
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis strategies and cancer therapy applications of PEDOT nanoparticles
Diogo
Dias
 ab,
Leonor
Resina
abc,
Frederico Castelo
Ferreira
ab,
Paola
Sanjuan-Alberte
*ab and
Teresa
Esteves
*ab
ab,
Leonor
Resina
abc,
Frederico Castelo
Ferreira
ab,
Paola
Sanjuan-Alberte
*ab and
Teresa
Esteves
*ab
aInstitute for Bioengineering and Biosciences, Department of Bioengineering, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: paola.alberte@tecnico.ulisboa.pt; teresa.esteves@tecnico.ulisboa.pt
bLaboratory i4HB - Institute for Health and Bioeconomy at Instituto Superior Técnico, Universidade de Lisboa, 1049-001, Lisbon, Portugal
cDepartament d’Enginyeria Química and Barcelona Research Center for Multiscale Science and Engineering, EEBE, Universitat Politècnica de Catalunya, C/Eduard Maristany 10-14, 08019, Barcelona, Spain
First published on 6th August 2024
Abstract
Cancer remains one of the leading causes of death, with traditional therapy approaches facing limitations such as nonspecific systemic toxicity and acquired resistance. As alternative and adjuvant treatment modalities, poly(3,4-ethylenedioxythiophene) (PEDOT) nanoparticles (NPs) leverage unique biocompatible, electrical, and thermal properties for combined imaging, controlled drug release and localised photothermal ablation. This review provides a comprehensive analysis of formulation of tailored PEDOT NPs as smart theragnostic agents toward precise, personalised nanomedicine. We outline common chemical and electrochemical synthesis techniques to control NP size, morphology, stability, and surface chemistry. Extensive structural and electrochemical characterisation relates polymerisation conditions to resultant properties. In particular, PEDOT NPs exhibit efficient near-infrared (NIR) absorption and photothermal conversion, enabling selective photothermal tumour ablation. Their intrinsic conductivity also enables electrical stimulation triggers to modulate the release of therapeutic payloads. While initial works confirm the potential of PEDOT NPs for spatiotemporal cancer treatment, clinical translation remains limited. Further efforts must focus on developing predictive preclinical models, scalable manufacturing methods and clinical partnerships to facilitate translation of these smart nanosystems from the laboratory to clinical use.
 Diogo Dias | Diogo Dias is a Biomedical Engineer who graduated from Instituto Superior Técnico (IST) – Universidade de Lisboa (Portugal) in 2023. During his Master's, he specialized in nanobioelectronic devices and developed skills in chemical synthesis, nanomaterials formulation, and cancer cell culture. He is currently a research assistant at the Institute for Bioengineering and Biosciences. His work focuses on creating innovative nanomaterials for healthcare applications, with a particular interest in anticancer therapies. |
 Leonor Resina | Leonor Resina is a PhD student at Instituto Superior Técnico (IST, Portugal) and Universitat Politècnica de Catalunya (Spain), working on electro-responsive materials for cancer treatment, developing smart electroconductive nanoparticles and transdermal devices. Leonor received her MSc in Biological Engineering from IST, where she performed the expansion and neural differentiation of human-induced pluripotent stem cells for her Master's thesis. Additionally, she worked as a research assistant on polymers for removing genotoxic impurities from active pharmaceutical ingredients and design, modeling, and economic analysis of processes for recovery of water and added value products from food industry wastewater streams. |
 Frederico Castelo Ferreira | Frederico C. Ferreira completed his PhD in 2004 from Imperial College London (IC, UK), followed by a collaborative industrial post-doc between IC and GlaxoSmithKline (2004–2006). He also holds a Master in Business and Administration (MBA) from New University of Lisbon (2008). Frederico is currently an Assistant Professor at the Instituto Superior Técnico and a member of the Institute of Bioengineering and Biosciences with diverse research interests including the development of externally responsive biomaterials, sustainable separations and biorefineries, and the production of sustainable and vegan scaffolds for cellular agriculture applications. |
 Paola Sanjuan-Alberte | Paola Sanjuan-Alberte is currently a “la Caixa” Junior Leader Fellow at the Institute for Bioengineering and Bioscience of Lisbon, Portugal, exploring nanobioelectronic systems for cancer therapy. She is a pharmacist by training who received an M.Pharm. degree in 2015 from the Universidad Santiago de Compostela, Spain. She was also awarded a PhD in 2019 from the University of Nottingham (UK) for her thesis on additive manufacturing of electrochemical systems and their application in bioelectronic medicine. |
 Teresa Esteves | Teresa Esteves is currently a Researcher at the Institute for Bioengineering and Bioscience in Lisbon, Portugal. She has a PhD in Inorganic Chemistry from the University of Lisbon. Her research activities at the Bioengineering Department of Instituto Superior Técnico are focused on new material development for process design tackling challenging separations. She has expertise in chemical synthesis, nanomaterials formulation and characterization, and downstream process design. Dr Esteves also develops smart targeting materials, responsive to external electrical and piezoelectrical stimuli, to address challenging unmet needs in cancer therapy. |
1. Introduction
Cancer is a multifaceted disease that remains one of the leading causes of death globally, with an estimated 10 million cancer deaths in 2020 according to the latest report by the World Health Organisation.1 Despite significant advancements in cancer research and treatment options over the past decades, current therapeutic approaches such as chemotherapy, radiotherapy and surgery still face major challenges. These are related to non-specific systemic toxicity, acquired drug resistance, limited therapeutic efficacy, tumour microenvironment complexity, and patient-to-patient clinical variations. Furthermore, cancer recurrence and metastasis also contribute to the difficulty of developing effective treatment options.2–4 The need to address these has led to the development of novel, complementary anticancer therapy strategies based on innovative approaches such as nanotechnology.The field of nanomedicine, which involves the application of nanotechnology for diagnosis, monitoring, prevention, and treatment of diseases is becoming more promising.5–9 Nanoparticles (NPs), which are particles in the size range of 1–100 nm, have unique properties including a higher surface area-to-volume ratio, a quantum nature in the 1–10 nm range and capacity for encapsulating or adsorbing and transporting different substances, which make them promising theragnostic tools for tumour diagnosis and treatment.10–12 Due to their small size, NPs can selectively accumulate in tumours via the enhanced permeability and retention (EPR) effect, resulting in passive targeted delivery to cancer tissues.13 This is an effect that causes nanosized materials to preferentially accumulate in tumours due to their leaky vasculature and lack of lymphatic drainage.14 On the other hand, by introducing specific ligands that are recognised by commonly expressed cell-surface receptors on tumour cells, NPs can actively target tumours. Additionally, NPs can be designed with additional functionalities, including sustained and triggered drug release, imaging contrast, and photothermal ablation. Various NPs made from organic and inorganic materials have been developed for targeted delivery of chemotherapeutic drugs, immunotherapy agents, siRNA, microRNA, and other anti-cancer therapeutics.15,16
NPs based on conductive polymers (CP) are an emerging class of organic nanomaterials, including polypyrrole (PPy), polydopamine (PDA), polyaniline (PANI), and poly(3,4-ethylenedioxythiophene) (PEDOT), which are among the highest attractive sensing materials due to their all-organic nature with intrinsic electrical conductivity, high signal transduction, optical transparency, mechanical flexibility, and chemical stability. These features primarily result from the fact that CPs contain a π-conjugated backbone, which is made up of alternating single (σ) and double (π) bonds along the polymer chain, enabling delocalised electron transfer.17,18 However, most CPs frequently need to be doped as the polymers lack charge carriers. The process of doping promotes the creation of radical cations/anions (polarons or bipolarons) in the backbone of the polymer, while counter-ions from the solution enter to balance the charge. The more extensive the doping process, the more electrically conductive CPs become.19,20
PEDOT, a polythiophene (PTh) derivative, was first developed in the late 1980s by scientists at the laboratories of Bayer AG, and is usually doped with polystyrene sulfonate (PSS) to improve its solubility in water, as the negative charge in the sulfonate group stabilises the delocalised positive charge in PEDOT. PEDOT:PSS is the most water-soluble CP, which is crucial since materials that dissolve easily in water present easier processability.21 When synthesised as NPs, PEDOT is typically doped with chloride ions or dodecyl benzenesulfonate, the latter acting as doping agent and surfactant simultaneously.22–27
PEDOT presents important advantages in comparison with other CPs like PPy and PANI, because of its biocompatibility, higher stability in biological environments, adjustable and higher electrical conductivity, electrochemical activity, thermoelectric behaviour, and high specific capacitance.23,28–30 For instance, unlike the most studied nontoxic CP, PPy, which is highly crosslinked, PEDOT's unique α,α linkages contribute to its superior electrochemical and electrical responses.25 All the unique properties stated above not only make PEDOT NPs more attractive than similar NPs formed by other CPs, but have drawn attention from various fields where PEDOT NPs present many of their potential applications: flexible electronics,31,32 sensors,33–36 energy storage devices,37–39 and tissue engineering (spinal cord,40 neural,41–43 muscle44).
Additionally, studies have shown that PEDOT NPs exhibit strong near-infrared (NIR) absorption, efficient photothermal conversion that is higher than commercially available gold-based photothermal ablation agents, and tumour targeting via the EPR effect.45–47 These properties make PEDOT NPs an attractive candidate as photothermal agents for minimally invasive ablation of cancer cells. Furthermore, the versatile surface chemistry of PEDOT allows functionalisation with various functional groups, targeting ligands, imaging agents, and therapeutic drugs, allowing for targeted drug delivery and multimodal imaging.28 These properties position PEDOT NPs at the forefront of personalised cancer nanomedicine, offering the potential for integrated therapy and diagnosis – a concept known as theragnostics.
Recent progress in the synthesis and application of PEDOT NPs has confirmed their efficacy in drug-delivery and photothermal therapy (PTT) for various types of cancer (Fig. 1). Therefore, a comprehensive review of PEDOT NP research focused on cancer therapy is justified.
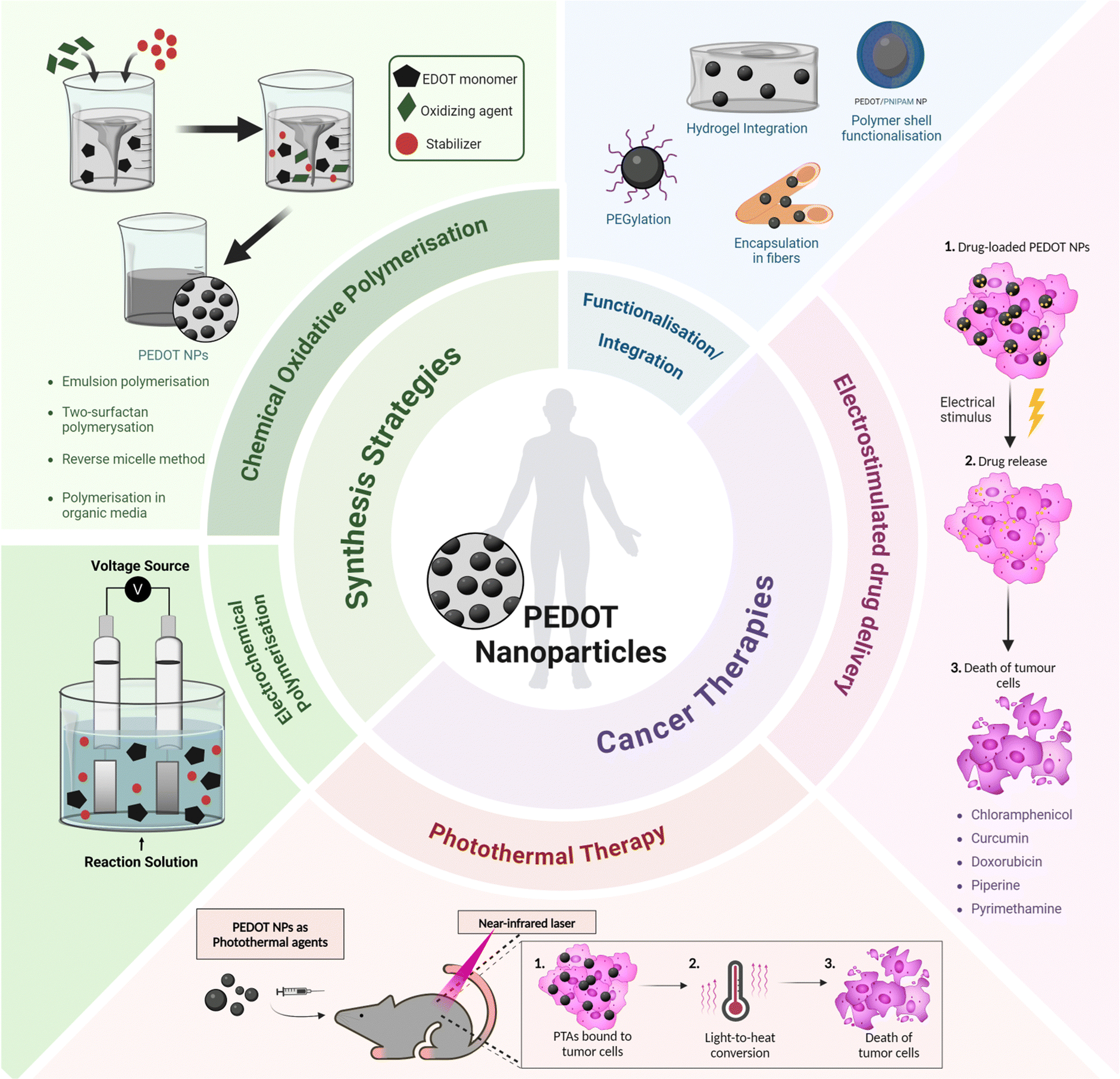 | ||
| Fig. 1 Overview of synthesis strategies and cancer therapy applications of PEDOT nanoparticles. PTA – photothermal agent. | ||
Integrated nanosystems based on PEDOT NPs aim to leverage their unique properties by combining them with other functional elements to create a versatile platform capable of combining therapeutic treatments, targeted drug delivery and diagnostic imaging. This review will explore some examples, such as stimuli responsive nanocomposites that integrate PEDOT NPs into hydrogels or multifunctional nano- and microfibres; core–shell NPs with a magnetic core coated with a PEDOT shell; and theragnostic nanomicelles comprising a PEDOT NP core surrounded by an amphiphilic polymer shell (Fig. 1). Thus, a comprehensive understanding of the synthesis, characterisation, and properties of PEDOT NPs is vital for designing and producing these integrated NP systems successfully. This knowledge will enable researchers to tailor the properties of PEDOT NPs to meet specific application requirements, ultimately enhancing the efficacy of cancer theragnostic platforms.
This review aims to provide a comprehensive exploration of PEDOT NPs, examining their synthesis strategies, characterisation, and applications in cancer treatment. Firstly, we address common techniques for synthesising PEDOT NPs of varying size, morphology, and surface chemistry along with associated characterisation techniques. We also analyse recent advancements in using PEDOT NPs for electrostimulated drug delivery, and photothermal ablation of tumours. Finally, we discuss the current challenges and prospects for clinical translation. We aim that this review supports further efforts dedicated at exploring the potential of these nanosystems for more precise and personalised cancer nanomedicine.
2. Synthesis and characterisation of PEDOT NPs
Comprehending polymerisation processes, oxidative environments, and colloidal stabilisation strategies is essential to successfully synthesise PEDOT NPs. Important variables that impact the final NP size distribution, electrical conductivity through doping efficiency, and dispersion stability, include the choice of oxidant, type of surfactant, polymerisation temperature, reaction time, and post-processing steps. This section summarises different polymerisation techniques and the use of various oxidants and stabilisers to synthesise PEDOT NPs, while discussing the advantages and disadvantages of each strategy.2.1. Synthesis approaches
Chemical oxidative polymerisation (using oxidant agents such as ferric chloride (FeCl3) and ammonium persulfate ((NH4)2S2O8, APS)) or electrochemical polymerisation (applying an oxidising potential through electrodes, where polymers deposit on the working electrode) are the two methods that can be used for generating CPs.21 Generally, electrochemical polymerisation is employed to fabricate CP films, as this method needs a solid conductive surface where the polymerisation can take place, resulting in a homogeneous layer of polymer. Regarding CP NPs, particularly PEDOT NPs, oxidants like FeCl3 and APS in aqueous medium are usually employed to induce oxidation and radical cation formation of EDOT (3,4-ethylenedioxythiophene). These radicals form dimers that subsequently get deprotonated. The oxidants initiate polymerisation into PEDOT chains, while contributing chloride (from FeCl3) or sulphate (from APS) anions act as PEDOT dopants to allow high electrical conductivity48 (Fig. 2).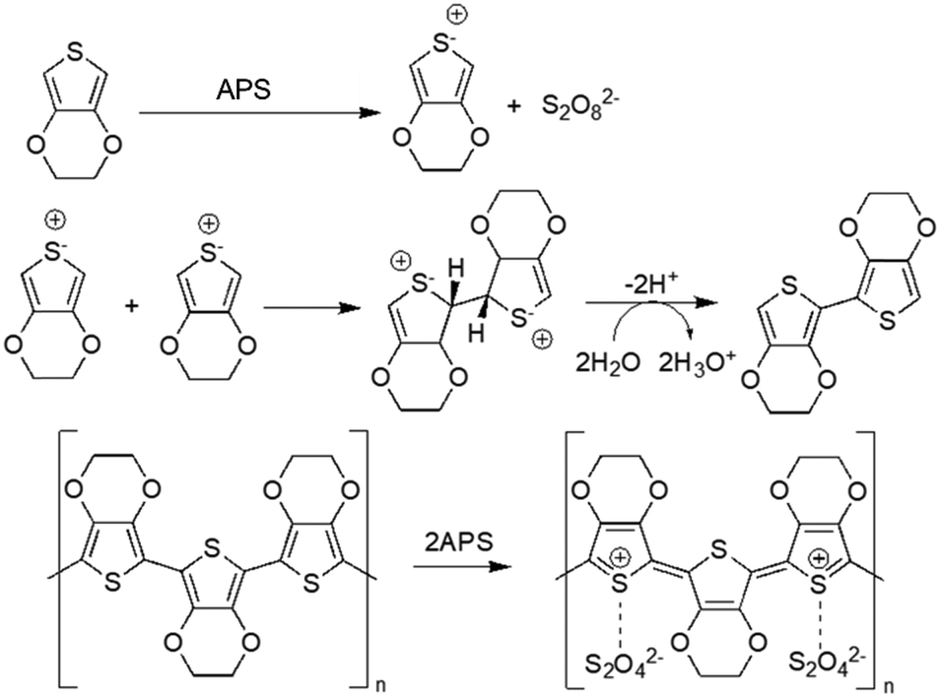 | ||
| Fig. 2 Oxidative polymerisation of EDOT using APS as oxidising agent49. | ||
Additionally, CPs can be synthesised by template-based and template-free approaches. When using hard template techniques, polymer nanostructures are created using moulds. Applying this method to NP synthesis, the polymer is precipitated or polymerised on the surface of the template, resulting in a core–shell structure. Hollow CP NPs can be obtained by removing the template. Gold,50 silica,51,52 and polystyrene NPs53–55 are some of the templates that have already been used to synthesise these nanostructures. Despite the fact that this technique is expected to give better control on the size and colloidal stability than soft templates, it has drawbacks, including the need for post-processing to eliminate the original template, which may have an impact on the nanostructures that are created, and the fact that this technique is too feeble for large-scale manufacturing.56,57
The soft template approach is the most commonly used for the generation of CP NPs, particularly PEDOT NPs. In this method, surfactants (also termed emulsifiers) are added to a solvent which typically consists of an aqueous solution to form a microemulsion, which enhances the colloidal stability and leads to a homogeneous size distribution of the NPs. As the polymerising PEDOT NPs gain form within the microemulsion, the amphiphilic surfactants adsorb onto their surface, providing steric stability that minimises particle aggregation, making it possible to better regulate the NPs’ size and morphology. Furthermore, each PEDOT NP has a surfactant coating that enables it to be suspended and dispersed in organic or aqueous solvents, resulting in high solvent processability. However, in comparison to pure forms, insulating surfactants tend to interfere with conjugation and also act as dopants in PEDOT, which lowers conductivity.20,21,58 Thus, to create self-stabilised PEDOT NPs with a colloidal morphology that permits solubility without the need for surfactants, some synthetic techniques are being employed, such as the use of binary organic solvent systems or electrochemical polymerisation, which will be discussed in detail in the following sections of this review.
Table 1 presents the literature published from 2002 to 2023 that reports different strategies to synthesise PEDOT NPs, including different methods used for polymerisation, or different combinations of oxidants, surfactants, and reaction media, resulting in PEDOT NPs with diverse sizes, morphologies, and electrical conductivities. Table 1 also includes information on the diameter, zeta potential, and electrical conductivity of the synthesised NPs, when available. These studies will be discussed in the next section, highlighting differences in NP properties depending on synthesis parameters.
| Synthesis method | Surfactant/stabiliser | Oxidising agent(s) | Reaction medium | Polymerisation time/temperature | Diameter (nm) | Zeta Potential (mV) | Electrical conductivity (S cm−1) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| DLS | SEM/TEM | ||||||||
| Abbreviations: AOT – sodium bis(2-ethylhexyl)sulfosuccinate; CL-PSS – cross-linked PSS; DBSA – dodecyl benzenesulfonic acid; (Fe(OTs)3) – iron(III) p-toluenesulfonate; H2O2 – hydrogen peroxide; o.n. – overnight; PEO – poly(ethylene oxide); PI-b-PMMA – polyisoprene-block-poly(methyl methacrylate); PSS-co-MA – PSS-co-(maleic acid); RT – room temperature; SDBS – sodium dodecyl benzenesulfonate; SDS – sodium dodecyl sulfate.a No surfactant used.b N/A: Not available or not reported in the referenced study. | |||||||||
| Chemical polymerisation | DBSA | FeCl3 | Water | 20 h/30 °C | N/Ab | 60–120 | N/Ab | <1 | 59 and 60 |
| APS | N/Ab | 35–60 | N/Ab | <50 | |||||
| PI-b-PMMA copolymers | FeCl3 | Cyclohexane and acetonitrile | 8 h/N/Ab | N/Ab | <30 | N/Ab | N/Ab | 61 | |
| α-EDOT-PEO | Fe(OTs)3 or APS | Methanol/water | 72 h/RT | 175–500 | 70–500 | N/Ab | 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × 10−2 × 10−2 |
62 | |
| AOT | FeCl3 | Hexane/water | 12 h/N/Ab | N/Ab | 30–100 | N/Ab | 16.7–33.7 | 63 | |
| DBSA and PSS-co-MA | FeCl3 and PSS-co-MA | Hexane/water | Few sec/N/Ab | N/Ab | 100–200 | N/Ab | 0.29 | 64 | |
| Chloroform/water | 1 h/N/Ab | 65 | <100 | −27 | N/Ab | 65 | |||
| 67.7 | <100 | −39 | N/Ab | 66 | |||||
| 50.6 | <100 | −74.4 | N/Ab | 67 | |||||
| DBSA | FeCl3 and H2O2 | Water | 48 h/RT | 90 | <90 | −52 | N/Ab | 68 | |
| DBSA or SDS | APS | Water | 72 h/N/Ab | N/Ab | 60–900 | N/Ab | <1 to 153 | 49 | |
| SDBS | FeCl3·6H2O | Methanol/H2SO4/water | 5 h/140 °C | N/Ab | 17.2 | N/Ab | N/Ab | 69 and 70 | |
| CL-PSS | Na2S2O8 and Fe2(SO4)3 | Water | 23 h/18 °C | 406–1100 | N/Ab | N/Ab | N/Ab | 71 | |
| Lutensol AT 50 | Fe(OTs)3 and H2O2 | Water | 24 h/45 °C | 90–150 | 30–40 | N/Ab | 2.1 × 10−6–2.6 | 47 | |
| —a | FeCl3 | Dichloromethane and acetonitrile | 24 h/0 °C | 50 | 45 | +33 | 1.6–220 | 72 | |
| DBSA | APS | Water | o.n./40 °C | 84 | 35 | −29.4 | N/Ab | 25 | |
| 157 | 99 | N/Ab | N/Ab | 23 | |||||
| 210 | 35 | −30 | N/Ab | 73 | |||||
| 215 | 109 | −26 | N/Ab | 22 and 26 | |||||
| 243 | 108 | N/Ab | N/Ab | 24 | |||||
| 24 h/30 °C | N/Ab | 49 | N/Ab | N/Ab | 74 | ||||
| SDBS | APS | Water | 18 h/40 °C | 157–235 | 111–146 | −30 | N/Ab | 27 | |
| Electrochemical polymerisation | —a | 1.15 V potential | Dichloromethane/water | N/Ab | N/Ab | 200–650 | N/Ab | N/Ab | 75 |
Firstly, seed polymerisation was used as a synthetic method to generate PEDOT NPs, using a hard template as seed particles that will serve as polymerisation nuclei. Using this approach, Armes and coworkers described the synthesis of PEDOT-coated poly(N-vinylpyrrolidone)-stabilised polystyrene (PVP-stabilised PS) latex particles.53 EDOT was polymerised in an aqueous solution using iron(III) tris(p-toluenesulfonate) (Fe(OTs)3) at 85 °C. This methodology has the disadvantage of needing high temperature for the polymerisation to occur, and also the use of iron is a debatable disadvantage as it may lead to cytotoxic effects. Other studies also synthesised PEDOT NPs with the assistance of hard templates, namely PS and silica particles.52,55 The use of PEDOT as a coating on several metallic NPs has already been extensively reviewed elsewhere,76 therefore, it will not be discussed in this review.
Combining a monomeric surfactant (sodium alkylnaphthalenesulfonate, NaANS) with iron(III) sulfate (Fe2(SO4)3) as an oxidant in an aqueous solution, Kudoh et al. produced PEDOT NPs with high conductivity (60 S cm−1), resulting in the formation of an insoluble surfactant–oxidant mixture.77 However, the morphologies and sizes of the particles as well as a description of how the oxidant–surfactant complex affects the high conductivity were not reported.
PEDOT NPs synthesis using the soft template approach was reported for the first time by emulsion polymerisation in dodecylbenzenesulfonic acid (DBSA) micellar solution by Oh et al.59 First, DBSA was added to deionised water to prepare a surfactant solution. Then, EDOT was added to the solution and dispersed under stirring. Finally, FeCl3 or APS were introduced into the reaction mixture to act as the oxidising agents and the mixtures were stirred for further 20 h at 30 °C, allowing enough time to polymerise the EDOT. The NPs were washed and subsequently dried, resulting in spherical NPs with diameters of 35–60 nm and 60–120 nm and conductivities <1 S cm−1 and 50 S cm−1, when prepared from DBSA–APS and from DBSA–FeCl3, respectively.60 Many other recent studies have already been based on this synthesis for the formulation of PEDOT NPs, as detailed in Table 1, optimising the protocol (polymerisation temperature of 40 °C), using APS as oxidant, and subsequently employing the NPs for drug delivery, for example.23–25,73 This same protocol was followed but using sodium dodecylbenzenesulfonate (SDBS) as surfactant instead of DBSA, thus performing the polymerisation at neutral pH.27 All these studies gave rise to spherical NPs with effective diameters not exceeding 100 nm.
Interestingly, Müller et al. used polyisoprene-block-poly(methyl methacrylate) (PI-b-PMMA) copolymers as surfactants in the synthesis of PEDOT NPs in a non-aqueous emulsion polymerisation approach.61 An emulsion system involving cyclohexane as the continuous phase and acetonitrile as the dispersed phase was described (Fig. 3A). The PI-b-PMMA formed micelles when added to cyclohexane, and the subsequent addition of acetonitrile led to the formation of PI-b-PMMA micelles with an acetonitrile core. The oxidative polymerisation of the EDOT monomer, which is soluble in both phases, was studied to produce PEDOT NPs inside the dispersed acetonitrile “nanoreactors”. It was assumed that polymerisation took place inside the acetonitrile droplets, where both the monomer and the oxidant were present, since FeCl3 is soluble in the acetonitrile phase but not in cyclohexane. Using several PI-b-PMMA surfactants, EDOT was successfully emulsion polymerised, producing PEDOT NPs with spherical morphology and diameters smaller than 30 nm.
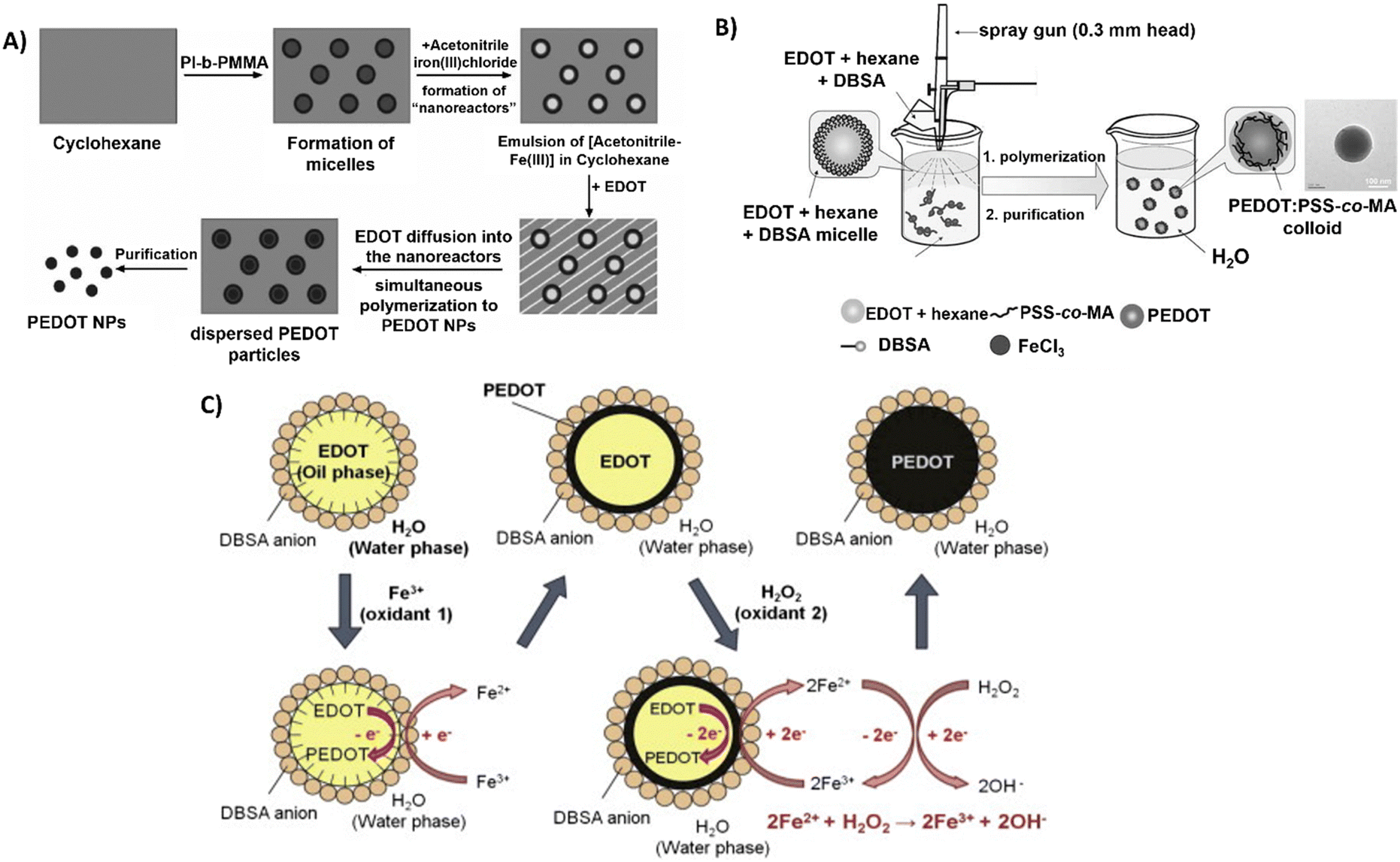 | ||
| Fig. 3 Different polymerisation synthesis approaches of PEDOT NPs. (A) PI-b-PMMA stabilised NPs in non-aqueous emulsion with cyclohexane and acetonitrile. Reproduced with permission from ref. 61 Copyright 2006 Wiley. (B) Spray emulsion polymerisation of PSS-co-MA stabilised PEDOT NPs. Reproduced with permission from ref. 64 Copyright 2010 Wiley. (C) Fe3+/H2O2 bi-oxidant system. Reproduced with permission from ref. 68 Copy right 2011 Elsevier. | ||
In a different study, spherical PEDOT NPs were synthesised in a methanol/water solution using poly(ethylene oxide) end-functionalised with an EDOT moiety (α-EDOT-PEO) as a reactive stabiliser, and APS or iron(III) p-toluenesulfonate hexahydrate (Fe(OTs)3·6H2O) as oxidants.62 Using APS as oxidant and 20–50 wt% stabiliser resulted in a yield of approximately 30% of PEDOT NPs after 72 h of reaction at room temperature (RT). Employing this method, highly spherical NPs with sizes ranging from 100 to 500 nm and conductivity up to 1.5 × 10−2 S cm−1 were successfully synthesised. The resulting NPs exhibited a significant improvement in colloidal stability compared to the NPs obtained in previous studies.
In another soft template approach, Zheng et al.63 prepared PEDOT NPs by a reverse micelle method, also called water-in-oil emulsion, consisting of nanosised water droplets stabilised by surfactants in an oil phase.78 An oxidant active point was formed at the reversed micelle's interface by mixing sodium bis(2-ethylhexyl)sulfosuccinate (AOT) with FeCl3, and the polymerisation reaction lasted 12 h at RT. The synthesised PEDOT NPs presented an electrical conductivity of 16.7 to 33.7 S cm−1 with particle sizes ranging from 30 to 100 nm. The dispersity of NP size shows the lack of control of particle sizes during synthesis, thus resulting in variability of results.
A novel spray emulsion polymerisation (SEP) technique was introduced by Han et al. to synthesise PEDOT NPs with the presence or absence of PSS-co-(maleic acid) (PSS-co-MA).64 An EDOT/DBSA/hexane solution was sprayed, using a spray gun, into an aqueous solution containing FeCl3 oxidant and PSS-co-MA, in the case of PEDOT:PSS-co-MA NPs (Fig. 3B). The functional groups of PSS-co-MA acted as oxidative polymerisation sites, dopants, and stabilisers of the core/shell morphology. Noncovalent interactions occurred between PSS-co-MA and PEDOT thiophene rings, with the polymerisation occurring within a few seconds, followed by magnetic stirring and posterior evaporation of the solvents. The resulting PEDOT and PEDOT:PSS-co-MA NPs exhibited sizes ranging from 100 to 200 nm, and conductivities of 3.2 and 0.29 S cm−1, respectively. This discrepancy indicates that PSS-co-MA created a shell around the PEDOT core, obstructing charge hopping between neighbouring particles and lowering the conductivity of the PEDOT:PSS-co-MA composite. This technique generated well-distributed aqueous-dispersible PEDOT NPs, where the stabiliser, PSS-co-MA, was essential in improving their water-solubility. Inspired by this work, Tania Betancourt's group synthesised PEDOT:PSS-co-MA NPs using a similar two-surfactant emulsion polymerisation method.65–67 The differences in the protocol concern the organic phase, where EDOT and DBSA were dissolved in chloroform instead of hexane, added dropwise to the aqueous phase containing PSS-co-MA, and only then the FeCl3 solution was added to the reaction mixture. The polymerisation lasted 1 h, upon stirring.
A different study used the bi-oxidant system, Fe(OTs)3 and hydrogen peroxide (H2O2), to synthesise PEDOT NPs by emulsion polymerisation.68 It was rationalised that H2O2's oxidative activity could restore Fe3+ (Fig. 3C). The cyclic oxidation–reduction process resulted in the sustained regeneration of these ions, oxidising more EDOT. The amount of iron salt added could be significantly reduced, conversion could be increased, and colloidal stability could be improved by adding this second oxidant, H2O2. Sequential additions of Fe(OTs)3 and H2O2 resulted in stable PEDOT NPs with sizes of approximately 100 nm. Based on the same principle, a miniemulsion polymerisation approach was reported for the successful synthesis of PEDOT NPs to investigate the function of the oxidising agents Fe(OTs)3 and H2O2 in particle synthesis efficiency and electrical properties.47 EDOT was emulsified in an aqueous phase with the nonionic surfactant Lutensol AT 50. In one set of experiments, varying the EDOT/Fe(OTs)3 ratio showed the conductivity increased with the increasing amount of Fe(OTs)3 being used as oxidiser. In a second set, keeping 1:1 EDOT/Fe(OTs)3 ratio constant, adding H2O2 after Fe(OTs)3 led to much more stable macroscopic dispersions and spherical NP morphology stable for months. Conversely, increasing H2O2 concentration decreased NPs’ conductivity due to side reactions with PEDOT chains. According to the findings, dispersions of approximately 30 nm PEDOT NPs with conductivities ranging from 2.1 × 10−6 to 2.6 S cm−1 could be achieved by adjusting the oxidant levels.
Paradee et al. studied the effects of oxidant concentration and surfactant type and concentration in the chemical oxidation polymerisation synthesis of PEDOT NPs.49 APS was used as the oxidising agent and DBSA or sodium dodecyl sulfate (SDS) as surfactants. Depending on the polymerisation conditions, different NP morphologies (plum, globular, orange-peel, coralliform, raspberry agglomerate, and irregular), sizes (60 to 900 nm) and electrical conductivities (<1 to 153 S cm−1) were obtained. Without surfactants, increasing APS concentration (from 0.15 M to 0.30 M) decreased particle size (from 212 to 70 nm) and increased conductivity (from 7.65 to 153.72 S cm−1). The smallest sizes and highest conductivity occurred without the presence of surfactants, which can be explained by the quantum size effect and fewer blocking surfactant aliphatic segments. DBSA and SDS surfactants facilitated morphology control but reduced conductivity.
Building upon these findings, as PEDOT NPs with diverse morphologies were obtained, Wailes et al. adapted these water-based protocols to synthesise PEDOT nanofibres (NF) and nanospheres (NS).79 This time, FeCl3 was used as an oxidant, and the shape and size of the NPs were controlled by the interaction between the surfactant (SDS) and the oxidant concentrations (FeCl3), as well as physical factors such as sonication or stirring during the synthesis process. As the concentration of FeCl3 increased, the particles became less reliant on one another for stability, while increasing surfactant concentrations favoured the production of fibres. Nonetheless, NS formed at very high surfactant concentrations. One important factor in determining the NPs' morphology is the ratio of SDS to FeCl3. SDS:FeCl3 ratios of around 2 were required for fibre formation, whereas ratios of around 3 were needed for longer and more dispersed fibres. Significantly higher ratios encouraged the formation of spherical NPs.
Using SDBS as a surfactant and FeCl3·6H2O as an oxidant, Zhou's group used a hydrothermal synthesis to create well-dispersed PEDOT NPs with an approximate diameter of 17.2 nm.69,70 EDOT's solubility in the dispersion medium was controlled using a mixture of deionised water, methanol, and H2SO4.
Moreover, through oxidative polymerisation of EDOT in the presence of cross-linked PSS (CL-PSS) as stabiliser and dopant, using divinylbenzene (DVB) as the crosslinker, and sodium persulfate (Na2S2O8) and iron(III) sulfate (Fe2(SO4)3) as oxidants in water, PEDOT/CL-PSS particles of 400 to 1100 nm were formulated.71 It was found that, in contrast to linear PEDOT:PSS, the PEDOT/CL-PSS complex's shape was an aggregated dispersion of stable particles because CL-PSS particles acted as seed particles.
Surfactants have significant disadvantages despite their crucial stabilisation properties and control over NP morphology during synthesis. Most surfactants’ insulating aliphatic chains reduce electrical conductivity by disrupting conjugation and ionic interactions along the polymer backbone. Apart from that, they may leave residues that may interfere with optical qualities, complicate purification, and remain integrated into the finished product. Additional NP aggregation can also be induced by higher surfactant concentrations.80,81 Thus, it is important to develop new methods that lessen these inherent constraints by encouraging the development of self-stabilised particles without relying on external surfactants.
On this basis, Cao and colleagues reported an approach to synthesise PEDOT NPs using a binary organic solvent system of dichloromethane and acetonitrile with FeCl3 as oxidant.72 The synthesis was similar to the one carried out by Müller et al.61 However, in this case, the polymerisation of EDOT in dichloromethane could form a regular spherical particle shape, which helps to provide processability and reduce the use of stabilisers. Optimising the polymerisation conditions including solvent ratios, oxidant levels, temperature, and time enabled obtaining 50 nm diameter PEDOT NPs, with a yield over 75%, and an electrical conductivity of 220 S cm−1. These NPs could directly disperse in organic solvents and water, without requiring surfactants, due to self-stabilisation, with stable conductivity over 30 days. Thus, showing the advantages of using organic solvent systems by increasing PEDOT NPs stability and homogeneity, as well as increasing their conductivity. However, the use of organic solvents may also increase NP cytotoxicity in case of poor purification methods, which is a critical point in therapeutic applications.
2.2. Characterisation of PEDOT NPs
A variety of microscopic, spectroscopic, electrochemical, and diffraction techniques are required for the physical and chemical evaluation of PEDOT NPs to guarantee that the synthesis is successful, thus resulting in NPs with optimal properties. A thorough comprehension of critical characteristics such as conductivity, colloidal stability, doping efficiency, and particle size distribution is necessary to create deterministic connections between NP behaviour and synthetic chemistry, hence directing intentional enhancements in performance. These insights are crucial for the effective engineering of PEDOT NPs aimed at their integration into more complex systems, as discussed earlier in the introduction. The set of characterisation parameters described in this section provides the critical insights needed to achieve this goal, and play a crucial role in determining the therapeutic potential of PEDOT NPs. Table 2 summarises the key parameters and their influence on the NPs' behaviour in biological systems, thereby affecting their effectiveness in cancer therapy.| Parameter | Impact on therapeutic potential | Measurement techniques | |
|---|---|---|---|
| Abbreviations: AFM – atomic force microscopy; DLS – dynamic light scattering; FTIR – Fourier-transform infrared spectroscopy; NMR – nuclear magnetic resonance; SEM – scanning electron microscopy; TEM – transmission electron microscopy; TGA – thermogravimetric analysis; XPS – X-ray photoelectron spectroscopy. | |||
| Size | Smaller size (10–200 nm) | – Enhanced tumour penetration | – DLS |
| – Improved cellular uptake | – TEM | ||
| – Longer circulation time | – SEM | ||
| Larger size (>200 nm) | – Potential for faster clearance | ||
| – Increased drug loading capacity | – DLS | ||
| Shape | Spherical | – Uniform cellular uptake | – TEM |
| Rod-like | – Better margination | – SEM | |
| – Enhanced tumour penetration | – AFM | ||
| – Higher protein corona formation | |||
| Surface charge | Positive | – Increased cellular uptake | – Zeta potential measurement (DLS) |
| – Potential for non-specific interactions | |||
| Negative | – Longer circulation time | ||
| – Reduced non-specific interactions | |||
| Neutral | – Reduced protein adsorption | ||
| – Improved stealth properties | |||
| Functionalisation | PEGylation | – Enhanced circulation stability and time | – FTIR |
| – Reduced immunogenicity | |||
| Targeting ligands | – Improved tumour-specific accumulation | – XPS | |
| – Enhanced cellular internalisation | |||
| Stimuli-responsive moieties | – Controlled drug release | – NMR | |
| – Improved therapeutic efficacy | |||
| Thermal Stability | Higher stability | – Sustained performance during PTT | – TGA |
| – Improved safety profile | |||
| Conductivity | Higher conductivity | – Enhanced photothermal conversion | – Four-point probe method |
| – Improved potential for electrically-triggered drug delivery | – Dielectric spectroscopy | ||
| – Improved potential for electrotherapy | |||
The biodistribution, cellular uptake, and clearance of NPs in the bloodstream are critically influenced by their physicochemical properties, particularly their size. NPs smaller than 10 nm, can be easily cleared by the kidneys, while larger NPs (>200 nm) may be cleared by phagocytic cells in the reticuloendothelial system (RES).82 Accordingly, therapeutic NPs with a size of <200 nm have longer circulation time in the bloodstream. Additionally, therapeutic NPs in 10–200 nm size exhibit enhanced tumour penetration due to the EPR effect and because they cannot be recognised by the RES. However, larger NPs may offer increased drug loading capacity and higher photothermal conversion efficiency, which can be advantageous for certain therapeutic approaches.8,83
Extravasation, which is the first step for a circulating NP to reach the target tissue,84 is also influenced not only by the size of the NPs, but also by their shape. Small NPs generally pass through capillary walls more easily than large NPs.85 Larger NPs are more likely to localise on the vessel walls, and non-spherical (ellipsoids, discoid shapes and nanorods with higher aspect ratios) particles exhibit better margination.84 Furthermore, rod-shaped NPs present enhanced tumour endosomal uptake than NPs of other shapes, suggesting that these NPs may be understood by immune system cells as rod-shaped bacteria.86
The surface charge of NPs affects their interaction with biological membranes, cellular uptake, and biodistribution. Positively charged NPs demonstrate increased cellular uptake due to electrostatic interactions with negatively charged cell membranes. However, this can also lead to non-specific interactions and a higher immune response compared to neutral or negatively charged NPs.85 Positively charged NPs are generally most rapidly cleared, followed by negatively charged NPs, while neutral and slightly negative NPs have the longest half-lives in circulation, because they can reduce protein adsorption.8 This phenomenon of non-specific adsorption of serum proteins forms a corona on the NP's surface, which can alter their physicochemical properties and reduce therapeutic efficacy. Specifically, studies report significantly larger amounts of protein attaching on the rod-like particles compared to the spheres.87
Functionalisation of PEDOT NPs offers opportunities to enhance their therapeutic potential. Surface modification of NPs with long-chain polymers such as polyethylene glycol (PEG) reduce the interactions with phagocytic cells of the RES, and minimise non-specific protein absorption onto the NP surface.8 Therefore, PEGylation increases the circulation time while shielding the NP surface from enzymes and antibodies that may prompt degradation, secretion and clearance.83,88 Addition of targeting ligands (e.g., antibodies, peptides) can improve tumour-specific accumulation and cellular internalisation. Incorporating stimuli-responsive moieties (e.g., pH-sensitive, redox-sensitive) enables controlled drug release and can significantly improve therapeutic efficacy.8
PEDOT NPs’ thermal stability is important for PTT, ensuring that their structural integrity and performance is maintained for consistent therapeutic efficacy/safety. Conductivity, ultimately one of the main properties of PEDOT, is not only a significant factor for efficient photothermal conversion, but it is also essential for potential application of these NPs for electrically-driven drug delivery, as well as potential applications for electrotherapy, paving the way for dual-mode cancer therapies. Therefore, optimising these key parameters will be critical for PEDOT NPs' in vivo efficacy, versatility and overall effectiveness for cancer therapy, and thus should be taken into consideration for the design and synthesis of these NPs.
The second approach relates to NP stability in solution, thus, not requiring sample drying. Dynamic light scattering (DLS) is used to determine the hydrodynamic size of NPs in solution by measuring time-dependent fluctuations in scattered light intensity.91 The obtained diameter values represent the particle and its surrounding solvent molecules, often yielding larger values due to this measurement. Due to its limitation in measuring the effective particle size, DLS serves mainly as a preliminary or reference method, often preceding microscopy examinations.
Regarding PEDOT NPs, studies in the literature focused mainly on the use of SEM/TEM and DLS to determine their diameters, which ranged between 30 and 400 nm, depending on the technique used. It was observed that, as expected, the diameter of the NPs determined by DLS was higher than that determined by SEM by approximately two-fold, as it can be seen in Table 1. The SEM and TEM images also allowed the visualisation of a typical spherical morphology of the PEDOT NPs in most studies.47,49,61,62,68,69,71,72,75 This implies that, in contrast to NPs with a rod-like shape, the great majority of NPs synthesised using the methods described above will exhibit less extravasation but less formation of protein corona when employed in in vivo experiments. Drug delivery studies using these NPs also reported an increase in the size of the loaded NPs due to the encapsulation of the drugs.20,22–27,73,92
Paradee et al.49 studied the effects of the oxidising agents and surfactants on the resultant PEDOT NPs sizes and morphologies. Based on this study and the results of the other studies discussed in Section 2.1, it is possible to state that increasing oxidant concentrations (particularly APS) lead to a decrease in the NP size. This effect is a result of the oxidising agent concentration having an impact on the initiation of the polymerisation and, consequently, leading to the formation of more active sites of the EDOT cations (Fig. 2). This leads to higher electro-repulsive forces between PEDOT polymeric cation radicals, and higher rate of polymerisation, thus resulting in smaller particle sizes.49 Surfactant concentrations have an interesting effect on particle size because, at concentrations below the critical micelle concentration (CMC, 0.29 × 10−2 M), PEDOT NPs appear larger and in a globular form due to the lack of micelles. Smaller PEDOT particles are produced when polymerisation within the micelle core takes place at concentrations higher than the CMC. However, when the concentration of surfactant increases further, PEDOT particles agglomerate as a result of micellar fusion, producing PEDOT NPs that are larger in size and take the form of globular clusters.49,81 Overall, one can conclude that for achieving PEDOT NPs with optimal size, the selection of appropriate surfactant and reaction initiators are key parameters to take into consideration.
The zeta (ζ)-potential is a measure of the effective electric charge at the surface of the NPs, and quantifies the charge stability of the colloidal particles, providing information about their tendency to aggregate or to remain discrete.89 In general, a ζ-potential above 25 mV (absolute value) indicates that the electrostatic repulsive forces exceed the attractive steric forces, implying colloidal stability, and the system is kept in a relatively stable dispersed state.22 The results of this measurement for the PEDOT NPs formulated in the studies listed in Table 1 and subsequent studies based on them revealed the synthesis of stable dispersed NPs in solution. Moreover, it is evident that in most formulations the PEDOT NPs exhibit a negative surface charge.22,25–27,65–68,73 This negative charge is crucial for maintaining colloidal stability and effective biodistribution due to longer circulation time, as indicated by the influence of surface charge on therapeutic potential in Table 2. In the case of synthesis in a bi-organic medium, the NPs show a positive surface charge of +33 mV.72 This shift is likely due to the adsorption of positively charged ions or molecules from the bi-organic medium onto the surface of the NPs. The positive surface charge can influence interactions with biological membranes and cells, potentially enhancing cellular uptake, thus affecting the therapeutic potential as also summarised in Table 2.
FTIR spectroscopy involves irradiating the sample with infrared radiation (12500 to 10 cm−1), with the absorbed radiation being converted into rotational or vibrational energy. FTIR is a crucial technique for chemical identification since it produces a spectrum with wavenumber that typically ranges from 4000 to 400 cm−1, representing the molecular fingerprint of the sample.93 Changes in the characteristic absorption band pattern point to a modification in the composition of the material. Thus, FTIR is particularly useful for confirming successful surface modifications.
In the FTIR spectrum of PEDOT NPs obtained using different synthesis conditions, key characteristic peaks corresponding to the PEDOT structure were observed. The vibrational bands around 1500–1400 cm−1 originated from the C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the quinoidal structure of the thiophene ring and the stretching of the thiophene ring, respectively. Peaks from 1200–1000 cm−1 are associated with C–O–C vibrations of the ethyleneoxy groups. The peaks of the C–S bond in the thiophene ring were observed from 680–950 cm−1. Additional smaller peaks between 2800 to 3000 cm−1 were present when the NPs were obtained using a surfactant. These peaks correspond to the aliphatic C–H stretching mode depending on the long alkyl tail of the surfactant (e.g. DBSA).23,25,27,49,60,66,68,69,72,94 When using APS as oxidant, the presence of a peak around 1680 cm−1 indicates the presence of a carbonyl group, which is associated with a side reaction like overoxidation, since APS is a strong oxidant.41 Additionally, in PEDOT NPs formulated with H2O2, a broad band in the range of 3300–3000 cm−1 is present, referable to the hydroxyl groups in the polymer chains.47 FTIR can also be employed for confirming the success of the drug loading process on PEDOT NPs. For instance, the characteristic curcumin (CUR) bands, which are identified at 3506 cm−1 (OH), 1594 cm−1 (CO) and 1269 cm−1 (enol C–O), were detected in the spectra of PEDOT/CUR NPs.24 The spectra of PEDOT NPs loaded with pyrimethamine (PYR) presented the bands associated with both PEDOT NPs and free PYR, attributed to the –NH2 group (3443 cm−1 and 3261 cm−1) and the CN group (between 1681–1409 cm−1) stretching vibration.22
C stretching of the quinoidal structure of the thiophene ring and the stretching of the thiophene ring, respectively. Peaks from 1200–1000 cm−1 are associated with C–O–C vibrations of the ethyleneoxy groups. The peaks of the C–S bond in the thiophene ring were observed from 680–950 cm−1. Additional smaller peaks between 2800 to 3000 cm−1 were present when the NPs were obtained using a surfactant. These peaks correspond to the aliphatic C–H stretching mode depending on the long alkyl tail of the surfactant (e.g. DBSA).23,25,27,49,60,66,68,69,72,94 When using APS as oxidant, the presence of a peak around 1680 cm−1 indicates the presence of a carbonyl group, which is associated with a side reaction like overoxidation, since APS is a strong oxidant.41 Additionally, in PEDOT NPs formulated with H2O2, a broad band in the range of 3300–3000 cm−1 is present, referable to the hydroxyl groups in the polymer chains.47 FTIR can also be employed for confirming the success of the drug loading process on PEDOT NPs. For instance, the characteristic curcumin (CUR) bands, which are identified at 3506 cm−1 (OH), 1594 cm−1 (CO) and 1269 cm−1 (enol C–O), were detected in the spectra of PEDOT/CUR NPs.24 The spectra of PEDOT NPs loaded with pyrimethamine (PYR) presented the bands associated with both PEDOT NPs and free PYR, attributed to the –NH2 group (3443 cm−1 and 3261 cm−1) and the CN group (between 1681–1409 cm−1) stretching vibration.22
Raman relies on inelastic scattering of monochromatic light rather than mid-IR light absorption as in FTIR, and this technique also identified characteristic ring and inter-ring vibrations in the range of 1500–500 cm−1. Transition intensities provide indications of effective conjugation and doping levels.24,47,64,75 The Raman fingerprints of PEDOT NPs appear at 992 cm−1, 1259 cm−1, 1368 cm−1, 1424 cm−1 and 1495 cm−1, which have been related to the oxyethylene ring, Cα–Cα’ inter-ring stretching, CβCβ stretching, symmetric CαCβ stretching and asymmetric CαCβ stretching vibrations, respectively.24 Increasing the amount of neutral π-bonds in the polymer backbone results in a high Raman signal. Thus, higher doping levels induce more charge carriers that shield the overall Raman scattering efficiency, thus reducing Raman peak intensities in the spectrum. Additionally, the Raman bands exhibit a small downshift to lower energies as doping helps promote quinoid resonance forms.47 For instance, Raman spectra of PEDOT NPs synthesised with different concentrations of Fe(OTs)3 showed no Raman signal at low Fe(OTs)3 concentrations, indicating a low polymerisation degree. At higher Fe(OTs)3 concentrations, the Raman signal decreases due to a higher doping level of the final material. Additionally, the intensity of the Raman signal decreases in the presence of H2O2, corresponding to a reduction in the number of neutral π-bonds along the polymer backbone, and a shift in the signal related to asymmetric CC stretching is observed. This shift indicates a decrease in electrical conductivity, attributed to changes in the conjugation of the PEDOT chains.47
In UV-Vis spectroscopy, the intensity of a UV-Vis light beam is measured both before and after it passes through the sample, which is positioned between a light source and a photodetector. The sample's maximum absorption level determines the plotted wavelength.89 The maximum absorption peak of PEDOT is in the 800–1000 nm range and is related to the polaronic and bipolaronic oxidised states. The peak position and intensity are positively correlated with the doping level of PEDOT. Generally, neutral PEDOT presents a broad absorption peak with lower intensity in the visible light region (400–600 nm), corresponding to the π–π* transition.22,23,47,66,72,73,75 Higher concentrations of Fe(OTs)3 cause an increase in absorption related to the polaronic and bipolaronic states (800–1000 nm), while lower concentrations cause an increase in absorption linked to the neutral π-bonds.47 UV-Vis spectra of PEDOT NPs synthesised with varying volumes of H2O2 show high oxidation levels in the polymer chains, evidenced by the absorption of oxidised states. However, the absorption related to the conjugated neutral π-bond decreases with more H2O2. This is likely because the radicals in the conjugated π-system interact with ˙OH radicals from H2O2, bonding to the polymer backbone and breaking the conjugation, which significantly reduces electrical conductivity by hindering charge propagation along the polymer chains.47
Moreover, UV-Vis spectroscopy is essential for characterising the optical properties of PEDOT NPs, which are crucial for their application in PTT. The absorption spectrum not only confirms the presence of PEDOT but also provides information on the NPs’ ability to absorb light in the NIR region. This is explored in more detail in Section 3.1.
Complementary to the vibrational characterisation of the molecular structure, X-ray diffraction (XRD) provides information on chain order, by evaluating the crystalline structure. This technique displays diffraction intensity over a range of angles, with peaks at specific 2θ values being related to lamellar sheet packing distances and π–π interchain stacking distances.95 The use of dopants and polymerisation conditions impact relative peak intensities and degree of crystallinity. The diffraction peaks of PEDOT NPs at 2θ of approximately 7°, 12°, and 18° correspond to a system in which the interchain packing is along a pseudo-orthorhombic a-axis. The sharper diffraction peak at 2θ of approximately 6° represents the interchain planar ring-stacking distance.49,60,64,72 Higher order in the PEDOT NP structure is induced by increasing the doping level with increasing oxidant concentration, namely APS. Additionally, the presence of surfactants reduces the degree of crystallinity of PEDOT NPs. Compared to PEDOT synthesised using DBSA, the diffraction peak of PEDOT generated using SDS is sharper. This effect is due to the bulky benzene ring structure of the DBSA causing PEDOT to have a more amorphous structure.49,66
While XPS measures the kinetic energy of electrons emitted from a sample during X-ray irradiation to characterise its composition, energy-dispersive X-ray (EDX) relies on the detection of X-rays emitted from the sample during electron beam bombardment, as it is performed in conjunction with SEM. XPS and EDX are complementary, but XPS provides additional chemical state information and is more surface-specific. Elemental analysis of NPs via EDX can confirm the presence of PEDOT elements (carbon, oxygen, sulphur). The degree of NP purification can also be determined by this analysis, since the absence of Fe, for instance in NPs made with FeCl3, suggests that Fe has been successfully removed from the NPs during purification.66
3. PEDOT NPs applications in cancer therapies
The unique properties of PEDOT NPs make them well-suited for a range of cancer therapy applications. In particular, the high NIR absorption and photothermal conversion efficiency enable their use in PTT. Additionally, PEDOT NPs can be developed as smart delivery systems for the targeted transport and delivery of chemotherapeutic drugs, owing to their facile surface modification and tuneable drug-loading capacity. A comprehensive overview of current developments in the use of PEDOT NPs in PTT and drug delivery approaches for cancer treatment is presented in this section (Table 3). We first address photothermal effects using PEDOT NP-based systems. Next, we highlight innovative PEDOT NP-based drug carriers designed for stimuli-responsive and targeted drug release.| Anticancer therapy | Configuration of PEDOT NPs-based system | Cancer model | Ref. |
|---|---|---|---|
| Abbreviations: Alg-g-PAA – poly(acrylic acid)-grafted sodium alginate; CAM – chloramphenicol; CUR – curcumin; Ce6 – chlorin e6; DOX – doxorubicin; HCPT – 10-hydroxycamptothecin; PES – phenylethynesulfonamide; PGGA – poly(γ-glutamic acid); PIP – piperine; PYR – pyrimethamine; SN-38 – 7-ethyl-10-hydroxycamptothecin.a These studies use core–shell NPs with PEDOT shell.b In these studies, only the cytotoxicity of the drug loaded PEDOT NPs was assessed, and not the effect of applying electrical stimulation to the cells with the NPs. | |||
| PTT | PEGylated PEDOT:PSS NPs (PEDOT:PSS-PEG) | 4T1 tumour-bearing mice (in vivo) | 88 |
| PEDOT:PSS-PEG loaded with DOX, SN38, and Ce6 | 4T1 mice breast cancer cells (in vitro) | 99 | |
| PEDOT:PSS NPs | RKO and HCT116 colorectal cancer cells (in vitro) | 100 | |
| PEDOT NPs encapsulated in a nanogel | HCT116 cervical cancer cells (in vitro) and HCT116 tumour-bearing mice (in vivo) | 70 | |
| PEDOT:PSS NPs | MDA-MB-231 breast cancer cells (in vitro) | 65 | |
| PEDOT NPs | MDA-MB-231 breast cancer cells (in vitro) | 66 | |
| PEDOT NPs embedded in a dynamic thiol-Michael hydrogel | MDA-MB-231 breast cancer cells (in vitro) | 67 | |
| Magnetic NPs with PEDOT coating loaded with HCPT (Fe2O3@PEDOT-HCPT)a | 4T1 breast, HeLa cervical cancer cells (in vitro) and 4T1 tumour-bearing mice (in vivo) | 101 | |
| Magnetic NPs with PEDOT:PSS coating (Fe3O4@PEDOT:PSS)a | MCF-7 tumour-bearing mice (in vivo) | 102 | |
| Magnetic NPs with PEDOT:PSS, Cyanine7, and 2-deoxyglucose-polyethylene glycol (MNP@PES-Cy7/2-DG)a | MCF-7 breast cancer cells (in vitro) and MCF-7 tumour-bearing mice (in vivo) | 103 | |
| Magnetic NPs with PEDOT coating loaded with siRNA (α-Fe2O3@PEDOT-siRNA)a | MCF-7 breast cancer cells (in vitro) and MCF-7 tumour-bearing mice (in vivo) | 97 | |
| PEDOT NPs loaded with CUR | PC3 prostate, MCF-7 breast cancer cells (in vitro)b | 25 | |
| PEDOT NPs and CUR loaded in poly(ε-caprolactone) (PCL) microfibres | MCF-7 breast cancer cells (in vitro)b | 23 | |
| PEDOT NPs and menadione integrated into a PGGA hydrogel | N/A | 74 | |
| Electrostimulated drug delivery | PEDOT NPs loaded with fibrin-homing peptides (CREKA and CR(NMe)EKA) | PC3 prostate cancer cells (in vitro)b | 73 |
| PEDOT NPs loaded with PYR | MG-63 osteosarcoma cells (in vitro)b | 22 | |
| PEDOT NPs loaded with CUR integrated into a PGGA hydrogel | N/A | 24 | |
| PEDOT NPs loaded with CAM integrated into a Alg-g-PAA hydrogel | HeLa cervical cancer cells (in vitro) | 27 | |
| PEDOT NPs loaded with CUR encapsulated inside coaxial poly(glycerol sebacate)/poly(caprolactone) (PGS/PCL) electrospun fibers | PC3 prostate, MCF-7 breast cancer cells (in vitro)b | 104 | |
| PEDOT NPs loaded with CR(NMe)EKA integrated into an injectable pH responsive phenylboronic acid grafted to chitosan (PBA-CS) hydrogel | MG-63 osteosarcoma, PC3 prostate cancer cells (in vitro)b | 105 | |
3.1. Photothermal therapy (PTT)
Among alternative approaches, that could lessen the limitations currently attached to established cancer therapy, is hyperthermic therapy (HT). Hyperthermia is a state in which the body is exposed to high temperatures (often >45 °C) and has been thoroughly researched in relation to cancer treatment since heat stress triggers the death of cancer cells.106 HT has the potential to be used alone or in combination with radiation therapy or chemotherapy.107 Localised hyperthermia triggered by NPs appears to be a promising strategy to treat cancer, since usual HT treatments are not tumour-focused, are invasive, and produce heat uniformly throughout the body,108 which may result in adverse effects. Recently, PTT, which uses light as a stimulus for thermal therapy, has received a lot of attention. PTT therapy is based on an approach where light (typically NIR) is used to excite PTT agents, which in turn release energy as heat. NIR light, with wavelength between 700 and 900 nm, is the most common source of excitation due to its low attenuation and deep penetration in biological tissues.109 PTT agents must fulfil some requirements, such as being dispersible in aqueous solutions; having a uniform shape and size (nanoscale); responding to NIR radiation; being sufficiently photostable to guarantee adequate diffusion time to reach tumours before losing their photosensitivity; and being biocompatible in living systems. CP NPs satisfy these conditions and have gained research interest due to their ability to be finely tuned to absorb NIR light, by chemically modifying the polymer backbone with functional groups or blended systems. The polymer chains also enable the coupling of drugs and biomarkers, allowing for applications like imaging and NIR-triggered drug delivery.110,111 To date, photothermal effects in cancer therapies have been described for PPy,112–120 PDA,121–130 PANI131–134 and PEDOT NPs, the latter being the focus of this review.In 2012, Cheng et al. reported for the first time the use of PEDOT:PSS as an effective and safe organic PTT agent for in vivo cancer treatment.88 The formulated PEDOT:PSS-PEG NPs were coated layer-by-layer with charged polymers before being conjugated with branched PEG, presenting a diameter of approximately 80 nm. The PEDOT:PSS-PEG NPs demonstrated high stability in a physiological environment, and a stealth-like behaviour following intravenous injection at a dose of 10 mg kg−1 in Balb/c mice. Their long blood circulation half-life of 21.4 ± 3.1 h allowed for significant accumulation of the NPs at tumour sites due to the EPR effect. Thus, longer blood circulation and stealth nature were conferred by surface PEGylation. The potential of these PEDOT:PSS-PEG NPs to eliminate tumours via NIR-mediated photothermal ablation (808 nm laser irradiation with a power density of 0.5 W cm−2 for 5 min) has been confirmed in experiments conducted on Balb/c mice bearing 4T1 tumours. Specifically, after laser irradiation, tumours on PEDOT:PSS-PEG-injected mice were completely eliminated one day post-irradiation, and these mice survived over 45 days without a single death. Furthermore, comprehensive blood tests and histological examination revealed no toxicity of the NPs to the treated mice within 40 days, indicating their safety profile at the tested dose (Fig. 4A).
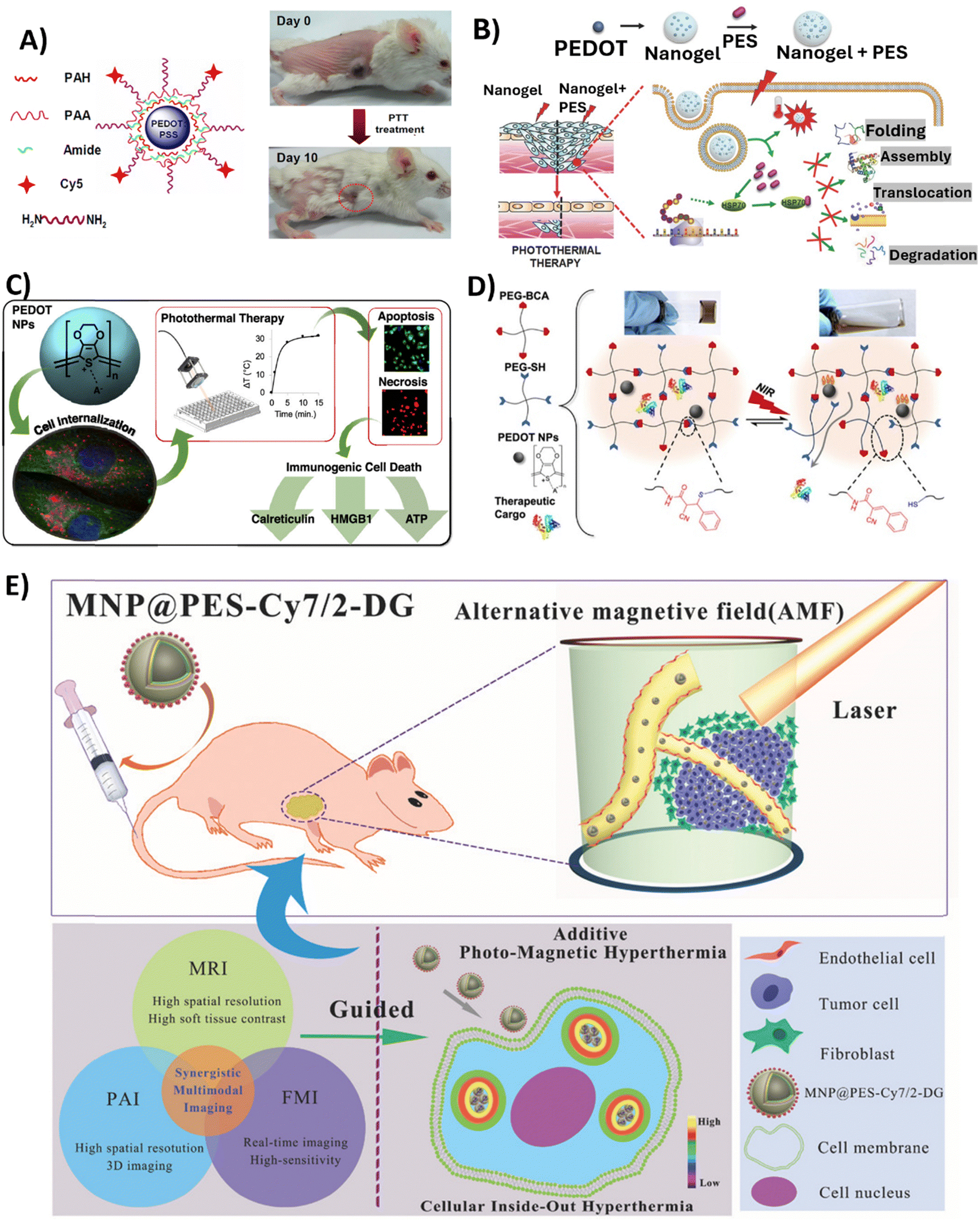 | ||
| Fig. 4 Overview of the studies employing PEDOT NPs-based systems in anticancer PTT. (A) PEDOT:PSS-PEG NPs structure (left); Representative photos of a PEDOT:PSS-PEG-injected 4T1 tumour-bearing mouse at day 0 before PTT treatment and at day 10 after treatment. Complete tumour elimination was achieved after PTT treatment (right). Reproduced with permission from ref. 88. Copyright 2012 American Chemical Society. (B) Schematic illustrating Nanogel + PES synthesis for the PTT of deeper cancer cells by using PES that has been released to inhibit HSP70 function. Reproduced with permission from ref. 70. Copyright 2016 Wiley. (C) Schematic of the study developed by Huff et al. Reproduced with permission from ref. 66. Copyright 2020 American Chemical Society. (D) Schematic showing the cargo-loaded Gel/PEDOT system (dynamic thiol-Michael cross-linking between 4-arm PEG-BCA and 4-arm PEG-SH in the presence of PEDOT NPs and BSA-FITC or DOX), and the photothermally modulated release of the therapeutic cargo by NIR light. Reproduced with permission from ref. 67. Copyright 2020 American Chemical Society. (E) Diagram of intracellular photomagnetic hyperthermia guided by trimodality molecular imaging under intravenous administration of MNP@PES-Cy7/2-DG. Reproduced with permission from ref. 103. Copyright 2018 Wiley. | ||
Encouraged by these results, Gong et al.99 conducted a follow-up study to further enhance the therapeutic potential of PEDOT:PSS-PEG NPs. Three aromatic medicinal compounds were loaded into the PEDOT:PSS-PEG NPs through π–π stacking and hydrophobic interactions.99 These molecules included the photosensitiser chlorin e6 (Ce6) and the chemotherapeutic medicines doxorubicin (DOX) and SN38. Remarkably, in vitro experiments with the 4T1 murine breast cancer cell line showed that loading the PEDOT:PSS-PEG NPs could enhance the solubility of water-insoluble drugs, such as SN38, while maintaining the drug's cytotoxicity. Furthermore, compared to free Ce6, Ce6-loaded NPs showed 5 to 6-fold higher cellular uptake and increased photodynamic therapy efficacy. Due to photothermal effects, DOX-loaded PEDOT:PSS-PEG NPs exposed to laser radiation (808 nm) resulted in a 5 times higher intracellular delivery of DOX. In comparison with individual treatments, the combination of laser and DOX-loaded NPs led to a substantial decrease in cancer cell viability and a synergistic effect on cancer cell death.
In another study involving PEGylation, Liu et al. reported the synthesis of PEDOT NPs with a size of 17.2 nm, and broad NIR absorption from 700–1250 nm.69 The NPs were modified with PSS, indocyanine green (ICG) dye, PEG, and glutaraldehyde (GTA) to obtain PEDOT:ICG@PEG-GTA NPs. Under 1064 nm laser irradiation, the photothermal conversion efficiency was 71.1% and a temperature of 76.6 °C was reached, indicating excellent photothermal properties. In vitro studies showed the NPs had low toxicity in bacterial (E. coli, and S. aureus) and human cancer cell lines (U87MG glioblastoma cell line, and HeLa cervical carcinoma cell line). By combining 808 nm and 1064 nm laser irradiation of NP-treated E. coli and S. aureus bacterial strains resulted in approximately 99% cell death, demonstrating a synergistic photothermal and photodynamic bactericidal effect. Although the effects of applying a PTT to the cancer cell lines used in this study have not been examined, the study's promising results point to the relevance of conducting such an experiment.
Other in vitro studies followed using PEDOT NPs as PTT agents. MacNeill et al. synthesised 100–200 nm size PEDOT:PSS NPs with elevated NIR absorption capacity.100 Studies performed on RKO and HCT116 colorectal cancer cell lines revealed negligible toxicity of the NPs, and photothermal ablation resulted in approximately 20% cell survival after NIR irradiation. Interestingly, 200–400 nm diameter and 4–10 μm length PEDOT nanotubes (NTs) showed stronger NIR absorption, higher temperature increase upon irradiation in comparison with NPs, and photothermal ablation that resulted in <5% cell survival. Therefore, PEDOT NTs appear to be superior agents over NPs for PTT of cancer. Thus, modifying the aspect ratio from a spherical to a tubular/rod shape may potentially be an effective way to improve photothermal performance of PEDOT NPs.100
Further works synthesised PEDOT:PSS-co-MA NPs with diameters <100 nm, strong NIR absorption (peak at 750 nm), high photothermal conversion efficiency of 53%, and significant photostability.65 Studies conducted in MDA-MB-231 breast cancer cells demonstrated the cytocompatibility of the NPs and showed that the presence of the NPs at the highest concentration of 500 μg mL−1 led to the ablation of approximately 80% of cancer cells upon 808 nm laser irradiation with a power density of 7 W cm−2 for 5 or 15min intervals.
To improve the depth at which PTT is effective for cancer treatment, another group developed a nanocomposite platform containing 17.2 nm PEDOT NPs as a photothermal agent along with the heat shock protein 70 (HSP70) inhibitor, phenylethynesulfonamide (PES), encapsulated in a thermoresponsive poly(N-isopropylacrylamide-co-acrylic acid) (PNIPAM-co-AAc) polymer shell.70 Upon NIR irradiation (808 nm, 0.1 W cm−2 for 10 min), the Nanogel + PES underwent a phase transition releasing PES to inhibit HSP70 function in HCT116 colon cancer cells and HCT116 tumour-bearing mice. In the latter case, Nanogel + PES with PEDOT NPs was administered at a dose of 10 mg kg−1. This led to increased susceptiveness to heat damage of cancer cells, as observed by the 2-fold increase in caspase-3 activity, and also led to an improvement in PTT depth effectiveness (Fig. 3B).
In 2020, Huff et al. developed PEDOT NPs to not only evaluate their potential as PTT agents, but also to investigate the causes of PTT-induced cell death.66 Therapies such as PTT offer a means to heat targeted cancer cells to temperatures that lead to the release of damage-associated molecular patterns (DAMPs), which are the key signals required to activate dendritic cells and tumour-targeting T cells. Specific DAMPs like surface-exposed calreticulin and secreted high mobility group box 1 (HMGB1) serve as adjuvants to stimulate antigen-presenting cells and modulate a strong anti-tumour immune response. Thus, PTT offers opportunities not only for direct cancer cell death through hyperthermia, but also for activation of immunogenic cell death pathways. The synthesised spherical PEDOT NPs, with a size of approximately 40 nm, presented the ability to strongly absorb NIR light between 700 and 900 nm. Upon irradiation with an 808 nm NIR laser at 3 W cm−2, 10 to 500 μg mL−1 of PEDOT NPs internalised in MDA-MB-231 cancer cells generated an increase in temperature of 32 °C. This level of photothermal heating was adequate to induce cell death after 5–15 min of laser exposure. It was observed that the NPs caused cell death through apoptosis and necrosis, depending on the concentration and incubation time. Apoptosis was more prominent with shorter incubation times, while necrosis was more common with longer incubation times (6–24 h). In addition, the results of this study showed that the emergence of DAMPs was produced by PEDOT NP-mediated PTT. Particularly, cells subjected to higher heat doses showed increased calreticulin translocation to the cell membrane, increased cytosolic HMGB1, and decreased internal ATP. Therefore, PTT employing PEDOT NPs has the potential for the selective killing of cancer cells through immunogenic cell death (Fig. 3C).
In a very recently published study,67 a dynamic thiol-Michael hydrogel with embedded PEDOT NPs was synthesised as a photothermally responsive material for controlled drug release,67 taking advantage of hydrogels’ capacity to absorb and retain a large amount of water and their tuneable characteristics and customisable manufacturing methods.135 In this study, spherical PEDOT NPs with a diameter of 50 nm and NIR optical absorption were prepared via oxidative emulsion polymerisation and incorporated in situ during hydrogel formation. These PEDOT-composite hydrogels (Gel/PEDOT) underwent a reversible gel-to-solution transition at 45–50 °C, which enabled thermoresponsive and NIR-laser-triggered release of the encapsulated model protein. For the release studies, bovine serum albumin labelled with fluorescein (BSA-FITC) was used, and pulsatile release was successfully demonstrated over 7 temperature cycles. Laser irradiation at 808 nm (2.65 W cm−2, 15 min) induced photothermal heating of the PEDOT NPs to trigger BSA-FITC release on demand. The cytocompatibility of Gel/PEDOT was confirmed in both MDA-MB-231 breast cancer cells and 3T3 fibroblasts. Additionally, DOX could be encapsulated and laser-triggered released, retaining bioactivity to induce breast cancer cell death (Fig. 4D).
The properties of PEDOT that make it an effective PTT agent can be utilised not only in the form of PEDOT NPs, but also as a coating applied onto the surface (shell) of other NP cores. This allows both taking advantage of PEDOT's excellent properties for photothermal ablation and good biocompatibility, as well as the intrinsic functionality of the NP core for applications like imaging or drug delivery. Based on that, iron oxide (Fe3O4) NPs doped with 10-hydroxycamptothecin (HCPT) drug nanorods (NR) coated with PEDOT were developed.101 Upon irradiation with an 808 nm NIR laser at a power density of 1 W cm−2 for 10 min, the nanostructures showed excellent photothermal heating, reaching a temperature of 45 °C. In vitro combination of chemotherapy and PTT led to less than 5% of 4T1 and HeLa cell viability. In vivo therapy studies involved intravenously injecting BALB/c mice bearing 4T1 tumours with the nanostructures at a dosage of 6 mg kg−1, and the PTT regimen resulted in complete tumour eradication, with 100% survival for over 48 days.
Similarly, Yan et al. also synthesised core–shell Fe3O4 NPs coated with PEDOT:PSS, Fe3O4@PEDOT:PSS.102In vivo multimodal imaging-guided hyperthermia was conducted in MCF-7 tumour-bearing mice intravenously injected with the NPs solution at a concentration of 0.8 mg mL−1, with each mouse receiving 200 μL of the solution, combining the optical and magnetic features of the synthesised NPs. On the second day of treatment, a remarkable decrease in tumour volume was observed, and tumours were completely eliminated after 16 days of treatment in the hyperthermia + NPs group. Next, the authors fabricated trimodal NPs by using a combination of Fe3O4 NPs, PEDOT:PSS, Cyanine7 (Cy7), and 2-deoxyglucose (2-DG)-polyethylene glycol (MNP@PES-Cy7/2-DG).103 Due to the inclusion of 2-DG, a glucose analogue, NPs uptake was enhanced for more selective targeting of tumour cells. With this strategy, MCF-7 cells showed significant intracellular uptake of MNP@PES-Cy7/2-DG, with combined NIR laser (808 nm with a power density of 0.75 W cm−2) and alternating magnetic field exposure leading to more than 96% of MCF-7 cells undergoing apoptosis. In MCF-7 tumour-bearing mice, intravenous injected NPs (0.15 mL at a concentration of 500 μg mL−1) exhibited long blood circulation (half-life of 20.38 ± 4.18 h) and high tumour accumulation. Photothermal-magnetic hyperthermia under the guidance of NIR fluorescence, photoacoustic, and magnetic resonance multimodal imaging, resulted in complete ablation of tumours with no recurrence or toxicity over 35 days (Fig. 4E). As such, one can say this study yielded a promising foundation for photomagnetic hyperthermia therapy guided by imaging in cancer treatment.
Odda et al.97 further expanded on the use of PEDOT-coated surface-engineered iron oxide NPs (α-Fe2O3@PEDOT) by loading them with siRNA for combination photothermal-gene therapy. The 65 nm NPs positively charged surfaces facilitated siRNA Bcl-2 (B-cell lymphoma-2) loading and PEDOT coating enhanced NIR absorption. Also, the formulated NPs demonstrated high photothermal conversion efficacy (54.3%) and photostability under NIR laser irradiation (808 nm laser with a power intensity of 1.0 W cm−2 for 10 min). Combination photothermal-gene therapy using Bcl-2 siRNA loaded particles (at concentrations up to 100 μg mL−1) showed a significant decrease in cell viability of MCF-7 (to 24%) and MDA-MB-231 (to 21%) cells compared to either therapy alone. Additionally, in mice xenograft models, α-Fe2O3@PEDOT-siRNA combined with NIR laser treatment resulted in almost complete tumour elimination demonstrating a highly effective gene/PTT synergistic antitumour effect.
Overall, the use of PEDOT NPs in PTT applications for cancer therapy appears to often involve stabilisation of the NPs with other polymer layers, with PSS and PEG being the most reported. Alternatively, encapsulation of PEDOT NPs within hydrogels, nanogels and polymeric shells appears to be an effective strategy. One can conclude that interactions with other polymeric matrices or chains do not appear to hinder PEDOT's capacity to respond to NIR, thus being a very promising PTT agent.
3.2. Electrically triggered drug delivery
To overcome the concerns related to currently administered chemotherapy, the development of new drug delivery strategies has been prompted. Particularly, the use of CP systems, including hydrogels, NPs, and nano- and microfibres, has shown the potential for delivering accurate dosages of drugs.20,136,137 Moreover, external stimuli have been used to trigger the delivery of drugs at specific locations and to control drug release rates. These stimuli can be endogenous (such as changes in pH, redox or enzymatic reactions) or exogenous (such as radiation, changes in temperature, ultrasounds, magnetic, and electric).10Electroresponsive devices of polymeric structure have shown great promise for on-demand drug release, with the potential to be used in localised treatment through wireless controls. The oxidation–reduction properties of CPs interfere in the electrostatic forces that exist between the drug and the charged polymer, thus applying external voltages may promote the formation or disruption of drug-polymer interactions.20 Furthermore, the electromechanical response of CPs allows for the intrinsic expansion and contraction of the polymers, which can be exploited to trigger the drug's mechanical release.23,138
PEDOT has been widely utilised to load multiple drugs, including those used in cancer therapy, for subsequent controlled release by applying electrical stimulation.139,140
Regarding the use of PEDOT NPs, Alemán's group has been focusing on these nanoplatforms for electrostimulated drug delivery of anticancer drugs. In an initial study, they investigated the controlled release of piperine (PIP) and curcumin (CUR) using PEDOT NPs as nanocarriers, with the drugs being loaded during emulsion polymerisation synthesis of NPs.25 CUR is particularly interesting as it exhibits a broad range of therapeutic features, including anticancer, antiviral, antifungal, antibacterial, and anti-inflammatory properties.141 Kinetics experiments revealed a significantly slower, more gradual release profile for CUR compared to PIP from the NPs, suggesting that CUR and PEDOT NPs had stronger polymer–drug interactions. CUR release of up to 38% of the loaded drug was significantly increased when a negative voltage potential of −1.25 V was applied to CUR/PEDOT NPs for 3 min. In contrast, PIP release was not enhanced, indicating that PIP does not have strong interactions with PEDOT, but the interactions between CUR and PEDOT NPs were responsive to electrical control due to charge changes along the polymeric matrix disrupting the interactions between CUR and PEDOT. Human prostate adenocarcinoma (PC3) and breast adenocarcinoma (MCF-7) cancer cell lines were subjected to increasing concentrations of free CUR and CUR/PEDOT NPs for 24 h to assess the cytotoxicity of the treatment. As a whole, the study showed that the electroresponsive CUR/PEDOT NPs system exhibits potential for spatiotemporally-regulated dosage, which is valuable for CUR-based anticancer therapy. In a follow-up study, a hybrid system was developed by incorporating CUR/PEDOT into a poly(γ-glutamic acid) (PGGA) hydrogel.24 The rationale was to leverage the biocompatibility and environmentally friendly nature of the PGGA hydrogel combined with the electrochemical properties of PEDOT NPs to further enhance the electrically controlled release of CUR. Passive diffusion-based release of hydrophobic CUR from the hydrogel/NP system was slow. However, applying −0.5 V electrical pulses for 15 min every 24 h significantly increased CUR release by more than 2-fold.
Despite their advantages, NPs present some limitations that make them less effective as nanocarriers, which are mostly associated with their propensity to aggregate under physiological conditions, causing them to lose their intended nanoscale properties.142 NP aggregation often hinders drug release from the particles, even when external stimuli are applied due to the difficulty of the drug to diffuse through polymer chains. Based on this observation, Alemán's group adopted a strategy to prevent this unwanted NP aggregation, developing a new electrosensitive bioplatform by dispersing PEDOT NPs in electrospun poly(ε-caprolactone) (PCL) microfibres (MFs),23 which act as a stabilising network to maintain nanoscale properties and facilitate controlled drug release from the embedded NPs.143 In this work, the PCL MFs were loaded with CUR and PEDOT NPs with a diameter of 99 ± 21 nm.23 Applying 1.0 V potential pulses (5 pulses of 60 s) electrically triggered significantly high CUR release from PCL/PEDOT/CUR MFs of up to 30%. This release was induced by volume changes in the PEDOT NPs that disrupted the PCL matrix, behaving as isotropic actuators upon electrostimulation (Fig. 5A). In our group, in a recent study using PEDOT/CUR NPs,104 adaptations were made to the NP manufacturing (earlier heating in the synthesis process, scale-up and prolonged reaction time), and to the electrical stimulation setup, which was based on carbon screen printed electrodes, instead of a conventional three-electrode system, allowing to design a more efficient system for the release of CUR from PEDOT NPs (65% of CUR release after applying a potential of −1.5 V for 180 s). Additionally, a wireless electrostimulation platform using these CUR/PEDOT NPs encapsulated inside coaxial poly(glycerol sebacate)/PCL (PGS/PCL) electrospun fibres was set up which promoted a decrease of 67% in cancer cell viability.104
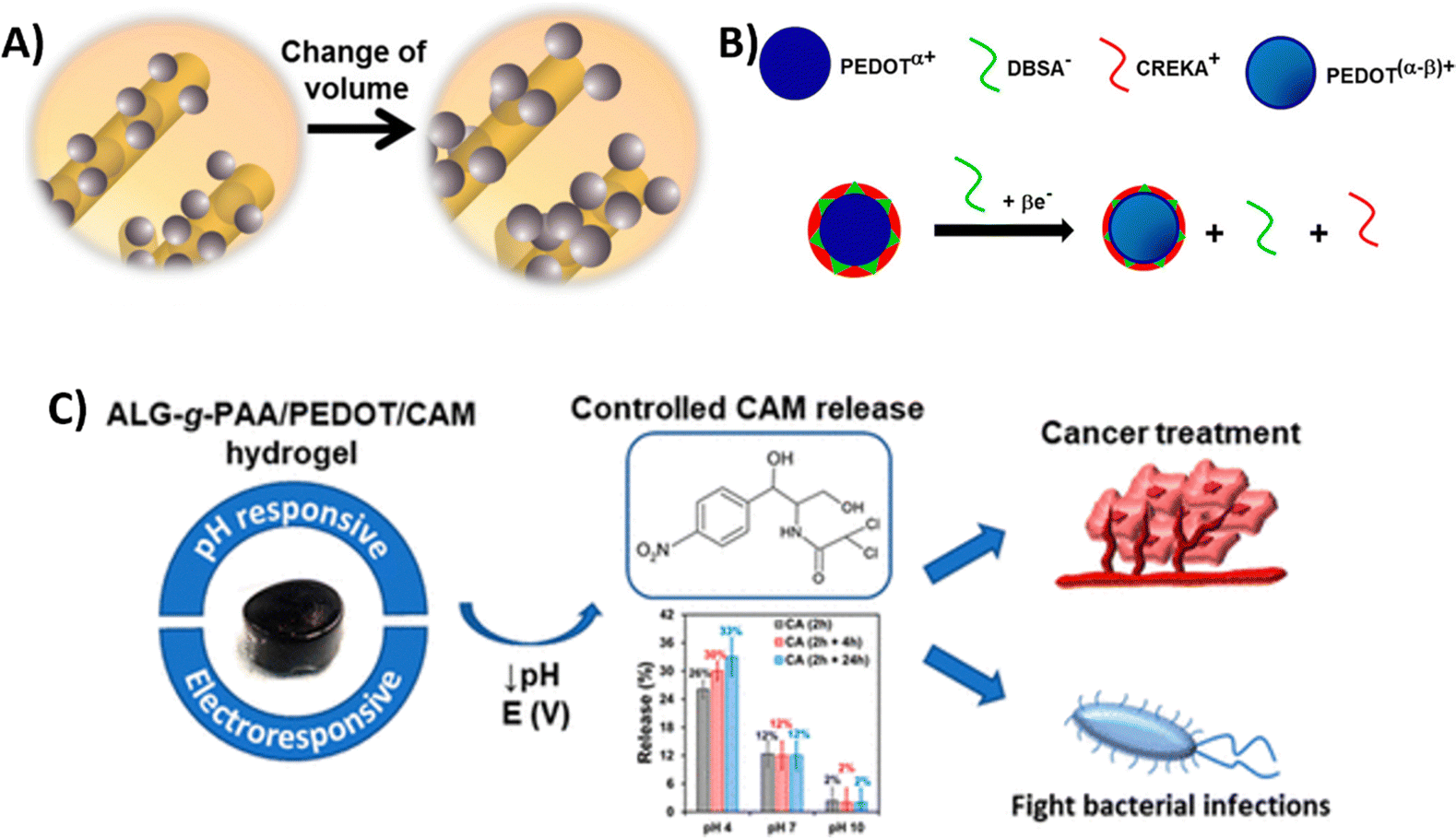 | ||
| Fig. 5 Overview of the studies employing PEDOT NPs-based systems in anticancer electrostimulated drug delivery. (A) Schematic representing the electroactuation mechanism followed by PCL/PEDOT/CUR MFs to release CUR upon electrostimulation. Reproduced with permission from ref. 23. Copyright 2018. American Chemical Society. (B) Schematic representing the mechanism of peptide release from CREKA/PEDOT NPs. Reproduced with permission from ref. 73. Copyright 2020. American Chemical Society. (C) Schematic representing the work developed by formulation of Alg-g-PAA/PEDOT/CAM system and electrochemical release of CAM.27 | ||
Another study developed 35–47 nm PEDOT NPs to load and achieve controlled, electrically-triggered release of the fibrin-targeting peptides Cys-Arg-Glu-Lys-Ala (CREKA) and CR(NMe)EKA. The latter being a CREKA analogue with enhanced peptide resistance against proteolysis, which is one of the major limitations of therapeutic peptide delivery.73 These pentapeptides interact with fibrin clots and have high targeting ability to fibrin–fibronectin complexes in animal tumour models, and these can be employed in cancer diagnosis and treatments.144 The results of the study showed that PC3 prostate cancer cells were more susceptible to peptide-loaded particles than healthy cells. Applying cyclic voltammetry electrical stimuli to peptide-loaded NPs caused significantly increased peptide release (up to 38% release after 100 cycles), when compared to absence of stimulation. The mechanism of peptide release can be explained by peptide···PEDOT interactions becoming weaker when polymer chains are reduced and oxidised by an external voltage (Fig. 5B). In a subsequent study,105 the group found that after loading CR(NMe)EKA/PEDOT NPs into an injectable pH responsive hydrogel, formed by phenylboronic acid grafted to chitosan (PBA-CS), the efficiency of the controlled peptide release increases approximately by a factor of 2.6.
The same group explored the use of PEDOT NPs to load in situ the pharmacological chaperone PYR,22 a powerful inducer of apoptosis in cancer cells, such as metastatic melanoma cells,145 and an antiparasitic drug against infections caused by protozoan parasites. In aqueous medium, passive PYR release from NPs was very slow, with only 1.6% released in 24 hours and 18% released in 80 days without electrical stimuli. Applying cyclic voltammetry, with a scan range from −0.5 V to 0.5 V, enhanced PYR release to approximately 50% after 30 min, while performing chronoamperometry at constant voltage of 1.0 V also increased the release to approximately 35% after 30 min. Biocompatibility assays showed that toxicity of loaded NPs in osteosarcoma MG-63 cells was very low in the absence of electrical stimulation.
Integrating PEDOT NPs coated with anodically polymerised poly(hydroxymethyl-3,4-ethylenedioxythiophene) (PHMeEDOT) chains into a (PGGA) biohydrogel allowed for the development of a vitamin K3 (or menadione) release system that could monitor the concentration of the drug being administered.74 Vitamin K3 molecules organised in shells surrounding PEDOT NPs agglomerates when the drug was added to the initial gelling solution, and were gradually released to a physiological medium. With vitamin K3 acting as the active principle in many diseases, including cancer,117 this integrated system holds great promise for the creation of theragnostic treatment systems.146
Very recently, researchers developed a hydrogel system for the controlled release of the antibiotic chloramphenicol (CAM).27 CAM, is also recognised as a potential option for cancer treatment, and has been reported to inhibit eukaryotic cells' mitochondrial function.147,148 PEDOT NPs loaded with CAM (PEDOT/CAM NPs) were incorporated into a pH-responsive poly(acrylic acid)-grafted sodium alginate (Alg-g-PAA) hydrogel. Passive CAM release from NPs was approximately 14% per hour but increased to (89 ± 5)% over 9 hours with chronoamperometric stimulation. Passive release from the Alg-g-PAA/PEDOT/CAM hydrogel was negligible after 24 hours at all different pH values tested, but 2 h electrostimulation resulted in substantially higher CAM release at pH 4 (30%), when compared to pH 7 (12%) and pH 10 (2%). Viability assays using HeLa cancer cells showed a concentration-dependent toxicity, confirming CAM bioactivity was retained after encapsulation and electrostimulated release. Using a dual electro-chemo stimulation, the combined conducting Alg-g-PAA/PEDOT/CAM hydrogel thus revealed its promise for cancer treatment by enabling regulated, targeted CAM distribution (Fig. 5C). It is interesting to note that both this study and a previous one conducted by the same group22 demonstrated that the drug-loaded PEDOT NPs' electrostimulated release of CAM inhibits bacterial growth, making it a potentially effective theragnostic system for the treatment of bacterial infections.26
Overall, we can conclude that PEDOT NPs are very versatile in terms of the choice of anticancer drug to be loaded into the particles, as seen from the wide variety of drugs discussed, from small molecules to peptides. It is also important to note that the diversity of electrostimulation techniques deals with voltages, that are safe for the human body, lowering potential risks for patients. Additionally, the integration of PEDOT NPs into other stimuli-responsive systems allows for a very highly controlled drug release, which is of utmost importance in cancer therapy.
Moreover, although the studies presented in this subsection do not yet evaluate in vivo responses to electrically controlled drug release, there are indeed studies in the literature demonstrating that drug-loaded conducting polymer NPs can be injected in vivo with drug release stimulated using microelectrodes.149 This suggests that the strategies employing PEDOT NPs described here could follow the same course. In the future, these NPs could be integrated with a wirelessly controlled, miniaturized implantable chip for precise drug delivery, where the NPs are localized within a hydrogel or semi-permeable membrane that allows selective drug passage, with the chip providing the necessary electrical stimulation for controlled drug release.
4. Challenges in the clinical application of PEDOT NPs & future directions
In the last few decades, there has been an increase in the amount of knowledge on nanoscale systems and development of NPs. Still, most of the research remains in the in vitro and in vivo phases, with very few advancing to clinical trials. Research on PEDOT NPs is no exception, and their translation into clinical applications faces several biological, technological, and legal challenges.150Regarding the biological part, understanding the pharmacokinetics (absorption, distribution, metabolism, and excretion) of the NPs as well as evaluating systemic toxicity and immunotoxicity are crucial to ensure that the NPs do not harm the overall health of the patient and do not cause inflammation or immunosuppression.10 Controlling the fate of PEDOT NPs in vivo remains difficult. While attempts have been made to modulate NPs to increase circulation time and tumour retention, risks of accumulation-related toxicity in liver, lungs, and kidneys persist.11 To address this challenge, future studies should explore biodegradable PEDOT formulations. Moreover, aggregation in biological fluids alters NP physicochemical properties and reduces therapeutic efficacy. Also, protein corona formation leads to uptake of the NPs by the mononuclear phagocytic system, which must be avoided.11,12 Developing stealth PEDOT NPs with reduced protein adsorption and improved stability in biological fluids represents a key area for future research.
Technological challenges include scale-up synthesis, forecasting clinical performance based on preclinical data and achieving scalable production of NPs verified by Good Manufacturing Practices (GMP). Clinical performance is further impacted by study design weaknesses, such as small sample sizes, and overreliance on animal models that do not translate effectively to humans.11,151
Finally, regulatory approval is another major bottleneck, as each specific formulated PEDOT NP system (which may present different synthesis technique, dopant, or functionalisation of the polymer) requires extensive safety and efficacy validation per FDA (USA) and CE (Europe) guidelines before marketing as a medical device. Manufacturers must demonstrate the goods’ short- and long-term safety and efficacy for the human body in order to receive regulatory clearance. Although this constitutes a costly and time-consuming process, which has hampered widespread clinical adoption so far,10–12,28 there are some clinically approved NP formulations being used to treat a variety of cancers at different stages.10
Therefore, to address these challenges, there is a necessity for advanced high-throughput preclinical platforms. Organ-on-a-chip technologies, ex vivo human tissue models, and 3D cell culture systems are a few examples of these animal-free platforms, which enable rapid, cost-effective, and ethically acceptable testing of PEDOT NPs, while having the potential to more closely mimic human physiological environments.152 For instance, organ-on-a-chip microfluidics devices and 3D in vitro models mimic important features of the tumour microenvironment and vasculature allowing a more thorough understanding of tumour extravasation of NPs in patients. By modelling the interactions among different tissue types, organ-on-a-chip microfluidics devices offer insights into potential off-target effects and biodistribution.153,154
Additionally, cancer patients who are predicted to have preferential tumour accumulation of nanomedicines, and thus are most likely to benefit from nanotherapeutics, can be stratified prior to the initiation of treatment by employing imaging agents in conjunction with EPR-predictive biomarkers.152
To improve scalability, future efforts should focus on developing continuous-flow synthesis methods for PEDOT NPs, which could offer better control over NP properties and facilitate large-scale production.155
Although not yet used specifically for PEDOT NPs, artificial intelligence (AI) emerges as a promising approach for advancing NP-based cancer therapies.10 AI algorithms have the potential to optimise multiple aspects of PEDOT NP design, including size, charge, drug encapsulation efficiency, and interactions with biological systems. AI enhances the precision of therapeutic payload delivery, improves selective targeting of cancer cells, and enables smart circulation of PEDOT NPs through complex tumour microenvironments through the use of machine learning and computational models.156 Furthermore, the development of new PEDOT NP formulations and chemical synthesis may be improved with the use of AI, potentially addressing the current challenges with reproducibility and scalability.157,158 As research in this field grows, the integration of AI technologies with PEDOT NP development represents a promising path forward for enhancing their efficacy and clinical translation in cancer treatment.
Close collaboration between researchers, regulatory authorities, and industry partners is key to overcoming regulatory hurdles. More specifically, the establishment of a regulatory framework specifically tailored to nanomedicine could simplify the approval process for PEDOT NPs and similar technologies. In addition, the development of standardised characterisation methods and quality control protocols for PEDOT NPs may help to enable regulatory compliance and increase reproducibility across research groups.
Therefore, interdisciplinary teamwork between researchers, clinicians, engineers and regulators, and the use of computational models and advanced preclinical technologies, rewards addressing biological, technological and regulatory challenges, thus improving the clinical translation of the use of PEDOT NPs for therapeutics against cancer.
5. Conclusions
The considerable potential of PEDOT NPs to serve as versatile anticancer theragnostic agents has been discussed in this review. We have outlined common techniques for synthesising NPs with size, morphology, electrical conductivity, and surface chemistry that are tailored by adjusting the temperature, reaction time, stabilisers, and oxidants. Furthermore, extensive structural, electrochemical, and thermal characterisation relates synthetic conditions to resultant NP behaviour and characteristics. Particularly, PEDOT NPs exhibit efficient NIR absorption and photothermal conversion, making them useful for targeted, minimally invasive tumour photothermal ablation. Electrical stimulation for triggering the on-demand release of therapeutic agents is further facilitated by their intrinsic conductivity and electrochemical activity.PEDOT NP-based platforms that combine drug delivery, photothermal heating, and imaging with temporal and spatial control have been designed in recent research. The potential of these nanosystems to concentrate in tumours, induce highly localised hyperthermia under NIR irradiation, and deliver anticancer drugs in response to electrical stimuli is confirmed by in vitro and in vivo research. Researchers must continue to systematically examine how slight modifications to the engineering of PEDOT NPs may affect therapeutic efficacy. The development of smart anticancer nanotheragnostics will be fuelled by the optimisation of synthesis procedures in conjunction with cytotoxicity testing and preclinical models. By addressing remaining problems in an interdisciplinary and cooperative manner, PEDOT NPs can fulfil their therapeutic potential and revolutionise cancer treatment.
We believe that our thorough examination of the fundamental concepts behind the synthesis, properties, and anticancer applications of PEDOT NPs has assisted in clearly illustrating these systems as powerful tools for personalised cancer nanomedicine. Future innovation should therefore concentrate on customising NP design for these highly tailored and targeted therapeutic modalities.
Author contributions
This review was conceptualised by FF, PS-A and TE. DD contributed to writing original draft. LR, FF, PS-A and TE contributed to the review and editing of this manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge funding from FCT—Portuguese Foundation for Science and Technology (FCT/MCTES), with dedicated funding from the project eOnco (2022.07252.PTDC), the PhD scholarship (SFRH/BD/145057/2019) and also through institutional funds to iBB (UIDB/04565/2020 and UIDP/04565/2020) and Associate Laboratory i4HB (LA/P/0140/2020). This project also received financial support from “la Caixa” Foundation (ID 100010434) LCF/BQ/PI22/11910025.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- L. Sapio and S. Naviglio, Int. J. Mol. Sci., 2022, 23, 5296 CrossRef PubMed.
- K. Nurgali, J. A. Rudd, H. Was and R. Abalo, Front. Pharmacol., 2022, 13, 1007762 CrossRef PubMed.
- R. Baghban, L. Roshangar, R. Jahanban-Esfahlan, K. Seidi, A. Ebrahimi-Kalan, M. Jaymand, S. Kolahian, T. Javaheri and P. Zare, Cell Commun. Signaling, 2020, 18, 59 CrossRef PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- M. Dessale, G. Mengistu and H. M. Mengist, Int. J. Nanomed., 2022, 17, 3735–3749 CrossRef PubMed.
- Y. Zheng, L. Michalek, Q. Liu, Y. Wu, H. Kim, P. Sayavong, W. Yu, D. Zhong, C. Zhao, Z. Yu, J. A. Chiong, H. Gong, X. Ji, D. Liu, S. Zhang, N. Prine, Z. Zhang, W. Wang, J. B.-H. Tok, X. Gu, Y. Cui, J. Kang and Z. Bao, Nat. Nanotechnol., 2023, 18, 1175–1184 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- J. Yin, S. Wang, T. Tat and J. Chen, Nature Rev. Bioeng., 2024, 2, 541–558 CrossRef.
- L. Sun, H. Liu, Y. Ye, Y. Lei, R. Islam, S. Tan, R. Tong, Y. B. Miao and L. Cai, Signal Transduction Targeted Ther., 2023, 8 Search PubMed.
- S. Gavas, S. Quazi and T. M. Karpiński, Nanoscale Res. Lett., 2021, 16, 173 CrossRef CAS PubMed.
- J. Baranwal, B. Barse, A. Di Petrillo, G. Gatto, L. Pilia and A. Kumar, Materials, 2023, 16, 5354 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- J. Wu, J. Pers. Med., 2021, 11, 771 CrossRef PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- R. Zeineldin and J. Syoufjy, 2017, pp. 3–12.
- T.-H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef PubMed.
- K. Namsheer and C. S. Rout, RSC Adv., 2021, 11, 5659–5697 RSC.
- J. Torras, J. Casanovas and C. Alemán, J. Phys. Chem. A, 2012, 116, 7571–7583 CrossRef CAS PubMed.
- A. Puiggalí-Jou, L. J. del Valle and C. Alemán, J. Controlled Release, 2019, 309, 244–264 CrossRef PubMed.
- Z. Rahimzadeh, S. M. Naghib, Y. Zare and K. Y. Rhee, J. Mater. Sci., 2020, 55, 7575–7611 CrossRef CAS.
- H. Enshaei, A. Puiggalí-Jou, N. Saperas and C. Alemán, Soft Matter, 2021, 17, 3314–3321 RSC.
- A. Puiggalí-Jou, A. Cejudo, L. J. Del Valle and C. Alemán, ACS Appl. Bio Mater., 2018, 1, 1594–1605 CrossRef PubMed.
- H. Enshaei, B. G. Molina, A. Puiggalí-Jou, N. Saperas and C. Alemán, Eur. Polym. J., 2022, 173, 111199 CrossRef CAS.
- A. Puiggalí-Jou, P. Micheletti, F. Estrany, L. J. del Valle and C. Alemán, Adv. Healthcare Mater., 2017, 6, 1700453 CrossRef PubMed.
- H. Enshaei, A. Puiggalí-Jou, L. J. del Valle, P. Turon, N. Saperas and C. Alemán, Adv. Healthcare Mater., 2021, 10, 2001636 CrossRef CAS PubMed.
- L. Resina, K. El Hauadi, J. Sans, T. Esteves, F. C. Ferreira, M. M. Pérez-Madrigal and C. Alemán, Biomacromolecules, 2023, 24, 1432–1444 CrossRef CAS PubMed.
- C. Boehler, Z. Aqrawe and M. Asplund, Bioelectron. Med., 2019, 2, 89–99 CrossRef.
- N. Gao, J. Yu, Q. Tian, J. Shi, M. Zhang, S. Chen and L. Zang, Chemosensors, 2021, 9, 79 CrossRef CAS.
- S. Stříteský, A. Marková, J. Víteček, E. Šafaříková, M. Hrabal, L. Kubáč, L. Kubala, M. Weiter and M. Vala, J. Biomed. Mater. Res., Part A, 2018, 106, 1121–1128 CrossRef PubMed.
- S. Ummartyotin, J. Juntaro, C. Wu, M. Sain and H. Manuspiya, J. Nanomater., 2011, 2011, 1–7 CrossRef.
- M. F. F. das Neves, J. P. V. Damasceno, O. D. L. Junior, A. J. G. Zarbin and L. S. Roman, Synth. Met., 2021, 272, 116657 CrossRef CAS.
- Z. Liu, B. Lu, Y. Gao, T. Yang, R. Yue, J. Xu and L. Gao, RSC Adv., 2016, 6, 89157–89166 RSC.
- R. Madhuvilakku, S. Alagar, R. Mariappan and S. Piraman, Anal. Chim. Acta, 2020, 1093, 93–105 CrossRef CAS PubMed.
- B. G. Molina, L. J. del Valle, P. Turon, E. Armelin and C. Alemán, J. Phys. Chem. C, 2019, 123, 22181–22190 CrossRef CAS.
- V. Chellappa, J. Annaraj and S. Suresh, Mater. Chem. Phys., 2024, 319, 129330 CrossRef CAS.
- J. W. Park, W. Na and J. Jang, J. Mater. Chem. A, 2016, 4, 8263–8271 RSC.
- I. Adam-Cervera, J. Huerta-Recasens, C. M. Gómez, M. Culebras and R. Muñoz-Espí, Polymers, 2023, 16, 100 CrossRef PubMed.
- M. Rajesh, C. J. Raj, B. C. Kim, R. Manikandan, S.-J. Kim, S. Y. Park, K. Lee and K. H. Yu, RSC Adv., 2016, 6, 110433–110443 RSC.
- A. Serafin, M. C. Rubio, M. Carsi, P. Ortiz-Serna, M. J. Sanchis, A. K. Garg, J. M. Oliveira, J. Koffler and M. N. Collins, Biomater. Res., 2022, 26, 63 CrossRef CAS PubMed.
- Y. Han, M. Sun, X. Lu, K. Xu, M. Yu, H. Yang and J. Yin, Composites, Part B, 2024, 273, 111241 CrossRef CAS.
- S. Guan, Y. Wang, F. Xie, S. Wang, W. Xu, J. Xu and C. Sun, Molecules, 2022, 27, 8326 CrossRef CAS PubMed.
- A. Escobar, A. Serafin, M. R. Carvalho, M. Culebras, A. Cantarero, A. Beaucamp, R. L. Reis, J. M. Oliveira and M. N. Collins, Adv. Compos. Hybrid Mater., 2023, 6, 118 CrossRef CAS.
- Y. Wang, Q. Wang, S. Luo, Z. Chen, X. Zheng, R. K. Kankala, A. Chen and S. Wang, Regener. Biomater., 2021, 8, rbab035 CrossRef PubMed.
- M. Culebras, B. Uriol, C. M. Gómez and A. Cantarero, Phys. Chem. Chem. Phys., 2015, 17, 15140–15145 RSC.
- M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J.-P. Simonato, Prog. Mater. Sci., 2020, 108, 100616 CrossRef CAS.
- M. Culebras, J. F. Serrano-Claumarchirant, M. J. Sanchis, K. Landfester, A. Cantarero, C. M. Gómez and R. Munoz-Espí, J. Phys. Chem. C, 2018, 122, 19197–19203 CrossRef CAS.
- S. Nie, Z. Li, Y. Yao and Y. Jin, Front. Chem., 2021, 9, 803509 CrossRef CAS PubMed.
- N. Paradee and A. Sirivat, Polym. Int., 2014, 63, 106–113 CrossRef CAS.
- S. M. Marinakos, D. A. Shultz and D. L. Feldheim, Adv. Mater., 1999, 11, 34–37 CrossRef CAS.
- L. Hao, C. Zhu, C. Chen, P. Kang, Y. Hu, W. Fan and Z. Chen, Synth. Met., 2003, 139, 391–396 CrossRef CAS.
- M. G. Han and S. H. Foulger, Chem. Commun., 2004, 2154–2155 RSC.
- M. A. Khan and S. P. Armes, Langmuir, 1999, 15, 3469–3475 CrossRef CAS.
- M. Martí, G. Fabregat, F. Estrany, C. Alemán and E. Armelin, J. Mater. Chem., 2010, 20, 10652–10660 RSC.
- S. Luo, H. Yu, A. C. A. Wan, Y. Han and J. Y. Ying, Small, 2008, 4, 2051–2058 CrossRef CAS PubMed.
- T. L. Kelly and M. O. Wolf, Chem. Soc. Rev., 2010, 39, 1526–1535 RSC.
- L. Xia, Z. Wei and M. Wan, J. Colloid Interface Sci., 2010, 341, 1–11 CrossRef CAS PubMed.
- N. Elgiddawy, N. Elnagar, H. Korri-Youssoufi and A. Yassar, Microorganisms, 2023, 11, 2006 CrossRef CAS PubMed.
- S.-G. Oh and S.-S. Im, Curr. Appl. Phys., 2002, 2, 273–277 CrossRef.
- J. W. Choi, M. G. Han, S. Y. Kim, S. G. Oh and S. S. Im, Synth. Met., 2004, 141, 293–299 CrossRef CAS.
- K. Müller, M. Klapper and K. Müllen, Macromol. Rapid Commun., 2006, 27, 586–593 CrossRef.
- M. Mumtaz, A. De Cuendias, J. L. Putaux, E. Cloutet and H. Cramail, Macromol. Rapid Commun., 2006, 27, 1446–1453 CrossRef CAS.
- H. Zheng, Y. Jiang, J. Xu and Y. Yang, Sci. China: Technol. Sci., 2010, 53, 2355–2362 CrossRef CAS.
- Y. Han, J. Yih, M. Chang, W. Huang, K. Ho, T. Hsieh and J. Lou, Macromol. Chem. Phys., 2011, 212, 361–366 CrossRef CAS.
- T. Cantu, K. Walsh, V. P. Pattani, A. J. Moy, J. W. Tunnell, J. A. Irvin and T. Betancourt, Int. J. Nanomed., 2017, 12, 615–632 CrossRef CAS PubMed.
- M. E. Huff, F. O. Gokmen, J. S. Barrera, E. J. Lara, J. Tunnell, J. Irvin and T. Betancourt, ACS Appl. Polym. Mater., 2020, 2, 5602–5620 CrossRef CAS.
- K. Thapa, T. M. FitzSimons, M. U. Otakpor, M. M. Siller, A. D. Crowell, J. E. Zepeda, E. Torres, L. N. Roe, J. Arts, A. M. Rosales and T. Betancourt, ACS Appl. Mater. Interfaces, 2023, 15, 52180–52196 CAS.
- C.-H. Wu, T.-M. Don and W.-Y. Chiu, Polymer, 2011, 52, 1375–1384 CrossRef CAS.
- L. Li, Y. Liu, P. Hao, Z. Wang, L. Fu, Z. Ma and J. Zhou, Biomaterials, 2015, 41, 132–140 CrossRef CAS PubMed.
- D. Liu, L. Ma, Y. An, Y. Li, Y. Liu, L. Wang, J. Guo, J. Wang and J. Zhou, Adv. Funct. Mater., 2016, 26, 4749–4759 CrossRef CAS.
- K. Muro, M. Watanabe, T. Tamai, K. Yazawa and K. Matsukawa, RSC Adv., 2016, 6, 87147–87152 RSC.
- G. Cao, S. Cai, Y. Chen, D. Zhou, H. Zhang and Y. Tian, Polymer, 2022, 252, 124952 CrossRef CAS.
- A. Puiggalĺ-Jou, L. J. Del Valle and C. Alemán, ACS Biomater. Sci. Eng., 2020, 6, 2135–2145 CrossRef PubMed.
- B. Molina, E. Domínguez, E. Armelin and C. Alemán, Gels, 2018, 4, 86 CrossRef CAS PubMed.
- M. W. Lee, D. J. Kwon, J. Park, J. C. Pyun, Y. J. Kim and H. S. Ahn, Chem. Commun., 2020, 56, 9624–9627 RSC.
- N. A. Shahrim, Z. Ahmad, A. Wong Azman, Y. Fachmi Buys and N. Sarifuddin, Mater. Adv., 2021, 2, 7118–7138 RSC.
- Y. Kudoh, K. Akami and Y. Matsuya, Synth. Met., 1998, 98, 65–70 CrossRef CAS.
- R. S. Chaurasiya and H. U. Hebbar, 2017, pp. 181–211.
- E. M. Wailes, C. M. MacNeill, E. McCabe and N. H. Levi-Polyachenko, J. Appl. Polym. Sci., 2016, 133, 43378 CrossRef.
- S. M. Shaban, J. Kang and D.-H. Kim, Compos. Commun., 2020, 22, 100537 CrossRef.
- T. Miyazawa, M. Itaya, G. C. Burdeos, K. Nakagawa and T. Miyazawa, Int. J. Nanomed., 2021, 16, 3937–3999 CrossRef PubMed.
- G. H. Zhu, A. B. C. Gray and H. K. Patra, Trends Pharmacol. Sci., 2022, 43, 709–711 CrossRef CAS PubMed.
- A. A. Yetisgin, S. Cetinel, M. Zuvin, A. Kosar and O. Kutlu, Molecules, 2020, 25, 2193 CrossRef CAS PubMed.
- M. Cooley, A. Sarode, M. Hoore, D. A. Fedosov, S. Mitragotri and A. Sen Gupta, Nanoscale, 2018, 10, 15350–15364 RSC.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- R. M. Visalakshan, L. E. G. García, M. R. Benzigar, A. Ghazaryan, J. Simon, A. Mierczynska-Vasilev, T. D. Michl, A. Vinu, V. Mailänder, S. Morsbach, K. Landfester and K. Vasilev, Small, 2020, 16, 2000285 CrossRef PubMed.
- L. Cheng, K. Yang, Q. Chen and Z. Liu, ACS Nano, 2012, 6, 5605–5613 CrossRef CAS PubMed.
- D. Titus, E. James Jebaseelan Samuel and S. M. Roopan, Green Synthesis, Characterization and Applications of Nanoparticles, Elsevier, 2019, pp. 303–319 Search PubMed.
- E. R. Fischer, B. T. Hansen, V. Nair, F. H. Hoyt, C. L. Schwartz and D. W. Dorward, Curr. Protoc. Microbiol., 2024, e1034 CrossRef PubMed.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- A. Puiggalí-Jou, S. Wedepohl, L. E. Theune, C. Alemán and M. Calderón, Mater. Sci. Eng., C, 2021, 119, 111598 CrossRef PubMed.
- F. Huth, A. Govyadinov, S. Amarie, W. Nuansing, F. Keilmann and R. Hillenbrand, Nano Lett., 2012, 12, 3973–3978 CrossRef CAS PubMed.
- C. Kvarnström, H. Neugebauer, S. Blomquist, H. J. Ahonen, J. Kankare and A. Ivaska, Electrochim. Acta, 1999, 44, 2739–2750 CrossRef.
- B. Ingham, Crystallogr. Rev., 2015, 21, 229–303 CrossRef.
- E. Korin, N. Froumin and S. Cohen, ACS Biomater. Sci. Eng., 2017, 3, 882–889 CrossRef CAS PubMed.
- A. H. Odda, T. Y. Cheang, H. F. Alesary, L. Liu, X. Qian, N. Ullah, G. Wang, Y. Pan and A. W. Xu, J. Mater. Chem. B, 2022, 10, 1453–1462 RSC.
- T.-H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef PubMed.
- H. Gong, L. Cheng, J. Xiang, H. Xu, L. Feng, X. Shi and Z. Liu, Adv. Funct. Mater., 2013, 23, 6059–6067 CrossRef CAS.
- C. M. MacNeill, E. M. Wailes and N. H. Levi-Polyachenko, J. Nanosci. Nanotechnol., 2013, 13, 3784–3791 CrossRef CAS PubMed.
- Y. Yang, X. Zhang, C. Yu, X. Hao, J. Jie, M. Zhou and X. Zhang, Adv. Healthcare Mater., 2014, 3, 906–915 CrossRef CAS PubMed.
- H. Yan, L. Zhao, W. Shang, Z. Liu, W. Xie, C. Qiang, Z. Xiong, R. Zhang, B. Li, X. Sun and F. Kang, Nano Res., 2017, 10, 704–717 CrossRef CAS.
- H. Yan, W. Shang, X. Sun, L. Zhao, J. Wang, Z. Xiong, J. Yuan, R. Zhang, Q. Huang, K. Wang, B. Li, J. Tian, F. Kang and S. S. Feng, Adv. Funct. Mater., 2018, 28, 1705710 CrossRef.
- L. Resina, F. F. F. Garrudo, C. Alemán, T. Esteves and F. C. Ferreira, Biomater. Adv., 2024, 160, 213830 CrossRef CAS PubMed.
- L. Resina, T. Esteves, S. Pérez-Rafael, J. I. H. García, F. C. Ferreira, T. Tzanov, S. Bonardd, D. D. Díaz, M. M. Pérez-Madrigal and C. Alemán, Biomater. Adv., 2024, 162, 213925 CrossRef CAS PubMed.
- A. Chicheł, J. Skowronek, M. Kubaszewska and M. Kanikowski, Rep. Pract. Oncol. Radiother., 2007, 12, 267–275 CrossRef.
- H. H. Kampinga, Int. J. Hyperthermia, 2006, 22, 191–196 CrossRef CAS PubMed.
- P. Kaur, M. L. Aliru, A. S. Chadha, A. Asea and S. Krishnan, Int. J. Hyperthermia, 2016, 32, 76–88 CrossRef CAS PubMed.
- S. Wang, C. Zhang, F. Fang, Y. Fan, J. Yang and J. Zhang, J. Mater. Chem. B, 2023, 11, 8315–8326 RSC.
- J. B. Vines, D. J. Lim and H. Park, Polymers, 2018, 10 Search PubMed.
- C. Yu, L. Xu, Y. Zhang, P. S. Timashev, Y. Huang and X. J. Liang, ACS Appl. Polym. Mater., 2020, 2, 4289–4305 CrossRef CAS.
- K. Yang, H. Xu, L. Cheng, C. Sun, J. Wang and Z. Liu, Adv. Mater., 2012, 24, 5586–5592 CrossRef CAS PubMed.
- M. Chen, X. Fang, S. Tang and N. Zheng, Chem. Commun., 2012, 48, 8934–8936 RSC.
- S. Lan, W. Xie, J. Wang, J. Hu, W. Tang, W. Yang, X. Yu and H. Liu, J. Nanopart. Res., 2018, 20, 300 CrossRef.
- J. Sun, S. Zhu, W. Xu and G. Jiang, Front. Bioeng. Biotechnol., 2022, 10, 1049437 CrossRef PubMed.
- H. Liu, W. Li, Y. Cao, Y. Guo and Y. Kang, J. Nanopart. Res., 2018, 20, 57 CrossRef.
- T. H. Tran, H. T. Nguyen, T. T. Phuong Tran, S. K. Ku, J.-H. Jeong, H.-G. Choi, C. S. Yong and J. O. Kim, Nanomedicine, 2017, 12, 1511–1523 CrossRef CAS PubMed.
- Y. Ma, J. Zhou, Z. Miao, H. Qian and Z. Zha, ACS Appl. Bio Mater., 2019, 2, 848–855 CrossRef CAS PubMed.
- T. E. Kim, H. J. Jang, S. W. Park, J. Wei, S. Cho, W. I. Park, B. R. Lee, C. D. Yang and Y. K. Jung, ACS Appl. Bio Mater., 2021, 4, 3453–3461 CrossRef CAS PubMed.
- R. M. Hathout, A. A. Metwally, S. H. El-Ahmady, E. S. Metwally, N. A. Ghonim, S. A. Bayoumy, T. Erfan, R. Ashraf, M. Fadel, A. I. El-Kholy and J. G. Hardy, J. Drug Delivery Sci. Technol., 2018, 47, 176–180 CrossRef CAS.
- Z. Zhu and M. Su, Nanomaterials, 2017, 7, 160 CrossRef PubMed.
- M. Suneetha, H. Kim and S. S. Han, Pharmaceutics, 2023, 15, 1281 CrossRef CAS PubMed.
- Y. Chen, M. Su, L. Jia and Z. Zhang, Nanomedicine, 2022, 17, 1115–1130 CrossRef CAS PubMed.
- W. Zhong, K. H. Wong, F. Xu, N. Zhao and M. Chen, Acta Biomater., 2022, 145, 135–145 CrossRef CAS PubMed.
- Y. Li, C. Jiang, D. Zhang, Y. Wang, X. Ren, K. Ai, X. Chen and L. Lu, Acta Biomater., 2017, 47, 124–134 CrossRef CAS PubMed.
- J. Xu, W. Xu, Z. Wang and Y. Jiang, J. Biomed. Mater. Res., Part A, 2023, 111, 158–169 CrossRef CAS PubMed.
- Q. Shu, J. Liu, Q. Chang, C. Liu, H. Wang, Y. Xie and X. Deng, ACS Biomater. Sci. Eng., 2021, 7, 5497–5505 CrossRef CAS PubMed.
- T. Du, T. Yang, L. Xu, X. Li, G. Yang and S. Zhou, Adv. NanoBiomed Res., 2022, 2, 2200076 CrossRef CAS.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- Y. Fu, L. Yang, J. Zhang, J. Hu, G. Duan, X. Liu, Y. Li and Z. Gu, Mater. Horiz., 2021, 8, 1618–1633 RSC.
- J. Zhou, Z. Lu, X. Zhu, X. Wang, Y. Liao, Z. Ma and F. Li, Biomaterials, 2013, 34, 9584–9592 CrossRef CAS PubMed.
- Y. Hong, W. Cho, J. Kim, S. Hwng, E. Lee, D. Heo, M. Ku, J.-S. Suh, J. Yang and J. H. Kim, Nanotechnology, 2016, 27, 185104 CrossRef PubMed.
- E. I. Yslas, L. E. Ibarra, M. A. Molina, C. Rivarola, C. A. Barbero, M. L. Bertuzzi and V. A. Rivarola, J. Nanopart. Res., 2015, 17, 389 CrossRef.
- Y. Zhang, F. Fang, Y. Chen, M. Li, L. Li, W. Li and J. Zhang, Chin. J. Chem. Eng., 2021, 38, 221–228 CrossRef CAS.
- W. Wang, R. Narain and H. Zeng, in Polymer Science and Nanotechnology, Elsevier, 2020, pp. 203–244 Search PubMed.
- N. K. Preman, R. R. Barki, A. Vijayan, S. G. Sanjeeva and R. P. Johnson, Eur. J. Pharm. Biopharm., 2020, 157, 121–153 CrossRef CAS PubMed.
- H. Idrees, S. Z. J. Zaidi, A. Sabir, R. U. Khan, X. Zhang and S. Hassan, Nanomaterials, 2020, 10, 1970 CrossRef CAS PubMed.
- S. Liu, Y. Fu, G. Li, L. Li, H. K. Law, X. Chen and F. Yan, Adv. Mater., 2017, 29, 1701733 CrossRef PubMed.
- D. Esrafilzadeh, J. M. Razal, S. E. Moulton, E. M. Stewart and G. G. Wallace, J. Controlled Release, 2013, 169, 313–320 CrossRef CAS PubMed.
- K. Krukiewicz, M. Cichy, P. Ruszkowski, R. Turczyn, T. Jarosz, J. K. Zak, M. Lapkowski and B. Bednarczyk-Cwynar, Mater. Sci. Eng., C, 2017, 73, 611–615 CrossRef CAS PubMed.
- A. Giordano and G. Tommonaro, Nutrients, 2019, 11, 2376 CrossRef CAS PubMed.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS PubMed.
- X. Hu, S. Liu, G. Zhou, Y. Huang, Z. Xie and X. Jing, J. Controlled Release, 2014, 185, 12–21 CrossRef CAS PubMed.
- B. Zhang, H. Wang, S. Shen, X. She, W. Shi, J. Chen, Q. Zhang, Y. Hu, Z. Pang and X. Jiang, Biomaterials, 2016, 79, 46–55 CrossRef CAS PubMed.
- C. Tommasino, L. Gambardella, M. Buoncervello, R. J. Griffin, B. T. Golding, M. Alberton, D. Macchia, M. Spada, B. Cerbelli, G. d’Amati, W. Malorni, L. Gabriele and A. M. Giammarioli, J. Exp. Clin. Cancer Res., 2016, 35, 137 CrossRef PubMed.
- C. V. Prasad, V. L. Nayak, S. Ramakrishna and U. V. Mallavadhani, Chem. Biol. Drug Des., 2018, 91, 220–233 CrossRef CAS PubMed.
- D. Killock, Nat. Rev. Clin. Oncol., 2015, 12, 190 CrossRef PubMed.
- F. Tian, C. Wang, M. Tang, J. Li, X. Cheng, S. Zhang, D. Ji, Y. Huang and H. Li, Oncotarget, 2016, 7, 51934–51942 CrossRef PubMed.
- N. Hosseini-Nassab, D. Samanta, Y. Abdolazimi, J. P. Annes and R. N. Zare, Nanoscale, 2017, 9, 143–149 RSC.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 CrossRef PubMed.
- M. A. Dobrovolskaia, P. Aggarwal, J. B. Hall and S. E. McNeil, Mol. Pharmaceutics, 2008, 5, 487–495 CrossRef CAS PubMed.
- D. Fan, Y. Cao, M. Cao, Y. Wang, Y. Cao and T. Gong, Signal Transduction Targeted Ther., 2023, 8, 293 CrossRef PubMed.
- A. Sontheimer-Phelps, B. A. Hassell and D. E. Ingber, Nat. Rev. Cancer, 2019, 19, 65–81 CrossRef CAS PubMed.
- L. Neufeld, E. Yeini, S. Pozzi and R. Satchi-Fainaro, Nat. Rev. Cancer, 2022, 22, 679–692 CrossRef CAS PubMed.
- R. Paliwal, R. J. Babu and S. Palakurthi, AAPS PharmSciTech, 2014, 15, 1527–1534 CrossRef CAS PubMed.
- O. Adir, M. Poley, G. Chen, S. Froim, N. Krinsky, J. Shklover, J. Shainsky-Roitman, T. Lammers and A. Schroeder, Adv. Mater., 2020, 32, 1901989 CrossRef CAS PubMed.
- D. Schletz, M. Breidung and A. Fery, J. Phys. Chem. C, 2023, 127, 1117–1125 CrossRef CAS.
- N. David, W. Sun and C. W. Coley, Nat. Comput. Sci., 2023, 3, 362–364 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |