
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamic driving forces for autoreduction of Cu sites in the zeolite SSZ-13
Daniel J.
Hutton
,
David H.
Lopez
and
Florian
Göltl
 *
*
The University of Arizona, Department of Biosystems Engineering, 1177 E 4th Street, 85719 Tucson, AZ, USA. E-mail: fgoeltl@arizona.edu
First published on 15th March 2024
Abstract
Cu-exchanged zeolites are widely used redox catalysts and the oxidation state of Cu is crucial in understanding their performance over a wide range of applications. Interestingly, a fraction of Cu sites in zeolites is reported to reduce at high temperatures in the absence of a reducing agent and as of today a detailed understanding of this process is still missing. In this contribution, we use first principles-based phase diagrams to explore thermodynamic driving forces for the autoreduction of Cu sites in the zeolite SSZ-13. We find that mainly monovalent Cu(II)–OH sites anchored at well-separated Al atoms and to a lesser degree di-hydroxyl Cu dimers drive the autoreduction of Cu sites in the zeolite SSZ-13. Using these insights, we can reproduce experimental trends in autoreduction reported in the literature. This work gives detailed insights into the autoreduction of Cu sites in SSZ-13 and demonstrates that the nature of Cu sites in the zeolite SSZ-13 depends on the exact conditions the material is exposed to. Optimizing these reaction conditions might allow to improve the performance of Cu-exchanged zeolites over a wide range of applications.
Introduction
Metal-exchanged zeolites are often used catalysts for redox reactions1–6 and in particular their Cu-exchanged forms have drawn significant attention over the past years.7–15 Their main applications include the selective catalytic reduction of nitrous oxides in the presence of NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3,4,8,16–19 and the partial oxidation of methane7,9,10,20–23 and other saturated alkanes. One of the key features in this context is the ability of the Cu site to change its oxidation state. Under oxidizing conditions, Cu can be found in an oxidation state of +2, while oxidation states of +1 or in rare cases 0 have been reported24 under reducing conditions.
3,4,8,16–19 and the partial oxidation of methane7,9,10,20–23 and other saturated alkanes. One of the key features in this context is the ability of the Cu site to change its oxidation state. Under oxidizing conditions, Cu can be found in an oxidation state of +2, while oxidation states of +1 or in rare cases 0 have been reported24 under reducing conditions.
Typically, the change in oxidation state is associated with the presence of an oxidizing or reducing agent. However, in the literature changes in the oxidation state of Cu sites have also been reported for the exposure of Cu-exchanged zeolites to inert conditions during the so-called Cu-autoreduction.25–34 Here, a fraction of Cu sites changes their oxidation state from +2 to +1 upon exposure of the system to inert He at high temperature. So far, several factors, such as the zeolite framework structure, the Si/Al ratio, and the Cu loading, have been shown to impact the autoreduction of Cu sites.25 Additionally, a phenomenological model for the reaction mechanism, which includes (i) the loss of H2O from 2 Cu(II)–OH sites and (ii) the desorption of ½ O2, has been suggested.25,32 So far, these insights are purely experimentally based and a theoretical model for these processes is still missing.
A zeolite that has gathered significant attention over the past years is Cu-exchanged SSZ-13, a medium- to high-silica zeolite in the chabazite framework structure. This material has been introduced as the industrial catalyst for the selective catalytic reduction of nitrous oxides3,4,19,35 and shows potential for the partial oxidation of methane to methanol.20,21,36–38 For this system, multiple studies have investigated the nature of the active sites and it was initially believed that mainly single Cu atoms in oxidation states of either +1 or +2 exist.16,24,39–43 More recent work, however, indicates that, depending on the exact encountered conditions, different Cu oxo- and hydroxyl dimers, ranging from Cu2O over Cu2OH to Cu2(OH)2, can also be present inside this material.21,36,37
Many of these insights have been obtained from first principles calculations-based, complex phase diagrams16,36,44,45 for the conversion of methane to methanol during the stepwise partial methane oxidation. These phase diagrams rely on the premise that if a system is exposed to specific conditions for an extended period, all chemical reactions are sufficiently fast that the system can reach its thermodynamic ground state at the given conditions. This precondition is met for a stepwise process in partial methane oxidation, where the system stays under the same conditions for hours, but also during autoreduction of Cu sites, where the system is kept at specific conditions for an extended time. Phase diagrams could therefore be used to gain detailed insights into changes in active site distribution in Cu-SSZ-13 during autoreduction.
Herein, we use phase diagrams to explore the oxidation state of Cu sites in the zeolite SSZ-13 under various conditions. We explore the impact of Al arrangements for monomer and dimer anchoring on Cu oxidation state and relate these insights to the autoreduction of Cu sites in SSZ-13 observed in experimental measurements.
Methods
The system
In this work we focus on SSZ-13, a zeolite in the chabazite framework. The chabazite framework is the zeolite framework with the smallest primitive unit cell, which only contains 12 symmetrically equivalent tetrahedral sites (T-sites) linked by O atoms (see Fig. 1). More specifically, the primitive unit cell consists of double six-O-membered ring units (d6R), which are linked by four-O-membered rings. In between the d6R structures, a pore is formed which is connected to adjacent pores via eight-O-membered rings. The small unit cell size and the high symmetry makes the chabazite framework the ideal model system to understand zeolites.46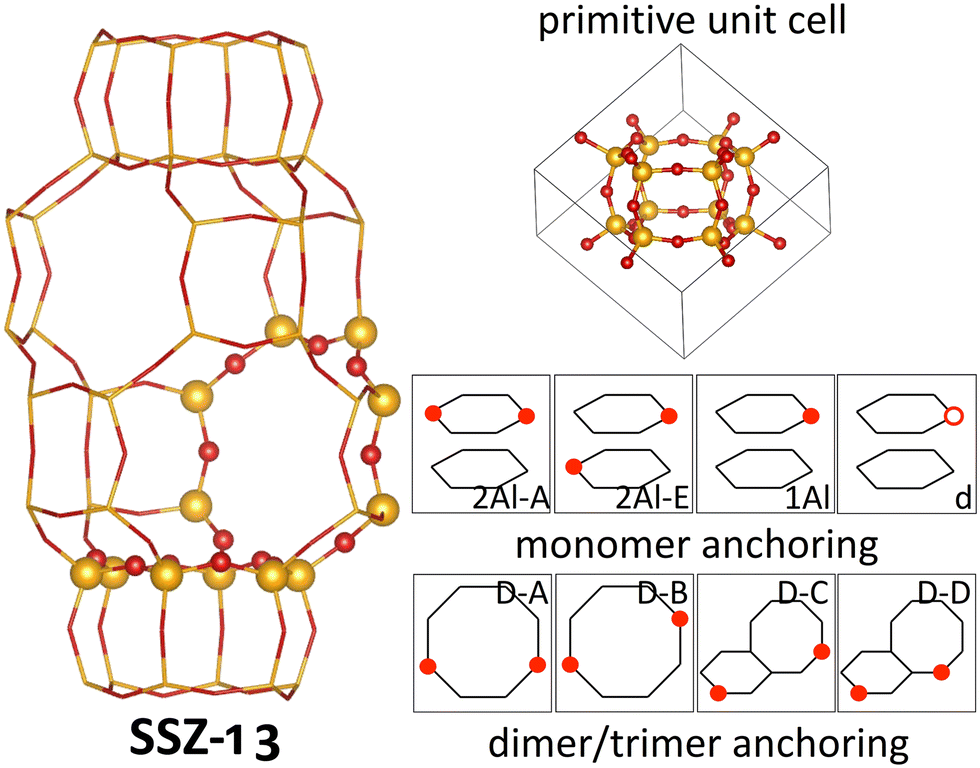 | ||
| Fig. 1 Main cavity (left) and primitive unit cell (right top) of the zeolite SSZ-13. In the atomistic representations, Si is shown in yellow, and O is shown in red. In the picture of the main cavity, the six- and eight-O-membered rings are highlighted, while the remaining framework is shown in a stick representation. Schematic representations of anchoring configurations for Cu monomers and dimers/trimers are shown in the bottom right. Here, Si(Al)–O bonds are shown as black lines, with Al positions marked by red circles. | ||
If all T-sites are occupied by Si, the system is chemically inert, but when T sites are occupied by Al, a local negative charge is generated which needs to be compensated by the presence of either a proton, another cation, or a larger, charged cluster. The exact nature of the exchanged cation depends on the anchoring configuration, i.e., the exact configuration of Al atoms surrounding it.
In the analysis in this work, we include four different Cu-monomer anchoring configurations,36,44,47 namely a configuration in which two Al atoms are on opposite sides of a six-O-membered ring (2Al-A), a configuration in which two Al atoms are positioned in different six-O-membered rings of the same primitive unit cell (2Al-E), and a configuration in which one Al atom is placed in one primitive unit cell (1Al). Additionally, we also include a silanol defect for monomer anchoring (d), which is obtained by removing one Si/Al atom from the framework and saturating the remaining bonds of four O atoms with H. These four monomer anchoring positions describe four different scenarios, namely a scenario in which Al atoms are paired in the same six-O-membered ring (2Al-A), a scenario in which Al atoms are close, but not in the same six-O-membered ring (2Al-E), a scenario in which all Al atoms are well separated (1Al), and a scenario in which framework defects are present in the material (d). All Al configurations are shown in Fig. 1. For these Al configurations, we add monovalent (Cu(I)) and divalent Cu monomers (Cu(II)) with up to six adsorbed water molecules. A schematic representation of the different Cu monomers before hydration is given in Scheme 1. Details on how these sites are generated and structural files for them are given in the literature.36,42,44 Upon water adsorption, the coordination of Cu to the framework in exchange positions 2Al-A, 2Al-E, and 1Al is reduced upon water adsorption until Cu sites are solvated and form a linear coordination to two water molecules for Cu(I) and a tetrahedral coordination to either four water molecules for 2Al-A and 2Al-E or three water molecules for the 1Al configuration. Additional water molecules form hydrogen bonds either with water coordinated to Cu or to Brønsted acid sites in the unit cell. These trends agree with other structures reported in the literature.16 For the defect site, Cu remains coordinated to the framework O atoms.
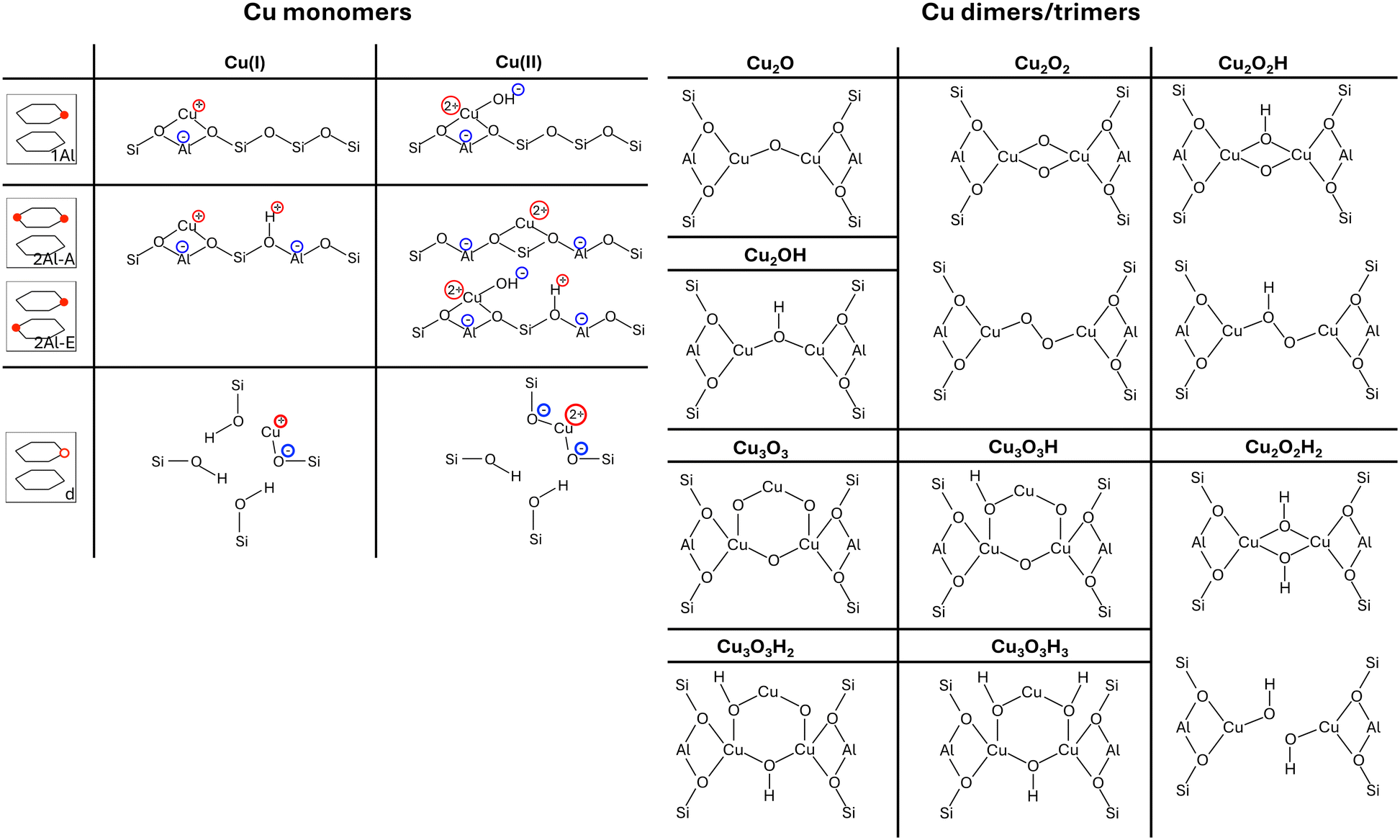 | ||
| Scheme 1 Schematic representation of all Cu(I) and Cu(II) monomers before hydration (left) and optimized dimer and trimer structures (right) included in our model. Monomer configurations are displayed for each exchange position, which is displayed schematically similar to Fig. 1. If multiple structures are displayed for the same dimer/trimer stoichiometry, the top structure is found for the D–A and D–B exchange position, while the bottom structure is found for the D–C and D–D exchange positions. | ||
Additionally, we consider four different Al configurations for the anchoring of dimer and trimer structures (D–A through D–D), two of which are in the eight-O-membered ring while the other two bridge a six-O-membered ring and an eight-O-membered ring. All configurations are displayed in Fig. 1. For these dimer exchange sites, we include Cu-oxo and Cu-hydroxyl dimers with one or two oxo or hydroxyl groups. For D–A and D–B, we also include Cu trimers with three oxo or hydroxyl groups. During optimization, different structural motifs were explored for all structures and structural files for these sites can be found in the literature.36 Furthermore, schematic representations of the different Cu dimers and trimers are shown in Scheme 1.
Throughout this work, we focus on calculations for a single, primitive, 12T chabazite unit cell, which shows a too high Al concentration (Si/Al = 5) compared to typical experimental measurements (12 < Si/Al < 20). However, past tests for dimers anchored in the D–C dimer exchange position have shown that these calculations reproduce results for a 24T chabazite unit cell (Si/Al = 11), within 0.06 eV.36
Phase diagrams
To compare the relative stability of different Cu sites, we will rely on phase diagrams. Here, we calculate the energy of each structure and extrapolate to the finite-temperature Gibbs free energies by adding zero-point energy corrections and vibrational, rotational, and translational entropies. Many of the Cu species have a different stoichiometry and we cannot directly compare their stabilities. We therefore calculate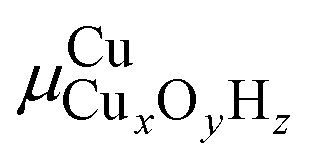 , the chemical potential of Cu for Cu site CuxOyHz, as36
, the chemical potential of Cu for Cu site CuxOyHz, as36for an Al configuration with two Al atoms in the primitive unit cell (2Al-A, 2Al-E, and all dimer anchoring configurations) and
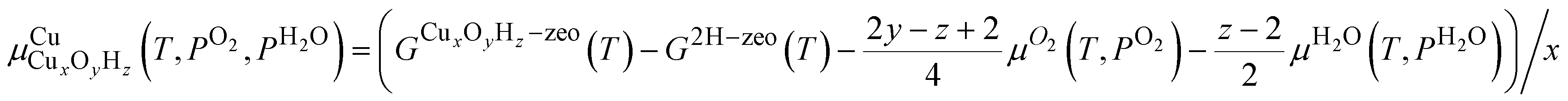
for the 1Al configuration, where only one Al atom is present in the unit cell. In these equations, GX refers to the Gibbs free energy of structure X, zeo is shorthand notation for the zeolite, which varies for each anchoring configuration (2Al-A, 2Al-E, 1Al, and d), and μO2 and μH2O refer to the chemical potentials of O2 and H2O in the gas phase, respectively. The gas phase chemical potentials furthermore depend on the gas phase pressures PO2 and PH2O, which are compared to a reference pressure P0 of one bar. Therefore, μCu is dependent on the temperature T as well as the gas phase pressures of O2 (PO2) and H2O (PH2O).
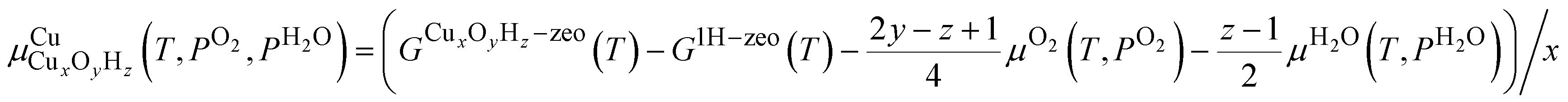
Overall, the most stable Cu configuration will always be associated with the lowest μCu. However, in a realistic zeolite a distribution of different anchoring points will be available. We will therefore study phase diagrams pairing one of the monomer exchange positions 2Al-A, 2Al-E, and 1Al with one dimer exchange position. To understand how the presence of framework defects impacts Cu site preference, we will study phase diagrams which include one monomer exchange position, the defect position, and, if explicitly mentioned, one dimer/trimer exchange position. If not stated otherwise, we will include all phase diagrams in the main text. More details considering the construction of phase diagrams can be found in the literature.36
Computational setup
Data used in this work was obtained using the Vienna Ab Initio Simulation Package (VASP),48,49a plane wave electronic structure code using PAW pseudopotentials.50,51 In all calculations we focused on modeling only the Γ point and we relied on a unit cell structure published in the literature52 and set unit cell volume to 830 Å. All structures were first optimized using the spin-polarized PBE-TS functional.53,54 For optimized structures, energies were calculated using RPA.55,56 Gibbs free energies were extrapolated to finite temperature based on the harmonic approximation from vibrational frequencies calculated at the PBE-TS level using Thermo.Pl.57 In this approach we assume that molecules lose all their translational entropy upon adsorption, but it has been suggested that water might only lose a fraction of translational entropy when binding to Cu in zeolites.16,58 Therefore, we expect that our approach underestimates the temperature for dehydration of Cu monomers. More details on the computational setup can be found in the literature.36Results and discussion
The goal of this work is to use phase diagrams to understand changes in the oxidation state of Cu sites under different conditions, such as Cu autoreduction. As discussed in the Methods section, the relative stability of the Cu sites depends on the temperature T and the gas phase pressures of O2 (PO2) and H2O (PH2O). Here, we will show PO2/T phase diagrams, but for each studied monomer/dimer/defect anchoring combination, we will show phase diagrams for three different water pressures, namely ln(PH2O/P0) values of −1, −8, and −15. These three scenarios correspond to exposure to water vapor (ln(PH2O/P0) = −1), an atmospheric water pressure (ln(PH2O/P0) = −8), and very low water pressure as could be encountered in a vacuum or helium atmosphere (ln(PH2O/P0) = −15).We begin our analysis with the oxidation states for phase diagrams where only Cu monomers are included (see Fig. 2). We immediately see that for 2Al-A, only Cu(II) sites are stabilized. Depending on the water pressure and temperature, Cu(II) coordinated to either five, one, or no water molecules are present, with a higher water coordination found at higher PH2O values and lower temperatures. For 2Al-E, we find that at low and high temperatures Cu(II) coordinated to five, three, or no H2O molecules are preferred. At an intermediate temperature range, however, Cu(I) coordinated to no H2O molecules are preferred. The temperature range where Cu(I) is found changes quite significantly between different H2O and O2 pressures. It can range between ~600 K and 1000 K for ln(PH2O/P0) = −1 to ∼325 K to ∼475 K for ln(PH2O/P0) = −15. We attribute the formation of Cu(I) at intermediate temperatures to the presence of only one Al atom per six ring. For the formation of Cu(II), the charges located at the framework and the Cu atom are separated, while the formation of Cu(I) requires the addition of a Brønsted proton. At intermediate temperatures and high PH2O, this will lead to the formation of Cu(I), while entropic penalties for the addition of a Brønsted proton will stabilize Cu(II) at higher temperatures. For 1Al, mainly Cu(I) coordinated to two, one or no H2O is preferred and only at high PO2, a small region of Cu(II)OH is preferred. This region shifts with PH2O and can be centered around 1100 K for ln(PH2O/P0) = −1 or around 500 K for ln(PH2O/P0) = −15.
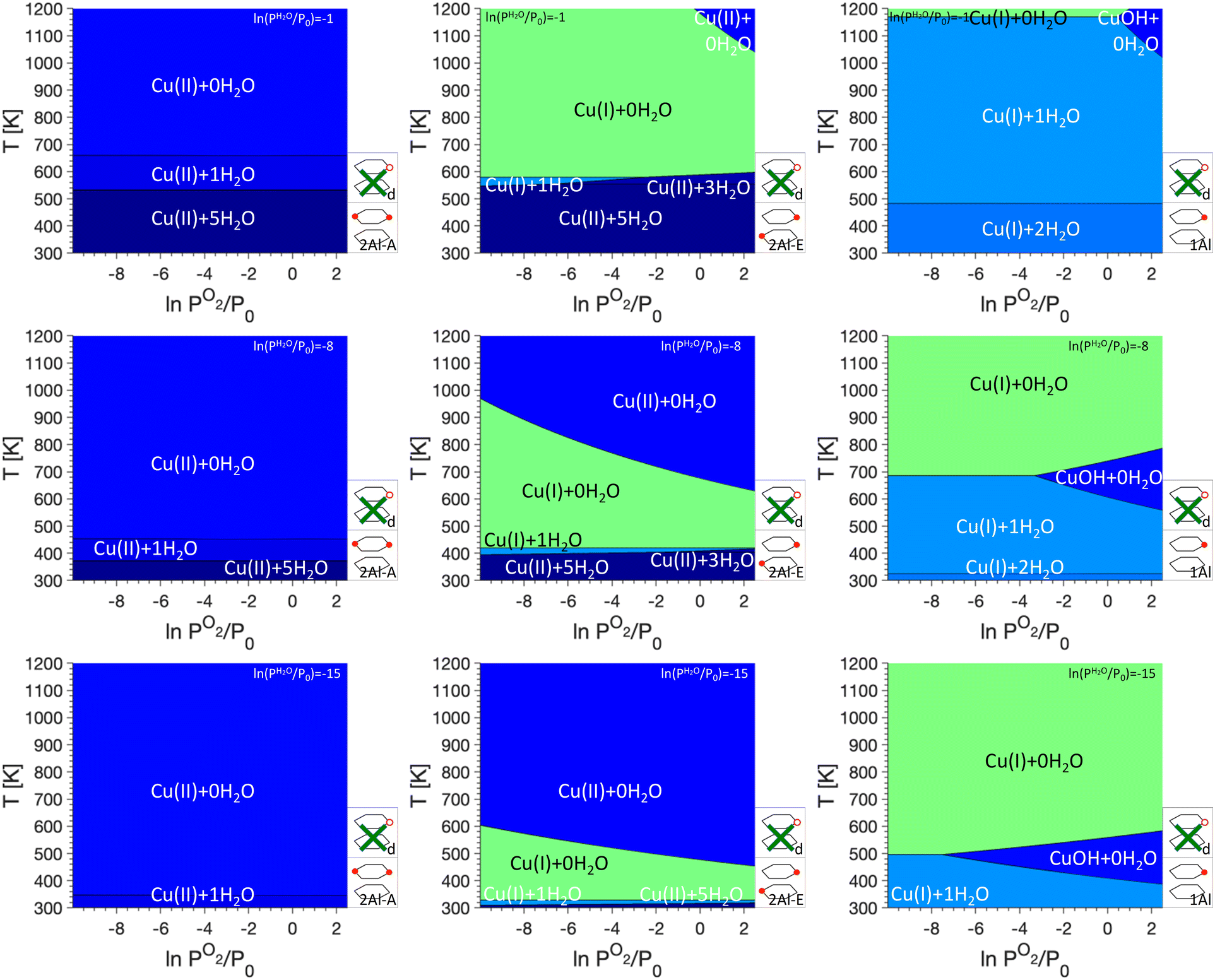 | ||
| Fig. 2 P O2/T phase diagrams for monomers anchored in 2Al-A (left), 2Al-E (middle), and 1Al at different PH2O values. Regions of uniform color correspond to the site indicated by the label. Schematics of the included anchoring configurations are shown in the bottom right, and green crosses indicate anchoring configurations that are not included. PH2O values are given in the legend on the top right. | ||
The introduction of framework defects by including monomer exchange positions and silanol defects for Cu anchoring in our model (see Fig. 3). This does not change the preferred Cu sites for 2Al-A. For 2Al-E and 1Al, however, the preferred sites change significantly, specifically at low PH2O. In both cases, a preference of Cu(II) that is not coordinated to water and anchored at a defect, is first seen at ln(PH2O/P0) = −8 and ln(PO2/P0) > 0 for 2Al-E and ln(PO2/P0) > −4 for 1Al, respectively. For 2Al-E, defect anchored Cu is found at intermediate temperatures of ∼600 K, while for 1Al, this Cu species is preferred at temperatures above 800 K. For ln(PH2O/P0) = −15, defect anchored Cu makes up a significant portion of the phase diagrams and for both anchoring configurations, almost the entire region where Cu(I) + 0H2O is preferred is replaced by a defect anchored Cu species.
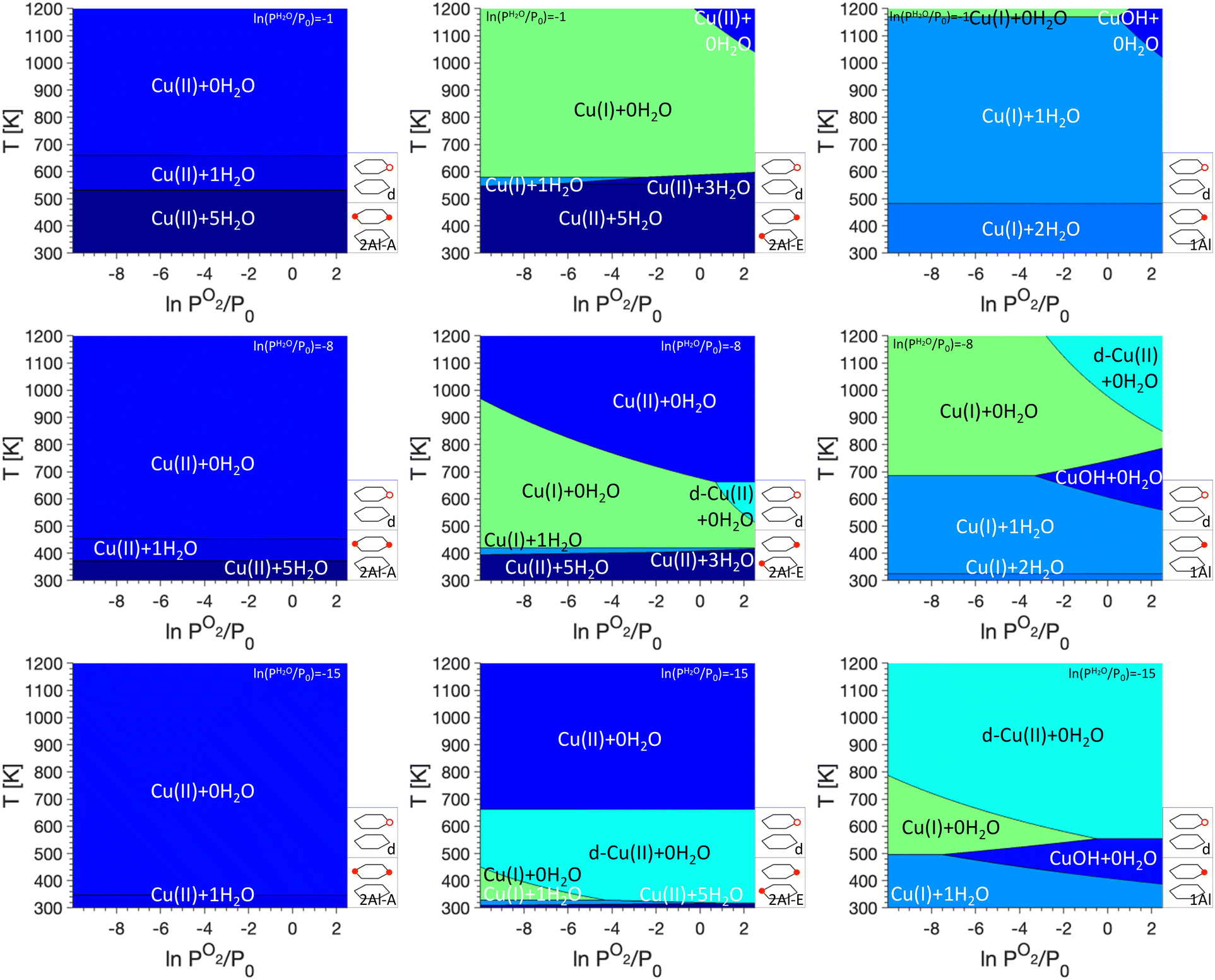 | ||
| Fig. 3 P O2/T phase diagrams for monomers anchored in 2Al-A (left), 2Al-E (middle), and 1Al with defects included at different PH2O values. Regions of uniform color correspond to the site indicated by the label. Schematics of the included anchoring configurations are shown in the bottom right and PH2O values are given in the legend on the top right. | ||
Next, we add dimer anchoring configurations in our analysis (Fig. 4). For 2Al-A, a small region in the phase diagrams shows preference for either Cu2O2H2 (D–A and D–B) or Cu2OH (D–A), which shifts to lower temperatures for lower PH2O. For D–C and D–D, dimers are never most stable. For 2Al-E, regions where dimers are preferred increase significantly and at high PH2O Cu(II) coordinated to five H2O is preferred at low temperature, while at lower PH2O Cu(II) without adsorbed water is found at high temperatures. For 1Al, the preference for Cu monomers almost completely disappears for D–A and D–B, while Cu(I) coordinated to one or two water molecules is present for D–C and D–D. Again, a preference for Cu(II) anchored to defects is seen at low PH2O. Besides the Cu dimers discussed in the literature, our phase diagrams also show a preference for Cu2O dimers at low PH2O for monomer anchoring configurations 1Al and to a lesser degree 2Al-E and dimer anchoring configurations D–B and D–D.
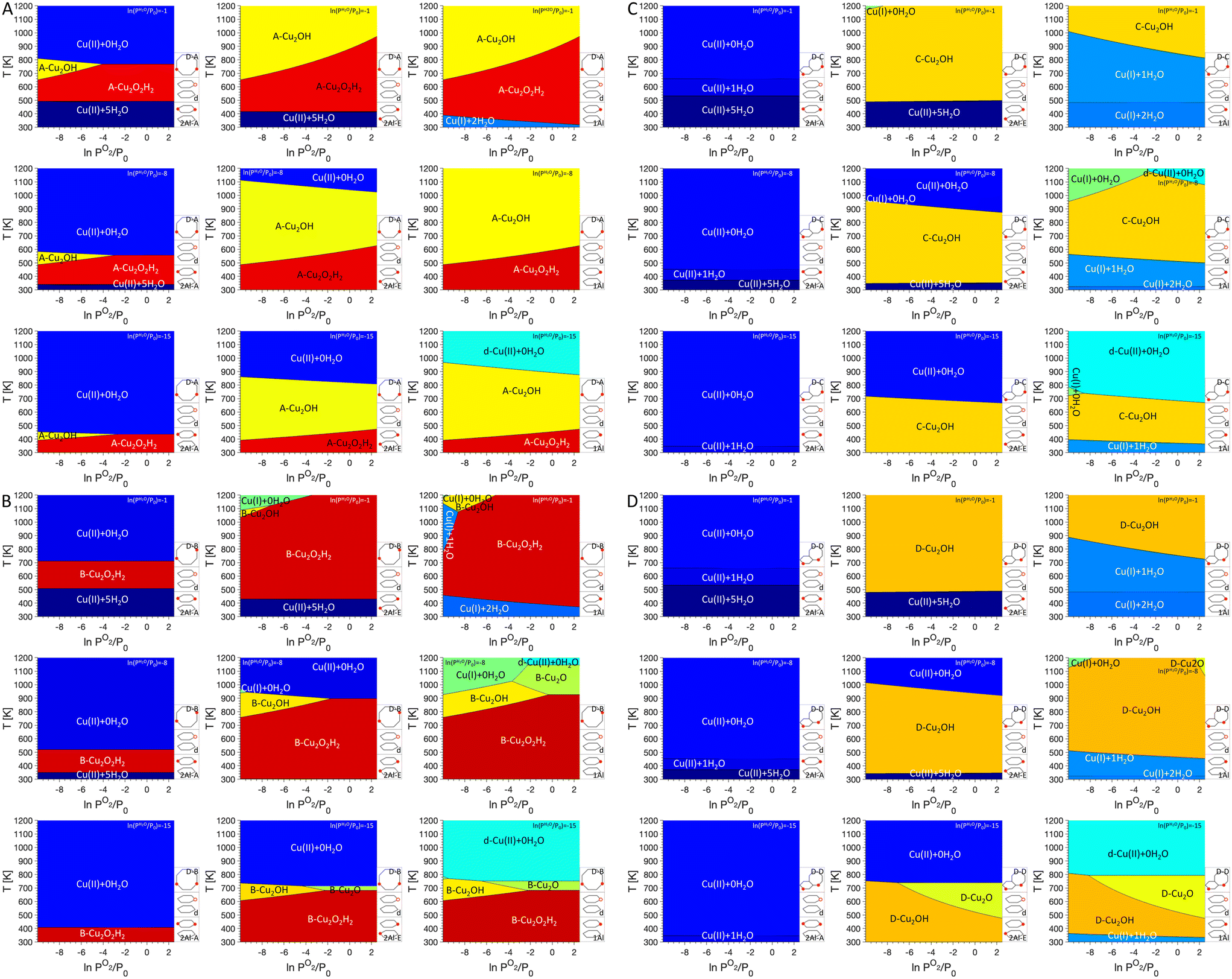 | ||
| Fig. 4 P O2/T phase diagrams for monomers anchored in 2Al-A (left), 2Al-E (middle), and 1Al and dimer/trimer anchoring configurations D–A (A), D–B (B), D–C (C), and D–D (D), with defects included at different PH2O values. Regions of uniform color correspond to the site indicated by the label. Schematics of the included anchoring configurations are shown in the bottom right and PH2O values are given in the legend on the top right. | ||
In this contribution we aim to understand how the charge state of Cu sites can change with different conditions. We want to assign a charge state to each of the preferred sites encountered in the phase diagrams. For Cu monomers, the situation is straightforward, Cu(I) is charged +1, while Cu(II) has a charge state of +2. Also, for the Cu2O2H2 sites a charge state of +2 can directly be assigned to each Cu atom. However, assigning an unambiguous charge state to Cu bound in Cu2OH sites is more complicated. This site is anchored at two Al atoms and contains one OH group. Therefore, the two Cu atoms should have a combined charge state of +3 or +1.5 per Cu atom. However, it is not clear if the charge is equally distributed over both Cu atoms or if one of the Cu atoms has a charge state of +2, while the other Cu has a charge state of +1. To resolve this problem, we study the charge distribution of the lowest unoccupied molecular orbital (LUMO) of the Cu2OH sites placed in all four dimer anchoring configurations (see Fig. 5). Analyzing these orbitals reveals that for D–A and D–B, the LUMO of the Cu2OH is delocalized over both Cu atoms. Here, it is not possible to unambiguously assign an integer charge state to each Cu atom and we will identify a [Cu2OH]2+ site. For D–C and D–D the situation is different. Here, the LUMO is almost exclusively located at the Cu atom in the six ring and therefore there is one Cu(II) atom located in the six-O-membered ring and one Cu(I) atom in the eight-O-membered ring.
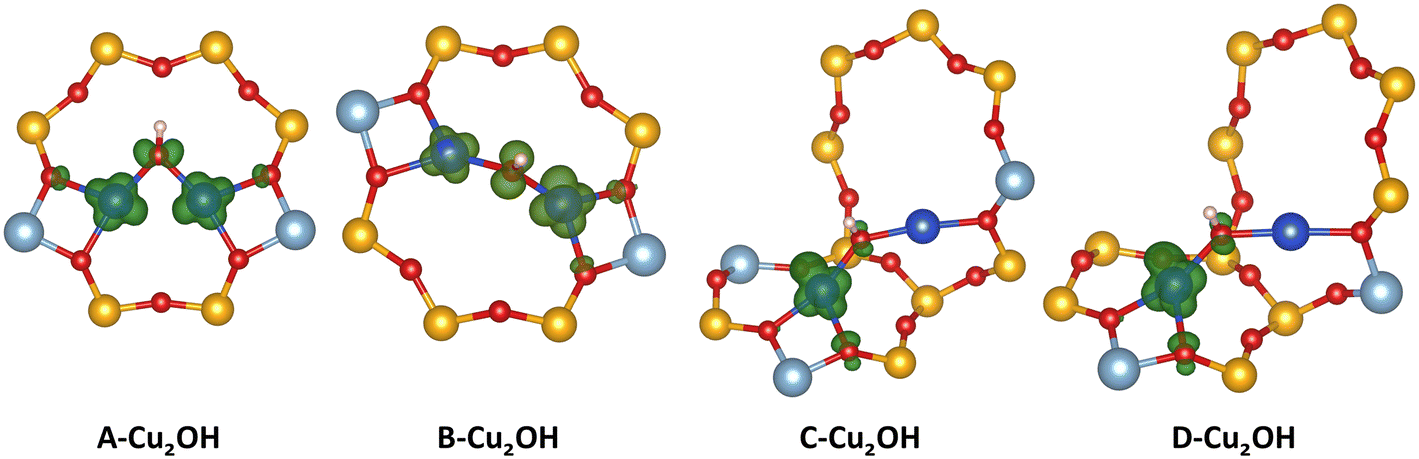 | ||
| Fig. 5 LUMO orbitals of Cu2OH sites in different dimer anchoring configurations. The LUMO charge isosurfaces are shown in green, Cu atoms in blue, Si atoms in yellow, O atoms in red, H atoms in white, and Al atoms in grey. | ||
Using this information, it is now possible to understand the changes in charge state based on the encountered conditions. Here, we will assume that the material is activated at 700 K for ln(PO2/P0) = 0 and ln(PH2O/P0) = −8. Subsequently, autoreduction is started at the same temperature but with a reduction in PO2 and PH2O to ln(PO2/P0) = −7 and ln(PH2O/P0) = −15. Such a reduction in pressure is consistent with exposing the system to a vacuum or a He atmosphere. Using these assumptions, we find that during activation, all monomers are in the +2 charge state, while dimers D–A, D–C, and D–D are occupied by Cu2OH sites and Cu2O2H2 is formed for D–B. Upon exposure to autoreduction conditions, only the 1Al monomers change charge state to +1, while the dimers anchored in D–B get reduced to Cu2OH. If defects are present in the material, Cu will migrate from the 1Al exchange position to the silanol defect and remain in the charge state +2.
Using these assumptions, we can now study several examples of hypothetical Cu-exchanged SSZ-13 samples (see Fig. 6). For all examples, we assume a distribution of exchange positions is available for Cu. Examples 1 and 2 are based on differences in high Si/Al ratio zeolites, which are synthesized with or without framework defects,44,59 examples 3 and 4 explore the impact of Al pairing for SSZ-13 samples with Si/Al = 15,60–62 while example 5 focuses on a zeolite with Si/Al = 8. We constructed these examples using the following assumptions: (i) the number of unit cells that contain two Al atoms will be lower at higher Si/Al ratios, (ii) if Al pairs in one six ring exist, they correspond to the 2Al-A configuration,62,63 and (iii) there is maximum ion exchange and there must be enough Al atoms in the framework to host all Cu during ion exchange (Cu/Al = 0.5). The distributions were then constructed in a way to match available experimental data.
 | ||
| Fig. 6 Five different examples for changes in charge states of hypothetical Cu-exchanged SSZ-13 samples upon autoreduction. In pie charts, Cu(II) is shown in red, Cu(I) in blue, and [Cu2OH]2+ in green. | ||
In example 1 we focus on a SSZ-13 zeolite with a Si/Al ratio of 70 without defects. For such a zeolite, we assume that all Al is well separated and only the 1Al anchoring configuration and no dimer anchoring configurations are present. Here, initially all Cu is in a +2 charge state during activation, and all Cu is reduced with the drop in pressure. In example 2, we choose a zeolite similar to example 1 but include enough framework defects to host all Cu atoms. Now, no Cu can be reduced when PO2 and PH2O are lowered, but Cu–OH migrates from extraframework positions to defect sites and loses the hydroxyl group. In example 3, we focus on a zeolite sample with a Si/Al ratio of 15. Here, we assume that some Al is paired in the six ring and 20% of all Cu can be exchanged in the 2Al-A configuration. Another 20% of Cu can be exchanged with Al atoms that are close, but not paired in the same six ring, which in our model corresponds to ion exchange in 2Al-E. Each dimer exchange position is present and can exchange 5% of all Cu, and the remaining 40% of Cu are exchanged in 1Al. For this zeolite, initially 90% of all Cu are in a +2 charge state during activation, while 5% of all Cu are present as [Cu2OH]2+ and Cu(I). Upon pressure reduction, we find a total of 45% as Cu(I) and Cu(II), with 10% being present as [Cu2OH]2+. In example 4, we focus on a zeolite with a Si/Al ratio of 15 but synthesized without Al pairs.60 Here, the exchange site distribution is comparable to example 3, but the 2Al-A sites are absent and 2Al-E and 1Al are increased by 10% compared to example 3. Accordingly, 10% more of the Cu can be reduced upon pressure reduction. In example 5 we focus on a zeolite with a Si/Al ratio of 8. Here, most of the Al is present in paired exchange positions, and we choose Cu in 2Al-A = 25% and Cu in 2Al-E = 25%. Additionally, all dimer exchange sites are present with 10%, which leaves 10% for Cu in the 1Al exchange position. For this material we find a Cu distribution of 80% Cu(II), 10% Cu(I), and 10% [Cu2OH]2+ after activation. When the pressure is lowered, only 10% of all sites are reduced from Cu(II) to Cu(I) and 10% of all sites are reduced from Cu(II) to [Cu2OH]2+.
These examples illustrate that our model predicts a significant impact of materials parameters on autoreduction. First, we find that lowering the Si/Al ratio reduces the amount of Cu sites that can be reduced during autoreduction. We also find that synthesis parameters can impact autoreduction in two ways, namely (i) silanol defects can be created, which are detrimental to autoreduction, and (ii) Al atoms can pair up in six rings, which again lowers the amount of Cu that can be reduced by a pressure change. In the previous paragraph we only studied examples for full ion exchange, where all exchange sites were occupied by Cu. When ion exchange levels are lowered, the least stable exchange positions will not be filled. In our case this is the 1Al position, which mainly contributes to autoreduction. Our model therefore predicts that fewer Cu atoms will be autoreduced for zeolites with lower ion exchange levels. The predictions of our model show excellent agreement with experimental measurements, which have shown that a smaller fraction of Cu atoms is autoreduced for zeolites with lower Si/Al ratios.25 However, our prediction of the impact of Cu exchange levels on autoreduction does not match experimental observations for the zeolite Mordenite. However, the Cu site distribution is expected to vary significantly with zeolite framework, which in turn will significantly impact changes upon exposure to autoreduction conditions. Additionally, the impact of synthesis protocol (i.e., the presence of defects) on autoreduction has been little studied so far and still requires experimental confirmation.
The work performed here also allows us to address one aspect that has been extensively discussed in the literature, namely the nature of the Cu dimers present in the zeolite SSZ-13 after activation in O2 for partial methane oxidation. In previous work, phase diagrams showed that for activation at 773 K hydroxylated dimers are present36 which were later characterized via UV-vis spectroscopy.37 At the same time, after activation in O2 at 873 K Rhoda et al. identified a Cu2O site which is active for methane activation.21 Here we again use phase diagrams to rationalize these different observations and a comparison of the four dimer sites is shown in Fig. 7. Since we are interested in changes for sites anchored in the different dimer exchange positions, 1Al was chosen as reference for Cu monomers and defects were not considered in the phase diagrams. In the specific activation procedure used by Rhoda et al.21 the material is first exposed to a He atmosphere at 973 K. This approach should allow for a thorough dehydration of the material, and we assume that the material will show a ln(PH2O/P0) of −15. Subsequently, the system is cooled to 823 K and exposed to O2, which we assume to be at ln(PO2/P0) of 0. As can be seen in Fig. 7, for D–B, D–C, and D–D, Cu2O sites are preferred and only for D–A, Cu2OH is found at the given conditions. In a conventional activation approach, the material is heated to temperatures between 723 K and 773 K and exposed to O2 for extended periods of time. A dehydration step is missing, and we therefore assume that this will lead to a ln(PH2O/P0) of −8. Under these conditions, Cu2OH is preferred for D–A, D–C, and D–D, while Cu2O2H2 is preferred for D–B.
 | ||
| Fig. 7 P O2/T phase diagrams for monomers anchored in 1Al and dimer/trimer anchoring configurations D–A, D–B, D–C, and D–D, without defects at different PH2O values. Regions of uniform color correspond to the site indicated by the label. Schematics of the included anchoring configurations are shown in the bottom right and PH2O values are given in the legend on the top right. Activation conditions for typical activation (AC1) and an activation protocol suggested by Rhoda et al.21 (AC2) are marked by black circles. | ||
Conclusions
In this contribution, we used phase diagrams to investigate the changes in charge state of Cu atoms in SSZ-13 under different conditions. We investigated various Al configurations for Cu anchoring and found that specifically Cu(II)–OH sites associated with well-separated Al atoms undergo autoreduction upon a reduction in pressure at activation conditions. At the same time, specific hydroxylated dimers can undergo a reduction. Using these insights, we were then able to reproduce trends for autoreduction previously observed in the literature. Overall, this work demonstrates that Cu sites in zeolites are highly sensitive to the applied conditions and the optimization of activation and reaction conditions is necessary to arrive at active sites with optimal outcomes for a specific application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Florian Göltl thanks the College of Agriculture and Life Science at the University of Arizona for support. We acknowledge computational time at the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy, contract DE-AC02-05CH11231.References
- P. Vanelderen, J. Vancauwenbergh, B. F. Sels and R. A. Schoonheydt, Coordination Chemistry and Reactivity of Copper in Zeolites, Coord. Chem. Rev., 2013, 257(2), 483–494 CrossRef CAS.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Recent Advances in Automotive Catalysis for NOx Emission Control by Small-Pore Microporous Materials, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- C. Paolucci, J. R. Di Iorio, W. F. Schneider and R. Gounder, Solvation and Mobilization of Copper Active Sites in Zeolites by Ammonia: Consequences for the Catalytic Reduction of Nitrogen Oxides, Acc. Chem. Res., 2020, 53, 1881–1892 CrossRef CAS PubMed.
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, The Active Site of Low-Temperature Methane Hydroxylation in Iron-Containing Zeolites, Nature, 2016, 536, 317–321 CrossRef CAS PubMed.
- M. L. Bols, B. E. R. Snyder, H. M. Rhoda, P. Cnudde, G. Fayad, R. A. Schoonheydt, V. Van Speybroeck, E. I. Solomon and B. F. Sels, Coordination and Activation of Nitrous Oxide by Iron Zeolites, Nat. Catal., 2021, 4(4), 332–340 CrossRef CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. Bokhoven, Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Dynamic Multinuclear Sites Formed by Mobilized Copper Ions in NOx Selective Catalytic Reduction, Science, 2017, 5630, 1–11 Search PubMed.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, The Direct Catalytic Oxidation of Methane to Methanol—A Critical Assessment, Angew. Chem., Int. Ed., 2017, 56(52), 16464–16483 CrossRef CAS PubMed.
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. Van Bokhoven, Misconceptions and Chellenges in Methane-to-Methanol over Transition-Metal-Exchanged Zeolites, Nat. Catal., 2019, 2, 485–494 CrossRef CAS.
- S. H. Krishna, A. Goswami, Y. Wang, C. B. Jones, D. P. Dean, J. T. Miller, W. F. Schneider and R. Gounder, Influence of Framework Al Density in Chabazite Zeolites on Copper Ion Mobility and Reactivity during NOx Selective Catalytic Reduction with NH3, Nat. Catal., 2023, 6(3), 276–285 CrossRef CAS.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, Methane to Methanol: Structure-Activity Relationships for Cu-CHA, J. Am. Chem. Soc., 2017, 139(42), 14961–14975 CrossRef CAS PubMed.
- D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed.
- L. Tao, E. Khramenkova, I. Lee, T. Ikuno, R. Khare, A. Jentys, J. L. Fulton, A. A. Kolganov, E. A. Pidko, M. Sanchez-Sanchez and J. A. Lercher, Speciation and Reactivity Control of Cu-Oxo Clusters via Extraframework Al in Mordenite for Methane Oxidation, J. Am. Chem. Soc., 2023, 145(32), 17710–17719 CrossRef CAS PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, Catalysis in a Cage: Condition-Dependent Speciation and Dynamics of Exchanged Cu Cations in SSZ-13 Zeolites, J. Am. Chem. Soc., 2016, 138(18), 6028–6048 CrossRef CAS PubMed.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis, J. Am. Chem. Soc., 2017, 139(13), 4935–4942 CrossRef CAS PubMed.
- J. Becher, D. F. Sanchez, D. E. Doronkin, D. Zengel, D. M. Meira, S. Pascarelli, J.-D. Grunwaldt and T. L. Sheppard, Chemical Gradients in Automotive Cu-SSZ-13 Catalysts for NOx Removal Revealed by Operando X-Ray Spectrotomography, Nat. Catal., 2021, 4(1), 46–53 CrossRef CAS.
- I. Bull, W. M. Xue, P. Burk, R. S. Boorse, W. M. Jaglowski, G. S. Kroemer, A. Moini, J. A. Patchett, J. C. Dettling and M. T. Caudle, Copper CHA Zeolite Catalysts, US Pat., 7601662B2, 2009 Search PubMed.
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, Formation of [Cu2O2]2+ and [Cu2O]2+ toward C-H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39, ACS Catal., 2017, 7(7), 4291–4303 CrossRef CAS.
- H. M. Rhoda, D. Plessers, A. J. Heyer, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Spectroscopic Definition of a Highly Reactive Site in Cu-CHA for Selective Methane Oxidation: Tuning a Mono-μ-Oxo Dicopper (II) Active Site for Reactivity, J. Am. Chem. Soc., 2021, 143, 7531–7540 CrossRef CAS PubMed.
- A. J. Heyer, D. Plessers, A. Braun, H. M. Rhoda, M. L. Bols, B. Hedman, K. O. Hodgson, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Methane Activation by a Mononuclear Copper Active Site in the Zeolite Mordenite: Effect of Metal Nuclearity on Reactivity, J. Am. Chem. Soc., 2022, 144(42), 19305–19316 CrossRef CAS PubMed.
- A. A. Latimer, A. Kakekhani, A. R. Kulkarni and J. K. Nørskov, Direct Methane to Methanol: The Selectivity-Conversion Limit and Design Strategies, ACS Catal., 2018, 8(8), 6894–6907 CrossRef CAS.
- D. W. Fickel and R. F. Lobo, Copper Coordination in Cu-SSZ-13 and Cu-SSZ-16 Investigated by Variable-Temperature XRD, J. Phys. Chem. C, 2010, 114(3), 1633–1640 CrossRef CAS.
- V. L. Sushkevich, A. V. Smirnov and J. A. Van Bokhoven, Autoreduction of Copper in Zeolites: Role of Topology, Si/Al Ratio, and Copper Loading, J. Phys. Chem. C, 2019, 123(15), 9926–9934 CrossRef CAS.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Composition-Driven Cu-Speciation and Reducibility in Cu-CHA Zeolite Catalysts: A Multivariate XAS/FTIR Approach to Complexity, Chem. Sci., 2017, 8(10), 6836–6851 RSC.
- Y. Li and W. K. Hall, Catalytic Decomposition of Nitric Oxide over Cu-Zeolites, J. Catal., 1991, 129(1), 202–215 CrossRef CAS.
- W. Keith Hall and J. Valyon, Mechanism of NO Decomposition over Cu-ZSM-5, Catal. Lett., 1992, 15(3), 311–315 CrossRef.
- H.-J. Jang, W. K. Hall and J. L. d'Itri, Redox Behavior of CuZSM-5 Catalysts: FTIR Investigations of Reactions of Adsorbed NO and CO, J. Phys. Chem., 1996, 100(22), 9416–9420 CrossRef CAS.
- M. Iwamoto, H. Yahiro, K. Tanda, N. Mizuno, Y. Mine, S. Kagawa and S. Kagawat, Removal of Nitrogen Monoxide through a Novel Catalytic Process. 1. Decomposition on Excessively Copper-Ion-Exchanged ZSM-5 Zeolites, J. Phys. Chem., 1991, 95(9), 3727–3730 CrossRef CAS.
- Y. Kuroda and M. Iwamoto, Characterization of Cuprous Ion in High Silica Zeolites and Reaction Mechanisms of Catalytic NO Decomposition and Specific N2 Adsorption, Top. Catal., 2004, 28(1), 111–118 CrossRef CAS.
- D. K. Lee, Kinetic Evaluation of Mechanistic Models for O2 Release from ZSM-5-Supported [Cu2+–O–Cu2+] Ions by Thermal Reduction or Chemical Interaction with Impinging N2O Molecules, Catal. Lett., 2005, 99(3), 215–219 CrossRef CAS.
- D.-J. Liu and H. J. Robota, In Situ XANES Characterization of the Cu Oxidation State in Cu-ZSM-5 during NO Decomposition Catalysis, Catal. Lett., 1993, 21(3), 291–301 CrossRef CAS.
- F. X. Llabrés i Xamena, P. Fisicaro, G. Berlier, A. Zecchina, G. T. Palomino, C. Prestipino, S. Bordiga, E. Giamello and C. Lamberti, Thermal Reduction of Cu2+−Mordenite and Re-Oxidation upon Interaction with H2O, O2, and NO, J. Phys. Chem. B, 2003, 107(29), 7036–7044 CrossRef.
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, A Complete Multisite Reaction Mechanism for Low-Temperature NH3-SCR over Cu-CHA, ACS Catal., 2020, 10(10), 5646–5656 CrossRef CAS.
- F. Göltl, S. Bhandari and M. Mavrikakis, Thermodynamics Perspective on the Stepwise Conversion of Methane to Methanol over Cu-Exchanged SSZ-13, ACS Catal., 2021, 11, 7719–7734 CrossRef.
- F. Göltl, S. Bhandari, E. Lebron-Rodriguez, J. Gold, S. I. Zones, I. Hermans, J. Dumesic and M. Mavrikakis, Identifying Hydroxylated Copper Dimers in SSZ-13 via UV-Vis-NIR Spectroscopy, Catal. Sci. Technol., 2022, 12, 2744–2748 RSC.
- A. R. Kulkarni, Z. J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites, ACS Catal., 2016, 6(10), 6531–6536 CrossRef CAS.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Isolated Cu2+ Ions: Active Sites for Selective Catalytic Reduction of NO, Chem. Commun., 2011, 47(2), 800–802 RSC.
- F. Göltl, P. Sautet and I. Hermans, Can Dynamics Be Responsible for the Complex Multipeak Infrared Spectra of NO Adsorbed to Copper(II) Sites in Zeolites?, Angew. Chem., Int. Ed., 2015, 54(27), 7799–7804 CrossRef PubMed.
- F. Göltl, R. E. Bulo, J. Hafner and P. Sautet, What Makes Copper-Exchanged SSZ-13 Zeolite Efficient at Cleaning Car Exhaust Gases?, J. Phys. Chem. Lett., 2013, 4, 2244–2249 CrossRef.
- F. Göltl, A. M. Love and I. Hermans, Developing a Thermodynamic Model for the Interactions Between Water and Cu in the Zeolite SSZ-13, J. Phys. Chem. C, 2017, 121, 6160–6169 CrossRef.
- U. Deka, A. Juhin, E. A. Eilertsen, H. Emerich, M. A. Green, S. T. Korhonen, B. M. Weckhuysen and A. M. Beale, Confirmation of Isolated Cu2+ Ions in SSZ-13 Zeolite as Active Sites in NH3-Selective Catalytic Reduction, J. Phys. Chem. C, 2012, 116(7), 4809–4818 CrossRef CAS.
- F. Göltl, S. Conrad, P. Wolf, P. Müller, A. M. Love, S. P. Burt, J. N. Wheeler, R. J. Hamers, K. Hummer, G. Kresse, M. Mavrikakis and I. Hermans, UV–Vis and Photoluminescence Spectroscopy to Understand the Coordination of Cu Cations in the Zeolite SSZ-13, Chem. Mater., 2019, 31, 9582–9592 CrossRef.
- F. Göltl, Three Grand Challenges for the Computational Design of Heterogeneous Catalysts, J. Phys. Chem. C, 2022, 126(7), 3305–3313 CrossRef.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Cu-CHA-a Model System for Applied Selective Redox Catalysis, Chem. Soc. Rev., 2018, 47(22), 8097–8133 RSC.
- F. Göltl, P. Sautet and I. Hermans, The Impact of Finite Temperature on the Coordination of Cu Cations in the Zeolite SSZ-13, Catal. Today, 2016, 267, 41–46 CrossRef.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Open-Shell Transition Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48(17), 13115–13118 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- F. Göltl and J. Hafner, Structure and Properties of Metal-Exchanged Zeolites Studied Using Gradient-Corrected and Hybrid Functionals. I. Structure and Energetics, J. Chem. Phys., 2012, 136(6), 064501 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko, R. A. Distasio, R. Car and M. Scheffler, Accurate and Efficient Method for Many-Body van Der Waals Interactions, Phys. Rev. Lett., 2012, 108(23), 1–5 CrossRef PubMed.
- J. Harl and G. Kresse, Cohesive Energy Curves for Noble Gas Solids Calculated by Adiabatic Connection Fluctuation-Dissipation Theory, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77(4), 1–8 CrossRef.
- F. Göltl and J. Hafner, Alkane Adsorption in Na-Exchanged Chabazite: The Influence of Dispersion Forces, J. Chem. Phys., 2011, 134(6), 064102 CrossRef PubMed.
- K. K. Irikura, Thermo.Pl, National Institute of Standards and Technology, 2002 Search PubMed.
- H. Li, C. Paolucci and W. F. Schneider, Zeolite Adsorption Free Energies from Ab Initio Potentials of Mean Force, J. Chem. Theory Comput., 2018, 14(2), 929–938 CrossRef CAS PubMed.
- F. Göltl, A. M. Love, S. C. Schuenzel, P. Wolf, M. Mavrikakis and I. Hermans, Computational Description of Key Spectroscopic Features of Zeolite SSZ-13, Phys. Chem. Chem. Phys., 2019, 21, 19065–19075 RSC.
- J. R. Di Iorio and R. Gounder, Controlling the Isolation and Pairing of Aluminum in Chabazite Zeolites Using Mixtures of Organic and Inorganic Structure-Directing Agents, Chem. Mater., 2016, 28, 2236–2247 CrossRef CAS.
- J. R. Di Iorio, C. T. Nimlos and R. Gounder, Introducing Catalytic Diversity into Single-Site Chabazite Zeolites of Fixed Composition via Synthetic Control of Active Site Proximity, ACS Catal., 2017, 7, 6663–6674 CrossRef CAS.
- J. R. Di Iorio, S. Li, C. B. Jones, C. T. Nimlos, Y. Wang, E. Kunkes, V. Vattipalli, S. Prasad, A. Moini, W. F. Schneider and R. Gounder, Cooperative and Competitive Occlusion of Organic and Inorganic Structure Directing Agents within Chabazite Zeolites Influences Their Aluminum Arrangement, J. Am. Chem. Soc., 2020, 142, 4807–4819 CrossRef CAS PubMed.
- R. E. Fletcher, S. Ling and B. Slater, Violation of Löwenstein's Rule in Zeolites, Chem. Sci., 2017, 8, 7483–7491 RSC.
| This journal is © The Royal Society of Chemistry 2024 |