Yahya and M. R. M. Rejab, Nanotechnol. Rev., 2022, 11, 1991–2014 CrossRef CAS .
- H.-Y. Wang, Y.-Y. Chen and Y.-Q. Zhang, Mater. Sci. Eng., C, 2015, 48, 444–452 CrossRef CAS PubMed .
- B. Niemczyk-Soczynska, J. Dulnik, O. Jeznach, D. Kolbuk and P. Sajkiewicz, Micron, 2021, 145, 103066 CrossRef CAS PubMed .
- A. G. B. Castro, M. C. Lo Giudice, T. Vermonden, S. C. G. Leeuwenburgh, J. A. Jansen, J. J. J. P. van den Beucken and F. Yang, ACS Biomater. Sci. Eng., 2016, 2, 2099–2107 CrossRef CAS PubMed .
- G. Liu and K. McEnnis, Polymers, 2022, 14, 993, DOI:10.3390/polym14050993 .
- S. Lappe, D. Mulac and K. Langer, Int. J. Pharm., 2017, 517, 338–347 CrossRef CAS PubMed .
- V. Lassalle, G. B. Galland and M. L. Ferreira, Bioprocess Biosyst. Eng., 2008, 31, 499–508 CrossRef CAS PubMed .
- R. Singh, P. Kesharwani, N. K. Mehra, S. Singh, S. Banerjee and N. K. Jain, Drug Dev. Ind. Pharm., 2015, 41, 1888–1901 CrossRef CAS PubMed .
- A. Wahab, G. M. Khan, M. Akhlaq, N. R. Khan, A. Hussain, M. F. Khan and H. Khan, Pharmazie, 2011, 66, 677–683 CAS .
- C.-C. Chen, C.-L. Fang, S. A. Al-Suwayeh, Y.-L. Leu and J.-Y. Fang, Int. J. Pharm., 2011, 415, 119–128 CrossRef CAS PubMed .
- J.-Y. Fang, S.-H. Hsu, Y.-L. Leu and J.-W. Hu, J. Biomater. Sci., Polym. Ed., 2009, 20, 1031–1047 CrossRef CAS PubMed .
- M. Shafique, M. Sohail, M. U. Minhas, T. Khaliq, M. Kousar, S. Khan, Z. Hussain, A. Mahmood, M. Abbasi, H. C. Aziz and S. A. Shah, Int. J. Biol. Macromol., 2021, 170, 207–221 CrossRef CAS PubMed .
- A. Zengin, J. P. O. Castro, P. Habibovic and S. H. van Rijt, Nanoscale, 2021, 13, 1144–1154 RSC .
- T. Wang, F. Zhang, R. Zhao, C. Wang, K. Hu, Y. Sun, C. Politis, A. Shavandi and L. Nie, Des. Monomers Polym., 2020, 23, 118–133 CrossRef CAS PubMed .
- M. A. Abd El-Ghaffar, M. S. Hashem, M. K. El-Awady and A. M. Rabie, Carbohydr. Polym., 2012, 89, 667–675 CrossRef CAS PubMed .
- J. Zhang, Q. Wang and A. Wang, Acta Biomater., 2010, 6, 445–454 CrossRef CAS PubMed .
- S. A. Shah, M. Sohail, S. A. Khan and M. Kousar, Mater. Sci. Eng., C, 2021, 126, 112169 CrossRef CAS PubMed .
- C. Branca, K. Khouzami, U. Wanderlingh and G. D’Angelo, J. Colloid Interface Sci., 2018, 517, 221–229 CrossRef CAS PubMed .
- Y.-S. Jung, W. Park, H. Park, D.-K. Lee and K. Na, Carbohydr. Polym., 2017, 156, 403–408 CrossRef CAS PubMed .
- E. Kushan and E. Senses, ACS Appl. Bio Mater., 2021, 4, 3507–3517 CrossRef CAS PubMed .
- A. Lupu, I. Rosca, V. R. Gradinaru and M. Bercea, Polymers, 2023, 15, 355, DOI:10.3390/polym15020355 .
- X. Li, E. Park, K. Hyun, L. Oktavia and M. Kwak, J. Rheol., 2018, 62, 107–120 CrossRef CAS .
- A. S. Giz, M. Berberoglu, S. Bener, S. Aydelik-Ayazoglu, H. Bayraktar, B. E. Alaca and H. Catalgil-Giz, Int. J. Biol. Macromol., 2020, 148, 49–55 CrossRef CAS PubMed .
- G. Liling, Z. Di, X. Jiachao, G. Xin, F. Xiaoting and Z. Qing, Carbohydr. Polym., 2016, 136, 259–265 CrossRef .
- Y. Yang, M. Li, G. Pan, J. Chen and B. Guo, Adv. Funct. Mater., 2023, 33, 2213089, DOI:10.1002/adfm.202214089 .
- C. Shao, H. Chang, M. Wang, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 28305–28318 CrossRef CAS PubMed .
- M. Li, Y. Dong, M. Wang, X. Lu, X. Li, J. Yu and B. Ding, Composites, Part B, 2023, 257, 110672 CrossRef CAS .
- L. Xu, Y. Chen, P. Zhang, J. Tang, Y. Xue, H. Luo, R. Dai, J. Jin and J. Liu, Biomater. Sci., 2022, 10, 5648–5661 RSC .
- S. Lü, C. Gao, X. Xu, X. Bai, H. Duan, N. Gao, C. Feng, Y. Xiong and M. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 13029–13037 CrossRef .
- H. Chen, R. Cheng, X. Zhao, Y. Zhang, A. Tam, Y. Yan, H. Shen, Y. S. Zhang, J. Qi, Y. Feng, L. Liu, G. Pan, W. Cui and L. Deng, NPG Asia Mater., 2019, 11, 3 CrossRef CAS .
- S. Maiz-Fernández, L. Pérez-Álvarez, L. Ruiz-Rubio, J. L. Vilas-Vilela and S. Lanceros-Mendez, Polymers, 2020, 12, 2261, DOI:10.3390/polym12102261 .
- A. Lupu, L. M. Gradinaru, D. Rusu and M. Bercea, Gels, 2023, 9, 719, DOI:10.3390/gels9090719 .
- M. H. Chen, L. L. Wang, J. J. Chung, Y.-H. Kim, P. Atluri and J. A. Burdick, ACS Biomater. Sci. Eng., 2017, 3, 3146–3160 CrossRef CAS PubMed .
- M. Li, R. Guo, J. Wei, M. Deng, J. Li, Y. Tao, M. Li and Q. He, Acta Biomater., 2021, 136, 546–557 CrossRef CAS PubMed .
- A. Carmignani, M. Battaglini, E. Sinibaldi, A. Marino, V. Vighetto, V. Cauda and G. Ciofani, ACS Appl. Nano Mater., 2022, 5, 1702–1713 CrossRef CAS .
- P. Nakielski, S. Pawłowska, C. Rinoldi, Y. Ziai, L. De Sio, O. Urbanek, K. Zembrzycki, M. Pruchniewski, M. Lanzi, E. Salatelli, A. Calogero, T. A. Kowalewski, A. L. Yarin and F. Pierini, ACS Appl. Mater. Interfaces, 2020, 12, 54328–54342 CrossRef CAS PubMed .
- Y. Zhou, H. Ye, Y. Chen, R. Zhu and L. Yin, Biomacromolecules, 2018, 19, 1840–1857 CrossRef CAS PubMed .
- Z. Khatun, M. Nurunnabi, M. Nafiujjaman, G. R. Reeck, H. A. Khan, K. J. Cho and Y. Lee, Nanoscale, 2015, 7, 10680–10689 RSC .
- D. Rybak, Y.-C. Su, Y. Li, B. Ding, X. Lv, Z. Li, Y.-C. Yeh, P. Nakielski, C. Rinoldi, F. Pierini and J. M. Dodda, Nanoscale, 2023, 15, 8044–8083 RSC .
- P. Wang, B. Chen, Y. Zhan, L. Wang, J. Luo, J. Xu, L. Zhan, Z. Li, Y. Liu and J. Wei, Pharmaceutics, 2022, 14, 2279, DOI:10.3390/pharmaceutics14112279 .
- S. Kaur, R. Tambat, V. Pathania, H. Nandanwar and S. Soni, J. Appl. Microbiol., 2022, 133, 569–578 CrossRef CAS .
- L. Shao, S. Majumder, Z. Liu, K. Xu, R. Dai and S. George, J. Photochem. Photobiol., B, 2022, 231, 112450 CrossRef CAS PubMed .
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L.-T. Weng, J. Xu, J. Weng, Y. Liu, F. Ren and X. Lu, NPG Asia Mater., 2017, 9, e372–e372 CrossRef CAS .
- Z. Guo, Z. Zhang, N. Zhang, W. Gao, J. Li, Y. Pu, B. He and J. Xie, Bioact. Mater., 2022, 15, 203–213 CAS .
- A. Omidinia-Anarkoli, S. Boesveld, U. Tuvshindorj, J. C. Rose, T. Haraszti and L. De Laporte, Small, 2017, 13, 1702207, DOI:10.1002/smll.201702207 .
- A. Singh and A. K. Dubey, ACS Appl. Bio Mater., 2018, 1, 3–20 CrossRef CAS .
- A. Oliveira, J. C. Sousa, A. C. Silva, L. D. R. Melo and S. Sillankorva, Front. Microbiol., 2018, 9, 1725 CrossRef .
- K. Turcheniuk, C.-H. Hage, J. Spadavecchia, A. Y. Serrano, I. Larroulet, A. Pesquera, A. Zurutuza, M. G. Pisfil, L. Héliot, J. Boukaert, R. Boukherroub and S. Szunerits, J. Mater. Chem. B, 2015, 3, 375–386 RSC .
- B. Guo, Y. Liang and R. Dong, Nat. Protoc., 2023, 18, 3322–3354 CrossRef CAS PubMed .
- S. Duan, R. Wu, Y.-H. Xiong, H.-M. Ren, C. Lei, Y.-Q. Zhao, X.-Y. Zhang and F.-J. Xu, Prog. Mater. Sci., 2022, 125, 100887 CrossRef CAS .
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, e1807333 CrossRef PubMed .
- M. Hippler, E. Blasco, J. Qu, M. Tanaka, C. Barner-Kowollik, M. Wegener and M. Bastmeyer, Nat. Commun., 2019, 10, 232 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2024 |

 a,
Jingtao
Du
b,
Mohammad Ali
Haghighat Bayan
a,
Jingtao
Du
b,
Mohammad Ali
Haghighat Bayan